

Abstract
Interleukin (IL)-6 and IL-11 are elevated in a variety of lung conditions and may impact on repair mechanisms in chronic inflammatory disorders. However, the mechanisms by which these cytokines influence fibroblast proliferation in normal and disease states have not been previously addressed. We examined the effect of these cytokines on proliferation and cell-cycle kinetics of primary human lung fibroblasts obtained from normal patients and patients with idiopathic pulmonary fibrosis (IPF). IL-6 inhibited the proliferation of normal fibroblasts due to the sustained phosphorylation of STAT-3 and production of the cyclin-dependent kinase inhibitor p19INK4D. In contrast IL-6 was mitogenic for IPF fibroblasts due to the sustained activation of MAPK, which in turn inhibited the production of p27Kip1, allowing activation of cyclin D1 and hyperphosphorylation of retinoblastoma protein. IL-11 was mitogenic for both normal and IPF fibroblasts. These results provide strong evidence for a fundamental abnormality in a cytokine-signaling pathway, as opposed to alterations in cytokine production, in the pathogenesis of IPF.
Idiopathic pulmonary fibrosis (IPF) is a primarily untreatable, progressive fibrotic lung disorder with 5-year survival ranging from 30 to 50%. 1,2 Morphologically, the disease is characterized by abnormal parenchymal tissue remodeling and re-epithelialization, increased collagen deposition, and angiogenesis. 3-5 The progression of fibrosis appears to be strongly correlated with the presence and number of fibroblastic foci in the injured lung. Fibroblasts within these foci assume a migratory, followed by a proliferative, and ultimately a profibrotic, myofibroblastic phenotype. 2,6
Myofibroblasts are characterized by the expression of markers of smooth muscle differentiation such as α-smooth muscle actin (α-SMA) and are thought to be primarily responsible for increased deposition of collagen within the lung and are a major source of cytokines, chemokines, and profibrogenic growth factors. 7,8 Under normal conditions, myofibroblasts disappear from wounds by apoptosis. 9,10 However, the increased number of myofibroblasts in IPF suggests that these cells may be hyperproliferative and/or resistant to apoptosis.
Interleukin (IL)-6 and IL-11 belong to a family of cytokines that include oncostatin M, ciliary neurotrophic factor, leukemia inhibitory factor, and cardiotrophin-1. 11,12 These cytokines are grouped together on the basis of a weak structural homology and the use of gp130-signaling subunit in their receptor complexes. IL-6 is released by a variety of cells including monocytes, macrophages, fibroblasts, and endothelial cells in response to inflammatory and profibrotic mediators such as IL-1β, tumor necrosis factor-α, platelet-derived growth factor, and transforming growth factor-β. 11,12 On this basis, it is not surprising that IL-6 mediates many inflammatory processes in the lung and its dysregulated release is implicated in the pathogenesis of a variety of respiratory conditions including interstitial lung diseases. 13 However, depending on the cell type, IL-6 can either stimulate or inhibit proliferation.
IL-11 is also released by a variety of cell types including fibroblasts and epithelial cells after stimulation by a number of factors central to lung fibrosis such as transforming growth factor-β1. 14,15 IL-11 attenuates the acute inflammatory response in the lung after exposure to hyperoxia 16 and radiation-induced injury 17 via pathways linked to the inhibition of apoptosis.
To more precisely define the role of these cytokines in the lung, mice genetically engineered to overexpress IL-6 and IL-11 in the lung have been developed. Mice that overexpressed IL-6 were shown to have a predominantly lymphocytic infiltration of the airways and alveolitis but minimal fibrosis, 18 whereas overexpression of IL-11 was associated with prominent airway remodeling, subepithelial fibrosis, and accumulation of fibroblasts, myofibroblasts, and myocytes as well as airway hyperresponsiveness. These studies provided the first evidence that although IL-6 and IL-11 use the same signal transducer these cytokines may have contrasting effects on cell function.
On this basis, we hypothesized that IL-6 would play a minor role in regulating fibroblast proliferation in chronic fibrotic lung diseases, whereas IL-11 would augment proliferation. We tested this hypothesis using primary cultures of human lung fibroblasts obtained from three normal patients (normal Fb) and three patients with IPF (IPF-Fb). Our results show that IL-6 inhibited proliferation of normal Fb. However, IL-6 was a potent mitogen for IPF-Fb. The switch in responsiveness corresponded with a shift from STAT-3-dependent signaling in normal Fb to ERK-dependent signaling in IPF-Fb. In contrast to IL-6, both normal Fb and IPF-Fb proliferated in response to IL-11.
Materials and Methods
Materials
Dulbecco’s modified Eagle’s medium (DMEM), RPMI 1640, fetal calf serum, penicillin, gentamicin, and amphotericin were purchased from Life Technologies (Melbourne, Australia), Monoclonal anti-gp130 antibodies and propidium iodide were purchased from Sigma Chemicals (St. Louis, MO). Coverslip chamber wells were obtained from Labtec (NUNC, Roskilder, Denmark). Genestein, wortmannin, C3 exoenzyme, pp2, and PD98059 were purchased from Biomol (Plymouth, PA). Antibodies against, IL-11, and IL-6 were purchased from Boehringer Ingelheim (Sydney, Australia). Antibodies against p27Kip1, p19INK4D, p21Cip1, cyclins D and E, and phosphorylated retinoblastoma protein (pRb), phosphorylated-STAT-3, phosphorylated-ERK 1/2 were purchased from Santa Cruz (La Jolla, CA). Tritiated thymidine was purchased from Bresagen (Adelaide, Australia).
Cell Culture
Primary cultures of fibroblasts were derived from normal human lung and patients with IPF as previously described. 19 Three primary lines of normal human alveolar lung fibroblasts were obtained from Clonetics (San Diego, CA), American Type Culture Collection (Manassas, VA) and lung transplant donor, respectively, and were used between passages 3 to 5. For comparison, primary cultures of lung fibroblasts harvested from three patients with UIP-IPF were used for all experiments. All IPF-Fb were used between passages 4 to 6. Fibroblasts were cultured in DMEM supplemented with 10% fetal calf serum, penicillin, gentamicin, and amphotericin. For all experiments, fibroblasts were quiesced by the addition of serum-free DMEM for 16 hours, before treatment with IL-6 or IL-11.
Anti-Sense Oligonucleotide Synthesis
Phosphothiorated 21-mer oligonucleotides were synthesized on an Applied Biosystem 394 synthesizer by means of β-cyanoethylphosphoramidite chemistry to minimize degradation by endogenous nucleotides. The anti-sense oligonucleotides (ASON) were directed against the translational site (AUG codon) and surrounding nucleotides of the human STAT-3 and STAT-5 genes. The sequence of the ASON to STAT-3 was 5′-CCATTGGGCCATCCTGTTTCT-3′ and the corresponding sense oligonucleotide sequence was 5′-AGAAACAGGATAACCCAATGG-3′. The sequence of the ASON to STAT-5 was 5′-AGCCCGCCAT-3′ and the corresponding sense oligonucleotide sequence was 5′-ATGGCGGGCT-3′. To examine the effect of ASON, cells were cultured at a density of 104 cells/well in a 96-well plate for 24 hours. Cells were incubated with 250 μl of fresh serum-free medium containing 12.5 μmol/L of either ASON or sense oligonucleotides for 24 hours. After incubation with oligonucleotides, fibroblasts were treated with IL-6 and IL-11 for 3, 6, 24, or 48 hours.
Assessment of Proliferation
Fibroblasts were seeded in DMEM supplemented with 10% fetal calf serum at 3 × 104 cells/well in 96-well plates and allowed to adhere for 24 hours before quiescing by replacing the medium with serum-free DMEM for a further 18 hours. Cells were then treated with IL-6 or IL-11 for either 24, 48, or 72 hours. When experiments were performed in the presence of pharmacological inhibitors, these were added 30 minutes before the addition of either cytokine. Tritiated thymidine (3H-Tdr, 1 μCu/well) was added to each well 12 hours before analysis. Cells were then rinsed with ice-cold phosphate-buffered saline (PBS) and treated with 5% trichloroacetic acid for 20 minutes at 4°C, followed by the addition of an equal volume of 0.5 N NaOH. Incorporation of 3H-Tdr was assessed by scintillation counting in a Packard Direct Counter (Canberra, NSW, Australia).
Cell Cycle Analysis
The effect of IL-6 or IL-11 on cell-cycle kinetics was assessed by incorporation of propidium iodide into DNA by fluorescence-activated cell sorting analysis. Briefly, 106 cells were induced into the log phase of growth and fixed in ice-cold 70% ethanol for 1 hour. After washing with PBS, ribonucleotide reductase (10 μl) and Triton X-100 (100 μl) were added to the cells for 30 minutes at 37°C. Propidium iodide (1 μg/ml) was added and incubated on ice for 10 minutes. Labeled cells were analyzed on a FACScalibur (Becton Dickinson, San Jose, CA) and quantified by CellQuest software. Ten to thirty thousand events were collected for analysis.
Flow Cytometry
For detection of intracellular gp130 staining, fibroblasts were permeabilized by treatment with 0.1% Triton X-100 for 3 minutes before incubation with a monoclonal gp130 antibody. After multiple washings, cells were incubated with a rabbit anti-mouse PE-conjugated secondary antibody. Labeled cells were analyzed by flow cytometry as described above. Ten to thirty thousand events were collected for analysis.
Western Blot Analysis
The expression of p27Kip1, p19INK4D, p21Cip1, cyclin D, cyclin E, pRb, phosphorylated-STAT3, and phosphorylated-ERK 1/2 were assessed by Western blot analysis as previously described. 20 Trypsinized cells (2 × 106) were suspended in lysis buffer (50 mmol/L Tris, 0.5 nmol/L EGTA, 150 mmol/L NaCl, 1% Triton X-100, pH 7.5) containing a cocktail of protease inhibitors and centrifuged at 10,000 rpm for 20 minutes. The protein content of the supernatant was determined by the Bradford method. Equal amounts of protein (40 μg) were added to sodium dodecyl sulfate-polyacrylamide gel electrophoresis buffer, boiled for 5 minutes, and electrophoresed on an 8 to 16% gradient gel and then transferred to nitrocellulose membranes. Membranes were incubated with Tris-buffered saline with 0.5% Tween-20 (TBST) containing 5% skim milk powder overnight to block nonspecific binding and then incubated with primary antibodies for a further 2 hours. After washing in TBST, membranes were incubated with horseradish peroxidase-conjugated secondary antibody in TBST, washed, and developed using the enhanced chemiluminescence detection system.
Confocal Microscopy
The expression of α-SMA by normal Fb and IPF-Fb and phospho-STAT3 translocation was visualized by confocal microscopy as described previously. 20 Briefly fibroblasts (1 × 104) were plated on coverslip chamber wells. After being quiesced for 12 hours in serum-free DMEM, cells were fixed in 4% paraformaldehyde for 15 minutes at room temperature and permeabilized by exposure to 1% Triton X-100 for 2 minutes. After PBS washes, fibroblasts were stained with a monoclonal α-SMA antibody for 1 hour. After further washes in PBS, slides were exposed to Alexa Fluor-488-conjugated secondary antibodies. Cells were counterstained with 4,6-diamidino-2-phenylindole to visualize nuclei. Fluorescent images were obtained using a confocal laser-scanning microscope (Bio-Rad MRC 1000) using COMOS software. Image processing was performed using Confocal Assistant software and Adope Photoshop.
Statistical Analysis
Data are expressed as mean ± SE of at least four experiments. Statistical comparisons of mean data were performed using one-way analysis of variance and Student’s t-test with Bonferroni correction performed posthoc to correct for multiple comparisons. A P value <0.05 was regarded as statistically significant.
Results
Cultures of IPF-Fb Are Predominantly α-SMA-Positive
Confocal microscopic analysis of fibroblast cultures at similar passage, revealed that 69 ± 5% of the IPF-Fb expressed high levels of α-SMA compared to 2 ± 0.2% normal Fb.
Cultures of IPF-Fb Proliferate Faster than Normal Fb in Response to Serum
The effect of 10% fetal calf serum on the rate of proliferation of IPF-Fb and normal fibroblasts was assessed by [3H]-thymidine incorporation. IPF-Fb proliferated faster than normal Fb (3500 ± 500 cpm versus 1500 ± 500 cpm, P < 0.01). The proliferative rates of the three IPF primary cultures were compared and found not to be significantly different (Table 1) ▶ .
Table 1.
Proliferation Rates of the Three Primary Lines of IPF-Fb
| Culture | IPF-Fb no. 1 | IPF-Fb no. 2 | IPF-Fb no. 3 |
|---|---|---|---|
| [3H]Thymidine incorporation (cpm) | 3600 ± 400 | 3410 ± 600 | 3540 ± 800 |
IPF-Fb were grown to 70% confluence before quiescing in serum-free media for 16 hours. Cells were then re-exposed to media containing 10% FCS for 48 hour and proliferation assessed by incorporation of [3H]thymidine as described in Materials and Methods. Because of the scarcity and limited life of IPF-Fb, results represent the mean ± sem of two experiments performed in triplicate.
IL-6 Inhibits Proliferation of Normal Fibroblasts but Is a Mitogen for IPF Fibroblasts
IL-6 inhibited the proliferation of normal Fb in a dose- and time-dependent manner to a maximum of 35 ± 2% (P < 0.01) at a concentration of 160 ng/ml after 48 hours (Figure 1a) ▶ . In contrast, this concentration of IL-6 was strongly mitogenic for IPF-Fb, increasing proliferation by 50% (Figure 1b) ▶ . Cell-cycle analysis showed that IL-6 induced cell-cycle arrest of normal Fb in the G0/1 phase of the cell cycle. In contrast, exposure of IPF-Fb to IL-6 allowed progression from the G0/1 phase to S phase (Figure 1c) ▶ .
Figure 1.
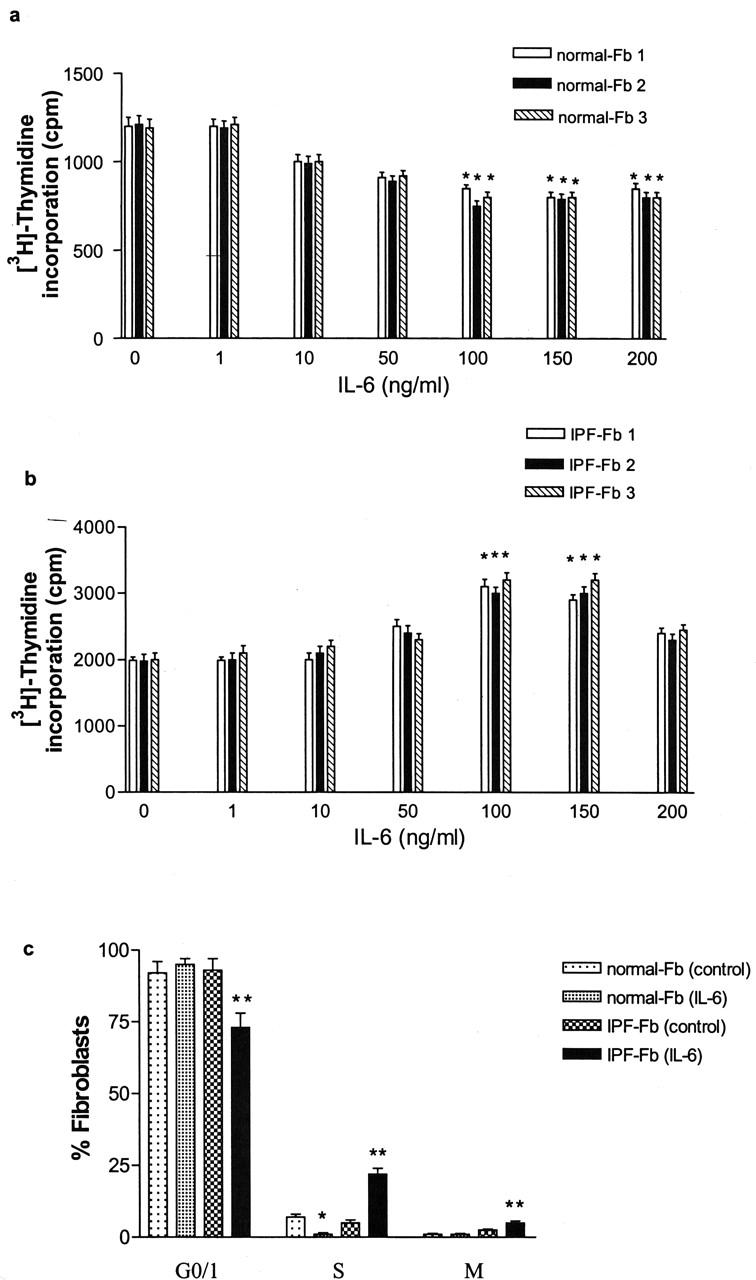
IL-6 inhibits proliferation of normal Fb but is a mitogen for IPF-Fb. Normal Fb (a) or IPF-Fb (b) were incubated with increasing concentrations of IL-6 for 24 hours and proliferation was assessed by incorporation of [3H]-thymidine as described in Materials and Methods. *, P < 0.01 compared with serum-free control (·). c: Exposure to IL-6 caused cell-cycle arrest of normal Fb in G0/1 (black bars), whereas in IPF-Fb, IL-6 induced a significant progression of cells into S phase (striped bars). *, P < 0.01 compared with serum-free normal Fb; **, P < 0.01 compared with serum-free IPF-Fb. Data shown represent mean ± SEM of four experiments performed in triplicate.
Expression of gp130 in Normal Fb Is Not Different to Expression in IPF-Fb
Both normal Fb and IPF-Fb expressed gp130 as evidenced by the rightward shift in mean fluorescence intensity (Figure 2) ▶ . However, expression of gp130 was not different between normal and diseased cells, with mean fluorescence intensity values of 52 ± 5 for normal Fb and 58 ± 6 for IPF-Fb (Figure 2) ▶ .
Figure 2.

Expression of gp130 is not different between normal Fb and IPF-Fb. Normal Fb (left) or IPF-Fb (right) were permeabilized and incubated with antibodies against gp130 and expression assessed by flow cytometry as described in Materials and Methods. Mean fluorescence intensity values were 52 ± 5 for normal Fb and 58 ± 6 for IPF-Fb. Data shown are representative of each of three normal and three IPF-Fb lines.
Reciprocal Signaling Requirements for IL-6 in Normal Fb and IPF-Fb
To dissect the signaling pathways mediating the effects of IL-6 in normal Fb and IPF-Fb, cells were incubated with selective signaling pathway inhibitors and proliferation assessed after 24 hours. Figure 3a ▶ shows the effect of ASON against STAT-3 and STAT5a/5b on IL-6-mediated growth arrest. ASON-STAT-3 abrogated the inhibitory effect of IL-6 on normal fibroblast proliferation whereas ASON-STAT5a/5b had no effect. In addition, exposure to PD98059 or nimesulide had no effect on IL-6-induced growth arrest (Figure 3b) ▶ , suggesting that ERK 1/2 and COX-2 were not involved.
Figure 3.

The inhibitory effect of IL-6 on normal Fb is STAT-3-dependent whereas the mitogenic effect on IPF-Fb is ERK-dependent. Normal Fb (a, b) or IPF-Fb (c, d) were incubated with ASON against STAT-3 or STAT 5a/b (a, c) or pharmacological inhibitors to signaling pathways (b, d) and proliferation was assessed by incorporation of [3H]-thymidine as described in Materials and Methods. Data shown are mean ± SEM of four experiments conducted in triplicate. *, P < 0.01 compared with serum-free normal Fb; **, P < 0.01 compared with serum free IPF-Fb.
In contrast to normal Fb, treatment of IPF-Fb with ASON-STAT-3 or ASON-STAT-5a/5b did not influence IL-6 induced mitogenic responses of IPF fibroblasts (Figure 3c) ▶ . However, in IPF-Fb, IL-6-induced proliferation was almost completely abrogated by PD98059. Addition of the COX-2 inhibitor nimesulide did not influence IL-6 responses, suggesting that PGE2 was not playing a significant role (Figure 3d) ▶ .
STAT 3 Induces the Expression of p19INK4D in Normal Fb but Represses p27Kip1 in IPF-Fb
The transition from the G0/1 to S phase of the cell cycle is coordinated by the activation and inactivation of CDKs. We investigated the interaction between STAT-3 phosphorylation and the expression of the CDK inhibitors p19INK4D, p21Cip1, and p27Kip1. In normal Fb, IL-6 induced the expression of p19INK4D and this effect was abrogated by ASON-STAT-3 (Figure 4a) ▶ . Accordingly, IL-6 did not induce the expression of either cyclin D1 or cyclin E1, which, in turn, inhibited the hyperphosphorylation of pRb, preventing the transition from G0/1 to the S phase.
Figure 4.
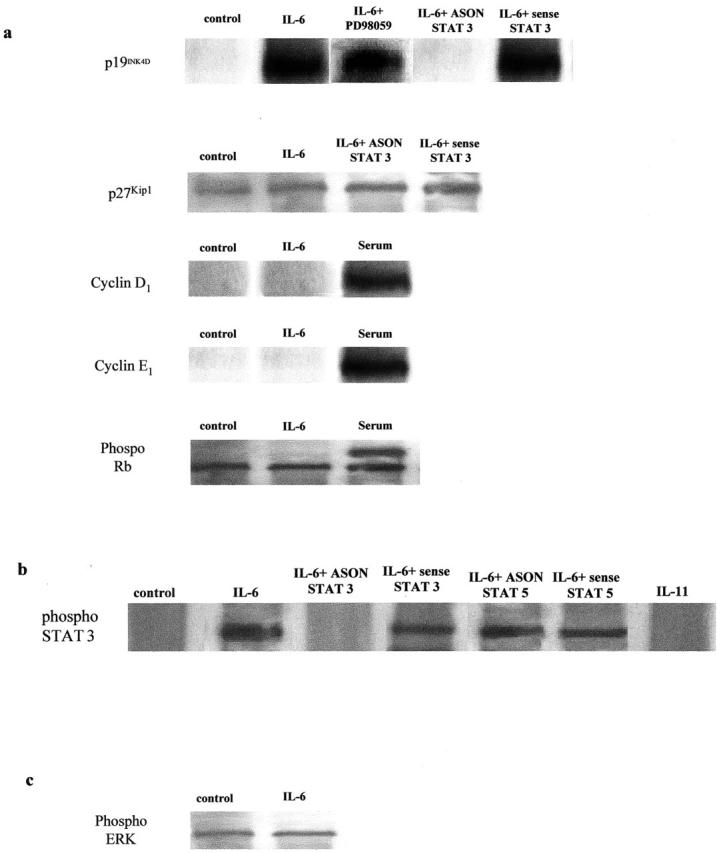
IL-6 induces the STAT-3-dependent expression the cdk inhibitor p19INK4D in normal Fb. a: Cells were exposed to IL-6 (100 ng/ml) in the presence or absence of ASON to STAT-3 and the expression of p19INK4D, cyclin D1, cyclin E1, and retinoblastoma protein assessed by Western blot analysis as described in Materials and Methods. The figure is representative of three separate experiments. b: Control blots confirming the specificity of STAT 3 phosphorylation induced by IL-6, but not IL-11. The effect of IL-6 on STAT 3 is blocked by ASON to STAT 3, but not sense oligonucleotides or ASON against STAT 5a/b. c: Control blot showing that exposure to IL-6 for 24 hours does not induce phosphorylation of ERK 1/2 above that seen in unstimulated control cells.
Expression of the CDK inhibitor p27Kip1 was elevated in IPF-Fb cultured in a serum-free media, consistent with cells in growth arrest (Figure 5) ▶ . The elevation in p27Kip1 was inhibited by IL-6. Treatment with PD98059 abrogated this effect, suggesting that the effects were dependent on the activation of MAPK (Figure 5) ▶ . Repression of p27Kip1 allowed phosphorylation of cyclin D1 and E1 and hyperphosphorylation of pRb resulting in progression from G0/1 to S phase (Figure 5) ▶ . Expression of p21Cip1 was not observed in either normal Fb or IPF-Fb in response to IL-6. Expression of p19INK4D was not observed in IPF-Fb in response to IL-6 or IL-11.
Figure 5.
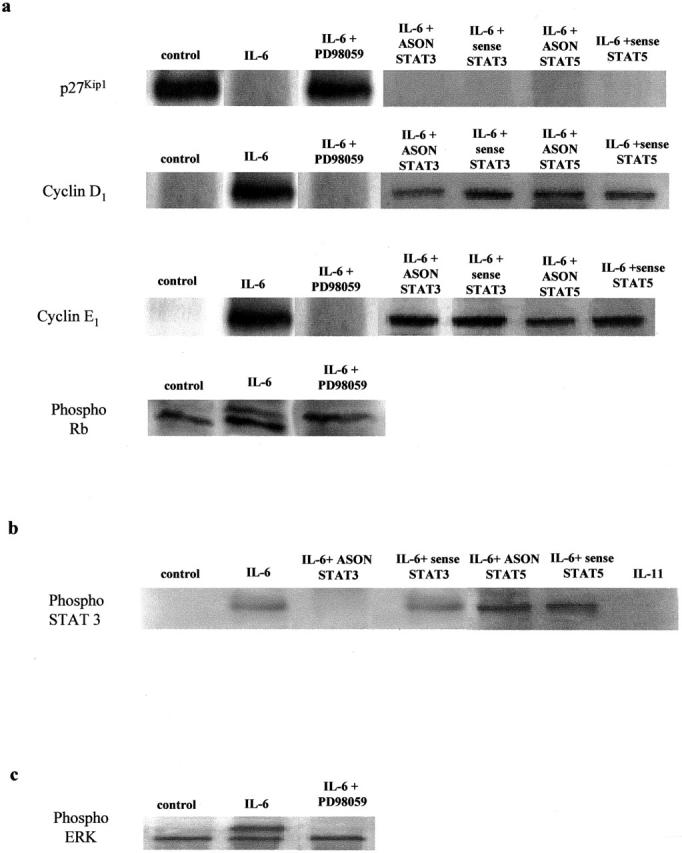
IL-6 inhibits the expression of cdk inhibitor p27Kip1 in IPF-Fb. a: Cells incubated in serum-free media were exposed to IL-6 (100 ng/ml) in the presence or absence of PD98059. Lysates were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis and the expression of p27Kip1, cyclin D1, cyclin E1, and phosphorylated retinoblastoma protein assessed by Western blot analysis as described in Materials and Methods. The figure is representative of three separate experiments. b: Control blots confirming the specificity of STAT 3 phosphorylation induced by IL-6, but not IL-11. The effect of IL-6 on STAT 3 is blocked by ASON to STAT 3, but not sense oligonucleotides or ASON against STAT 5a/b. c: Control blot showing that exposure to IL-6 for 24 hours induces phosphorylation of ERK 1/2 compared to unstimulated control cells. Phosphorylation of ERK 1/2 is reduced to basal levels after exposure to PD98059 (50 μmol/L).
The Kinetics of ERK 1/2 and STAT-3 Activation Differ in Normal Fb and IPF-Fb
Figure 6a ▶ shows that in normal Fb, phosphorylated ERK 1/2 is only transiently expressed after IL-6 exposure, whereas phosphorylated STAT-3 was detected for at least 6 hours. In contrast, in IPF-Fb phosphorylated ERK 1/2 was detected for up to 12 hours, whereas expression of phosphorylated STAT-3 was transient in nature, being detected for up to 3 hours (Figure 6) ▶ .
Figure 6.

Sustained STAT-3 signaling in normal Fb is replaced by sustained ERK signaling in CFF-Fb in response to IL-6. Normal Fb (a) or IPF-Fb (b) were exposed to IL-6 (100 ng/ml) for periods of time up to 24 hours. Lysates were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis and the duration of phosphorylation (activation) of ERK 1/2 (top) or STAT-3 (bottom) assessed by Western blot analysis. The figure is representative of three separate experiments.
IL-11 Is a Mitogen for Normal and IPF Fibroblasts
In contrast to IL-6, IL-11 was mitogenic for both normal Fb and IPF-Fb (Figure 7, a and b) ▶ increasing the rate of proliferation by 100% above control in both cell types. The mitogenic effect of IL-11 was associated with a significant increase in the number of cells progressing from G0/1 to the S phase.
Figure 7.
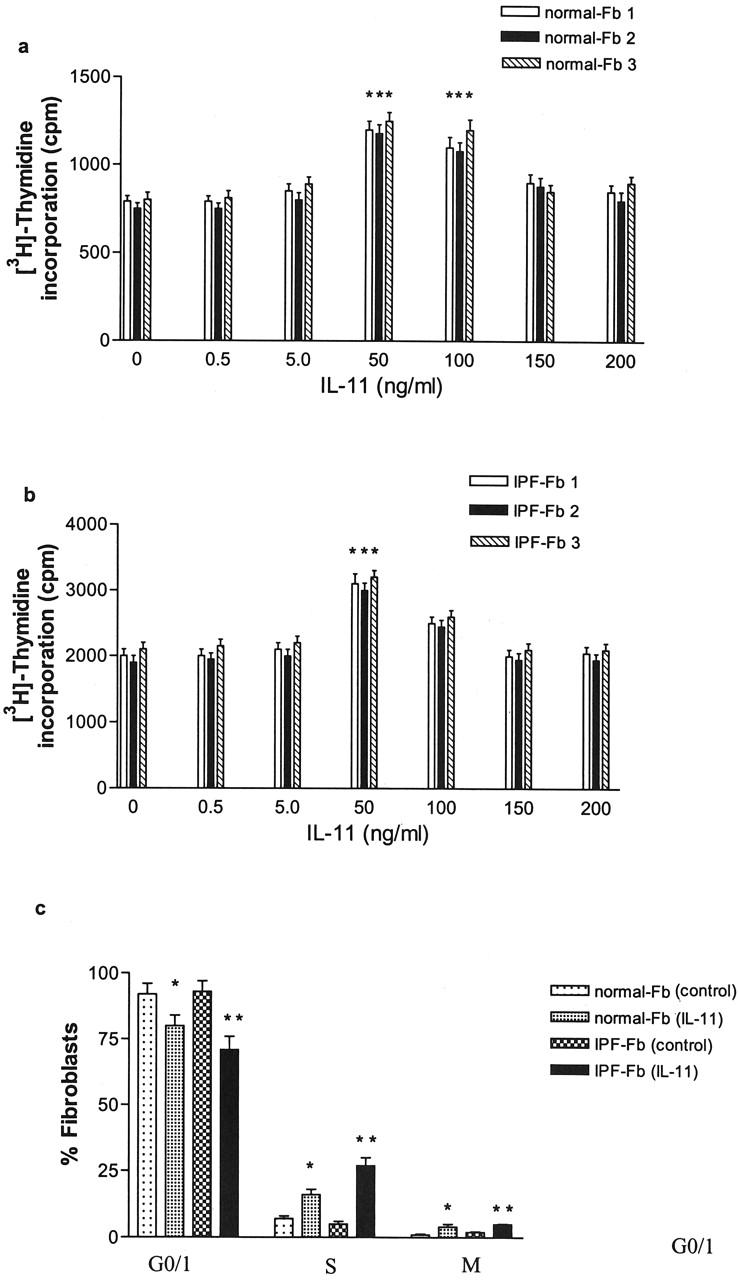
IL-11 is a mitogen for both normal Fb and IPF-Fb. Normal Fb (a) or IPF-Fb (b) were incubated with increasing concentrations of IL-11 for 24 hours and proliferation assessed by incorporation of [3H]-thymidine as described in Materials and Methods. *, P < 0.01 compared with serum-free control (·). *, P < 0.01 compared with serum-free normal Fb, **, P < 0.01 compared with serum-free IPF-Fb. c: Exposure to IL-11 induced a significant progression of both normal Fb (white bars) and IPF-Fb (striped bars) into S phase. Data shown represent mean ± SEM of four experiments performed in triplicate.
The IL-11-Dependent Proliferation of Fibroblasts Is Mediated by p42/p44 MAPK
Transfection of ASON to STAT-3 or STAT-5a/b did not affect the proliferative response to IL-11 in either normal Fb or IPF-Fb (Figure 8, a and c) ▶ . Having demonstrated that the IL-11-dependent proliferation of both normal and IPF fibroblasts were not STAT-dependent, we investigated the role of ERK 1/2 in mediating this effect. Figure 7b ▶ shows that pretreatment of cells with PD98059 inhibited mitogenic responses to IL-11 in both normal Fb. A similar response was observed in IPF-Fb (Figure 8d) ▶ . Inhibition of COX-2 activity by nimesulide did not influence proliferation.
Figure 8.

The mitogenic effects of IL-11 on normal Fb and IPF-Fb are ERK-dependent. Normal Fb (a, b) or IPF-Fb (c, d) were incubated with IL-11 (50 ng/ml) in the presence or absence of ASON against STAT-3 or STAT 5a/b (a, c) or pharmacological inhibitors to ERK, COX-2, or PKC (b, d) and proliferation assessed by incorporation of [3H]-thymidine as described in Materials and Methods. Data shown are mean ± SEM of four experiments conducted in triplicate. *, P < 0.01 compared with serum-free normal Fb; **, P < 0.01 compared with serum-free IPF-Fb.
IL-11 Inhibits p27Kip1 by the Activation of the MAPK Pathway in Both Normal Fb and IPF-Fb
Exposure to IL-11 inhibited the expression of p27Kip1 in both normal Fb and IPF-Fb (Figure 9) ▶ . However, pretreatment of these cells with PD98059, abrogated the inhibition of p27Kip1, suggesting a direct role for ERK 1/2 in this effect. Down-regulation of p27Kip1 allowed the expression and activation of cyclins D1 and E1 and hyperphosphorylation of pRb.
Figure 9.
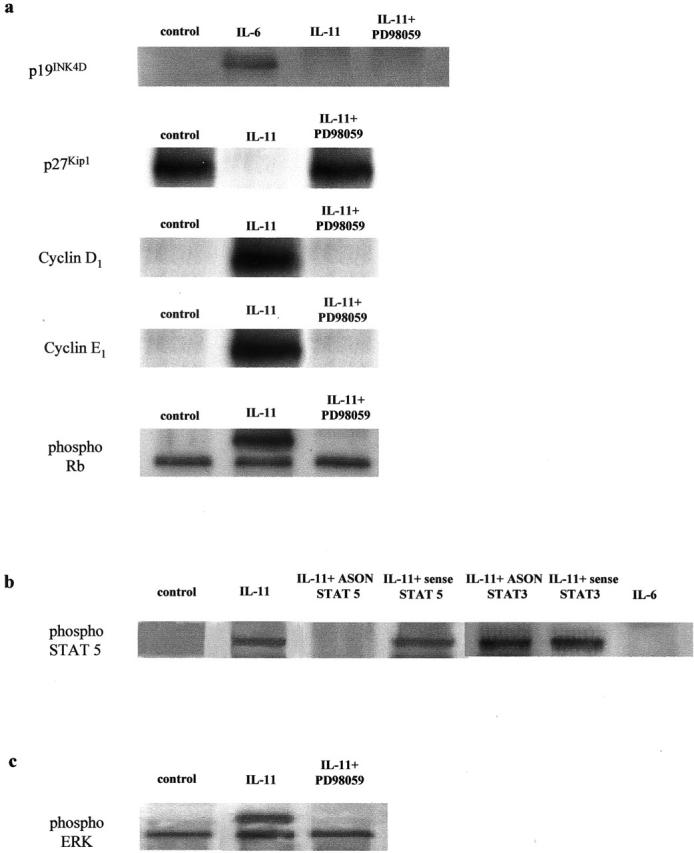
IL-11 inhibits expression of ERK-dependent p27Kip1 expression in normal Fb and IPF-Fb. Cells were incubated in serum-free media and exposed to IL-11 (50 ng/ml) in the presence or absence of PD98059. Lysates were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis and the expression of p27Kip1, cyclin D1, cyclin E1, and phosphorylated retinoblastoma protein assessed by Western blot analysis as described in Materials and Methods. The figure is representative of three separate experiments.
IL-11 Induces Sustained Activation of ERK 1/2 in Both Normal Fb and IPF-Fb
Because IL-11 was a mitogen for fibroblasts, we examined the duration of activation of ERK 1/2 after IL-11 in both normal Fb and IPF-Fb. In both cell types, ERK 1/2 demonstrated sustained activation with phosphorylated protein being detected for more than 6 hours in a similar pattern to that observed for IL-6 exposure in IPF-Fb (data not shown).
Discussion
In this study, we have examined the effect of IL-6 and IL-11, two cytokines belonging to the IL-6/gp130 family on the proliferation and cell-cycle kinetics of normal Fb and IPF-Fb. Our study has yielded several major findings: 1) in normal Fb, IL-6 is anti-proliferative, whereas in IPF-Fb IL-6 is strongly proproliferative. 2) In normal Fb, IL-6 induces sustained activation (>6 hours) of STAT-3, but only transient activation (1 hour) of ERK 1/2, resulting in the induction of the cdk4/6 inhibitor p19INK4D, inhibition of cyclin D1 and E1 expression, and inhibition of phosphorylation of pRb, culminating in cell-cycle arrest. 3) In IPF-Fb, IL-6 induced transient activation (1 hour) of STAT-3 but sustained activation (>12 hours) of ERK 1/2, down-regulation of the cdk inhibitor p27Kip1 resulting in the induction of cyclin D1 and E1 expression as well as hyperphosphorylation of pRb forcing progression through the cell cycle and proliferation. 4) IL-11 was mitogenic for both normal Fb and IPF-Fb and in both cell types and proliferation was mediated via p42/44 MAPK-dependent down-regulation of p27Kip1. 5) surface expression of gp130 did not appear to differ between normal Fb and IPF-Fb. Our results provide strong evidence for a fundamental abnormality in a cytokine signaling pathway, as opposed to alterations in cytokine production, in the pathogenesis of IPF.
It is now increasingly recognized that the pathogenesis of IPF is directly related to the presence and activity of myofibroblasts. 7,21 We have shown that there are a significantly higher number of α-SMA-positive staining cells (myofibroblasts) within the IPF-Fb population compared to normal Fb (69% versus 3%). Our findings are in general agreement with the proportions observed by Ramos and colleagues 22 (63% versus 15%). Hadden and Henke 23 also reported a high percentage of α-SMA-positive cells (55%) in their fibrotic cultures but did not report the number of α-SMA-positive cells in control cultures. The increased number of myofibroblasts in this condition implies that these cells may be hyperproliferative and/or resistant to apoptosis. However, whether IPF-Fb proliferate faster than normal Fb has been a source of controversy. Jordana and colleagues 24 reported that fibroblasts derived from areas of fibrosis proliferated faster than cells derived from histologically normal areas of lung tissue. In contrast, Ramos and colleagues 22 showed that the growth rate of IPF-Fb was significantly slower than normal Fb. Our data, using primary cultures obtained from three different patients, clearly shows that IPF-Fb proliferate faster than normal Fb under standard culture conditions.
IL-6 and IL-11 belong to a family of closely related pleiotropic cytokines that are produced by a variety of cells in response to inflammatory stimuli. 11 The actions of these cytokines are mediated through specific cell-surface receptors, which consist of a unique α chain and the shared signal transducing subunit, gp130. 25,26 IL-6 has a variety of well-documented direct effects that are potentially relevant to pulmonary inflammation and fibrosis and levels of IL-6 are also elevated in a variety of chronic lung diseases such as asthma, sarcoidosis, 13 and scleroderma. 27 Mitogenic responses of fibroblasts to IL-6 show marked tissue and species heterogeneity. For example, IL-6 inhibits proliferation of dermal fibroblasts and fibroblastic synovial cells from patients with rheumatoid arthritis, 28 whereas it is mitogenic for murine lung fibroblasts. 29
Our results demonstrate that in primary cultures of normal Fb, exposure to IL-6 results in growth arrest in the G0/1 phase of the cell cycle. However, in the presence of ASON to STAT-3, cell-cycle arrest was abrogated and proliferation returned to control levels, suggesting a critical role for this transcription factor in regulating this response. Indeed, we have shown that cell-cycle arrest in response to IL-6 is associated with sustained activation of STAT-3 and concomitant, but transient activation of ERK 1/2. Activation of STAT-3 was associated with the induction of the cdk4/6 inhibitor p19INK4D and as a consequence, down-regulation of cyclin D1 and cyclin E1, which ultimately leads to hypophosphorylation of Rb, preventing cells from transiting through G0/1 to S phase. In contrast to our findings, Fukada and colleagues, 30 using transformed pro-B cells, showed that under normal conditions, gp130-mediated STAT-3 signaling induced G1 to S phase transition. The reasons for the discrepancy are not clear, although it is highly likely that gp130-mediated signaling can exert distinct biological activities on different target cells. Consistent with this, Nakajima and colleagues, 31 reported that gp130-stimulation could simultaneously induce growth enhancing and suppressing signals in M1 cells. Another reason may relate to our use of a single cytokine (IL-6) to activate gp130, compared to chronic, cytokine-independent activation of gp130 used by Fukada and co-workers. 30 Given that gp130 is the common signal-transducing subunit for a family of cytokines, the final effect observed may be dependent on which cytokine binds and activates the receptor complex. Indeed, as highlighted in the Results section, we have shown that IL-11 is a potent mitogen for both normal Fb and IPF-Fb.
The most striking observation of this study was that in contrast to normal Fb, treatment of IPF-Fb with IL-6 induced progression through G0/1 to S phase and proliferation. In addition, the responsiveness of IPF-Fb to IL-6, was resistant to treatment with ASON to STAT-3, but were inhibited by PD98059, suggesting a role for ERK in mediating the proliferative response. Indeed, IL-6 induced a sustained activation of ERK 1/2 (>12 hours) accompanied by only a transient activation of STAT-3. Further investigation revealed that sustained ERK 1/2 activity was associated with down-regulation of p27Kip1 induction of cyclin D1 and E1 and hyperphosphorylation of Rb. Previous studies have shown a correlation between the activation of ERK 1/2 and proliferation. For example, ERK 1/2 is a major negative regulator of p27Kip1 and has also been shown to stimulate cyclin D1 promoter activity. 32
As is the case for IL-6, little is known about the effects of IL-11 on fibroblast proliferation, although transgenic mice with targeted overexpression of IL-11 in the lung, demonstrate an increase in the number of myofibroblasts, suggesting that IL-11 is a mitogen. 33,34 Our data support this finding by demonstrating that IL-11 induces a strong proliferative response in both normal Fb and IPF-Fb. Furthermore we have shown that the mode of action, namely ERK 1/2 activation, down-regulation of p27Kip1 and consequent induction of cyclin D1 and E1, and hyperphosphorylation of Rb, is similar between the two different cell types.
One potential reason as to why IL-6 has differing effects on normal Fb compared to IPF-Fb may relate to the observation that fibroblasts derived from patients with IPF are composed of heterogeneous subpopulations that display differing sensitivities to cytokines and growth factors. 24 This duality may be because of sites in the lung where IPF-Fb are harvested from, because the magnitude of inflammation and fibrosis are well known to be heterogeneous in distribution and areas of active fibrosis may yield rapidly proliferative fibroblasts compared to areas of established fibrosis in which cells may be hypoproliferative. 2 However, this argument does not adequately address the dichotomy observed for IL-6 and IL-11 on normal Fb proliferation.
Similarly, eicosanoids such as PGE2 have been shown to regulate fibroblast proliferation. 35,36 Wilborn and colleagues 35 reported that IPF-Fb have decreased expression of cyclooxygenase and PGE2 production compared to normal Fb, allowing unrestricted proliferation. However, in our study we did not observe evidence of aberrant synthesis of PGE2 in response to IL-6 because the COX-2 selective inhibitor nimesulide did not influence the proliferative rate in either normal Fb or IPF-Fb.
In conclusion, we have provided initial evidence for a fundamental signaling abnormality involving IL-6 activation of gp130 in IPF. Specifically, we have demonstrated that IPF-Fb proliferate in response to IL-6, whereas in normal Fb, this cytokine is anti-mitogenic. The mechanism involves a shift in signaling dependency from STAT-3 in normal Fb to ERK in IPF-Fb. In contrast, IL-11 was mitogenic for both types of fibroblasts. The contrasting roles of ERK and STAT in healthy and IPF fibroblasts however, provide interesting insights into possible phenotypic changes that occur in IPF. Results from animal models have demonstrated that overexpression of IL-6 does not induce fibrosis per se. From these data and ours, it is compelling to suggest that in normal tissue, IL-6 does not initiate fibrosis. Indeed, under normal conditions, IL-6 may inhibit fibrosis. In stark contrast, in diseases such as IPF, characterized by phenotypic differences in fibroblasts, IL-6 seems likely to promote fibroblast proliferation, which may ultimately contribute to the development of fibrosis.
Acknowledgments
We thank Dr. Paul Rigby from the Lotteries Commission Biomedical Confocal Microscopy Centre, UWA, for assistance with the confocal microscopy, Dr. Marie Bogoyevitch from the Department of Biochemistry and Molecular Biology, UWA for helpful comments and advice, and Ms. Joanne Ballard for her expert assistance with the preparation of the manuscript.
Footnotes
Address reprint requests to Dr. Darryl Knight, Asthma and Allergy Research Institute, Ground Floor, “E” Block, Sir Charles Gairdner Hospital, Verdun St., Nedlands, Western Australia, 6009. E-mail: dknight@cyllene.uwa.edu.au.
Supported by grants from the National Health and Medical Research Council of Australia and Asthma WA.
YM is a recipient of the South African Pulmonology Society/3M Respiratory Fellowship.
References
- 1.Bjoraker JA, Ryu JH, Edwin MK, Myers JL, Tazelaar HD, Schroeder DR, Offord KP: Prognostic significance of histopathologic subsets in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 1998, 157:199-203 [DOI] [PubMed] [Google Scholar]
- 2.Selman M, King TE, Pardo A: Idiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann Intern Med 2001, 134:136-151 [DOI] [PubMed] [Google Scholar]
- 3.Selman M, Pardo A: Idiopathic pulmonary fibrosis: an epithelial/fibroblastic cross-talk disorder. Respir Res 2002, 3:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mason RJ, Schwarz MI, Hunninghake GW, Musson RA: NHLBI Workshop Summary. Pharmacological therapy for idiopathic pulmonary fibrosis. Past, present, and future. Am J Respir Crit Care Med 1999, 160:1771-1777 [DOI] [PubMed] [Google Scholar]
- 5.Gauldie J: Inflammatory mechanisms are a minor component of the pathogenesis of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2002, 165:1205-1206 [DOI] [PubMed] [Google Scholar]
- 6.Coker RK, Laurent GJ: Pulmonary fibrosis: cytokines in the balance. Eur Respir J 1998, 11:1218-1221 [DOI] [PubMed] [Google Scholar]
- 7.Kuhn C, McDonald JA: The roles of the myofibroblast in idiopathic pulmonary fibrosis. Ultrastructural and immunohistochemical features of sites of active extracellular matrix synthesis. Am J Pathol 1991, 138:1257-1265 [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang K, Rekhter MD, Gordon D, Phan SH: Myofibroblasts and their role in lung collagen gene expression during pulmonary fibrosis. A combined immunohistochemical and in situ hybridization study. Am J Pathol 1994, 145:114-125 [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang HY, Gharaee-Kermani M, Phan SH: Regulation of lung fibroblast alpha-smooth muscle actin expression, contractile phenotype, and apoptosis by IL-1beta. J Immunol 1997, 158:1392-1399 [PubMed] [Google Scholar]
- 10.Zhang HY, Phan SH: Inhibition of myofibroblast apoptosis by transforming growth factor beta(1). Am J Respir Cell Mol Biol 1999, 21:658-665 [DOI] [PubMed] [Google Scholar]
- 11.Taga T: gp130 and the interleukin-6 family of cytokines. Annu Rev Immunol 1997, 15:797-819 [DOI] [PubMed] [Google Scholar]
- 12.Taga T: The signal transducer gp130 is shared by interleukin-6 family of haematopoietic and neurotrophic cytokines. Ann Med 1997, 29:63-72 [DOI] [PubMed] [Google Scholar]
- 13.Shahar I, Fireman E, Topilsky M, Grief J, Kivity S, Spirer Z, Ben Efraim S: Effect of IL-6 on alveolar fibroblast proliferation in interstitial lung diseases. Clin Immunol Immunopathol 1996, 79:244-251 [DOI] [PubMed] [Google Scholar]
- 14.Elias JA, Wu Y, Zheng T, Panattieri R: Cytokine- and virus-stimulated airway smooth muscle cells produce IL-11 and other IL-6-type cytokines. Am J Physiol 1997, 273:L648-L655 [DOI] [PubMed] [Google Scholar]
- 15.Elias JA, Zheng T, Whiting NL, Trow TK, Merrill WW, Zitnik R, Ray P, Alderman EM: IL-11 and transforming growth factor-beta regulation of fibroblast-derived IL-11. J Immunol 1994, 152:2421-2429 [PubMed] [Google Scholar]
- 16.Waxman AB, Einarsson O, Seres T, Knickelbein RG, Warshaw JB, Johnston R, Homer RJ, Elias JA: Targeted lung expression of interleukin-11 enhances murine tolerance of 100% oxygen and diminishes hyperoxia-induced DNA fragmentation. J Clin Invest 1998, 101:1970-1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Orazi A, Du X, Yang Z, Kashai M, Williams DA: Interleukin-11 prevents apoptosis and accelerates recovery of small intestinal mucosa in mice treated with combined chemotherapy and radiation. Lab Invest 1996, 75:33-42 [PubMed] [Google Scholar]
- 18.DiCosmo BF, Geba GP, Picarella D, Elias JA, Rankin JA, Stripp BR, Whitsett JA, Flavell RA: Airway epithelial cell expression of interleukin-6 in transgenic mice. Uncoupling of airway inflammation and bronchial hyperreactivity. J Clin Invest 1994, 94:2028-2035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Keerthisingam CB, Jenkins RG, Harrison NK, Hernandez-Rodriguez NA, Booth H, Laurent GJ, Hart SL, Foster ML, McAnulty RJ: Cyclooxygenase-2 deficiency results in a loss of the anti-proliferative response to transforming growth factor-beta in human fibrotic lung fibroblasts and promotes bleomycin-induced pulmonary fibrosis in mice. Am J Pathol 2001, 158:1411-1422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scaffidi AK, Moodley YP, Weichselbaum M, Thompson PJ, Knight DA: Regulation of human lung fibroblast phenotype and function by vitronectin and vitronectin integrins. J Cell Sci 2001, 114:3507-3516 [DOI] [PubMed] [Google Scholar]
- 21.Katzenstein AL, Myers J: Idiopathic pulmonary fibrosis. Clinical relevance of pathologic classification. Am J Respir Crit Care Med 1998, 157:1301-1315 [DOI] [PubMed] [Google Scholar]
- 22.Ramos C, Montano M, Garcia-Alvarez J, Ruiz V, Uhal BD, Selman M, Pardo A: Fibroblasts from idiopathic pulmonary fibrosis and normal lungs differ in growth rate, apoptosis, and tissue inhibitor of metalloproteinases expression. Am J Respir Cell Mol Biol 2001, 24:591-598 [DOI] [PubMed] [Google Scholar]
- 23.Hadden H, Henke C: Induction of lung fibroblast apoptosis by soluble fibronectin peptides. Am J Respir Crit Care Med 2000, 162:1553-1560 [DOI] [PubMed] [Google Scholar]
- 24.Jordana M, Schulman J, McSharry C, Irving L, Newhouse M, Jordana G, Gauldie J: Heterogenous proliferative characteristics of human adult lung fibroblast lines and clonally derived fibroblasts from control and fibrotic tissue. Am Rev Respir Dis 1988, 137:579-584 [DOI] [PubMed] [Google Scholar]
- 25.Heinrich PC, Behrmann I, Muller-Newen G, Schaper F, Graeve L: Interleukin 6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem J 1998, 334:297-314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kishimoto T, Akira S, Narazaki M, Taga T: Interleukin-6 family of cytokines and gp130. Blood 1995, 86:1243-1254 [PubMed] [Google Scholar]
- 27.Kawaguchi Y, Hara M, Wright TM: Endogenous IL-1alpha from systemic sclerosis fibroblasts induces IL-6 and PDGF-A. J Clin Invest 1999, 103:1253-1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nishimoto N, Ito A, Ono M, Tagoh H, Matsumoto T, Tomita T, Ochi T, Yoshizaki K: IL-6 inhibits the proliferation of fibroblastic synovial cells from rheumatoid arthritis patients in the presence of soluble IL-6 receptor. Int Immunol 2000, 12:187-293 [DOI] [PubMed] [Google Scholar]
- 29.Fries KM, Felch ME, Phipps RP: Interleukin-6 is an autocrine growth factor for murine lung fibroblast subsets. Am J Respir Cell Mol Biol 1994, 11:552-560 [DOI] [PubMed] [Google Scholar]
- 30.Fukada T, Ohtani T, Yoshida Y, Shirogane T, Nishida K, Nakajima K, Hibi M, Hirano T: STAT3 orchestrates contradictory signals in cytokine-induced G1 to S cell-cycle transition. EMBO J 1998, 17:6670-6677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nakajima K, Yamanaka Y, Nakae K, Kojima H, Ichiba M, Kiuchi N, Kitaoka T, Fukada T, Hibi M, Hirano T: A central role for Stat3 in IL-6-induced regulation of growth and differentiation in M1 leukemia cells. EMBO J 1996, 15:3651-3658 [PMC free article] [PubMed] [Google Scholar]
- 32.Terada Y, Inoshita S, Nakashima O, Kuwahara M, Sasaki S, Marumo F: Regulation of cyclin D1 expression and cell cycle progression by mitogen-activated protein kinase cascade. Kidney Int 1999, 56:1258-1261 [DOI] [PubMed] [Google Scholar]
- 33.Tang W, Geba GP, Zheng T, Ray P, Homer RJ, Kuhn CR, Flavell RA, Elias JA: Targeted expression of IL-11 in the murine airway causes lymphocytic inflammation, bronchial remodeling, and airways obstruction. J Clin Invest 1996, 98:2845-2853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zheng T, Zhu Z, Wang J, Homer RJ, Elias JA: IL-11: insights in asthma from overexpression transgenic modeling. J Allergy Clin Immunol 2001, 108:489-496 [DOI] [PubMed] [Google Scholar]
- 35.Wilborn J, Crofford LJ, Burdick MD, Kunkel SL, Strieter RM, Peters-Golden M: Cultured lung fibroblasts isolated from patients with idiopathic pulmonary fibrosis have a diminished capacity to synthesize prostaglandin E2 and to express cyclooxygenase-2. J Clin Invest 1995, 95:1861-1868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McAnulty RJ, Hernandez-Rodriguez NA, Mutsaers SE, Coker RK, Laurent GJ: Indomethacin suppresses the anti-proliferative effects of transforming growth factor-beta isoforms on fibroblast cell cultures. Biochem J 1997, 321:639-643 [DOI] [PMC free article] [PubMed] [Google Scholar]