

Abstract
Cartilage oligomeric matrix protein (COMP) is a large pentameric extracellular glycoprotein found in cartilage, tendon, and synovium, and plays structural roles in cartilage as the fifth member of the thrombospondin family. Familial mutations in type 3 repeats of COMP are known to cause pseudoachondroplasia (PSACH) and multiple epiphyseal dysplasia (EDM1). Although such mutations induce enlarged rough endoplasmic reticulum (rER) as a morphological change, the metabolic trafficking of mutated COMP remains unclear. In transfected COS7 cells, wild-type COMP was rapidly secreted into culture medium, while the great majority of COMP with the type 3 repeats mutation (D472Y) remained in the cells and a small portion of mutated COMP was secreted. This finding was followed up with a confocal study with an antibody specific to COMP, which demonstrated mutated COMP tightly associated with abnormally enlarged rER. Phosphorylated eIF2α, an ER stress protein, was expressed as a pathological reaction in virtually all COS7 cells expressing mutated but not wild-type COMP. Moreover, COS7 cells expressing mutated COMP exhibited significantly more apoptotic reaction than those expressing wild-type COMP. Pathological accumulation of COMP in rER and apoptosis in COS7 cells that were induced by the mutation (D472Y) in COMP imply that COMP mutations play a role in the pathogenesis of PSACH.
Pseudoachondroplasia (PSACH) and multiple epiphyseal dysplasia (EDM1) are closely related to each other and autosomal dominant skeletal dysplasia. PSACH patients clinically exhibit disproportionately short stature, joint laxity, and early-onset osteoarthritis, 1-3 while EDM1 patients exhibit mildly short stature and joint pain, particularly in the hips, in addition to early-onset osteoarthritis. 2-4 Brigg et al 5 and Hecht et al 6 examined these two types of human skeletal dysplasia and independently found mutations in the COMP gene on chromosome 19p12–13.1.
Cartilage oligomeric matrix protein (COMP) was initially termed the high-molecular-weight cartilage matrix glycoprotein that was isolated from articular cartilage 7,8 and characterized later as a territorial homopentameric matrix protein with a subunit size of 100 to 110 kd. 9 Interestingly, COMP proved to be the fifth member of the thrombospondin family, members of which have a coiled-coil region responsible for multimerization and interchain disulfide bonds, 10 four epidermal growth factor-like type 2 repeats, seven highly conserved type 3 repeats that consist of 13 calcium-binding loops, and a COOH-terminal globular domain. 11-13 Most mutations identified in the COMP gene are located within the exons encoding the calcium-binding type 3 repeats 2,14,15 and are postulated to cause qualitative defects in the protein, induce COMP conformational change, and reduce calcium-binding activity. 16-18
COMP is primarily synthesized in cells and then secreted as an extracellular territorial matrix around chondrocytes 9 and can also be found in synovium, tendons, and dermal fibroblasts. 19-22 In ultrastructural studies, chondrocytes from PSACH and EDM1 patients have been found to be characterized by enormous vesicles formed from rough endoplasmic reticulum (rER) that have a unique lamellar appearance with alternating electron-lucent and electron-dense layers. 23-25 Extracellular matrix components, including aggrecan and type IX collagen, have been shown to be retained in these enlarged vesicles. 25-29 The retention appeared to be cell type-specific since COMP is secreted normally from patient tendon and ligament in vitro and patient chondrocytes cultured in monolayers. 27,28,30 However, it is still unclear how these mutations affect COMP trafficking and its pathway of secretion to cause disease phenotypes.
In this study, we examined the effect of a mutation (D472Y) in the calcium-binding domain previously identified for severe PSACH 6 on the process of secretion in cultured COS7 cells with newly prepared antibodies specific for COMP. This mutation was found to affect an early stage of the membrane trafficking of COMP at the rER before the Golgi apparatus and plasma membrane. Recently, it has been shown that a variety of toxic insults, including calcium ionophores, inhibitors of glycosylation, chemical toxicants, and oxidative stress, can cause ER stress and ultimately lead to cell death. 31-39 We found that significant ER stress and resultant apoptosis occurred in cells expressing mutated COMP compared with wild-type COMP and discuss the significance of this in PSACH.
Materials and Methods
Preparation of Human Normal COMP cDNA
We isolated periosterial cells from five normal subjects without any clinical disorders related to PSACH or EDM1 at surgical operation and used them for isolation of cDNA to encode COMP under informed consent. Cells were grown with 10 ng/ml TGF-β, 10% fetal calf serum, and Dulbecco’s modified Eagle’s medium (DMEM). Total RNA was extracted from cultured human synovial cells and used as the template for amplification of full-length COMP cDNA according to standard protocols with Pyrobest DNA Polymerase (Takara Bio Inc., Kyoto, Japan). The full-length COMP cDNA amplified with primers F-NdeI (5′-cagatccatgcggccgccatggtccccgacaccgcct gcgtt-3′) and R-NdeI (5′-ggtcttcacgcggccgcctaggcttgccgcagctgatgggt-3′) was introduced into the bacterial expression vector pET3a (Invitrogen, CA) for preparation of anti-COMP antibodies. Three COMP clones were established from each of the five individuals. All of the 15 clones of wild-type COMP cDNA were sequenced at every step and confirmed with an ABI Prism 310 DNA Sequencer (Applied Biosystems, CA) and found to possess the same 11 SNPs (Accession No: AB086984 in DDBJ (DNA Data Bank of Japan) database (http://www.ddbj.nig.ac.jp/)). The full-length COMP cDNA in pET3a vector was re-amplified with primers F-NotI (5′-cagatccatgcgg ccgccatggtccccgacaccgcctgcgtt-3′) and R-NotI (5′-ggtcttcacgcggccgcctaggcttgccgcagctgatgg gt-3′) and transferred into the mammalian expression vector pCMV (Invitrogen, CA) for transfection.
Mutagenesis of COMP cDNA
The point mutation (G1414A) found for a patient with severe PSACH 6 was introduced into the wild-type COMP cDNA by PCR. The PCR primers used for the mutagenesis were the mutagenesis primers 1414-F (5′- ggaggactcagaccacgatggccagggtgatgcctgcgacgacgactacgacaat-3′) and 1414-R (5′-attgtcgtagtcgtcgtcgcaggcatcaccctggccatcgtggtctgagtcctcc −3′), in addition to the two primers F-NotI and R-NotI. Mutated COMP cDNA was introduced into pCMV vector. It was confirmed by sequencing that the correct mutation had been introduced and that other PCR-generated errors were not incorporated. All of the COMP cDNA and its derivative clones were sequenced at every step and confirmed with an ABI Prism 310 DNA Sequencer.
Preparation of Anti-COMP Antibodies and Their Characterization
The antigens were the N-terminal and C-terminal fragments of COMP. Both fragments of COMP were recombinant proteins that were prepared from the truncated COMP cDNA. The two cDNAs were generated using full-sized COMP cDNA as the template by PCR with the two following primer sets. The N-terminal fragment (residue number: 89–290) primers were NF (5′-cttgccaggcatatgctccactgcgcgcccggcttctgc-3′) and NR (5′-gagtcatc gcatatgctagtccttacggcactgcggctccgg-3′), and the C-terminal fragment (residue number: 525–757) primers were CF (5′-cttgccacccatatggaagtcacgctcaccgacttcagg-3′) and CR (5′-tccggccatatgtcaccc tggtccctaggcttgccg-3′). Each COMP fragment cDNA was introduced into the bacterial expression vector pET3a and used for transformation of E. coli BL21 (DE3) (Novagen, NY). The transformed cells were homogenized with a teflon homogenizer and sonicated in 50 mmol/L Tris-HCl (pH 7.6), containing protease inhibitors and 2% 2-mercaptoethanol. After centrifugation at 15,000 × g for 20 minutes, the pellets were resuspended in the same buffer containing 1% Triton-X100. After centrifugation at 15,000 × g for 20 minutes again, the pellets were resuspended in the same buffer containing 8 mol/L urea. After centrifugation again, the resultant supernatants were applied to a Resource Q ion-exchange column (Amersham Pharmacia Biotech, NJ) pre-equilibrated with 50 mmol/L NaCl/8 mol/L urea/50 mmol/L Tris-HCl (pH 7.6). Elution was performed with a linear gradient of 50 mmol/L to 1000 mmol/L NaCl in 8 mol/L urea/50 mmol/L Tris-HCl (pH 7.6). Aliquots from fractions were analyzed by Coomassie brilliant blue R-250 dye staining following SDS-PAGE. The eluates containing COMP fragments were collected and dialyzed against 20 mmol/L ammonium bicarbonate buffer (pH 7.5), followed by lyophilization. Lyophilized material was resuspended in 50 mmol/L Tris-HCl (pH 7.6), and injected s.c. in rabbits and BALB/c mice every 2 weeks with Freund’s incomplete adjuvant after the first immunization with Freund’s complete adjuvant (Difco Laboratory, DM). From each species, two antibodies to the N-terminal and C-terminal fragments, respectively, were established. A rabbit anti-COMP polyclonal antibody to the N-terminal fragment whose immnoreactivity was stronger than the other antibodies in Western blotting was chosen for use in experiments thereafter.
To evaluate the specificity of the antibody, we performed an adsorption experiment and immunohistochemistry. For the former, the primary antibody was incubated with or without 10 μg/ml recombinant N-terminal COMP fragment. The protocol is described in detail for Western blot analysis below.
For immunohistochemistry, murine growth plates of the knee including chondrocytes were taken at 2 weeks after birth. Samples were fixed for 12 hours in fresh 4% paraformaldehyde in phosphate-buffered saline (PBS) at 4°C, embedded in paraffin, and serially sectioned (5-μm thick). Sections adsorbed to silanized over-dried glass slides were deparaffinized and hydrated. After incubation with a blocking buffer containing 20% calf serum in PBS for 30 minutes at room temperature, sections were incubated overnight at 4°C with the newly developed anti-COMP antibodies followed by biotinylated secondary antibodies for 30 minutes at room temperature. After washing, sections were incubated with avidin biotin-peroxidase complex (Vectastain ABC Kit, Vector Laboratories, CA) for 10 minutes at room temperature. Immnoreactivity was visualized by incubation with diaminobenzidine and 0.03% hydrogen peroxide solution for 5 minutes at room temperature.
Cell Culture and Transfection
COS7 cells were cultured with DMEM containing 10% fetal calf serum, penicillin, and streptomycin. Cells were grown to approximately 70% confluence in 60-mm dishes for 24 hours before transfection. The DNA construct of mammalian expression vector pCMV (3 μg) containing either wild-type COMP, mutated COMP, or vector only was mixed with 12 μl of LipofectAMINE (Gibco BRL, MD) in 2 ml of OPTI-MEM serum-free medium (Gibco BRL, MD) and added to cells, and incubated for 3 hours. The cells were further cultured in 2.5 ml of DMEM containing 10% fetal calf serum in 5% CO2.
Endoglycosidase H Digestion
The transfected cells were harvested, washed twice with PBS and dissolved in 0.5%SDS/1% 2-mercaptoethanol/50 mmol/L sodium acetate (pH 5.5), containing protease inhibitors. After protein content was determined, the same amount of each lysate was boiled for 10 minutes. Endoglycosidase H (Endo H) (Sigma, MO) (0.25 m unit/5 μl) in 50 mmol/L sodium acetate (pH 5.5), was added to each sample and incubated for 3 hours at 37°C. Endo H digestions were terminated by heating at 95°C.
Staurosporine Treatment of COS7 Cells
To prepare a positive control for ER stress and DNA fragmentation, COS7 cells were treated with staurosporine. When cells were grown to 80% confluence, staurosporine was added at a final concentration of 1 mmol/L to culture with DMEM containing 10% fetal calf serum, and the cells were incubated for 3 hours for ER stress and 12 hours for DNA fragmentation at 37°C in 5% CO2.
Western Blot Analysis
The transfected cells were harvested and then washed twice with PBS at 4°C, and the pellets were dissolved in 0.5% SDS/150 mmol/L NaCl/50 mmol/L Tris-HCl (pH 7.6), containing protease inhibitors. Samples of cell lysates and conditioned media were mixed with 4% SDS/20% glycerol/160 mmol/L Tris-HCl (pH 6.8), with or without 4% 2-mercaptoethanol, boiled for 5 minutes, centrifuged, and applied to 5% polyacrylamide gel. After electrophoresis, proteins on the gel were electroblotted to PVDF membrane (Millipore, Bedford, MA). The membranes were blocked in 3% nonfat milk/1%BSA/0.05% Tween 20/150 mmol/L NaCl/50 mmol/L Tris-HCl (pH 7.6), for 2 hours and were then incubated with the primary antibody to N-terminal COMP (1:10,000). After four washes with 0.05% Tween 20/150 mmol/L NaCl/50 mmol/L Tris-HCl (pH 7.6), the membranes were incubated with a horseradish peroxidase (HRP)-linked secondary antibody (1:10,000) (Bio-Rad Lab, CA), and immunoreactivity was detected by the ECL Plus Western Blotting Detection System (Amersham Pharmacia Biotech, NJ). An anti-G3PDH antibody (Trevigen Inc., MD) was used for immunoblotting to normalize protein contents of cell lysate samples. The densities of COMP in conditioned media and cell lysates were measured using a densitometer (Bio-Rad Lab). The secreted COMP/intracellular COMP ratio was calculated for three independent experiments. In ER stress experiments, membranes were incubated with a primary antibody to eIF2α (1:1000) or Phospho-eIF2α (1:1000) (Cell Signaling Technology Inc., MA) followed by HRP-conjugated secondary antibody.
Confocal View Analysis
COS7 cells grown on 1% gelatin-coated coverslips were transfected, as described above. After 2-day culture, cells were washed in PBS and fixed with cooled 90% methanol for 10 minutes at 4°C followed by acetone for 10 minutes at 4°C. The fixed cells were hydrated once with PBS, and washed with 0.05% Tween-containing PBS to ensure permeabilization for antibody staining. After washing again with PBS, the cells were incubated with blocking buffer containing 20% calf serum in PBS for 20 minutes at room temperature. The cells were then incubated with the primary antibodies followed by rhodamine (TRITC)- and fluorescein isothiocyanate (FITC)-conjugated secondary antibodies (Jackson Immunoresearch Labs Inc., PA). The stained specimens were mounted and examined under a confocal laser microscope (Zeiss LSM 510, Jena). The primary antibodies included the polyclonal antibody to N-terminal COMP, a monoclonal antibody to Grp78, a representative marker of endoplasmic reticulum (obtained from Stressgen Biotechnologies Corp, CA), a monoclonal antibody to Golgi 58K protein, a representative marker of the Golgi apparatus (obtained from Sigma), and a polyclonal antibody to Phospho-eIF2α, a marker of ER stress (obtained from Cell Signaling Technology Inc).
Terminal dUTP Nick-End Labeling Assay
COS7 cells grown on coverslips were transfected with either wild-type COMP, mutated COMP, or vector only. After 48 hours of incubation, the transfected cells were washed and fixed as described above. The cells were then washed twice with PBS and incubated with 50 μl terminal dUTP nick-end labeling (TUNEL) reaction mixture (Roche Diagnostic, IN) in a humid chamber for 60 minutes at 37°C. For subsequent double-staining, the cells were washed three times again and stained with the anti-COMP antibody as described above. The specimens were examined under a confocal microscope (Zeiss LSM 510, Jena). Six fields were randomly selected, in which total, TUNEL-positive, and COMP-positive cells were counted. The counts of the six fields were summed for each transfection. Each field contained approximately 200 cells. The TUNEL-positive cells/total cells and TUNEL- and COMP-positive cells/COMP-positive cells ratios were calculated. Transfection was performed three times.
Analysis of DNA Fragmentation
Transfected cells and staurosporine-treated cells (2 × 106) were incubated in 100 μl ice-cold lysis buffer (10 mmol/L Tris-HCl (pH 7.4), 10 mmol/L EDTA, 0.5% SDS) at 4°C for 10 minutes. The lysates were treated with 40 mg RNase A at 37°C for 1 hour, and then with 40 mg of proteinase K at 50°C for 1 hour. To precipitate genomic DNA, the mixture was incubated with 1 volume of isopropanol and 0.2 volumes of 3 mol/L sodium acetate at −20°C for 12 hours and centrifuged at 10,000 × g for 20 minutes. Pellets were air-dried and dissolved in 100 μl of Tris EDTA (TE) buffer. The DNA samples were subjected to 1.5% agarose gel electrophoresis using 40 mmol/L Tris acetate (pH 8.0) and 1 mmol/L EDTA as a running buffer. The gel was stained with 0.1 mg/ml of Syber Green (BioWhittaker Molecular Applications, ME) and observed under ultraviolet light.
Statistical Analysis
We used the unpaired t-test for statistical analysis. Statistical significance was confirmed when P values were <0.05.
Results
Mutated COMP Exhibited the Same Pentamer Structure as Wild-Type COMP but Its Secretion Was Markedly Disturbed
To evaluate the specificity of our newly developed anti-COMP antibody, we stained murine growth plates of knee, including chondrocytes, 2 weeks after birth with this antibody. The immunohistochemical distribution was very similar to that in a previous study performed with a different COMP-specific antibody 40 (Figure 1A–C ▶ ). Immnoreactivity associated with anti-COMP antibody was intracellularly observed in the proliferating zone of the growth plate while it was pronounced in the pericellular and territorial matrix of the hypertrophic zone. The specificity of the present anti-COMP antibody was then further examined by Western blotting analysis. As shown in Figure 1D ▶ , the COMP protein synthesized in chondrocytes was specifically recognized by anti-COMP antibody, and the resulting immunoreaction was completely absorbed with recombinant COMP fragment used as immunogen (see Materials and Methods). Thus, the present anti-COMP antibody was confirmed to specifically stain COMP protein in cartilage tissue.
Figure 1.
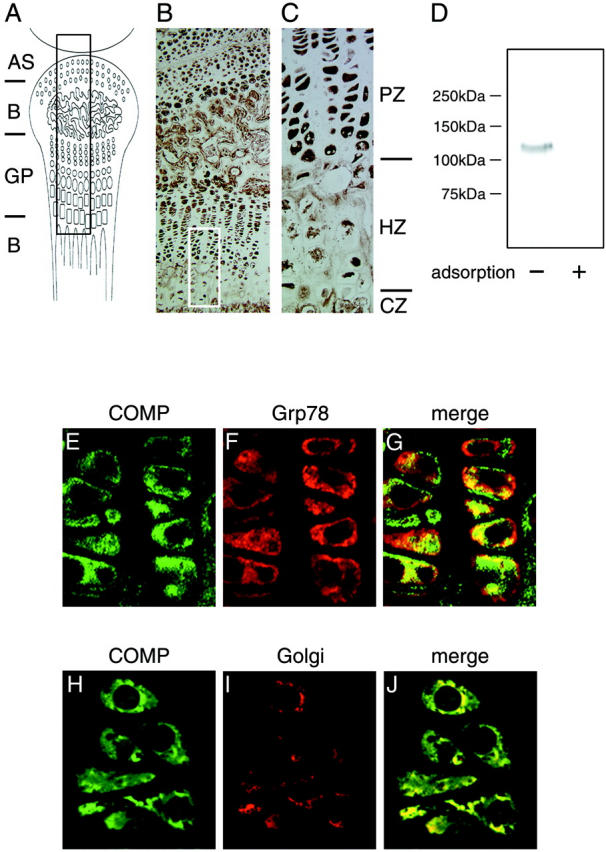
Characterization of a newly-prepared anti-COMP antibody. Murine growth plates of the knee including chondrocytes at 2 weeks after birth were fixed and embedded in paraffin. Sections were stained with a newly prepared anti-COMP antibody and detected by diaminobenzidine and 0.03% hydrogen peroxide. A: Schema of sagittal section of murine knee joint including a growth plate (GP). AS and B represent articular surface and bone, respectively. B: Stained section of murine knee joint with anti-COMP antibody that represents the black boxed area in A (Magnification, ×10). C: The white boxed area in B at higher magnification (Magnification, ×40). Note that the distribution of immunoreactivity with this antibody was similar to that with a COMP-specific antibody described in a previous study. 40 PZ, HZ, and CZ represent the proliferating zone, hypertrophic zone, and calcification zone, respectively. D: Western blot showing that anti-COMP antibody specifically recognized native COMP protein synthesized in chondrocytes. The symbols (+) and (-) indicate with or without recombinant COMP fragment used for the adsorption experiment as described in Materials and Methods. E–J: Confocal view to examine the subcellular distribution of COMP in chondrocytes of cartilage. The cartilage tissue section was stained with anti-COMP antibody (E, H) and anti-ER antibody (F) or anti-Golgi antibody (I) followed by relevant FITC- and rhodamine-labeled secondary antibodies. G and J are merged images of E and F, and H and I, respectively. Experimental details are provided in Materials and Methods.
Furthermore, subcellular distribution of COMP was immunohistochemically studied by confocal analysis using anti-COMP antibody (Figure 1, E and H) ▶ and anti-ER antibody (Grp78) (Figure 1F) ▶ or anti-Golgi antibody (Figure 1I) ▶ . As shown in the merged images (Figure 1, G and J) ▶ , a portion of COMP was clearly colocalized with ER and the Golgi apparatus. This in vivo subcellular distribution of COMP in native chondrocytes of cartilage was identical to that in COS cells transfected with wild-type COMP cDNA as described below.
COS7 cells were used here as host cells because of their low background expression of COMP protein, as clearly demonstrated in Figure 2 ▶ (lanes 3, 6, 9, and 12). The human COMP cDNA used here was cloned from normal subjects and its wild-type sequence was confirmed by sequencing as described in Materials and Methods. 2,5 The pathogenic mutation (D472Y) used here was initially identified as the causal mutation for a familial case with severe clinical manifestations. 6 Thus, this important mutation was introduced into the wild-type COMP cDNA. COS7 cells were transfected with either wild-type COMP cDNA or the mutated COMP cDNA and examined biochemically by Western blot analysis with newly prepared anti-COMP antibodies. As seen in Figure 2 ▶ , both the wild-type and mutated COMP were detected in both cell lysates and culture media as a single band with a molecular weight (Mr) of 100 to 110 kd under reducing conditions. The apparent molecular weight of COMP in conditioned media was similar to that of cellular COMP, although it was slightly larger than expected on the basis of its amino acid composition because of glycosylation, as described below. The molecular weight of COMP in in vitro-cultured cells was found to be identical to that of COMP in in vivo human cartilage and tendon tissue (unpublished). Both wild-type and mutated COMP were found to migrate as one band with a high Mr of about 500 kd representing a pentamer complex in the gel under non-reducing conditions (Figure 2) ▶ . Both the mutated and normal COMP were secreted into the culture media, but the velocities and amounts of their secretion were markedly different (Figure 2) ▶ . As clearly shown in the time-course experiment at 12, 24, and 48 hours after transfection, the ratio of secreted COMP to cellular COMP was much less with the mutated COMP than with wild-type COMP (Figure 3,A and B) ▶ , suggesting disturbance of trafficking of COMP for transport or secretion. All results obtained with an anti-COMP antibody recognizing the N-terminal fragment of COMP were reproducibly confirmed with another anti-COMP antibody recognizing the C-terminal fragment of COMP (data not shown).
Figure 2.

Oligomerization of wild-type and mutated COMP. COS7 cells were transiently transfected with wild-type COMP (lanes 1, 4, 7 and 10), mutated COMP (lanes 2, 5, 8 and 11), or vector alone (lanes 3, 6, 9 and 12) and cultured for 48 hours at 37°C under serum-free conditions. Cell lysates (1–3 and 7–9) and conditioned media (4–6 and 10–12) were analyzed by immunoblotting with the anti-COMP antibody to recognize the N-terminal fragment of COMP. The samples were treated with SDS sample buffer in the (1–6) absence or (7–12) presence of 2-mercaptoethanol (ME).
Figure 3.

Comparison of processes of secretion of wild-type and mutated COMP. COS7 cells were transfected with wild-type or mutated COMP and cultured at 37°C. The cells and conditioned media were saved at 12 hours, 24 hours, and 48 hours after transfection and analyzed by immunoblotting with the anti-COMP antibody. The protein content of each cell lysate was normalized based on that of G3PDH. Samples were adjusted to contain equal amounts of proteins. A: Top and middle columns represent wild-type COMP and mutated COMP, respectively. The bottom column represents G3PDH as an authentic housekeeping protein. B: Time course of changes in the secreted COMP/cellular COMP ratio. The means ± SEM of the ratios obtained from three independent transfections are shown. Open and closed columns represent wild-type and mutant COMP, respectively.
Intracellular COMP Is a Glycoprotein with High-Mannose Oligosaccharides That Are Sensitive to Endo H
To biochemically characterize cellular COMP, we examined the sensitivity of COMP to Endo H, an enzyme cleaving N-linked high-mannose oligosaccharides but not complex oligosaccharides. As shown in Figure 4 ▶ , both wild-type COMP and mutated COMP were synthesized as a glycoprotein with Mr of 100 to 110 kd and were sensitive to treatment with Endo H. Since it has been shown that glycoproteins with high-mannose oligosaccharides but not complex oligosaccharides, such as chondroitin sulfate proteoglycan, were sensitive to treatment with Endo H, 41 our results indicate that intracellular COMP possesses high-mannose-type oligosaccharides.
Figure 4.

Intracellular COMP is an Endo H-sensitive glycoprotein. COS7 cells were transfected with wild-type or mutated COMP and cultured at 37°C. The cells were saved at 24 hours and 48 hours and solubilized with 0.5%SDS/1% 2-mercaptoethanol/50 mmol/L sodium acetate (pH 5.5), containing protease inhibitors after washing with PBS. The samples were incubated in the presence or absence of Endo H for 3 hours. Treated samples were analyzed by immunoblotting with the anti-COMP antibody.
Mutated COMP Was Located in rER and Not Transferred into Golgi Apparatus
To investigate the effect of mutation of COMP on its subcellular localization, we studied COMP with confocal microscopy. As shown in Figure 5 ▶ , wild-type COMP was diffusely located in the cytoplasm (Figure 5A) ▶ , and partially colocalized with Grp78, an ER marker (Figure 5, B and C) ▶ . COMP expressed in transfected cells exhibited subcellular localization very similar to that of endogenous COMP in native chondrocytes (Figure 1, A–C ▶ ). In contrast, mutated COMP was tightly colocalized with Grp78 (Figure 5F) ▶ . Strikingly, mutated COMP was induced in a lump of abnormally enlarged and somewhat swollen or gnarled ER (Figure 5, D and E) ▶ . Notably, the immunofluorescence representing mutated COMP was stronger than that representing wild-type COMP (Figure 5, A and D) ▶ .
Figure 5.
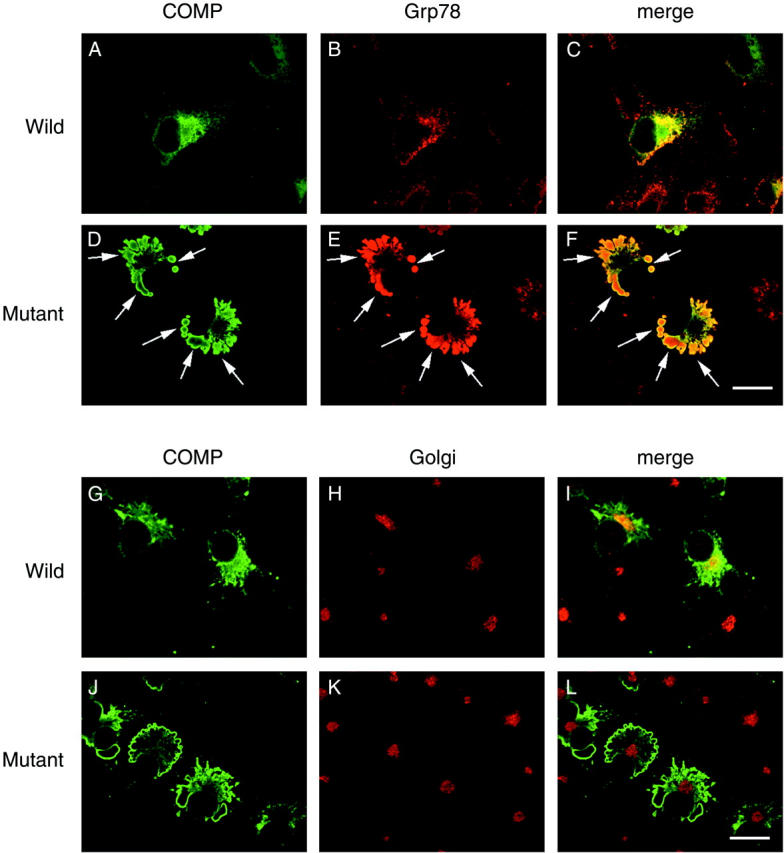
Confocal analysis of subcellular localization of COMP. COS7 cells were transfected with wild-type (A, B, C, G, H, and I) or mutated COMP (D, E, F, J, K, and L) and were cultured for 48 hours at 37°C. The cells were fixed and incubated with the primary antibodies described below followed by FITC- and rhodamine-labeled secondary antibodies. Primary antibodies were the anti-COMP antibody (A, D, G, and J), an anti-Grp78 (an ER maker) antibody (B and E) and an anti-Golgi 58K protein (a Golgi apparatus maker) antibody (H and K). C and F are merged images of COMP and Grp78 staining, while J and L are those of COMP and Golgi 58K protein staining. Virtually all COMP staining in cells with mutated COMP overlapped Grp78 staining (arrows). No COMP staining was detected in untransfected cells. Wild-type COMP was partially colocalized with the Golgi apparatus, whereas mutated COMP was not associated with the Golgi apparatus. Bar, 20 μm.
We also examined the relationship between COMP and the Golgi apparatus. Small amounts of wild-type COMP (Figure 5G) ▶ were colocalized with the Golgi apparatus (Figure 5, H and I) ▶ . However, little mutated COMP (Figure 5J) ▶ was colocalized with the Golgi apparatus (Figure 5, K and L) ▶ , indicating that disturbance had occurred before the trans-Golgi networks in the trafficking route.
ER Stress Occurred in Cells Expressing Mutated COMP
ER is known to be sensitive to alteration of homeostasis by a variety of different stimuli such as glucose deprivation, perturbation of calcium homeostasis, exposure to free radicals, perturbation of protein folding, and accumulation of malfolded proteins. 42 These alterations could induce a pathological reaction termed ER stress that elicits two major cellular-protecting responses, the attenuation of protein synthesis and the up-regulation of genes encoding chaperones that facilitate the process of protein folding in the ER. These responses reduce the accumulation and aggregation of malfolded proteins in the compartments of cells. 43 Double-stranded RNA-dependent kinase (PKR) and heme-regulated inhibitor kinase (HRI) phosphorylation of eukaryotic initiation factor 2 α (elF2α) is a well-characterized mechanism of down-regulation of protein synthesis under conditions of stress. 44 Phosphorylation of eIF2α occurs in response to various stressful stimuli, including nutrient deprivation, heat shock, viral infection, and treatment with compounds that deplete endoplasmic reticular calcium levels. 44,45 Therefore, we examined cells expressing mutated COMP to determine whether they exhibited ER stress using anti-phospho-eIF2α antibody.
Phosphorylated eIF2α, an ER stress protein, was only minimally detected in cells expressing wild-type COMP (Figure 6B) ▶ , but was clearly detected in cells expressing mutated COMP (Figure 6E) ▶ . Phosphorylated eIF2α was completely colocalized with mutated COMP in the latter cells (Figure 6, D and F) ▶ . Western blot analysis showed that phosphorylated eIF2α was more abundant in cells expressing mutated COMP than those expressing wild-type COMP (Figure 6G) ▶ . These results indicate that the mutation of COMP dominantly induced ER stress.
Figure 6.

Induction of ER stress in cells expressing mutated COMP. COS7 cells were transfected with wild-type (A-C) or mutated COMP (D-F) and cultured for 48 hours at 37°C. Cells were fixed and double-immunostained with the anti-COMP antibody (A and D) and an anti-phospho-eIF2α antibody (B and E), a probe for ER stress, followed by FITC- and rhodamine-labeled secondary antibodies. C and F are merged images of COMP and phospho-eIF2α staining. Note the induction of ER stress in cells expressing mutated COMP but not in those expressing wild-type COMP. Bar, 20 μm. G: Transfected COS7 cells were cultured for 24 hours at 37°C and analyzed by immunoblotting with an anti-eIF2α or phospho-eIF2α antibody. As a positive control, untransfected cells were treated with 1 mmol/L staurosporine (STS) for 3 hours at 37°C. Note that phosphorylated eIF2α of cells expressing mutated COMP were more abundant than those expressing wild-type COMP.
Apoptosis Was Induced in Cells Expressing Mutated COMP
The occurrence of ER stress in cells expressing mutated COMP led us to examine the possibility that cells expressing mutated COMP underwent apoptotic reaction. As a critical and final indicator of such a pathogenic reaction, we performed the TUNEL and DNA ladder assay. As clearly shown in Figure 7A and B ▶ , we observed stronger TUNEL-positive reaction in cells expressing mutated COMP than in those expressing wild-type COMP. The ratio of apoptosis in cells transfected with vector alone was slightly lower than in those expressing wild-type COMP (not significant, data not shown). When we focused on transfected cells expressing either wild-type or mutated COMP, the difference in ratio of apoptosis between cells with wild-type and mutated COMP proved to be significant (P < 0.005) (Figure 7C) ▶ . In DNA ladder assay, cells expressing mutated COMP exhibited DNA fragmentation, whereas cells expressing wild-type COMP did not (Figure 7D) ▶ .
Figure 7.

Induction of apoptotic reaction in cells expressing mutated COMP. COS7 cells were transfected with wild-type (A) or mutated COMP (B) and cultured for 48 hours at 37°C. The cells were double-stained with a TUNEL assay kit (Roche Diagnostic, IN) (green) and an anti-COMP antibody followed by a rhodamine-labeled secondary antibody (red). Note that apoptosis occurred exclusively in cells expressing mutated COMP. Arrows show both TUNEL- and COMP-positive cells. Bar, 20 μm. C: The TUNEL- and COMP-positive cells/COMP-positive cells ratios were calculated. Apoptosis was significantly more prominent in cells expressing mutated COMP (14.6 ± 2.1%) than in those expressing wild-type COMP (4.6 ± 0.6%) (*, P < 0.005). The means ± SEM of the ratios obtained from three independent transfections are shown. D: Transfected COS7 cells were cultured for 96 hours at 37°C. As a positive control, untransfected cells were treated with 1 mmol/L STS for 12 hours at 37°C. Each genomic DNA was precipitated as described in Materials and Methods. The DNA samples were subjected to 1.5% agarose gel electrophoresis, stained with 0.1 μg/ml of Syber Green and visualized under ultraviolet light. Note that cells expressing mutated COMP exhibited DNA fragmentation, whereas those expressing wild-type COMP did not. The left lane represents 100-bp DNA size marker (Promega, WI).
Discussion
COMP is synthesized at the rER and processed on the trafficking and secretion route. 16,27-29,46 Secreted COMP observed in the circulation may play a role in storage and delivery of hydrophobic hormones and vitamins such as retinoic acid and vitamin D to target organs. 47 COMP is widely believed to play a structural role in the morphogenesis of cartilage with other matrix proteins. Here we focused on the process of trafficking of wild-type and mutated COMP. In our transient expression system, confocal study clearly revealed that mutated COMP abnormally accumulated almost exclusively in rER but not in Golgi networks, resulting in swollen rER structures. As shown in Figure 2 ▶ , the pentamer-forming ability of mutated COMP in cells and culture media appeared to be virtually intact, suggesting that the pentamerizing domain in the amino-terminal region of COMP was locally and functionally independent of and not interfered with by the calcium-binding domain, where the mutation (D472Y) was located. The most prominent effect of the COMP mutation was reported to be remarkable reduction of the binding of calcium ion. 16-18
Other matrix proteins such as aggrecan and type IX, type XI, and type XII collagens are known to bind COMP and to accumulate together with mutated COMP. 18,26,29,46,48 Interaction of these proteins is assumed to require the intact calcium-binding domain of COMP. It is likely that this domain enables COMP to undergo appropriate protein folding and to bind with other matrix proteins to be secreted together. 16-18 This appears to explain the dominant-interference mechanism of the COMP mutation in the region of the type 3 calcium-binding repeats that causes disease through disturbance of an interaction of mutated COMP with COMP-binding proteins, as described above. This could be further evidence that other matrix proteins also accumulate together with mutated COMP in abnormal cis-ternae of swollen rER with a unique lamellar structure inpatient cartilage.
The recent report that COMP-deficient mice exhibited no anatomical or histological abnormalities and no clinical signs of PSACH or EDM1 indicates that the phenotypes in PSACH/EDM1 cartilage disorders were not due to reduced amounts of COMP in cartilage. 49 Since COMP is thought to play an important role in cartilage, it may be functionally compensated for in a redundant fashion by similar matrix proteins in such mice. These presumptive matrix proteins have yet to be identified, but they seem not to be members of the thrombospondin family. 49
The ER is a principal site of synthesis and folding for membrane proteins as well as secreted proteins and also serves as a cellular storage site for calcium ion. 50,51 Inhibition of N-glycosylation by tunicamycin or alteration of intracellular calcium homeostasis resulted in accumulation of unfolded proteins in ER and led to the induction of chaperone proteins and elevation of intracellular calcium, changes that have been collectively termed unfolded protein response or ER stress. 52,53 Recently, it was demonstrated that a novel ER-specific apoptosis pathway is mediated by caspase-12, and it was shown that procaspase-12 is predominantly located in the ER and specifically activated by disturbances of ER homeostasis such as ER stress and mobilization of intracellular calcium ion stores. 54 The observation of abnormal ER bearing massively accumulated mutated COMP (Figure 5) ▶ suggested examination to determine whether cells expressing mutated COMP suffered ER stress and apoptosis. As shown in Figure 6 ▶ , it was noteworthy that phosphorylated eIF2α, an ER stress protein, was detected as much stronger signals in cells expressing mutated COMP than in those expressing wild-type COMP. In addition, such cells expressing mutated COMP exhibited marked apoptosis at higher frequency than cells expressing wild-type COMP (Figure 7) ▶ . These new findings were compatible with the in vitro finding of reduced survival of cells obtained from PSACH patients 55 and with the in vivo observation of narrowed and irregular columns in the proliferating or hypertrophic zone and reduced numbers of chondrocytes in the growth plates of PSACH patients. 56,57 Since the present experiments were mainly performed with cultured COS7 cells, we could not simply extend our conclusion to in vivo effects of the mutation. However, it is very likely that other interacting or related proteins such as type IX collagen and aggrecan play partner roles in the secretion and function of COMP. 18,25,26,29,46,48
In summary, mutation of COMP was shown to slow and to reduce the process of secretion and to induce accumulation of mutated COMP in rER. Cells expressing mutated COMP but not wild-type COMP exhibited apoptosis as well as ER stress. Further study is needed to clarify such in vivo function and interaction of COMP with other extracellular matrix proteins and cell surface molecules.
Acknowledgments
We thank Dr. T. Kudo (Osaka University Medical School) for his valuable comments on the apoptosis experiment. We also thank Drs. H. Takuma and T. Yamashita for their technical advice and Ms. Y. Hasegawa for her technical assistance.
Footnotes
Address reprint requests to H. Mori, Ph.D., Institute of Gerontology, Osaka City University Medical School, 1–4-3 Asahimachi, Abenoku, Osaka 545-8585, Japan. E-mail: mori@med.osaka-cu.ac.jp.
Supported in part by a grant-in-aid for Scientific Research on Priority Areas (C)-Advanced Brain Science Project- from the Ministry of Education, Culture, Sports, Science, and Technology, Japan (to H.M.). This study was also supported by grants-in-aid from the Uehara Memorial Foundation, the Nakatomi Foundation, and the Osaka Gas Aging Foundation.
References
- 1.McKeand J, Rotta J, Hecht JT: Natural history study of pseudoachondroplasia. Am J Med Genet 1996, 63:406-410 [DOI] [PubMed] [Google Scholar]
- 2.Ikegawa S, Ohashi H, Nishimura G, Kim KC, Sannohe A, Kimizuka M, Fukushima Y, Nagai T, Nakamura Y: Novel and recurrent COMP (cartilage oligomeric matrix protein) mutations in pseudoachondroplasia and multiple epiphyseal dysplasia. Hum Genet 1998, 103:633-638 [DOI] [PubMed] [Google Scholar]
- 3.Beighton P, Giedion ZA, Gorlin R, Hall J, Horton B, Kozlowski K, Lachman R, Langer LO, Maroteaux P, Poznanski A: International Working Group on Constitutional Diseases of Bone. International classification of osteochondrodysplasias. Am J Med Genet 1992, 44:223-229 [DOI] [PubMed] [Google Scholar]
- 4.Berg PK: Dysplasia epiphysialis multiplex: a case report and review of the literature. Am J Roentgenol 1966, 97:31-38 [DOI] [PubMed] [Google Scholar]
- 5.Briggs MD, Hoffman SMG, King LM, Olsen AS, Mohrenweiser H, Rimoin DL, Gaines ES, Cekleriak JA, Knowlton RG, Cohn DH: A mutation in the calcium binding domain of the cartilage oligomeric matrix protein (COMP) results in pseudoachondroplasia. Nat Genet 1995, 10:330-336 [DOI] [PubMed] [Google Scholar]
- 6.Hecht JT, Nelson LD, Crowder E, Wang Y, Elder FFB, Harrison WR, Harrison WR, Francomano CA, Prange CK, Lennon GG, Deere M, Lawler J: Mutations in exon 17B of cartilage oligomeric matrix protein (COMP) cause pseudoachondroplasia. Nat Genet 1995, 10:325-329 [DOI] [PubMed] [Google Scholar]
- 7.Fife RS, Brandt KD: Identification of a high-molecular-weight (greater than 400,000) protein in hyaline cartilage. Biochim Biophys Acta 1984, 802:506-514 [DOI] [PubMed] [Google Scholar]
- 8.Franzen A, Heinegård D, Solursh M: Evidence for sequential appearance of cartilage matrix proteins in developing mouse limbs and in cultures of mouse mesenchymal cells. Differentiation 1987, 36:199-210 [DOI] [PubMed] [Google Scholar]
- 9.Hedbom E, Antonsson P, Hjerpe A, Aeschlimann D, Paulsson M, Rosa-Pimentel E, Sommarin Y, Wendel M, Oldberg Å, Heinegard D: Cartilage oligomeric matrix protein: an acidic oligomeric protein (COMP) detected only in cartilage. J Biol Chem 1992, 267:6132-6136 [PubMed] [Google Scholar]
- 10.Efimov VP, Lustig A, Engel J: The thrombospondin-like chains of cartilage oligomeric matrix protein are assembled by a five-stranded α-helical bundle between residues 20 and 83. FEBS Lett 1994, 341:54-58 [DOI] [PubMed] [Google Scholar]
- 11.Oldberg Å, Antonsson P, Lindblom K, Heinegård D: COMP (cartilage oligomeric matrix protein) is structurally related to the thrombospondins. J Biol Chem 1992, 267:22346-22350 [PubMed] [Google Scholar]
- 12.DiCesare PE, Mörgelin M, Mann K, Paulsson M: Cartilage oligomeric matrix protein and thrombospondin 1: purification from articular cartilage, electron microscopic structure, and chondrocyte binding. Eur J Biochem 1994, 223:927-937 [DOI] [PubMed] [Google Scholar]
- 13.Newton G, Weremowicz S, Morton CC, Copeland NG, Gilbert DJ, Jenkin NA, Lawler J: Characterization of human and mouse cartilage oligomeric matrix protein. Genomics 1994, 24:435-439 [DOI] [PubMed] [Google Scholar]
- 14.Susic S, McGrory J, Cole WG: Multiple epiphyseal dysplasia and pseudoachondroplasia due to novel mutations in the calmodulin-like repeats of cartilage oligomeric matrix protein. Clin Genet 1997, 51:219-224 [DOI] [PubMed] [Google Scholar]
- 15.Briggs MD, Mortier GR, Cole WG, King LM, Golik SS, Bonaventure J, Nuytinck L, De Paepe A, Leroy JG, Biesecker L, Lipson M, Wilcox WR, Lachman RS, Rimoin DL, Knowlton RG, Cohn DH: Diverse mutations in the gene for cartilage oligomeric matrix protein in the pseudoachondroplasia-multiple epiphyseal dysplasia disease spectrum. Am J Hum Genet 1998, 62:311-319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen H, Deere M, Hecht DJ, Lawler J: Cartilage oligomeric matrix protein, and a mutation in its type 3 repeats causes conformational changes. J Biol Chem 2000, 275:26538-26544 [DOI] [PubMed] [Google Scholar]
- 17.Maddox BK, Mokashi A, Keene DR, Bächinger HP: A cartilage oligomeric matrix protein mutation associated with pseudoachondroplasia changes the structural and functional properties of the type 3 domain. J Biol Chem 2000, 275:11412-11417 [DOI] [PubMed] [Google Scholar]
- 18.Thur J, Rosenberg K, Nitsche DP, Pihlajamaa T, Ala-Kokko L, Heinegård D, Paulsson M, Maurer P: Mutation in cartilage oligomeric matrix protein causing pseudoachondroplasia and multiple epiphyseal dysplasia affect binding of calcium and collagen 1, 2, 9. J Biol Chem 2001, 276:6083-6092 [DOI] [PubMed] [Google Scholar]
- 19.DiCesare PE, Hauser N, Lehman D, Pasumarti S, Paulsson M: Cartilage oligomeric matrix protein (COMP) is an abundant component of tendon. FEBS Lett 1994, 354:237-240 [DOI] [PubMed] [Google Scholar]
- 20.DiCesare PE, Carlson CS, Stollerman ES, Chen FS, Leslie M, Perris R: Expression of cartilage oligomeric matrix protein by human synovium. FEBS Lett 1997, 412:249-252 [DOI] [PubMed] [Google Scholar]
- 21.Smith RK, Zunino L, Webbon PM, Heinegård D: The distribution of cartilage oligomeric matrix protein (COMP) in tendon and its variation with tendon site, age, and load. Matrix Biol 1997, 16:255-271 [DOI] [PubMed] [Google Scholar]
- 22.Dodge GR, Hawkins D, Boesler E, Sakai L, Jimenez SA: Production of cartilage oligomeric matrix protein (COMP) by cultured human dermal and synovial fibroblasts. Osteoarthritis Cartilage 1998, 6:435-440 [DOI] [PubMed] [Google Scholar]
- 23.Cooper RR, Ponsetti IV, Maynard JA: Pseudoachondroplastic dwarfism: a rough-surfaced endoplasmic recticulum storge disorder. J Bone Joint Surg Am 1973, 55:475-484 [PubMed] [Google Scholar]
- 24.Pedrini-Mille A, Maynard JA, Pedrini VA: Pseudoachondroplasia: biochemical and histochemical studies of cartilage. J Bone Joint Surg Am 1984, 66:1408-1414 [PubMed] [Google Scholar]
- 25.Stanescu R, Stanescu V, Muriel MP, Maroteaux P: Multiple epiphyseal dysplasia, Fairbank type: morphogic and biochemical study of cartilage. Am J Med Genet 1993, 45:501-507 [DOI] [PubMed] [Google Scholar]
- 26.Rimoin DL, Rasmussen IM, Briggs MD, Roughley PJ, Gruber HE, Warman ML, Olsen BR, Hsia YE, Yuen J, Reinker K, Garber AP, Grover J, Lachman RS, Cohn DH: A large family with features of pseudoachondroplasia and multiple epiphyseal dysplasia: exclusion of seven candidate gene loci that encode proteins of the cartilage extracellular matrix. Hum Genet 1994, 93:236-242 [DOI] [PubMed] [Google Scholar]
- 27.Maddox BK, Keene DR, Sakai LY, Charbonneau NL, Morris NP, Ridgway CC, Boswell BA, Sussman MD, Horton WA, Bächinger HP, Hecht JT: The fate of cartilage oligomeric matrix protein is determined by cell type in the case of a novel mutation in pseudoachondroplasia. J Biol Chem 1997, 272:30993-30997 [DOI] [PubMed] [Google Scholar]
- 28.Delot E, Brodie SG, King LM, Wilcox WR, Cohn DH: Physiological and pathological secretion of cartilage oligomeric matrix protein by cells in culture. J Biol Chem 1998, 273:26692-26697 [DOI] [PubMed] [Google Scholar]
- 29.Vranka J, Mokashi A, Keene DR, Tufa Sara, Corson G, Sussman M, Horton WA, Maddox K, Sakai L, Bächinger HP: Selective intracellular retention of extracellular matrix proteins and chaperones associated with pseudoachondroplasia. Matrix Biol 2001, 20:439-450 [DOI] [PubMed] [Google Scholar]
- 30.Hecht JT, Deere M, Putnam E, Cole W, Vertel B, Chen H, Lawler J: Characterization of cartilage oligomeric matrix protein (COMP) in human normal and pseudoachondroplasia musculoskeletal tissues. Matrix Biol 1998, 17:269-278 [DOI] [PubMed] [Google Scholar]
- 31.Li LJ, Li X, Ferrario A, Rucker N, Liu ES, Wong S, Gomer CJ, Lee AS: Establishment of a Chinese hamster ovary cell line that expresses grp78 antisense transcripts and suppresses A23187 induction of both GRP78 and GRP94. J Cell Physiol 1992, 153:575-582 [DOI] [PubMed] [Google Scholar]
- 32.Chatterjee S, Cheng MF, Berger SJ, Berger NA: Induction of M(r) 78,000 glucose-regulated stress protein in poly(adenosine diphosphate-ribose) polymerase- and nicotinamide adenine dinucleotide-deficient V79 cell lines and its relation to resistance to the topoisomerase II inhibitor etoposide. Cancer Res 1994, 54:4405-4411 [PubMed] [Google Scholar]
- 33.Perez-Sala D, Mollinedo F: Inhibition of N-linked glycosylation induces early apoptosis in human promyelocytic HL-60 cells. J Cell Physiol 1995, 163:523-531 [DOI] [PubMed] [Google Scholar]
- 34.Shao RG, Shimizu T, Pommier Y: Brefeldin A is a potent inducer of apoptosis in human cancer cells independently of p53. Exp Cell Res 1996, 227:190-196 [DOI] [PubMed] [Google Scholar]
- 35.McCormick TS, McColl KS, Distelhorst CW: Mouse lymphoma cells destined to undergo apoptosis in response to thapsigargin treatment fail to generate a calcium-mediated grp78/grp94 stress response. J Biol Chem 1997, 272:6087-6092 [DOI] [PubMed] [Google Scholar]
- 36.Guo H, Tittle TV, Allen H, Maziarz RT: Brefeldin A-mediated apoptosis requires the activation of caspases and is inhibited by Bcl-2. Exp Cell Res 1998, 245:57-68 [DOI] [PubMed] [Google Scholar]
- 37.Liu H, Miller E, van de Water B, Stevens JL: Endoplasmic reticulum stress proteins block oxidant-induced Ca2+ increases and cell death. J Biol Chem 1998, 273:12858-12862 [DOI] [PubMed] [Google Scholar]
- 38.Lee J, Bruce-Keller AJ, Kruman Y, Chan SL, Mattson MP: 2-Deoxy-D-glucose protects hippocampal neurons against excitotoxic and oxidative injury: evidence for the involvement of stress proteins. J Neurosci Res 1999, 57:48-61 [DOI] [PubMed] [Google Scholar]
- 39.Yu Z, Luo H, Fu W, Mattson MP: The endoplasmic reticulum stress-responsive protein GRP78 protects neurons against excitotoxicity and apoptosis: suppression of oxidative stress and stabilization of calcium homeostasis. Exp Neurol 1999, 155:302-314 [DOI] [PubMed] [Google Scholar]
- 40.Fang C, Carison CS, Leslie MP, Tulli H, Stolerman E, Perris R, Ni L, DiCesare PE: Molecular cloning, sequencing, and tissue and developmental expression of mouse cartilage oligomeric matrix protein (COMP). J Orthop Res 2000, 18:593-603 [DOI] [PubMed] [Google Scholar]
- 41.Vertel BM, Hitti Y: Biosynthetic precursors of cartilage chondroitin sulfate proteoglycan. Coll Relat Res 1987, 7:57-75 [DOI] [PubMed] [Google Scholar]
- 42.Kim PS, Arvan P: Endocrinopathies in the family of endoplasmic reticulum (ER) storage diseases: disorders of protein trafficking and the role of ER molecular chaperones. Endocr Rev 1998, 19:173-202 [DOI] [PubMed] [Google Scholar]
- 43.Kaufman RJ: Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev 1999, 13:1211-1233 [DOI] [PubMed] [Google Scholar]
- 44.de Haro C, Mendez R, Santoyo J: The eIF-2α kinases and the control of protein synthesis. EMBO J 1996, 10:1378-1387 [DOI] [PubMed] [Google Scholar]
- 45.Prostko CR, Dholakia JN, Brostrom MA, Brostrom CO: Activation of the double-stranded RNA-regulated protein kinase by depletion of endoplasmic reticular calcium stores. J Biol Chem 1995, 270:6211-6215 [DOI] [PubMed] [Google Scholar]
- 46.Hecht JT, Hayes E, Snuggs M, Decker G, Montufar-Solis D, Doege K, Mwalle F, Poole R, Stevens J, Duk PJ: Calreticulin, PDI, Grp94, and BiP chaperone protein are associated with retained COMP in pseudoachondroplasia chondrocytes. Matrix Biol 2001, 20:251-262 [DOI] [PubMed] [Google Scholar]
- 47.Guo Y, Bozic D, Malashkevich VN, Kammerer RA, Schulthess T, Engel J: All-trans retinol, vitamin D, and other hydrophobic compounds bind in the axial pore of the five-stranded coiled-coil domain of cartilage oligomeric matrix protein. EMBO J 1998, 17:5265-5272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Holden P, Meadows RS, Chapman KL, Grant ME, Kadler KE, Briggs MD: Cartilage oligomeric matrix protein interacts with type IX collagen, and disruptions to these interactions identify a pathogenetic mechanism in a bone dysplasia family. J Biol Chem 2001, 276:6046-6055 [DOI] [PubMed] [Google Scholar]
- 49.Svensson L, Aszodi A, Heinegård D, Hunziker EB, Reinholt FP, Fässler R, Oldberg Å: Cartilage oligomeric matrix protein-deficient mice have normal skeletal development. Mol Cell Biol 2002, 22:4366-4371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sambrook JF: The involvement of calcium in transport of secretory proteins from the endoplasmic reticulum. Cell 1990, 61:197-199 [DOI] [PubMed] [Google Scholar]
- 51.Rapoport TA: Transport of proteins across the endoplasmic reticulum membrane. Science 1992, 258:931-936 [DOI] [PubMed] [Google Scholar]
- 52.Buckley BJ, Whorton AR: Tunicamycin increases intracellular calcium levels in bovine aortic endothelial cells. Am J Physiol 1997, 273:1298-1305 [DOI] [PubMed] [Google Scholar]
- 53.Sidrauski C, Chapman R, Walter P: The unfolded protein response: an intracellular signalling pathway with many surprising features. Trends Cell Biol 1998, 8:245-249 [DOI] [PubMed] [Google Scholar]
- 54.Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, Yuan J: Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-β. Nature 2000, 403:98-103 [DOI] [PubMed] [Google Scholar]
- 55.Hecht JT, Montufar-Solis D, Decker G, Lawler J, Daniels K, Duke J: Retention of cartilage oligomeric matrix protein (COMP) and cell death in redifferentiated pseudoachondroplasia chondrocytes. Matrix Biol 1998, 17:625-633 [DOI] [PubMed] [Google Scholar]
- 56.Stanescu V, Stanescu R, Maroteaux P: Pathogenesis of pseudoachondroplasia and diastrophic dysplasia. Prog Clin Biol Res 1982, 104:385-394 [PubMed] [Google Scholar]
- 57.Stöss H, Pesch HJ, Spranger J: Different morphologic findings and genetic heterogeneity in pseudoachondroplasia: light and electron microscopic observation in iliac crest bioptic material. Prog Clin Biol Res 1982, 104:379-383 [PubMed] [Google Scholar]