

Abstract
Interstitial myofibroblasts are α-smooth muscle actin-positive cells that play a crucial role in the accumulation of excess extracellular matrix during renal interstitial fibrogenesis. Despite their importance in the pathogenesis of renal fibrosis, relatively little is known about the regulators and the mechanism controlling the activation of renal interstitial myofibroblasts in disease conditions. Here, we show that hepatocyte growth factor (HGF) acts as a potent inhibitor of the transforming growth factor (TGF)-β1-mediated myofibroblastic activation from normal rat renal interstitial fibroblasts (NRK-49F). Simultaneous incubation of HGF abolished TGF-β1-induced de novo α-smooth muscle actin expression, F-actin reorganization, and interstitial collagen I overproduction in a dose-dependent manner. To decipher the mechanism underlying HGF antagonizing TGF-β1’s action, we examined the effects of HGF on TGF-β1-mediated Smad signaling. HGF neither inhibited Smad-2/3 phosphorylation and their association with Smad-4 induced by TGF-β1, nor significantly affected inhibitory Smad-6 and -7 expression and cellular abundance of Smad transcriptional co-repressors in NRK-49F cells. However, pretreatment with HGF markedly attenuated activated Smad-2/3 nuclear translocation and accumulation. This action of HGF was apparently dependent on HGF-mediated extracellular signal-regulated kinase-1 and -2 (Erk-1/2) phosphorylation and activation. Inhibition of Erk-1/2 activation by Mek kinase inhibitor PD98059 restored TGF-β1-mediated Smad-2/3 nuclear accumulation and myofibroblast activation. In vivo, HGF selectively blocked Smad-2/3 nuclear accumulation in renal interstitial cells in the fibrotic kidneys induced by unilateral ureteral obstruction. Therefore, HGF suppresses TGF-β1-mediated renal interstitial myofibroblastic activation; and this action of HGF is likely related to a mitogen-activated protein kinase-dependent blockade of Smad nuclear translocation.
Interstitial myofibroblasts, characterized by α-smooth muscle actin (α-SMA) expression, are a unique cell population that is primarily responsible for the overproduction and deposition of interstitial matrix as seen in the fibrotic kidneys. 1-3 In response to injurious stimuli, appropriate activation of fibroblasts is necessary and perhaps important for tissue repair and wound healing. However, dysregulation of myofibroblastic activation after persistent, chronic renal injury could instigate excess accumulation of extracellular matrix that ultimately leads to interstitial scarring and end-stage renal failure. 2,4-6 Because interstitial myofibroblasts are virtually absent in normal kidney, their activation is possibly one of the decisive events in the pathogenesis of progressive renal fibrotic diseases. 2,7,8 In this regard, myofibroblastic activation is often considered as a reliable predictor for the progression and prognosis of chronic renal insufficiency in a variety of experimental animal models and in patients. 5,9,10
Although it is widely accepted that myofibroblastic activation plays a critical role in the progression of renal fibrotic lesions and dysfunctions, relatively little is known about the factors and the underlying mechanisms that regulate its activation in the diseased kidneys. Several lines of evidence suggest a critical role of peptide growth factors in the regulation of myofibroblastic activation. 3,11-13 For instance, transforming growth factor (TGF)-β1 has been demonstrated to activate fibroblast cells to become myofibroblasts and to produce large amount of matrix proteins. 14-17 This observation is consistent with in vivo studies showing that TGF-β1 is implicated in tissue fibrogenesis in a variety of organs after chronic injury. 18,19 Blockage of TGF-β1 signaling, through various manipulations, prevents myofibroblastic activation and consequently mitigates renal interstitial fibrosis. 20-22 However, despite TGF-β1 being recognized as a positive regulator of myofibroblasts, factors with negative regulation of myofibroblastic activation remain poorly defined.
Earlier studies in our laboratory have identified hepatocyte growth factor (HGF) as a potent inhibitor of myofibroblastic transdifferentiation from tubular epithelial cells triggered by TGF-β1. 11,23 In accordance with this, administration of HGF protein or its gene prevents renal interstitial fibrosis in numerous animal models of renal diseases. 24-27 These observations suggest that HGF is an anti-fibrotic factor that counteracts profibrotic TGF-β1 actions in tubular epithelial cells, in which HGF receptor, c-met, is abundantly expressed. 28,29 Nonetheless, it remains unknown whether HGF also suppresses myofibroblastic activation from interstitial fibroblast cells; and if so, what mechanism accounts for HGF’s action.
In the present study, we demonstrate that HGF markedly blocks TGF-β1-mediated myofibroblastic activation of renal interstitial fibroblasts. The action of HGF is likely related to the blockade of nuclear translocation and accumulation of activated Smad-2/3 protein triggered by TGF-β1. Our results indicate that HGF specifically antagonizes the profibrotic action of TGF-β1 in interstitial fibroblasts in a mitogen-activated protein kinase-dependent manner.
Materials and Methods
Antibodies and Reagents
The mouse monoclonal anti-α-SMA (clone 1A4), anti-vimentin (clone V9), and rabbit polyclonal anti-extracellular signal-regulated kinase-1 and -2 (Erk-1/2) were purchased from Sigma (St. Louis, MO). The phospho-specific Erk-1/2 antibody was obtained from Cell Signaling Technology, Inc. (Beverly, MA). The phospho-specific Smad-2 antibody as well as total Smad-2 antibody was purchased from Upstate (Charlottesville, VA). Rabbit anti-phosphoserine antibody was obtained from Zymed Laboratories Inc. (South San Francisco, CA). The goat polyclonal anti-Type I collagen antibody was obtained from Southern Biotechnology Associates, Inc. (Birmingham, AL). The anti-Smad-7 (sc-7004), anti-Smad-6 (sc-13048), anti-Smad-4 (sc-7066), anti-Smad-2/3 (sc-6032), anti-Sp1 (sc-420), anti-c-Ski (sc-9140), anti-SnoN (sc-9595), anti-TGIF (sc-9826), and anti-actin (sc-1616) antibodies were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Affinity-purified secondary antibodies were purchased from Jackson ImmunoResearch Laboratories, Inc. (West Grove, PA). Recombinant human TGF-β1, epidermal growth factor (EGF), insulin-like growth factor-I, platelet-derived growth factor, and monocyte chemotactic protein-1 were purchased from R & D Systems (Minneapolis, MN). Recombinant human HGF protein was provided by Genentech Inc. (South San Francisco, CA). PD98059 (Mek inhibitor), myristoylated protein kinase A inhibitor (PKAI) and Ro-31-8220 (PKC inhibitor) were purchased from Calbiochem (La Jolla, CA). Cell culture media, fetal bovine serum, and supplements were obtained from Life Technologies, Inc. (Grand Island, NY). All other chemicals were of analytic grade and were obtained from Sigma or Fisher {Pittsburgh, PA) unless otherwise indicated.
Cell Culture and Treatment
Normal rat kidney interstitial fibroblast cells (NRK-49F) and human embryonic kidney 293 (HEK-293) cells were obtained from American Type Culture Collection (Manassas, VA). Cells were maintained in Dulbecco’s modified Eagle’s medium/F12 medium supplemented with 10% fetal bovine serum. The NRK-49F cells were seeded on six-well culture plates to ∼60 to 70% confluence in the complete medium containing 10% fetal bovine serum for 16 hours, and then changed to serum-free medium after washing twice with medium. Recombinant human TGF-β1 was added to the culture at a final concentration of 2 ng per ml except as otherwise indicated. For dose-dependent studies, TGF-β1 was used at the concentrations of 0.01, 0.05, 0.1, 0.5, 1, and 5 ng/ml, respectively. Recombinant human HGF was also added at the same time at the concentration as indicated. For some experiments, the cells were incubated with vehicle (phosphate-buffered saline, PBS) or 0.2 nmol/L of various cytokines. The cells were typically incubated for 48 hours after addition of cytokines except when indicated otherwise, before harvesting and subjecting to Western blot or immunofluorescence staining, respectively.
Western Blot Analysis
NRK-49F cells and cytokine-treated cells were lysed with sodium dodecyl sulfate (SDS) sample buffer (62.5 mmol/L Tris-HCl, pH 6.8, 2% SDS, 10% glycerol, 50 mmol/L dithiothreitol, and 0.1% bromphenol blue). Samples were heated at 100°C for ∼5 to 10 minutes before loading and separated on precasted 10% or 5% SDS-polyacrylamide gels (Bio-Rad, Hercules, CA). The proteins were electrotransferred to a nitrocellulose membrane (Amersham, Arlington Heights, IL) in transfer buffer containing 48 mmol/L Tris-HCl, 39 mmol/L glycine, 0.037% SDS, and 20% methanol at 4°C for 1 hour. Nonspecific binding to the membrane was blocked for 1 hour at room temperature with 5% Carnation nonfat milk in TBS buffer (20 mmol/L Tris-HCl, 150 mmol/L NaCl, and 0.1% Tween 20). The membranes were then incubated for 16 hours at 4°C with various primary antibodies in blocking buffer containing 5% milk at the dilutions specified by the manufacturers. After extensive washing in TBS buffer, the membranes were then incubated with horseradish peroxidase-conjugated secondary antibody (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA) for 1 hour at room temperature in 5% nonfat milk dissolved in TBS. Membranes were then washed with TBS buffer and the signals were visualized using the enhanced chemiluminescence system (ECL, Amersham), as described previously. 23
Immunofluorescence Staining
Indirect immunofluorescence staining was performed using an established procedure. 23,30 Briefly, control or cytokine-treated NRK-49F cells cultured on coverslips were washed with cold PBS twice, and fixed with cold methanol:acetone (1:1) for 10 minutes at −20°C. After extensive washing with PBS containing 0.5% bovine serum albumin for three times, the cells were blocked with 20% normal donkey serum in PBS buffer for 30 minutes at room temperature, and then incubated with the specific primary antibodies described above. For immunostaining of Smad-2/3 protein, NRK-49F cells after various treatments were fixed with 4% paraformaldehyde at 4°C for 20 minutes, followed by permeabilization with PBS containing 0.2% Triton X-100 at room temperature for 30 minutes. The slides were blocked with 1% horse serum and 0.15% glycine in PBS for 30 minutes at room temperature. The primary anti-Smad-2/3 antibody was added at the dilution of 1:500, followed by incubation at 4°C for 16 hours. To visualize the primary antibodies, cells were stained with fluorescein isothiocyanate-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories). For visualizing F-actin, cells were stained with tetramethyl-rhodamine isothiocyanate-conjugated phalloidin (Sigma). For some samples, cells were double-stained with 4′,6-diamidino-2-phenylindole, HCl, to visualize the nuclei. Stained cells were mounted with anti-fade mounting medium (Vector Laboratories, Burlingame, CA) and viewed with a Nikon Eclipse E600 Epi-fluorescence microscope equipped with a digital camera (Nikon, Melville, NY).
Nuclear Protein Extraction
NRK-49F fibroblast cells were subjected to various treatments with different cytokines for 30 minutes except where otherwise indicated. For HGF blockage of TGF-β1 signaling, cells were treated with 40 ng/ml of HGF 30 minutes before addition of 2 ng/ml of TGF-β1. Likewise, for blocking HGF-triggered Erk1/2 activation, cells were pretreated with 10 μmol/L of PD98059 30 minutes before HGF treatment. Cell nuclei were isolated by procedures described previously. 31 Briefly, cells were washed with cold PBS and scraped off the plate with a rubber policeman. Cells were collected by centrifugation and cell pellets were resuspended in 4 vol of buffer A containing 20 mmol/L Hepes, pH 7.9, 0.5 mol/L sucrose, 1.5 mmol/L NaCl, 60 mmol/L KCl, 0.15 mmol/L spermidine, 0.5 mmol/L spermine, 0.5 mmol/L ethylenediaminetetraacetic acid, 1 mmol/L dithiothreitol, 0.2 mmol/L phenylmethyl sulfonyl fluoride, plus 5 μg/ml of leupeptin, soybean trypsin inhibitor, antipain, and chymostatin. An equal volume of buffer A containing 0.6% Nonidet P-40 was added with gentle mixing to lyse the cells. Immediately after lysis, the solution was diluted with 8 vol of buffer A, and the integrity of cell nuclei were examined under microscope. After being collected by centrifugation at 5000 × g for 30 minutes at 4°C, the nuclei were lysed with SDS sample buffer and subjected to Western blot analysis as described above.
Immunoprecipitation
Immunoprecipitation was performed essentially according to the procedures described previously. 32-34 NRK-49F cells grown on a 100-mm plate were lysed on ice in 1 ml of RIPA buffer containing 1× PBS, 1% Nonidet P-40, 0.1% SDS, 10 μg/ml phenylmethyl sulfonyl fluoride, 1 mmol/L sodium orthovanadate, and 1% protease inhibitors cocktail (Sigma). Whole cell lysates were clarified by centrifugation at 12,000 × g for 10 minutes at 4°C and the supernatants were transferred into a fresh tube. To preclear cell lysates, 0.25 μg of normal rabbit IgG and 20 μl of protein A/G Plus-Agarose (Santa Cruz) were added into 1 ml of whole cell lysates. After incubation for 1 hour at 4°C, supernatants were collected by centrifugation at 1000 × g for 5 minutes at 4°C. Lysates were immunoprecipitated overnight at 4°C with 1 μg of anti-Smad-2/3, followed by precipitation with 20 μl of protein A/G Plus-Agarose for 3 hours at 4°C. After four washes with RIPA buffer, the immunoprecipitates were boiled for 5 minutes in SDS sample buffer. The resulting precipitated complexes were separated on SDS-polyacrylamide gels and blotted with anti-Smad-4, anti-phosphoserine, and anti-Smad2/3, respectively, as described above.
Animal Model
To study the effects of HGF on Smad nuclear accumulation in renal interstitial cells in vivo, indirect immunofluorescence staining was performed on the cryosections of the fibrotic kidneys induced by unilateral ureteral obstruction (UUO), as described previously. 27 Male CD-1 mice weighing at ∼18 to 22 g were purchased from Harlan Sprague Dawley (Indianapolis, IN). UUO was performed using an established procedure. 27 The recombinant human HGF expression plasmid (pCMV-HGF) and the empty expression plasmid vector (pcDNA3) were administrated into mice by rapid injection of naked plasmid solution through the tail vein, as previously described. 27 Mice were injected with plasmids before (day −1) and after (day 3) UUO, respectively. Groups of mice (n = 5) were sacrificed at 7 days after UUO and the kidneys were removed. Kidney cryosections were immunostained for Smad-2/3 as described above. For distinguishing the tubular versus interstitial compartments, cryosections were also stained with fluorescein-conjugated lectin from Tetragonolobus purpureas (Sigma) for localizing the proximal tubules. Cell nuclei were visualized by staining with 4′,6-diamidino-2-phenylindole. Stained sections were mounted with anti-fade mounting medium and viewed with a Nikon Eclipse E600 Epi-fluorescence microscope. The Smad-2/3-positive nuclei were counted in the interstitial regions and expressed as percentages of Smad-positive nuclei per total nuclei, which were determined on five nonoverlapping cortical fields per mouse, five mice per group.
Statistical Analysis
All data examined were expressed as mean ± SE. Statistical analysis of the data were performed using SigmaStat software (Jandel Scientific, San Rafael CA). Comparison between groups was made using one-way analysis of variance followed by Student-Newman-Keuls test. A P value of less than 0.05 was considered to be statistically significant.
Results
HGF Abrogates TGF-β1-Induced Myofibroblastic Activation
Because α-SMA expression is the hallmark for myofibroblasts, we investigated the myofibroblastic activation from renal interstitial fibroblast cells by examining the α-SMA protein expression after incubation with various agents. As shown in Figure 1 ▶ , among numerous agents tested, only TGF-β1 markedly induced de novo α-SMA expression in renal interstitial fibroblast NRK-49F cells. Other cytokines at the same molar concentration (0.2 nmol/L) failed to induce α-SMA expression after 2 days of incubation (Figure 1A) ▶ . The induction of α-SMA by TGF-β1 in renal interstitial fibroblasts occurred in a dose-dependent manner. Appreciable induction of α-SMA expression was observed when TGF-β1 was added at a concentration as low as 0.5 ng/ml (Figure 1B) ▶ . These results suggest that TGF-β1 is a potent profibrogenic cytokine capable of inducing myofibroblastic activation from renal interstitial fibroblasts in vitro.
Figure 1.

TGF-β1 induces de novo expression of α-SMA in renal interstitial fibroblast NRK-49F cells. A: NRK-49F cells were incubated for 2 days without (control) or with the same molar concentration (0.2 nmol/L) of various cytokines. The cell lysates were immunoblotted with specific antibodies against α-SMA and actin, respectively. B: Dose-dependent induction of α-SMA expression by TGF-β1 in NRK-49F cells. The cells were incubated with TGF-β1 at the specific concentrations as indicated for 2 days. The α-SMA expression was detected by Western blot analysis.
Because HGF has been demonstrated to possess anti-fibrotic activities in vivo, 11,23 we reasoned whether HGF blocks myofibroblastic activation from renal interstitial fibroblasts. As shown in Figure 2 ▶ , incubation of NRK-49F cells with HGF alone did not affect α-SMA expression. However, simultaneous incubation of NRK-49F cells with HGF dramatically repressed TGF-β1-initiated α-SMA expression in a dose-dependent manner (Figure 2A) ▶ . At the concentration of 40 ng/ml (∼0.42 nmol/L), HGF almost completely abolished the α-SMA expression induced by 2 ng/ml (∼0.16 nmol/L) of TGF-β1. This result was independently confirmed by an indirect immunofluorescence staining for α-SMA in NRK-49F cells. As shown in Figure 2C ▶ , TGF-β1 induced de novo expression of α-SMA that was assembled into abundant α-SMA-positive microfilament fibers in the cytoplasm of NRK-49F cells. Simultaneous incubation with HGF primarily abolished the α-SMA staining induced by TGF-β1 (Figure 2E) ▶ . Figure 2F ▶ shows the percentages of α-SMA-positive cells after various treatments. Of note, the difference in α-SMA expression appears not because of an altered cell density after different treatments, because neither TGF-β1 nor HGF significantly affected NRK-49F cell growth rate (Figure 2G) ▶ . Collectively, these results indicate that HGF elicits its anti-fibrotic actions by suppressing myofibroblastic activation of renal interstitial fibroblasts initiated by profibrotic cytokine TGF-β1.
Figure 2.

HGF abrogates the expression of α-SMA in renal interstitial fibroblast NRK-49F cells. A: Western blot analysis demonstrates that HGF blocked α-SMA expression induced by TGF-β1 in a dose-dependent manner in NRK-49F cells. Cells were incubated with a fixed amount of TGF-β1 (2 ng/ml) and increasing amounts of HGF as indicated for 2 days. The cell lysates were probed with antibodies against α-SMA and actin, respectively. B to E: Representative photographs of the α-SMA visualized by indirect immunofluorescence staining in NRK-49F cells after various treatments for 2 days. B: Control; C: 2 ng/ml of TGF-β1; D: 40 ng/ml of HGF; E: TGF-β1 plus HGF. C: The α-SMA-positive microfilaments were evident in TGF-β1-treated cells. F: Graphic presentation of the percentage of α-SMA-positive cells in various groups. *, P < 0.01 versus control; **, P < 0.05 versus TGF-β1 group (n = 3). G: Neither TGF-β1 nor HGF significantly affected renal interstitial fibroblast cell growth. NRK-49F cells were treated with 2 ng/ml of TGF-β1, 40 ng/ml of HGF, or both in serum-free medium for 3 days. Cell numbers were counted and presented as means ± SE. No statistically significant difference in cell numbers was found in various groups (n = 3). Scale bar, 10 μm.
HGF Blocks Interstitial Matrix Accumulation and Deposition
Myofibroblastic activation of renal interstitial fibroblasts was accompanied with significant morphological transformations. After incubation with TGF-β1 for 2 days, NRK-49F cells became elongated in shape and displayed activated myofibroblastic appearances under phase-contrast microscope (Figure 3B) ▶ . Incubation of NRK-49F cells with HGF blocked this morphological transformation (Figure 3D) ▶ . Because actin cytoskeleton provides the architectural basis for cell morphology, we examined the alterations in actin structure during myofibroblastic activation induced by TGF-β1. As illustrated in Figure 3, E to H ▶ , TGF-β1 induced actin reorganization in fibroblast NRK-49F cells. The activated cells displayed abundant stress fibers assembled by actin (Figure 3F) ▶ . Consistently, HGF primarily blocked actin reorganization in NRK-49F cells triggered by TGF-β1 (Figure 3H) ▶ . The expression of vimentin was also studied by Western blot analysis and immunostaining in NRK-49F cells after various treatments. Western blot showed that total vimentin protein levels were not significantly altered after incubation of NRK-49F cells with TGF-β1, HGF, or both (data not shown). However, TGF-β1 induced architectural reorganization of cytoplasmic vimentin in NRK-49F cells to form long intermediate filaments, resembling the overall morphology of activated myofibroblasts (Figure 3J) ▶ . HGF also blocked the alteration in vimentin architecture induced by TGF-β1 (Figure 3L) ▶ .
Figure 3.

HGF blocks TGF-β1-induced morphological transformation, F-actin, and vimentin reorganization in renal interstitial fibroblast NRK-49F cells. NRK-49F cells were incubated without (control) (A, E, I) or with 2 ng/ml of TGF-β1 (B, F, J), 40 ng/ml of HGF (C, G, K), or both (D, H, L) for 2 days. A to D: TGF-β1 induced morphological transformation of NRK-49F cells into a myofibroblastic appearance (B). HGF blocked this transformation (D). E to L: Representative micrographs of tetramethyl-rhodamine isothiocyanate-conjugated phalloidin staining (E–H) and vimentin staining (I–L) showing F-actin and vimentin reorganization in NRK-49F cells induced by TGF-β1 (F, J). HGF primarily abolished TGF-β1-induced actin and vimentin reorganization (H, L). Scale bar, 20 μm.
We next examined the expression and extracellular deposition of interstitial matrix components in activated myofibroblast cells. As shown in Figure 4 ▶ , NRK-49F cells at basal conditions expressed trivial amount of collagen I. However, after myofibroblastic activation by TGF-β1, the NRK-49F cells became activated, principal matrix-producing cells that expressed a striking amount of interstitial collagen I. Immunofluorescence staining revealed a remarkable amount of the assembled collagen I deposited in the extracellular compartment (Figure 4B) ▶ . HGF by itself did not affect collagen I expression in NRK-49F cells (Figure 4C) ▶ ; nevertheless, it completely blocked the expression of collagen I induced by TGF-β1 (Figure 4D) ▶ . Thus, on activation by TGF-β1, renal interstitial fibroblast cells are transformed into activated myofibroblasts that produce large amounts of interstitial matrix; and HGF completely blocks this process of myofibroblastic activation and its subsequent matrix accumulation and deposition.
Figure 4.

HGF suppresses TGF-β1-induced collagen I expression and its extracellular assembly in renal interstitial fibroblast NRK-49F cells. Immunofluorescence staining shows the distribution and abundance of collagen I in NRK-49F cells after various treatments. A: Control; B: 2 ng/ml of TGF-β1; C: 40 ng/ml of HGF; D: TGF-β1 plus HGF. B: TGF-β1 induced interstitial matrix component collagen I expression and its extracellular assembly. D: HGF abrogated TGF-β1-initiated collagen I expression and extracellular assembly. Scale bar, 10 μm.
HGF Does Not Inhibit Smad-2/3 Phosphorylation and Their Association with Smad-4, but Blocks Activated Smad’s Nuclear Translocation
To elucidate the mechanism by which HGF blocks myofibroblastic activation and interstitial matrix accumulation induced by TGF-β1, we investigated the potential inhibition of TGF-β1 signaling by HGF in interstitial fibroblast cells. It is well documented that TGF-β1, on binding to its receptors, initiates phosphorylation of intermediate signal molecules Smads. 35,36 Thus, we examined the phosphorylation and activation of Smad-2 by TGF-β1 in NRK-49F cells by Western blot using phospho-specific Smad-2 antibody. Figure 5A ▶ shows the kinetics of Smad-2 activation after incubation with TGF-β1 in NRK-49F cells. Smad-2 phosphorylation was detected as early as 10 minutes, reached the peak at 30 minutes, and sustained to 3 hours after addition of TGF-β1 (Figure 5A) ▶ . After 6 hours with TGF-β1 incubation, phosphorylated Smad-2 levels returned toward baseline. To investigate whether HGF interferes with Smad-2 phosphorylation and activation triggered by TGF-β1, NRK-49F cells were pretreated with HGF for 30 minutes before incubation with TGF-β1. As shown in Figure 5B ▶ , HGF did not block TGF-β1-initiated Smad-2 phosphorylation in renal interstitial fibroblast NRK-49F cells.
Figure 5.
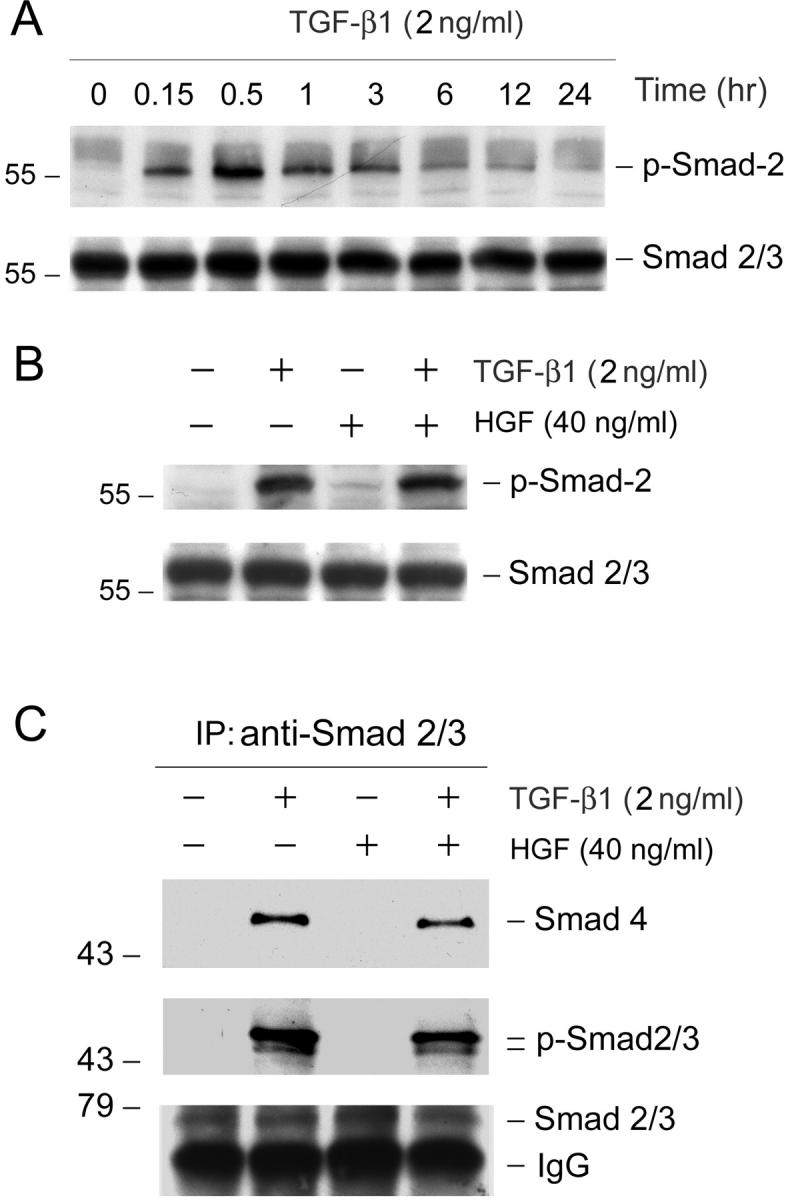
HGF neither inhibits TGF-β1-initiated Smad-2/3 phosphorylation nor affects their association of Smad-4 in renal interstitial fibroblast NRK-49F cells. A: Kinetics of Smad-2 phosphorylation and activation after TGF-β1 incubation in NRK-49F cells. Cells were treated with 2 ng/ml of TGF-β1 for various periods of time as indicated. The cell lysates were probed with antibodies against phospho-specific Smad-2 and total Smad-2, respectively. B: HGF did not inhibit Smad-2 phosphorylation triggered by TGF-β1. Cells were pretreated with 40 ng/ml of HGF for 30 minutes and followed by incubation with 2 ng/ml of TGF-β1 for an additional 30 minutes. Activation of Smad-2 was assessed by using phospho-specific Smad-2 antibody. C: HGF does not affect Smad-2/3 phosphorylation and their association with Smad-4 induced by TGF-β1. NRK-49F cells were pretreated with 40 ng/ml of HGF for 30 minutes and followed by incubation with 2 ng/ml of TGF-β1 for an additional 30 minutes. Cell lysates were immunoprecipitated with antibody against Smad-2/3. The precipitated complexes were immunoblotted with antibodies against Smad-4, phosphoserine, and Smad-2/3, respectively.
We further investigated the phosphorylation of Smad-2 and Smad-3 and their association with Smad-4 in NRK-49F cells after TGF-β1 stimulation by co-immunoprecipitation. As shown in Figure 5C ▶ , both Smad-2 and -3 were phosphorylated after TGF-β1 treatment, as demonstrated by the presence of phosphoserine-specific bands in the complexes immunoprecipitated by anti-Smad-2/3 antibody. Of note, the predominant form of receptor-regulated Smads (R-Smads) in NRK-49F cells was Smad-2 (Figure 5C) ▶ . Co-immunoprecipitation also revealed that the phosphorylated Smad-2/3 were physically associated with Smad-4 after TGF-β1 stimulation in NRK-49F cells (Figure 5C) ▶ . However, pretreatment of HGF did not significantly affect Smad-2/3 phosphorylation and their association with Smad-4 (Figure 5C) ▶ .
Once phosphorylated and bound to Smad-4, Smad-2/3 have to translocate into the nuclei to control the transcription of their targeted genes. 35 We examined whether HGF interferes with this nuclear translocation process of activated Smad-2/3 in renal interstitial fibroblast cells. To this end, cell nuclei were isolated after various treatments and the nuclear accumulation of phosphorylated Smad-2 was analyzed by Western blot. As demonstrated in Figure 6 ▶ , abundant phosphorylated Smad-2 accumulated in cell nuclei after incubating with TGF-β1 for 30 minutes in NRK-49F cells. HGF treatment alone did not induce nuclear accumulation of Smad-2; however, pretreatment with HGF markedly attenuated TGF-β1-mediated nuclear translocation of activated Smad-2 (Figure 6, A and B) ▶ . Consistent with the biochemical analysis, immunostaining for Smad-2/3 proteins also revealed that TGF-β1 induced Smad-2/3 nuclear translocation in NRK-49F cells after incubation for 30 minutes (Figure 6D) ▶ and that pretreatment with HGF prevented TGF-β1-initiated Smad-2/3 nuclear translocation and accumulation (Figure 6F) ▶ . These results indicate that although HGF does not inhibit TGF-β1-initiated Smad-2/3 phosphorylation and their association with Smad-4, it blocks Smad-2/3-mediated gene transcription primarily by preventing activated Smad-2/3 nuclear translocation and accumulation.
Figure 6.

HGF blocks activated Smad-2 nuclear translocation and accumulation in renal interstitial fibroblast NRK-49F cells. A: Representative Western blot demonstrates that HGF attenuated nuclear accumulation of activated Smad-2 triggered by TGF-β1. Cell nuclei were isolated from NRK-49F cells after various treatments as indicated and nuclear accumulation of phospho-Smad-2 was examined by Western blot. The samples were also probed with ubiquitous transcription factor Sp1 for normalization of nuclear proteins. B: Graphic presentation of the relative abundance of nuclear phospho-Smad-2 protein normalized to Sp1 after various treatments. Data are presented as mean ± SE of three independent experiments. *, P < 0.01 versus control. †, P < 0.01 versus TGF-β1. C to F: Representative micrographs show the cellular localization of Smad-2/3 by indirect immunofluorescence staining in NRK-49F cells after various treatments for 45 minutes. C: Control; D: 2 ng/ml of TGF-β1; E: 40 ng/ml of HGF; F: TGF-β1 plus HGF. D: Smad-2/3 nuclear accumulation was clearly evident in TGF-β1-treated cells. F: Pretreatment of NRK-49F cells with HGF for 30 minutes prevented Smad-2/3 nuclear translocation initiated by TGF-β1. Scale bar, 10 μm.
HGF Blockage of Smad-2/3 Nuclear Translocation Is Dependent on Erk-MAP Kinase Activation
To decipher the mechanism underlying HGF inhibition of interstitial myofibroblastic activation, we examined the signal pathway leading to the blockade of TGF-β1-initiated Smad-2/3 nuclear translocation in NRK-49F cells. As shown in Figure 7A ▶ , incubation of NRK-49F cells with HGF, but not TGF-β1, induced a dramatic activation of extracellular signal-regulated kinase-1 and -2 (Erk-1/2), members of mitogen-activated protein (MAP) kinase family, in NRK-49F cells. This activation was abolished by PD98059, a specific inhibitor of Erk-1/2 upstream kinase Mek1 (Figure 7B) ▶ , but not by other kinase inhibitors such as protein kinase A (PKA) or PKC inhibitors (data not shown). To demonstrate whether Erk-1/2 activation is responsible for blockade of Smad-2 nuclear translocation, we assessed the nuclear accumulation of phosphorylated Smad-2 after pretreatment of NRK-49F cells with PD98059 to block HGF-mediated Erk-1/2 signaling. As shown in Figure 7 ▶ , although HGF blocked TGF-β1-initiated Smad-2 nuclear translocation, pretreatment of NRK-49F cells with PD98059 abolished HGF’s action and restored the nuclear accumulation of the phosphorylated Smad-2 (Figure 7, C and G) ▶ . Of note, PD98059 by itself did not affect either basal or TGF-β1-induced Smad-2 nuclear accumulation (data not shown). Consistent with this result, inhibition of HGF-mediated Erk-1/2 signaling by PD98059 also primarily restored TGF-β1-induced α-SMA expression in renal interstitial fibroblast NRK-49F cells (Figure 7D) ▶ , suggesting that Erk-1/2 activation is required for HGF suppression of interstitial myofibroblastic activation.
Figure 7.

HGF blockade of Smad-2 nuclear translocation is dependent on mitogen-activated protein (MAP) kinase activation. A: HGF induced Erk-MAP kinase activation in renal interstitial fibroblast NRK-49F cells. Cells were treated with either 2 ng/ml of TGF-β1 or 40 ng/ml of HGF for various periods of time as indicated. Cell lysates were probed with antibodies against phospho-specific Erk-1/2 and total Erk1/2, respectively. B: Erk-1/2 phosphorylation triggered by HGF was completely abolished by preincubating with Mek1 inhibitor. Cells were treated with Mek1 inhibitor PD98059 (10 μmol/L) or vehicle (DMSO) for 30 minutes, followed by incubation with 40 ng/ml of HGF for an additional 30 minutes. C: Inhibition of Erk-1/2 phosphorylation restored phospho-Smad-2 nuclear translocation. NRK-49F cells were pretreated with PD98059 (10 μmol/L) for 30 minutes, followed by incubating with 2 ng/ml of TGF-β1, and 40 ng/ml of HGF for an additional 30 minutes. Cell nuclei were isolated from NRK-49F cells after various treatments as indicated and nuclear accumulation of activated Smad-2 was examined by using phospho-specific Smad-2 antibody. The samples were also probed with nuclear protein Sp1. D to G: Representative photographs show the cellular localization of Smad-2/3 in NRK-49F cells after various treatments for 30 minutes. D: Control; E: 2 ng/ml of TGF-β1; F: 2 ng/ml of TGF-β1 plus 40 ng/ml of HGF; G: 10 μmol/L PD98059 plus TGF-β1 and HGF. H: Inhibition of Erk-1/2 phosphorylation abolished HGF inhibition of α-SMA expression triggered by TGF-β1 in renal fibroblasts. The α-SMA expression was examined in NRK-49F cells after various treatments for 16 hours as indicated. Scale bar, 10 μm.
To further support this notion, we investigated the effects of Erk-1/2 activation elicited by other growth factors such as EGF on Smad’s nuclear accumulation and α-SMA expression. As presented in Figure 8 ▶ , EGF induced Erk-1/2 activation in renal interstitial fibroblast NRK-49F cells (Figure 8A) ▶ . Likewise, EGF also blocked the nuclear accumulation of activated Smad-2/3 triggered by TGF-β1 (Figure 8E) ▶ ; and inhibition of Erk-1/2 by PD98059 restored TGF-β1-induced Smad-2/3 nuclear translocation (Figure 8F) ▶ . Similar to HGF, EGF suppressed α-SMA expression and myofibroblastic activation of interstitial fibroblast NRK-49F cells (Figure 8G) ▶ .
Figure 8.

Erk-1/2 activation by EGF also blocks Smad-2 nuclear translocation and α-SMA expression induced by TGF-β1 in renal interstitial fibroblasts. A: EGF induced Erk-1/2 phosphorylation and activation in renal interstitial fibroblasts. NRK-49F cells were treated with 2 ng/ml of TGF-β1 and 40 ng/ml of EGF for 30 minutes and Erk-1/2 activation was detected by using phospho-specific Erk1/2 antibody. Pretreatment of NRK-49F cells with PD98059 (10 μmol/L) abolished EGF-induced Erk1/2 phosphorylation. B: EGF blocked Smad-2 nuclear translocation in an Erk-1/2-dependent manner. Pretreatment of NRK-49F cells with 10 μmol/L of PD98059 restored Smad-2 nuclear translocation. Cells were pretreated with 10 μmol/L of PD98059 for 30 minutes, followed by incubation with 2 ng/ml of TGF-β1 or/and 40 ng/ml of EGF for an additional 30 minutes. Cell lysates were probed with phospho-Smad-2 and Sp1, respectively. C to F: Immunofluorescence staining shows the cellular localization of Smad-2/3 in NRK-49F cells after various treatments for 30 minutes. C: Control; D: 2 ng/ml of TGF-β1; E: 2 ng/ml of TGF-β1 plus 40 ng/ml of EGF; F: 10 μmol/L of PD98059 plus TGF-β1 and EGF. G: EGF blocked TGF-β1-initiated α-SMA expression in renal interstitial fibroblast NRK-49F cells. The α-SMA expression was examined in NRK-49F cells after various treatments for 16 hours as indicated. Scale bar, 10 μm.
HGF Neither Affects Inhibitory Smad-6 and -7 Expression nor Modulates the Abundance of Smad Transcriptional Co-Repressors
One potential pathway for HGF inhibition of Smad signaling could be mediated by inducing inhibitory Smads (Smad-6 and -7) expression, which in turn blocks TGF-β1 signaling. 36-39 To address this issue, we examined the effects of HGF on Smad-6, -7, and -2 expression in renal interstitial fibroblast NRK-49F cells. As shown in Figure 9 ▶ , neither TGF-β1 nor HGF significantly affect Smad-6, -7, and -2 protein expression in NRK-49F cells. Simultaneous incubation of NRK-49F cells with both HGF and TGF-β1 also did not alter the abundance of these Smad proteins. In addition, other agents also did not significantly modulate Smad-6, Smad-7, and Smad-2 protein expression in interstitial fibroblast NRK-49F cells (Figure 9A) ▶ .
Figure 9.

HGF does not affect Smad-2 and inhibitory Smad-6 and -7 expression in renal interstitial fibroblast NRK-49F cells. A: NRK-49F cells were incubated with the same molar concentration (0.2 nmol/L) of various cytokines for 2 days and the cell lysates were probed with antibodies against Smad-2, Smad-7, Smad-6, and actin, respectively. B: NRK-49F cells were treated without or with 2 ng/ml of TGF-β1, 40 ng/ml of HGF, or both for 2 days as indicated. The protein levels of Smad-2, Smad-7, Smad-6, and actin were examined by Western blot analysis.
We also explored the possibility whether HGF increases the abundance of Smad transcriptional co-repressors and thereby, inhibiting TGF-β1-initiated Smad signaling. To this end, we examined the effects of HGF on the protein levels of Smad transcriptional co-repressors TGIF, c-Ski, and SnoN in NRK-49F cells. As shown in Figure 10 ▶ , the protein levels of TGIF, c-Ski, and SnoN were very low in renal fibroblasts, comparing to HEK-293 cells. HGF apparently did not affect the abundance of these Smad transcriptional co-repressors (Figure 10) ▶ .
Figure 10.

HGF does not increase the protein levels of Smad transcriptional co-repressors in renal interstitial fibroblasts. NRK-49F cells were treated with 40 ng/ml of HGF for various periods of time as indicated. Whole-cell lysates were immunoblotted with specific antibodies against c-Ski, SnoN, TGIF, and actin, respectively. Human embryonic kidney 293 (HEK-293) cell lysate was loaded on an adjacent lane to serve as a positive control for co-repressor expression.
HGF Selectively Blocks Smad-2/3 Nuclear Accumulation in Renal Interstitial Cells in the Fibrotic Kidney
Earlier studies indicate that delivery of HGF gene induced intrarenal Erk-1/2 phosphorylation and prevented renal interstitial fibrosis in a mouse model of renal disease caused by UUO. 27 To investigate whether HGF blocks Smad-2/3 nuclear accumulation in renal interstitial cells in vivo, we examined the Smad-2/3 localization in the obstructed kidneys receiving either control pcDNA3 or pCMV-HGF plasmids at 7 days after UUO. As shown in Figure 11 ▶ , in the obstructed kidney receiving empty vector pcDNA3, interstitial expansion was clearly evident with a widened space between renal tubules. The majority of the interstitial cells exhibited a positive nuclear staining for Smad-2/3 (Figure 11, A and C) ▶ . However, in the kidney receiving pCMV-HGF plasmid injection, much less interstitial expansion was observed at 7 days after ureteral obstruction. More importantly, most interstitial cells displayed no or little Smad-2/3 accumulation in their nuclei (Figure 11, B and D) ▶ , suggesting HGF specifically blocks Smad-2/3 nuclear accumulation in the interstitial cells in vivo. Figure 11E ▶ shows the percentages of Smad-2/3-positive nuclei in the interstitium of the kidneys receiving either pcDNA3 or pCMV-HGF plasmids. Of note, HGF did not significantly affect Smad-2/3 nuclear accumulation in the tubular epithelial cells. This suggests that HGF blocks Smad-2/3 nuclear translocation in the interstitium of the fibrotic kidneys in a cell-type-specific manner.
Figure 11.
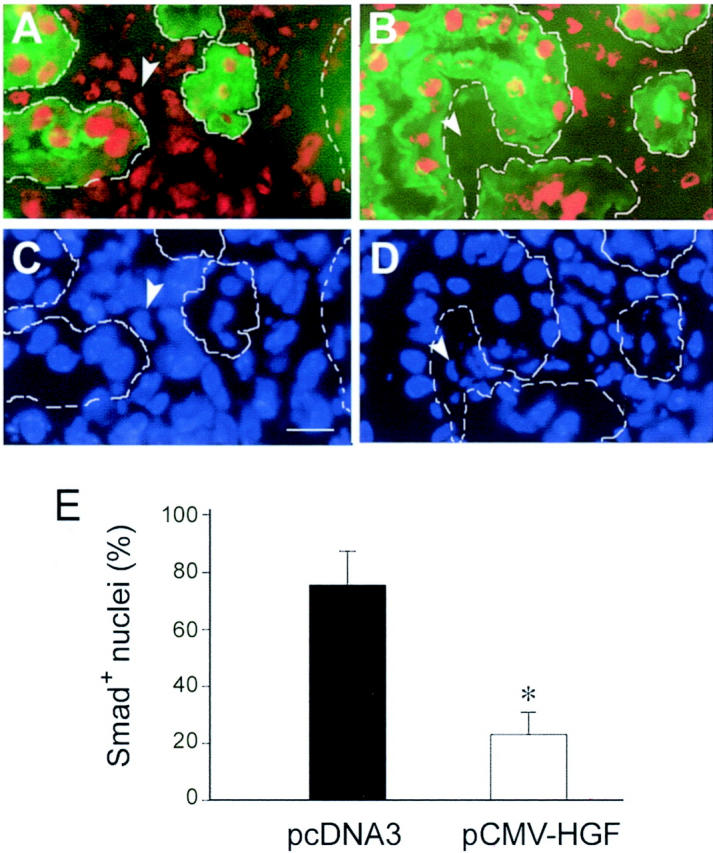
HGF selectively blocks Smad-2/3 nuclear accumulation in renal interstitial cells in obstructive nephropathy. A to D: Representative micrographs show Smad-2/3 nuclear accumulation in the fibrotic kidneys induced by UUO. Cryosections of the obstructed kidneys from mice receiving either empty pcDNA3 plasmid (A, C) or pCMV-HGF plasmid (B, D) were stained for Smad-2/3 (red) and proximal tubules with fluorescein-conjugate lectin from T. purpureas (green), and cell nuclei with 4′,6-diamidino-2-phenylindole (blue). Tubular compartments were depicted by broken lines along renal tubules. Smad-2/3-positive nuclei were counted in the widened interstitium and expressed as percentages of Smad-2/3-positive nuclei (red) per total nuclei (blue). Arrowheads indicate the corresponding cell nuclei either positive (A, C) or negative (B, D) for Smad-2/3 in renal interstitium. E: Graphical presentation of the percentage of Smad-2/3-positive cell nuclei in the interstitial areas of the obstructed kidneys. Data were presented as mean ± SE from five animals per group. *, P < 0.01 versus pcDNA3 group.
Discussion
The present study was undertaken to address two specific issues, ie, whether HGF suppresses TGF-β1-mediated renal interstitial myofibroblastic activation from quiescent fibroblasts, and if so, how HGF antagonizes the profibrotic action of TGF-β1 in fibroblasts. Using rat renal interstitial fibroblast NRK-49F cells as a model system, we have demonstrated that HGF efficiently blocks myofibroblastic activation from interstitial fibroblast cells triggered by TGF-β1. This action of HGF is neither dependent on a potential interruption of Smad-2/3 phosphorylation and their association with Smad-4, nor results from alterations in inhibitory Smad-6 and -7 expression or in cellular abundance of Smad transcriptional co-repressors. Rather, HGF elicits its action by attenuating nuclear translocation and accumulation of the activated Smad-2/3. This, in turn, is dependent on Erk-1/2 activation initiated by HGF in renal interstitial fibroblast cells. Hence, HGF suppression of renal interstitial myofibroblastic activation is associated with the interception of Smad signaling, which results in a disruption of profibrotic TGF-β1 signal transduction. Our findings provide significant insights into understanding the mechanism by which HGF counteracts the profibrotic actions of TGF-β1.
In response to tissue injury, renal interstitial fibroblasts undergo an activation process to become α-SMA-positive myofibroblast cells. Profibrotic TGF-β1 is the prime stimulator of this phenotypic activation, as shown herein and in previous studies. 40 These observations underscore the vital importance of TGF-β1 in initiating myofibroblastic activation from interstitial fibroblast cells. In accordance with this, up-regulation of TGF-β1 expression is found in most, if not all, forms of chronic renal fibrotic diseases in animal models and in patients, which often precedes interstitial myofibroblast activation and matrix accumulation. 13,19 TGF-β1 not only induces myofibroblastic activation from interstitial quiescent fibroblasts, but also initiates myofibroblastic transdifferentiation from tubular epithelial cells at the advanced stage of chronic renal diseases. 41-43 HGF has been previously demonstrated to block myofibroblastic transdifferentiation from tubular epithelial cells. Together with the present study, these results indicate that HGF can completely prevent renal myofibroblastic activation in the diseased kidney, regardless of the potential sources: whether from interstitial fibroblasts or tubular epithelia. These observations highlight the effectiveness and efficacy of HGF as a potential therapeutic agent for inhibiting myofibroblast accumulation and interstitial fibrogenesis in the diseased kidneys.
HGF blockade of myofibroblastic activation from interstitial fibroblast cells is likely mediated by antagonizing TGF-β1 signaling. TGF-β1, on binding to its type II and type I receptors that contains a cytoplasmic serine/threonine kinase domain, initiates a cascade of signaling transduction events involving intermediate mediator Smad proteins. 35,36 There are three classes of Smads: the receptor-regulated Smads (R-Smads), the common Smad (Co-Smad), and the inhibitory Smads (I-Smads), each of which have distinct functions. 44,45 Earlier studies reveal that TGF-β1 specifically initiates Smad-2 and -3 phosphorylation, which in turn bind to Co-Smad-4 and translocate into the nuclei, where they control the transcription of TGF-β1-responsive genes. 35,46,47 Thus, disruption of any steps in this cascade of signal transduction processes may potentially lead to interception of TGF-β1 signaling that results in blockage of myofibroblastic activation. In addition, induction of inhibitory Smad proteins could lead to suppression of R-Smad signaling by competitively binding to TGF-β receptors. 37,38,48 However, the fact that HGF does not significantly affect I-Smads (Smad-6 and -7) expression (Figure 9) ▶ , R-Smads (Smad-2 and -3) phosphorylation (Figure 5) ▶ , and Co-Smad (Smad-4) association with R-Smads (Figure 5C) ▶ underscores that it suppresses myofibroblastic activation by a mechanism independent of these signaling events. Thus, blockage of activated R-Smad’s nuclear translocation and accumulation (Figure 6) ▶ plays a decisive role in mediating HGF inhibition of Smad signaling. Such intercepting action by HGF in the Smad-signaling circuit will undoubtedly attenuate the availability of activated R-Smads for directing gene transcription necessary for myofibroblastic activation. This view is further strengthened by a previous report demonstrating a similar mechanism for blocking TGF-β1 signaling by oncogenic Ras and EGF receptor in another cell type. 49
Because phosphorylated Smads assemble complexes with transcriptional co-activators or co-repressors, cellular co-repressors abundance likely dictates the outcome of a TGF-β response. 46,47 Three Smad transcriptional co-repressors, namely TGIF, c-Ski, and SnoN, have been found; and they exhibit both histone deacetylase-dependent and -independent transcriptional repression to the Smad complex. 50-52 Recent studies demonstrate that EGF signaling via the Ras-Mek pathway triggers the phosphorylation of TGIF, which leads to TGIF stabilization and favors the formation of Smad-2-TGIF co-repressor complexes in response to TGF-β in human keratinocyte HaCaT cells. 47 However, no significant alterations in cellular abundance of Smad transcriptional co-repressors TGIF, c-Ski, and SnoN by HGF was found in NRK-49F fibroblast cells in the present study (Figure 10) ▶ , suggesting that cellular context may play an important role in dictating an appropriate signaling route.
Blockade of Smad nuclear translocation by HGF is primarily dependent on Erk-MAP kinase activation in renal interstitial fibroblast cells (Figure 7) ▶ . This is consistent with a previous observation that activated Erk directly phosphorylates Smad-2/3 in the linker region between the DNA-binding domain (MH1) and the receptor-interaction/transcriptional domain (MH2). Such Erk-mediated phosphorylation in the linker region of Smad-2/3 has been shown to prevent their nuclear translocation in Mv1Lu mink lung epithelial cells. 49 The potential of Erk-1/2 activation for inhibition of Smad signaling suggests that other growth factors may be active in blocking myofibroblastic activation, in light of their ability in inducing Erk-1/2 activation. Indeed, this speculation was confirmed by the observations that receptor tyrosine kinases activated by EGF significantly blocked myofibroblastic activation from interstitial fibroblasts (Figure 8) ▶ . It is of interest to note that earlier studies have demonstrated that administration of either EGF or insulin-like growth factor-I also attenuated renal interstitial fibrosis in obstructive nephropathy. 53,54 However, controversy exists regarding the role of EGF in renal fibrogenesis. For instance, contradicting to a beneficial role of exogenous EGF in obstructive nephropathy in neonatal rats, transgenic mice overexpressing dominant-negative EGF receptor manifested an attenuated interstitial fibrosis after renal injury, suggesting that functional inactivation of EGF receptor in proximal tubular epithelial cells reduces the progression of chronic renal failure. 55 In accordance with this, EGF has also been shown to synergistically act with TGF-β1 to promote tubular epithelial to mesenchymal transition. 41 Therefore, it appears clear that while EGF blocks myofibroblastic activation from interstitial fibroblasts (Figure 8) ▶ , it actually promotes myofibroblastic transdifferentiation from tubular epithelial cells. This may help to reconcile the discrepancy regarding the role of EGF in renal interstitial fibrogenesis.
The anti-fibrotic potential of HGF has recently been tested in numerous animal models of chronic renal diseases. 23-27 However, much attention in earlier studies is understandably directed to the tubular epithelial cells, 23,56 where the HGF receptor is robustly expressed. 28,29 The novelty of this study is the demonstration of HGF blockade of renal myofibroblastic activation from interstitial fibroblasts, a key event in interstitial fibrogenesis. Perhaps less recognized, renal interstitial fibroblasts express c-met receptor and respond to HGF stimulation. 33 Hence, HGF may have much broader implications in renal structure and functions than previously thought. The observation that HGF prevents both myofibroblastic activation from interstitial fibroblasts and myofibroblastic transdifferentiation from tubular epithelial cells reinforces the therapeutic efficacy of HGF in chronic interstitial fibrotic diseases.
Our study also provides evidence for a selective blockade of Smad nuclear accumulation by HGF in renal interstitial cells of the fibrotic kidneys in vivo (Figure 11) ▶ . Earlier studies have showed a marked activation of Erk-1/2 in the obstructed kidney after delivery of HGF plasmid, 27 which is consistent with a role for Erk-1/2 activation in blocking Smad nuclear translocation. Although the identity of the cells in the expanded interstitium remain elusive, it is reasonable to assume that a majority of the interstitial cells in the fibrotic kidneys are likely to be activated myofibroblasts, because myofibroblasts predominate in the interstitium at 7 days after obstructive injury. 57 Hence, our in vivo data virtually recapitulate in vitro cell signaling events and suggest that HGF specifically suppresses interstitial myofibroblast activation by blocking Smad nuclear accumulation in vivo as well. Of interest, HGF does not block Smad nuclear translocation in tubular epithelia, implying that its inhibition of Smad nuclear accumulation is cell type-dependent. Because HGF is able to block TGF-β1-induced myofibroblastic transformation from tubular epithelia, 23 further studies are warranted to dissect the signaling pathways by which HGF antagonizes TGF-β1 action in tubular epithelial cells both in vitro and in vivo.
The interruption of TGF-β1 signaling by HGF sheds new lights on the mechanism for its therapeutic potential for chronic renal fibrosis. These results uncover a pathway that HGF blocks TGF-β1 actions primarily via a mechanism independent of modulation of TGF-β1 expression and/or activation. Rather, HGF intercepts the pathogenic signaling triggered by TGF-β1 in a MAP kinase-dependent manner. This type of signal transduction therapy specifically targets the hyperactive TGF-β1 pathways that cause myofibroblastic activation and interstitial fibrogenesis in the first place. In view of its ability in blocking matrix-producing myofibroblasts from various origins, either from interstitial fibroblasts or tubular epithelial cells, HGF may have the real potential to block the onset and progression of chronic interstitial fibrosis and renal insufficiency.
Footnotes
Address reprint requests to Youhua Liu, Ph.D., Department of Pathology, University of Pittsburgh School of Medicine, S-405 Biomedical Science Tower, 200 Lothrop St., Pittsburgh, PA 15261. E-mail: liuy@msx.upmc.edu.
Supported by the National Institutes of Health (grants DK54944, DK61408, and DK64005) and the American Heart Association Pennsylvania-Delaware Affiliate (postdoctoral fellowships to J. Y. and C. D.).
References
- 1.Eddy AA: Molecular basis of renal fibrosis. Pediatr Nephrol 2000, 15:290-301 [DOI] [PubMed] [Google Scholar]
- 2.Muchaneta-Kubara EC, el Nahas AM: Myofibroblast phenotypes expression in experimental renal scarring. Nephrol Dial Transplant 1997, 12:904-915 [DOI] [PubMed] [Google Scholar]
- 3.Powell DW, Mifflin RC, Valentich JD, Crowe SE, Saada JI, West AB: Myofibroblasts. I. Paracrine cells important in health and disease. Am J Physiol 1999, 277:C1-C9 [DOI] [PubMed] [Google Scholar]
- 4.Klahr S: Urinary tract obstruction. Semin Nephrol 2001, 21:133-145 [DOI] [PubMed] [Google Scholar]
- 5.De Heer E, Sijpkens YW, Verkade M, den Dulk M, Langers A, Schutrups J, Bruijn JA, van Es LA: Morphometry of interstitial fibrosis. Nephrol Dial Transplant 2000, 15:72-73 [DOI] [PubMed] [Google Scholar]
- 6.Fogo AB: Pathology of progressive nephropathies. Curr Opin Nephrol Hypertens 2000, 9:241-246 [DOI] [PubMed] [Google Scholar]
- 7.Essawy M, Soylemezoglu O, Muchaneta-Kubara EC, Shortland J, Brown CB, el Nahas AM: Myofibroblasts and the progression of diabetic nephropathy. Nephrol Dial Transplant 1997, 12:43-50 [DOI] [PubMed] [Google Scholar]
- 8.Remuzzi G, Bertani T: Pathophysiology of progressive nephropathies. N Engl J Med 1998, 339:1448-1456 [DOI] [PubMed] [Google Scholar]
- 9.Badid C, Vincent M, Fouque D, Laville M, Desmouliere A: Myofibroblast: a prognostic marker and target cell in progressive renal disease. Renal Fail 2001, 23:543-549 [DOI] [PubMed] [Google Scholar]
- 10.Roberts IS, Burrows C, Shanks JH, Venning M, McWilliam LJ: Interstitial myofibroblasts: predictors of progression in membranous nephropathy. J Clin Pathol 1997, 50:123-127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu Y: Hepatocyte growth factor and the kidney. Curr Opin Nephrol Hypertens 2002, 11:23-30 [DOI] [PubMed] [Google Scholar]
- 12.Tang WW, Ulich TR, Lacey DL, Hill DC, Qi M, Kaufman SA, Van GY, Tarpley JE, Yee JS: Platelet-derived growth factor-BB induces renal tubulointerstitial myofibroblast formation and tubulointerstitial fibrosis. Am J Pathol 1996, 148:1169-1180 [PMC free article] [PubMed] [Google Scholar]
- 13.Border WA, Noble NA: TGF-beta in kidney fibrosis: a target for gene therapy. Kidney Int 1997, 51:1388-1396 [DOI] [PubMed] [Google Scholar]
- 14.Bottinger EP, Bitzer M: TGF-β1 signaling in renal disease. J Am Soc Nephrol 2002, 13:2600-2610 [DOI] [PubMed] [Google Scholar]
- 15.Yokoi H, Sugawara A, Mukoyama M, Mori K, Makino H, Suganami T, Nagae T, Yahata K, Fujinaga Y, Tanaka I, Nakao K: Role of connective tissue growth factor in profibrotic action of transforming growth factor-beta: a potential target for preventing renal fibrosis. Am J Kidney Dis 2001, 38:S134-S138 [DOI] [PubMed] [Google Scholar]
- 16.Lewis MP, Norman JT: Differential response of activated versus non-activated renal fibroblasts to tubular epithelial cells: a model of initiation and progression of fibrosis? Exp Nephrol 1998, 6:132-143 [DOI] [PubMed] [Google Scholar]
- 17.Heusinger-Ribeiro J, Eberlein M, Wahab NA, Goppelt-Struebe M: Expression of connective tissue growth factor in human renal fibroblasts: regulatory roles of RhoA and cAMP. J Am Soc Nephrol 2001, 12:1853-1861 [DOI] [PubMed] [Google Scholar]
- 18.Schnaper HW, Hayashida T, Hubchak SC, Poncelet AC: TGF-beta signal transduction and mesangial cell fibrogenesis. Am J Physiol 2003, 284:F243-F252 [DOI] [PubMed] [Google Scholar]
- 19.Cotton SA, Gbadegesin RA, Williams S, Brenchley PE, Webb NJ: Role of TGF-beta1 in renal parenchymal scarring following childhood urinary tract infection. Kidney Int 2002, 61:61-67 [DOI] [PubMed] [Google Scholar]
- 20.Isaka Y, Akagi Y, Ando Y, Tsujie M, Sudo T, Ohno N, Border WA, Noble NA, Kaneda Y, Hori M, Imai E: Gene therapy by transforming growth factor-beta receptor-IgG Fc chimera suppressed extracellular matrix accumulation in experimental glomerulonephritis. Kidney Int 1999, 55:465-475 [DOI] [PubMed] [Google Scholar]
- 21.Isaka Y, Tsujie M, Ando Y, Nakamura H, Kaneda Y, Imai E, Hori M: Transforming growth factor-beta 1 antisense oligodeoxynucleotides block interstitial fibrosis in unilateral ureteral obstruction. Kidney Int 2000, 58:1885-1892 [DOI] [PubMed] [Google Scholar]
- 22.Ziyadeh FN, Hoffman BB, Han DC, Iglesias-De La Cruz MC, Hong SW, Isono M, Chen S, McGowan TA, Sharma K: Long-term prevention of renal insufficiency, excess matrix gene expression, and glomerular mesangial matrix expansion by treatment with monoclonal antitransforming growth factor-beta antibody in db/db diabetic mice. Proc Natl Acad Sci USA 2000, 97:8015-8020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang J, Liu Y: Blockage of tubular epithelial to myofibroblast transition by hepatocyte growth factor prevents renal interstitial fibrosis. J Am Soc Nephrol 2002, 13:96-107 [DOI] [PubMed] [Google Scholar]
- 24.Mizuno S, Matsumoto K, Kurosawa T, Mizuno-Horikawa Y, Nakamura T: Reciprocal balance of hepatocyte growth factor and transforming growth factor-beta 1 in renal fibrosis in mice. Kidney Int 2000, 57:937-948 [DOI] [PubMed] [Google Scholar]
- 25.Liu Y, Rajur K, Tolbert E, Dworkin LD: Endogenous hepatocyte growth factor ameliorates chronic renal injury by activating matrix degradation pathways. Kidney Int 2000, 58:2028-2043 [DOI] [PubMed] [Google Scholar]
- 26.Mizuno S, Matsumoto K, Nakamura T: Hepatocyte growth factor suppresses interstitial fibrosis in a mouse model of obstructive nephropathy. Kidney Int 2001, 59:1304-1314 [DOI] [PubMed] [Google Scholar]
- 27.Yang J, Dai C, Liu Y: Systemic administration of naked plasmid encoding hepatocyte growth factor ameliorates chronic renal fibrosis in mice. Gene Ther 2001, 8:1470-1479 [DOI] [PubMed] [Google Scholar]
- 28.Liu Y, Tolbert EM, Sun AM, Dworkin LD: In vivo and in vitro evidence for increased expression of HGF receptor in kidney of diabetic rat. Am J Physiol 1996, 271:F1202-F1210 [DOI] [PubMed] [Google Scholar]
- 29.Rabkin R, Fervenza F, Tsao T, Sibley R, Friedlaender M, Hsu F, Lassman C, Hausmann M, Huie P, Schwall RH: Hepatocyte growth factor receptor in acute tubular necrosis. J Am Soc Nephrol 2001, 12:531-540 [DOI] [PubMed] [Google Scholar]
- 30.Yang J, Chen S, Huang L, Michalopoulos GK, Liu Y: Sustained expression of naked plasmid DNA encoding hepatocyte growth factor in mice promotes liver and overall body growth. Hepatology 2001, 33:848-859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu Y, Beedle AB, Lin L, Bell AW, Zarnegar R: Identification of a cell-type-specific transcriptional repressor in the promoter region of the mouse hepatocyte growth factor gene. Mol Cell Biol 1994, 14:7046-7058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nissen RM, Yamamoto KR: The glucocorticoid receptor inhibits NFkappaB by interfering with serine-2 phosphorylation of the RNA polymerase II carboxy-terminal domain. Genes Dev 2000, 14:2314-2329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang X, Li Y, Dai C, Yang J, Mundel P, Liu Y: Sp1 and Sp3 transcription factors synergistically regulate HGF receptor gene expression in kidney. Am J Physiol 2003, 284:F82-F94 [DOI] [PubMed] [Google Scholar]
- 34.Yang L, Lin HK, Altuwaijri S, Xie S, Wang L, Chang C: APPL suppresses androgen receptor transactivation via potentiating Akt activity. J Biol Chem 2003, 278:16820-16827 [DOI] [PubMed] [Google Scholar]
- 35.Massague J: How cells read TGF-beta signals. Nat Rev Mol Cell Biol 2000, 1:169-178 [DOI] [PubMed] [Google Scholar]
- 36.Massague J, Chen YG: Controlling TGF-beta signaling. Genes Dev 2000, 14:627-644 [PubMed] [Google Scholar]
- 37.Li JH, Zhu HJ, Huang XR, Lai KN, Johnson RJ, Lan HY: Smad7 inhibits fibrotic effect of TGF-beta on renal tubular epithelial cells by blocking Smad2 activation. J Am Soc Nephrol 2002, 13:1464-1472 [DOI] [PubMed] [Google Scholar]
- 38.Schnaper HW, Hayashida T, Poncelet AC: It’s a Smad World: regulation of TGF-beta signaling in the kidney. J Am Soc Nephrol 2002, 13:1126-1128 [DOI] [PubMed] [Google Scholar]
- 39.Patil S, Wildey GM, Brown TL, Choy L, Derynck R, Howe PH: Smad7 is induced by CD40 and protects WEHI 231 B-lymphocytes from transforming growth factor-beta-induced growth inhibition and apoptosis. J Biol Chem 2000, 275:38363-38370 [DOI] [PubMed] [Google Scholar]
- 40.Strutz F, Zeisberg M, Renziehausen A, Raschke B, Becker V, van Kooten C, Muller G: TGF-beta 1 induces proliferation in human renal fibroblasts via induction of basic fibroblast growth factor (FGF-2). Kidney Int 2001, 59:579-592 [DOI] [PubMed] [Google Scholar]
- 41.Okada H, Danoff TM, Kalluri R, Neilson EG: Early role of Fsp1 in epithelial-mesenchymal transformation. Am J Physiol 1997, 273:F563-F574 [DOI] [PubMed] [Google Scholar]
- 42.Fan JM, Ng YY, Hill PA, Nikolic-Paterson DJ, Mu W, Atkins RC, Lan HY: Transforming growth factor-beta regulates tubular epithelial-myofibroblast transdifferentiation in vitro. Kidney Int 1999, 56:1455-1467 [DOI] [PubMed] [Google Scholar]
- 43.Yang J, Liu Y: Dissection of key events in tubular epithelial to myofibroblast transition and its implications in renal interstitial fibrosis. Am J Pathol 2001, 159:1465-1475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wrana JL: Regulation of Smad activity. Cell 2000, 100:189-192 [DOI] [PubMed] [Google Scholar]
- 45.Mehra A, Wrana JL: TGF-beta and the Smad signal transduction pathway. Biochem Cell Biol 2002, 80:605-622 [DOI] [PubMed] [Google Scholar]
- 46.Wotton D, Massague J: Smad transcriptional corepressors in TGF beta family signaling. Curr Top Microbiol Immunol 2001, 254:145-164 [PubMed] [Google Scholar]
- 47.Lo RS, Wotton D, Massague J: Epidermal growth factor signaling via Ras controls the Smad transcriptional co-repressor TGIF. EMBO J 2001, 20:128-136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Terada Y, Hanada S, Nakao A, Kuwahara M, Sasaki S, Marumo F: Gene transfer of Smad7 using electroporation of adenovirus prevents renal fibrosis in post-obstructed kidney. Kidney Int 2002, 61(Suppl 1):94-98 [DOI] [PubMed] [Google Scholar]
- 49.Kretzschmar M, Doody J, Timokhina I, Massague J: A mechanism of repression of TGFbeta/ Smad signaling by oncogenic Ras. Genes Dev 1999, 13:804-816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stroschein SL, Wang W, Zhou S, Zhou Q, Luo K: Negative feedback regulation of TGF-beta signaling by the SnoN oncoprotein. Science 1999, 286:771-774 [DOI] [PubMed] [Google Scholar]
- 51.Sun Y, Liu X, Eaton EN, Lane WS, Lodish HF, Weinberg RA: Interaction of the Ski oncoprotein with Smad3 regulates TGF-beta signaling. Mol Cell 1999, 4:499-509 [DOI] [PubMed] [Google Scholar]
- 52.Wotton D, Lo RS, Lee S, Massague J: A Smad transcriptional corepressor. Cell 1999, 97:29-39 [DOI] [PubMed] [Google Scholar]
- 53.Chevalier RL, Goyal S, Thornhill BA: EGF improves recovery following relief of unilateral ureteral obstruction in the neonatal rat. J Urol 1999, 162:1532-1536 [PubMed] [Google Scholar]
- 54.Chevalier RL, Goyal S, Kim A, Chang AY, Landau D, LeRoith D: Renal tubulointerstitial injury from ureteral obstruction in the neonatal rat is attenuated by IGF-1. Kidney Int 2000, 57:882-890 [DOI] [PubMed] [Google Scholar]
- 55.Terzi F, Burtin M, Hekmati M, Federici P, Grimber G, Briand P, Friedlander G: Targeted expression of a dominant-negative EGF-R in the kidney reduces tubulo-interstitial lesions after renal injury. J Clin Invest 2000, 106:225-234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu Y: Hepatocyte growth factor promotes renal epithelial cell survival by dual mechanisms. Am J Physiol 1999, 277:F624-F633 [DOI] [PubMed] [Google Scholar]
- 57.Yang J, Shultz RW, Mars WM, Wegner RE, Li Y, Dai C, Nejak K, Liu Y: Disruption of tissue-type plasminogen activator gene in mice reduces renal interstitial fibrosis in obstructive nephropathy. J Clin Invest 2002, 110:1525-1538 [DOI] [PMC free article] [PubMed] [Google Scholar]