

Abstract
The high frequency of mutation, deletion, and promoter silencing of the gene encoding p16INK4A (p16) in premalignant dysplasias and squamous cell carcinomas (SCC) of epidermis and oral epithelium classifies p16 as a tumor suppressor. However, the point during neoplastic progression at which this protein is expressed and presumably impedes formation of an SCC is unknown. Induction of p16 has been found to be responsible for the senescence arrest of normal human keratinocytes in culture, suggesting the possibility that excessive or spatially abnormal cell growth in vivo triggers p16 expression. We examined 73 skin and oral mucosal biopsy specimens immunohistochemically to test this hypothesis. p16 was not detectable in benign hyperplastic lesions, but instead was expressed heterogeneously in some dysplastic and carcinoma in situ lesions and consistently at areas of microinvasion and at superficial margins of advanced SCCs. p16-positive cells in these regions coexpressed the γ2 chain of laminin 5, identified previously as a marker of invasion in some carcinomas. Normal keratinocytes undergoing senescence arrest in culture proved to coordinately express p16 and γ2 and this was frequently associated with increased directional motility. Keratinocytes at the edges of wounds made in confluent early passage cultures also coexpressed p16 and γ2, accompanying migration to fill the wound. These results have identified the point during neoplastic progression in stratified squamous epithelial at which the tumor suppressor p16 is expressed and suggest that normal epithelia may use the same mechanism to generate non-dividing, motile cells for wound repair.
Squamous cell carcinoma (SCC) is the malignancy of the oral mucosal epithelium, the epidermis, and other stratified squamous epithelia. SCCs arise within areas of abnormal, pre-invasive cell growth (dysplasia), which may take months to many years to progress to invasive cancer. 1-3 A consistent feature of SCC is loss of the ability to express functional p16INK4A (p16) by mutation, deletion, or promoter hypermethylation. 4-14 p16 is a specific inhibitor of the cyclin D1-dependent kinases cdk4 and cdk6. Normally, in the absence of p16, cyclin D1/cdk4 and cyclin D1/cdk6 complexes phosphorylate and inactivate the Rb protein, permitting E2F-dependent transcription of genes encoding a set of proteins necessary to initiate chromosome replication and ultimately another round of cell division. 15 Most cervicogenital SCCs 16,17 and a fraction of head and neck SCCs 18 contain human papilloma viruses (HPV) and viral DNA integration as a feature of their neoplastic progression. These tumors and their premalignant dysplasias typically have unaltered p16 alleles and often express p16 protein, presumably as a response to inhibition of Rb function by the HPV E7 viral oncoprotein.
Humans and mice that inherit a heterozygous or homozygous loss of function mutation in the p16 gene 19,20 or that express a p16-insensitive mutant form of cdk4 21 are predisposed to a variety of spontaneous and carcinogen-induced cancers, but they undergo normal development and form structurally and functionally normal stratified squamous epithelia. These results confirm the tumor suppressor function of p16 and also are consistent with the finding that p16 protein is not expressed as a feature of normal stratified squamous epithelial renewal or differentiation. 17,22,23 Importantly, the point during neoplastic progression toward SCC at which p16 protein becomes expressed and functions as a tumor suppressor has remained unknown.
Western blot analysis of normal human oral and epidermal keratinocytes in culture has identified an increase in p16 levels with serial passage, as cultures approach the end of their finite replicative lifespan. 24-28 Immunocytochemical analysis of such cultures has revealed that p16 expression occurs heterogeneously and abruptly, followed closely by growth arrest, and that the probability that a cell will express p16 increases steadily with each passage until all cells in the culture are p16-positive and senescent. 26,29 Primary keratinocytes engineered to express TERT, thereby acquiring the ability to stabilize their telomeres, still undergo p16-enforced senescence. 25,26,29 This fate may be relieved by mutational or epigenetic loss/reduction of p16 expression in the TERT-transfected cell population, yielding immortalized lines. 25,26,29,30 The mechanism responsible for inducing serial passage-related p16 expression in keratinocytes has not been characterized, although inadequate culture conditions hasten their p16-related aging. 31 However, p16-dependent senescence in keratinocytes is clearly distinct from the telomere-sensitive, p53/p21cip1-dependent replicative aging mechanism that enforces the replicative lifespan limit of human dermal fibroblasts and several other cell types in culture. 29,32-34
The goal of this study was to determine the setting in which p16 is expressed and functions as a tumor suppressor in stratified squamous epithelia in vivo. We began with the hypothesis that abnormal keratinocyte growth places excessive replication demands on keratinocyte stem cell populations, similar to the effects of long-term serial culture. Presuming this, both conditions may trigger p16 expression and growth arrest. We characterized p16 expression immunohistochemically in a range of normal, benign hyperplastic, pre-invasive, and invasive epithelial tissue specimens from the skin and oral mucosa. We report here that p16 is not associated with benign hyperplasia, but rather occurs heterogeneously in cells of some premalignant lesions and consistently in areas of microinvasion and at superficial margins of invasive SCC. Such regions of p16 expression proved to overexpress the γ2 subunit of the basement membrane protein laminin 5, which had been previously described as a marker of invasion in epithelial cancers. 35-37 In addition, we have found that senescing keratinocytes and experimentally wounded early-passage keratinocytes in culture co-express p16 and γ2, associated with growth arrest and increased directional motility, suggesting that there are important shared elements between the p16 tumor suppressor mechanism and the wound response in stratified squamous epithelia.
Materials and Methods
Tissue Sections
Formalin-fixed, paraffin-embedded biopsy specimens were obtained from the archives of the Dermatopathology service at Brigham and Women’s Hospital and from Pathology Services Inc., Cambridge, MA. Hematoxylin and eosin (H&E) sections of each specimen were evaluated by light microscopy using World Health Organization histopathological staging criteria. Tissue specimens were classified by the most advanced neoplastic stage identified in sections, but most carcinoma in situ (CIS) and tumor specimens also contained adjacent regions of dysplasia and normal epithelium, permitting immunohistochemical comparison of different regions within the same section. Skin specimens having regions of normal epidermis, benign hyperplasia, seborrheic keratosis, dysplastic actinic keratosis, CIS, and microinvasive and advanced SCC were identified and diagnosed by P.M.K. and M.S., who are specialists in dermatopathology. Oral mucosal specimens having regions of normal epithelium, benign hyperplasia (benign alveolar ridge hyperkeratosis), mild, moderate, and severe dysplasia, CIS, and microinvasive and advanced SCC were identified and diagnosed by S.B.W., who is a specialist in oral and maxillofacial pathology.
Cell Lines and Culture Methods
Human oral and epidermal keratinocyte cell lines previously characterized for p16 protein expression by this laboratory 26,29 as either heterogeneous (strain N, N/TERT-2G, OKB8), homogenously strongly positive (N/p53DD/cdk4R/TERT, OKF4/p53DD/cdk4R/TERT), or completely negative (POE-9n/TERT-1, N/TERT-1, SCC-13), were used as controls to test the specificity of p16 antibodies, to optimize fixation, processing, and immunostaining conditions, and for cell migration and wounding experiments.
Cells were cultured in a nutritionally optimized, keratinocyte serum-free medium (K-sfm; Invitrogen, Carlsbad, CA) 38 supplemented, as previously described, 26,29,39 with 25 μg/ml bovine pituitary extract, 0.2 ng/ml epidermal growth factor, and 0.4 mmol/L CaCl2. For experiments to evaluate correlations between proliferative state and p16 or γ2 expression, cultures received 0.1 mmol/L bromodeoxyuridine (BUdR) 24 hours before fixation to label cells that had entered S phase during this time period. To generate confluent, monolayer cultures, cells were grown to about 40% confluence in K-sfm, then re-fed with a medium consisting of a 1:1 (vol:vol) mixture of calcium-free Dulbecco’s modified Eagle’s medium (Invitrogen) and 0.4 mmol/L CaCl2-supplemented complete K-sfm, which permitted the formation of healthy, confluent, unstratified cultures. Linear, ∼2-mm-wide “wounds” were made in such cultures by dragging the point of a sterile Eppendorf pipette tip across the monolayer culture, after which the cultures were returned to the incubator for 24 to 48 hours until fixation.
Antibodies
The p16-specific mouse monoclonal antibody (MoMAb) G175–405 (PharMingen, San Diego, CA) was used at 2 μg/ml for all experiments. The results were confirmed using the p16-specific MoMAb JC2 22 (provided by J. Koh, University of Vermont) at 4 μg/ml. Rabbit polyclonal anti-p16 antibody C20 (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) was used in some initial experiments at 2 μg/ml. The human involucrin-specific MoMAb SY5 40 (Research Diagnostics, Inc., Flanders, NJ) was used at 1:100 dilution as a positive control in each experiment to test the overall quality of the immunostain procedure. A MoMAb specific for non-immune IgG1, κ isotype (PharMingen) was used as a negative control. MoMAb 19562, specific for the γ2 chain of laminin 5 41 (Chemicon International, Temecula, CA), was used at 10 μg/ml. Rabbit polyclonal antibody J20, specific for γ2 42 (provided by J. Jones, Northwestern University Medical School, Chicago, IL), was used at 1:40 dilution. The BUdR-specific rat monoclonal antibody BU1/75 (ICR1), IgG2a isotype, specific for BUdR (Accurate Chemical & Scientific Corp., Westbury, NY) was used at 1:50 dilution.
Optimization of Fixation, Processing and p16 Antigen Retrieval Methods
Cell lines N/p53DD/cdk4R/TERT and POE-9n/TERT-1 were grown to confluence in separate p100 dishes and treated with 25 μg/ml dispase (Invitrogen) to detach the cells as a coherent sheet. 43 The detached sheets were rinsed with PBS, compressed and shaped into elongated masses using a rubber policeman, placed together in a meshed plastic fixation chamber, fixed in 10% neutral buffered formalin (Fisher Scientific Co., Fair Lawn, NJ) for either 1 or 3 days. These cultured cell masses were processed by the Brigham and Women’s Hospital Dermatopathology service in the same way as clinical specimens. A similar preparation was made from OKF4/p53DD/cdk4R/TERT and N/TERT-1 cultures. Four-μm-thick paraffin sections were cut and baked onto glass slides at 37°C for 30 minutes. For one experiment, a set of sections was baked onto slides at 55°C for 30 minutes to examine the effects of heat on p16 immunoreactivity. Slides with tissue sections were de-paraffinized in 100% xylene and rehydrated in graded ethanol and PBS.
We compared four commonly used antigen retrieval methods 44 to enhance detectability of p16: 10 minutes in PBS preheated to 90°C, 10 minutes in 0.2N HCl preheated to 90°C, 15 minutes in 0.01 mol/L sodium citrate buffer (pH 6.0) heated in a microwave oven, and 15 minutes in 0.01 mol/L sodium citrate buffer (pH 6.0) autoclaved at 120°C under 15 lbs of pressure. The latter method proved to yield maximal p16 staining of known positive specimens and little or no background staining of known negative specimens, so we used this as part of our standard protocol.
Immunohistochemical Staining
Each experiment immunostaining clinical specimens included control sections cut from blocks containing known p16-positive and -negative cultured cell lines. Four-μm sections of formalin-fixed, paraffin-embedded tissue were baked on glass slides at 37°C for 30 minutes, deparaffinized in 100% xylene, and rehydrated in graded ethanol and PBS. Slides were then immersed in 0.01 mol/L sodium citrate buffer (pH 6.0), heated to 120°C under pressure in an autoclave for 15 minutes, cooled to room temperature, and rinsed in PBS. Endogenous peroxidase was inactivated by immersion in 0.3% H2O2 in PBS for 10 minutes. Non-specific antigens were blocked by incubating slides in 10% horse serum in PBS for 25 minutes. Slides were then incubated with primary antibody for 2 hours at 4°C, with biotinylated secondary antibody (1:100 dilution) for 30 minutes, and with avidin/biotin/peroxidase complex (ABC) reagent (Vectastain Elite ABC kit, Vector Laboratories, Burlingame, CA) for 45 minutes, with a PBS rinse following each step. Sections were then incubated with the peroxidase substrate 3,3-diaminobenzidine tetrahydrochloride (Vector Laboratories) for 1.5 minutes, rinsed in water, and mounted with coverslips using GEL/MOUNT (Biomeda Corp., Foster City, CA).
Immunostaining of Cultured Cells
Cells growing in p100 dishes (Falcon/BD Biosciences, Bedford, MA) were fixed in freshly-prepared 4% paraformaldehyde-PBS for 30 minutes and permeabilized with 0.1% Triton X-100 in PBS for 5 minutes. Some cultures had received 0.1 mmol/L BUdR for the final 24 hours before fixation to label cells that were in S phase during this time period. After fixation and permeabilization, BUdR-treated cultures were incubated for 1 hour with 0.2 mol/L HCl and then neutralized with 0.1 mol/L borate buffer (pH 8.5) to make DNA containing BUdR accessible to the antibody, as previously described. 29 Before immunostaining, cultures were blocked with 10% normal horse serum in PBS (before ABC peroxidase staining) or 10% normal goat serum in PBS (before indirect immunofluorescence staining). For single-label ABC peroxidase staining, cultures were incubated with primary antibody for 45 minutes, biotinylated secondary antibody for 30 minutes, ABC reagent for 45 minutes, each interspersed with PBS rinses before exposure to NovaRed color substrate for 1.5 to 2 minutes. For double-label immunofluorescence staining, the two primary antibodies were mixed and incubated with fixed cultures. This was followed by incubation with a mixture of two species-appropriate Alexa Fluor fluorescent dye-conjugated secondary antibodies (Molecular Probes Inc.): A11029-green anti-mouse IgG, A11032-red anti-mouse IgG, A11037-red anti-rabbit IgG, and A11006-green anti-rat IgG. Images were captured on a NIKON E600 Microscope with a SPOT2 digital camera using SPOTcam v.3.5.5 software (Digital Instruments, Inc.).
HPV Testing
A subset of specimens were screened for human papillomavirus by polymerase chain reaction (PCR) amplification of DNA extracted from deparaffinized sections, using primers specific for conserved HPV sequences as previously described. 17 Briefly, extracted DNA was amplified by PCR using primers designed to anneal to the L1 region of a wide range of HPV types. 45 Amplification was performed in the presence of radio-labeled nucleotides and reaction products were digested with restriction enzymes, resolved by polyacrylamide gel electrophoresis, and exposed to X-ray film. 46 HPV types were assigned by comparison to restriction digestion product profiles of known HPV types. The positive control included 1 pg of HPV16 genomic DNA; the negative control consisted of protein digestion and PCR reaction components without specimen extract. The integrity of input DNA was verified by the presence of co-amplified human genomic DNA sequences.
Results
Optimization of Immunohistochemical Staining Protocol
We first sought to confirm the specificity of our antibodies and determine the effects of varying specimen fixation and slide processing conditions. We examined the p16 immunostaining patterns of formalin-fixed, Triton-permeabilized monolayers and paraffin sections of human keratinocyte cell lines previously determined to be either completely p16-deficient, heterogeneous for p16 expression, or homogeneous high level expressers of p16. 26,29 We also examined formalin-fixed newborn foreskin epidermis, which we expected from previous reports 22 to be p16-negative. As expected, 26,29 mid-lifespan cultures of strain N epidermal keratinocytes (Figure 1, a and b) ▶ and OKF4 floor-of-mouth keratinocytes (data not shown) were heterogeneous populations of small, proliferative, p16-negative and larger, senescent, p16-positive cells. Immunodetectable p16 protein was distributed homogenously throughout the cytoplasm, with little nuclear staining. Cells of the p16-deficient, epidermal SCC line SCC-13 showed no p16 staining (Figure 1, c and d) ▶ . Our optimized protocol, described in Methods, yielded strong staining of sections prepared of the p16-expressing N/p53DD/cdk4R/TERT cell line 29 and no background staining of the p16 gene-deleted keratinocyte line POE9n/TERT 26 (Figure 1e) ▶ . However, variations in tissue fixation, slide preparation, and antigen retrieval conditions proved to have marked effects on the staining results. Baking sections at 55°C, a common histopathology laboratory procedure to improve adhesion of sections to slides, resulted in substantially reduced p16 immunostaining of p16-positive cells (Figure 1f) ▶ . Formalin fixation for 1 day before embedding resulted in accurate detection of p16-positive and negative cells (Figure 1, e and g) ▶ , whereas fixation for three days resulted in a general low-to-moderate background stain of all epithelial elements in normal skin (Figure 1h) ▶ .
Figure 1.

Optimization of the p16 immunostaining protocol. a and b: Phase contrast (a) and brightfield (b) views of normal primary epidermal keratinocyte strain N cells stained for p16 (brown). Note that small, dividing cells are p16-negative and large, senescence-arrested cells are p16-positive, with predominantly cytoplasmic staining, as previously described. 26,29 c and d: Phase contrast (c) and brightfield (d) views of epidermal SCC line SCC-13 cells stained for p16. Note absence of p16 staining. e and f: p16 immunostaining (brown) of formalin-fixed, paraffin-embedded cultured cells of the p16-expressing line N/p53DD/cdk4R/TERT (N/P/C/TERT) and the p16 gene-deleted line POE-9n/TERT, following baking of sections onto slides for 30 minutes at 37°C (e) or at 55°C (f). Note that baking at 37°C yielded accurate p16 staining of these lines, while baking at 55°C resulted in virtually no p16 staining of N/p53DD/cdk4R/TERT. g and h: p16 immunostaining (brown) of human foreskin specimen fixed in 10% formalin for 1 day (g) or 3 days (h) before embedding. Section shown in g was counterstained with hematoxylin. Note accurate absence of p16 staining of epidermis fixed for 1 day and artifactual homogenous staining of all living layers of the epidermis after fixation for 3 days. i and j: Benign papillary hyperplasia and hyperkeratosis of alveolar ridge epithelium stained with H&E (i) or for p16 (j). Section shown in j was counterstained with hematoxylin. Note absence of p16 staining.
Initial Survey of p16 Immunostaining in Normal and Pathological Skin and Oral Tissue Specimens
As described above, up-regulation of p16 causes senescence arrest in normal, primary human keratinocytes grown in culture, restricting their expansion potential. This suggested the possibility that excessive or spatially abnormal cell division in vivo, as in epithelial hyperplasia, dysplasia, and CIS, would trigger p16 expression. To test this hypothesis, we examined two normal skin specimens and a set of 15 dermatopathologic clinical specimens. Thirty-five different areas identified within these specimens as histologically normal, hyperplastic, dysplastic, CIS, or SCC were scored for p16 immunostaining (Table 1 ▶ ; Figure 2 ▶ ). The normal epidermal and seborrheic keratosis specimens were either negative or showed faint, homogenous staining of the entire epithelium and some components of the stroma (Figure 2, a to c) ▶ . A similar pattern had been previously reported for some archival specimens of normal ectocervical epithelium, and interpreted as non-specific background staining. 17 Since this pattern was identical to that of normal epidermis following prolonged formalin fixation (Figure 1h) ▶ , we judged these and other specimens that showed similar overall background to be p16-negative. The p16 staining patterns of some of the premalignant lesions were very different. Two of six areas of actinic keratosis showed a variegated pattern of intensely stained and unstained cells, the former which we scored as p16-positive (Figure 2, d to f) ▶ . Four of five areas of CIS contained p16-positive cells, including most of the cells extending upward from the basal layer to the surface (Figure 2, g to i) ▶ . Some regions of dysplastic and microinvasive epithelium overlying tumors (Figure 2, j and k) ▶ and at tumor margins displayed variegated p16 staining. As we had found previously in studies of keratinocytes in culture 26,29 (Figure 1, b and f) ▶ , the p16 staining in these pathological tissue specimens was predominantly cytoplasmic (Figures 2 and 3) ▶ . Deeply invasive, advanced SCCs typically contained few or no p16-positive cells (Figure 2, j and k) ▶ .
Table 1.
p16 Immunostaining of Skin Specimens, Set 1
| Specimen diagnosis | Region and p16 immunostaining | |||||
|---|---|---|---|---|---|---|
| NOR | HYP | AK | CIS | EISC | SCC | |
| Normal epidermis | N | |||||
| Normal epidermis | N | |||||
| Seborrheic keratosis | N | |||||
| Seborrheic keratosis | N | |||||
| Seborrheic keratosis | N | |||||
| Seborrheic keratosis | N | |||||
| Seborrheic keratosis | N | |||||
| Actinic keratosis | N | |||||
| Carcinoma in situ | N | P | ||||
| Carcinoma in situ | P | |||||
| Carcinoma in situ | N | P | ||||
| SCC arising in AK | N | N | N | N | ||
| SCC arising in AK | N | N | N | |||
| SCC arising in AK | N | N | N | |||
| SCC arising in AK | N | V | V | N | ||
| SCC arising in CIS | N | V | N | V | R | |
| SCC arising in CIS | N | P | N | |||
NOR, Normal epithelium; HYP, Hyperplastic epithelium; AK, Actinic keratosis/dysplasia; CIS, Carcinoma in situ; EISC, Microinvasion or superficial invasive margin of squamous cell carcinoma; SCC, Deeply invasive squamous cell carcinoma; N, Negative; V, Variegated positive (>25% positive cells); P, All or most cells positive; R, Rare positive cells.
Underlined specimens were tested for HPV (all in this table were negative).
Figure 2.
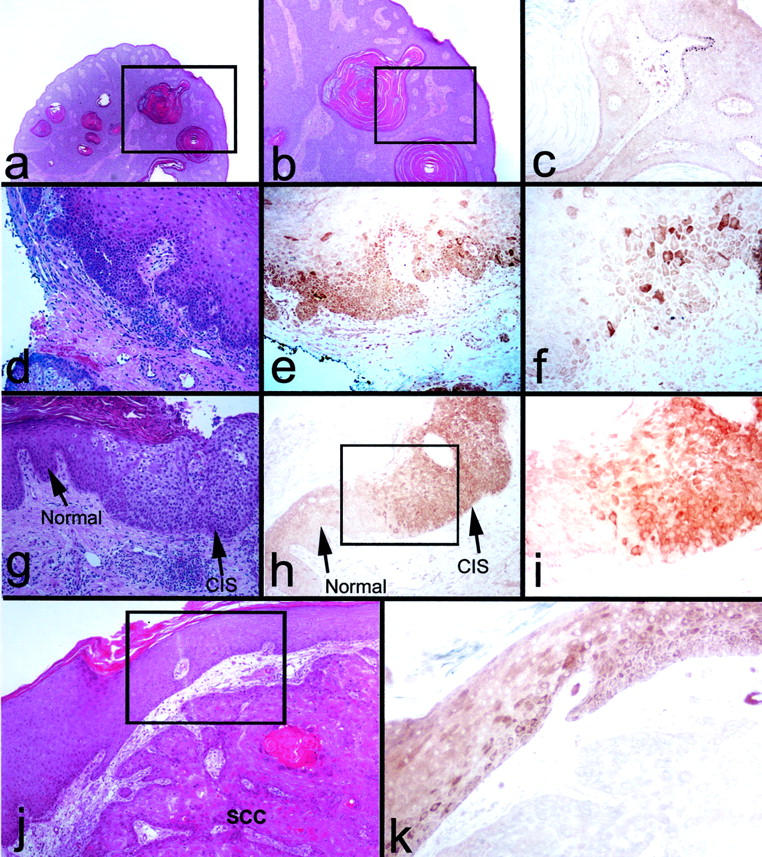
p16 expression in epidermal dysplasia and CIS. a to c: Seborrheic keratosis, exhibiting hyperkeratosis and benign hyperplasia. a and b: H&E. b: p16 immunostain (brown). b: Magnification of area marked in a. c: Magnification of area marked in b. Note weak, homogenous staining present throughout the epithelium (dark spots in some cells in the basal layer are melanin in melanocytes) and in cells of the underlying dermis, interpretable as non-specific background staining caused by prolonged formalin fixation of the specimen. d–f: Dysplastic epithelium with budding into connective tissue, adjacent to invasive SCC (not shown in field). d: H&E. e and f: p16 immunostain (brown). Note heterogeneous, cytoplasmic p16 staining of CIS. g–i: CIS adjacent to normal-appearing epithelium. g: H&E. (h and i) p16 immunostain (brown). i: Magnification of area marked in h. Note variegated p16 staining of CIS. j and k: SCC with overlying dysplastic/atypical epithelium. j: H&E. k: p16 immunostain (brown). k: Magnification of area marked in j. Note that the SCC is p16-negative, while the overlying dysplastic/atypical epithelium exhibits heterogeneous p16 staining.
Thirty-three areas in 18 oral histopathological clinical specimens were identified in H&E-stained sections and analyzed for p16 expression (Table 2 ▶ , Figure 3 ▶ ). The results were similar to those for the skin specimens described above. Three of eleven areas of moderate dysplasia, all three of severe dysplasia, all three areas of CIS and an early invasive area of SCC exhibited variegated p16 staining (Figure 3, a to i) ▶ . One of five areas of deeply invasive SCC contained several scattered p16-positive cells (Figure 3l) ▶ . All eight areas of normal oral epithelium, both benign hyperplasias (Figure 1, i and j) ▶ , and the one mild dysplasia examined were p16-negative.
Table 2.
p16 Immunostaining of Oral Specimens, Set 1
| Specimen diagnosis | Region and p16 immunostaining | |||||||
|---|---|---|---|---|---|---|---|---|
| NOR | HYP | MLD | MOD | SEV | CIS | EISC | SCC | |
| Hyperplasia | N | |||||||
| Mod dysplasia | N | N | ||||||
| Mod dysplasia | N | N | ||||||
| Mod dysplasia | N | |||||||
| Mod dysplasia | N | N | ||||||
| Mod dysplasia | N | V | ||||||
| Mod dysplasia | N | |||||||
| Mod dysplasia | N | N | ||||||
| Mod dysplasia | N | |||||||
| Mod dysplasia | N | V | ||||||
| Severe dysplasia | V | P | ||||||
| Carcinoma in situ | N | P | P | |||||
| Carcinoma in situ | N | N | P | P | ||||
| SCC | N | |||||||
| SCC | R | |||||||
| SCC | N | |||||||
| SCC | N | P | V | N | ||||
| SCC | N | |||||||
NOR, normal epithelium; HYP, hyperplastic epithelium; MLD, mild dysplasia; MOD, moderate dysplasia; SEV, severe dysplasia; CIS, carcinoma in situ; EISC, microinvasion or superficial invasive margins of SCC; SCC, deeply invasive SCC; N, negative; V, variegated positive (>25% positive cells); P, all or most cells positive; R, rare positive cells.
Underlined specimens were tested for HPV (all in this table were negative).
Figure 3.

p16 expression in oral dysplasia, CIS, and superficially invasive SCC. a–c, d–f, g–i, and j–l: Four pathological epithelial specimens stained with H&E (a,d,e,g,h,j) and for p16 (b,c,f,i,k,l). a–c: Moderate dysplasia. c: Magnification of the area demarcated in b. Note variegated p16 staining patterns. d–f and g–i: Two cases of CIS adjacent to normal-appearing epithelium. e and h: Magnifications of the areas demarcated in d and g, respectively. Lines in e and h show the border between histologically normal epithelium and CIS. Note variegated p16 staining in CIS areas in f and homogenous p16 expression in CIS areas in i, with absence of p16 in intervening areas of normal-appearing epithelium. Specimen (g–i) was found by PCR analysis to contain HPV31 DNA (see Results). j–l: Advanced SCC. The upper left corner of j shows the external surface of the tumor and the arrow indicates the point below which islands of SCC cells are growing beneath the normal thickness of the mucosal connective tissue. l: A region of the specimen to the lower right and outside the area shown in j. Note variegated p16 staining in superficial regions and paucity of p16-positive cells in deeper regions of the tumor.
We tentatively concluded from these results that p16 expression in stratified squamous epithelia in vivo does not result from chronic hyperplasia/excessive cell division but, instead, is up-regulated heterogeneously in cells that, by conventional histopathological criteria, are premalignant or at an early stage of invasion into underlying connective tissue.
Increased Incidence of p16-Positive Cells in Areas of Dysplasia, CIS, and Early Invasive SCC
Based on the above results, we tested the hypothesis that p16 expression occurs specifically in dysplastic keratinocytes that have progressed to a stage immediately preceding invasion. We examined a new set of 21 skin and 19 oral specimens, containing a total of 94 histopathologically distinct regions. As summarized in Tables 3 and 4 ▶ , p16-positive cells were most frequently found in regions of CIS (6 of 8) and in epithelial cells at the connective tissue interface of superficial invasion (15 of 18). Five of 17 regions of actinic keratosis also contained p16-positive cells, 4 of these 5 being adjacent to an invasive SCC. Five of 15 deeply invasive regions of SCC contained p16-positive cells. None of 23 normal or hyperplastic epithelial regions expressed p16. These results were in complete agreement with those of our first set of specimens. The combined results from all specimens examined are summarized graphically in Figure 4 ▶ . We concluded that p16 expression is not induced in stratified squamous epithelia as a result of hyperplasia, but is selectively expressed in advanced dysplastic and CIS lesions, typically adjacent to and involved with early connective tissue invasion.
Table 3.
p16 and Laminin 5γ2 Staining of Skin Specimens, Set 2
| Specimen diagnosis | Region and p16 immunostaining | Lam 5γ2 | |||||
|---|---|---|---|---|---|---|---|
| NOR | HYP | AK | CIS | EISC | SCC | ||
| Psoriasiform hyperplasia | N | ||||||
| Actinic keratosis | N | ||||||
| Actinic keratosis | N | ||||||
| Actinic keratosis | N | N | |||||
| Actinic keratosis | N | ||||||
| Actinic keratosis | N | ||||||
| Actinic keratosis | N | ||||||
| Actinic keratosis | N | N | |||||
| Actinic keratosis | V | ||||||
| Carcinoma in situ | N | P | PL | ||||
| SCC arising in AK | N | N | N | N | |||
| SCC arising in AK | N | V | N | N | |||
| SCC arising in AK | N | V | V | R | |||
| SCC arising in AK | N | V | V | N | P | ||
| SCC arising in AK | N | N | V | N | N | ||
| SCC arising in AK | N | N | V | R | P | ||
| SCC arising in AK | N | N | V | N | |||
| SCC arising in AK | N | V | N | ||||
| Early invasive SCC | N | N | V | P | |||
| SCC arising in CIS | N | V | N | P | |||
| Papillary SCC | R | N | |||||
NOR, normal epithelium; HYP, hyperplastic epithelium; AK, actinic keratosis/dysplasia; CIS, carcinoma in situ; EISC, microinvasion or superficial invasive margin of SCC; SCC, deeply invasive SCC; N, negative; V, variegated positive (>25% positive cells); P, all or most cells positive; R, rare positive cells; L, lack of complete colocalization with p16.
Underlined specimens were tested for HPV (all in this table were negative).
Figure 4.

Incidence of p16-positive cells in skin and oral tissue specimens exhibiting various degrees of neoplasia. A: Skin specimens. B: Oral specimens. As described in Results, staining was evaluated for histologically distinct areas within 36 skin and 37 oral specimens, yielding a total of 85 areas in skin specimens and 77 in oral specimens. Numbers on bars indicate the number of areas having the indicated p16 phenotype. Note that normal and benign hyperplastic epithelium was p16-negative, while a large majority of advanced dysplasias and CIS lesions contained p16-positive cells. Microinvasive regions and superficially invading regions at the lateral margins of SCCs were also p16-positive, while deeper regions of SCCs were mostly p16-negative with rare p16-positive cells located primarily in terminally differentiated whorls.
It has been well documented that a fraction of non-cervicogenital SCCs are associated with high-risk and other HPV types. 10,18 Because ectocervical CIS lesions containing DNA of high-risk HPV types have been found to exhibit high levels of p16 expression, 16,17,23 we analyzed a subset of our specimens for the presence of HPV DNA. As shown in Tables 1 to 4 ▶ , only one specimen, an oral CIS, contained detectable HPV DNA (HPV type 31). We concluded that most of the lesions and tumors in our study were not HPV-related and, therefore, that the p16 up-regulation we observed resulted from mechanisms other than the consequence of E7 oncoprotein expression.
Laminin 5γ2 Expression During Neoplastic Progression in Epidermis and Oral Epithelium
The distribution of p16-positive cells at the early invasive regions of SCCs somewhat resembled the expression pattern previously reported for the γ2 chain of the basement membrane protein laminin 5 in SCCs of the skin, and of the oral, esophageal, and cervical mucosa. 35-37,47,48 We immunohistochemically stained sequential sections to determine whether the same cells that express p16 also express γ2. The results for 8 skin and 19 oral mucosal specimens, which included regions ranging from normal epithelium to deeply invasive SCC, are summarized in Tables 3 and 4 ▶ . Fifteen of 17 areas of dysplasia, microinvasion, and superficial margins of advanced SCCs that expressed p16 also expressed γ2, with 13 of these 15 showing clear co-localization of γ2 and p16 (Figure 5, a to f) ▶ . Interestingly, cells at the epithelial-stromal interfaces of deeply invasive SCCs typically expressed neither protein. We concluded that p16 and γ2 typically becomes expressed coordinately in cells at locations just preceding and at the time of initial invasion into connective tissue.
Figure 5.
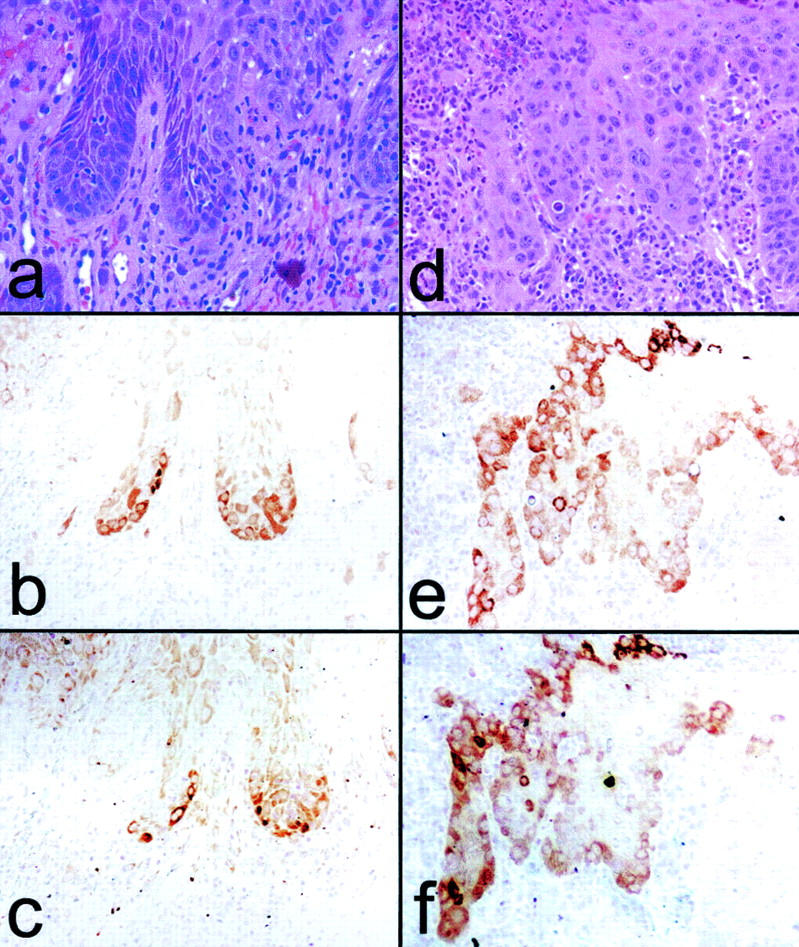
p16 and laminin 5γ2 coexpression in dysplasias and microinvasive SCC. a and d: H&E. b and e: p16 staining. c and f: γ2 staining. a–c: Area of mild-to-moderate dysplasia in oral epithelium adjacent to invasive SCC (outside the area shown). d–f: superficially invasive SCC. Note variegated p16 and γ2 co-expression in epithelial cells at the connective tissue interface at sites of incipient or early invasion.
Expression Pattern of Laminin 5 γ2 in Culture and Correlation with p16 Expression
We next sought to determine whether up-regulation of γ2 expression accompanies that of p16 in cultured keratinocytes. Cessation of division at the end of the replicative lifespan of primary human keratinocytes in culture is tightly linked to the expression and accumulation of p16, such that keratinocytes exhibit an inverse correlation between immunostaining for BUdR incorporation and p16 expression. 29 As shown in Figure 6, a to f ▶ , double-immunofluorescence staining of normal primary human epidermal and oral keratinocytes for p16 and γ2 revealed a perfect correlation between p16 expression and elevated γ2 expression. All keratinocytes displayed some γ2 expression, but p16-positive cells displayed substantially higher intracellular levels of γ2, in a particulate and perinuclear (presumably rough endoplasmic reticulum) location (Figure 6, d to f) ▶ , as expected for a protein destined for secretion. The p16/γ2-positive cells were growth arrested (Figure 6, g to j) ▶ , as expected from previous studies of p16-related keratinocyte senescence in culture. 26,29
Figure 6.
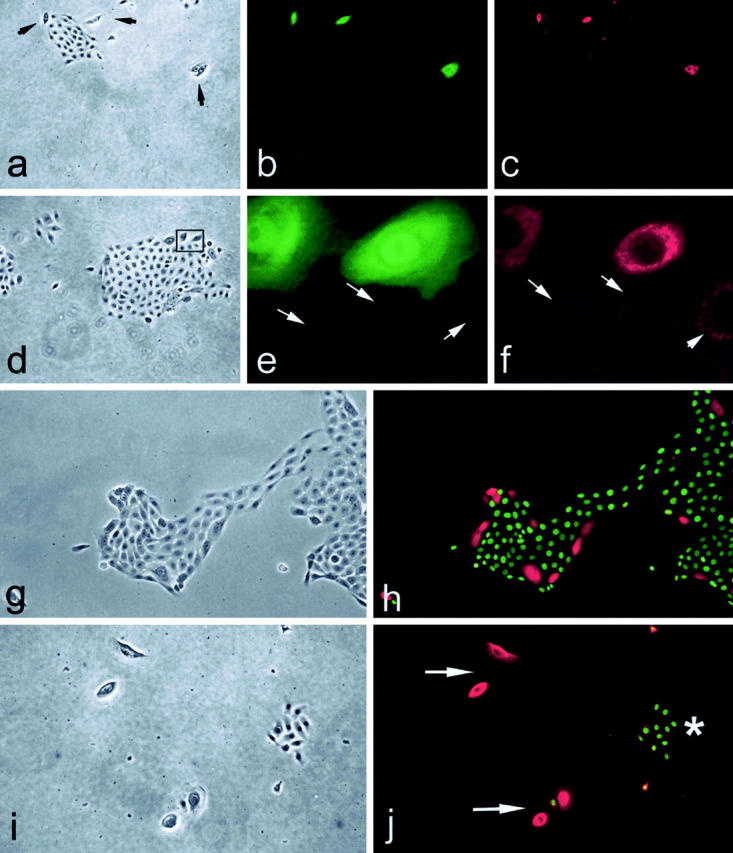
Coexpression of p16 and laminin 5γ2 by senescence-arrested cells in cultures of primary human keratinocytes. Four areas (a–c, d–f, g and h, and i and j) in cultures of strain N keratinocytes. a: Phase contrast. b: p16. c: γ2. Note coexpression of p16 and γ2 by three large cells. d: Phase contrast. e: p16 staining. f: γ2 staining of magnified area in d. Note coexpression of p16 and high levels of γ2 by large cells with adjacent smaller cells (arrows in e and f) being p16-negative and with low levels of γ2. g: Phase contrast. h: Double immunostaining for p16 (red) and BUdR (green). Note inverse correlation between cell division and p16 expression, as previously described. 29 i: Phase contrast. j: Double immunostaining for γ2 (red) and BUdR (green). Note inverse correlation between cell division and γ2 expression, with small, dividing keratinocytes shown with asterisk and large, growth-arrested keratinocytes shown by arrows.
Migrating keratinocytes in culture leave tracks of laminin 5 behind them on the substratum, identifiable by immunostaining with laminin 5-specific antibodies. 49-51 We therefore examined γ2 and p16 expression and motility in cultures of the normal primary oral keratinocyte line OKB8, the normal primary epidermal keratinocyte line strain N, and the TERT-immortalized strain N line N/TERT-2G, all of which heterogeneously produce p16-positive, growth-arrested cells during serial passage ( 26 and our unpublished results). As shown in Figure 7 ▶ , isolated p16/γ2-positive cells, as well as small colonies of p16-negative cells that contained at least one p16/γ2-positive cell, exhibited a great increase in directional motility compared to the rather static behavior of p16-negative individual cells and colonies. The p16-positive cells in motile colonies tended to be at the leading edge of the colony (Figure 7, d to i) ▶ , as expected if these cells were providing the force for and determining the direction of migration. We concluded that p16/γ2-coexpression also occurs in non-neoplastic normal primary or TERT-immortalized keratinocytes in culture, associated with growth arrest and increased directional motility. This result demonstrated that the mechanism responsible for inducing this phenotype is not restricted to neoplastic progression and can be triggered by mechanisms functional in normal keratinocytes.
Figure 7.

Coexpression of p16 and laminin 5γ2 by keratinocytes in culture; correlation with directional motility. Panels show OKB8 (a–c), strain N (d–f, j–l, m–o), and N/TERT-2G (g–i) keratinocyte cultures immunostained for p16 (a,d,g,j,o) or γ2 (b,c,e,f,h,i,k,l,n). m: Phase contrast view of the field shown in n. Inset in o: Phase contrast view of the field shown in o. Panels c, f, i, and l are overexposed, grayscale versions of b, e, h, and k, respectively, revealing the migration history of cells by their deposition of γ2 on the culture dish surface. Note coexpression of p16 and γ2 and substantial directional motility of single p16/γ2-positive cells and small colonies that contain one or more p16/γ2-positive cells. Note up-regulation of γ2 and p16 by the cells at the edge of experimental wounds made in a confluent monolayer of early passage keratinocytes.
Expression of Laminin 5γ2 and p16 by Keratinocytes at Wound Edges in Culture
Keratinocytes near the edge of a wound in vivo express high levels of laminin 5. 49,52-55 We sought to determine whether keratinocytes responding to a wound in culture up-regulate γ2 expression and whether p16 expression is temporally associated with this. We created ∼2-mm-wide wounds in confluent cell sheets of early passage, p16-negative primary keratinocytes, and immunostained the cultures 24 hours later for p16 and γ2. The first, and sometimes second, row of cells at the edge of the wound displayed up-regulation of p16 and γ2 (Figure 7, m to o) ▶ . We concluded that induction of expression of both of these proteins can occur abruptly as the result of a change in spatial organization of a region of epithelium, unassociated either with replicative senescence or genetic changes related to cancer progression. These results are consistent with the possibility that the microenvironment of early invasion triggers in neoplastic keratinocytes the same mechanism that is activated in normal keratinocytes in response to a wound.
Discussion
These experiments have revealed the timing and location of p16 expression during neoplastic progression in the epidermis and oral mucosal epithelium. Our results show clearly that p16 expression is not induced by chronic hyperplasia but instead is induced in cells that have progressed to the stage of incipient or initial invasion into underlying connective tissue. The finding of coordinate p16 and γ2 expression at sites of severe dysplasia and microinvasion in vivo, as well as in senescent keratinocytes and young wound-edge keratinocytes in culture, suggests an unexpected and interesting connection between a biological response of normal keratinocytes and that of keratinocytes in lesions making the transition from premalignant dysplasia to invasive carcinoma.
We found no p16-positive cells in five seborrheic keratoses and four alveolar ridge hyperkeratoses, two conditions that are considered to be almost invariably benign. 56 Our initial hypothesis, that the inevitable induction of p16 expression and consequent permanent growth arrest observed in normal keratinocytes after 25 to 60 population doublings in culture is a manifestation of a p16-dependent mechanism to limit stem cell expansion potential in vivo, was not supported. Rather, p16 expression was tightly associated with neoplasia. We initially suspected that the homogenous p16 expression in several oral and epidermal CIS lesions may have been the consequence of HPV involvement, as reported recently for cervical CIS lesions 16,17,23 and for verrucous carcinomas of the skin. 57 However, we found HPV DNA in only 1 of 6 oral and skin CIS specimens. Thus, the pattern of p16 expression we observed resulted from cellular mechanisms, unconnected to HPV viral oncogene activity.
Consistent with previous p16 allelic analyses of head-and-neck SCCs, 8,13,14 we found a complete absence of, or at most very rare, p16-positive cells in deeply invasive SCCs, even at the epithelial-stromal interface. In contrast, we found p16 expression in many cells at the superficial circumferential margins of advanced SCCs. Such p16-positive cells may either be premalignant cells of the field from which the adjacent SCC had arisen, or normal keratinocytes admixed with invasive SCC cells at the expanding tumor margin that are responding to the destabilized basement membrane by activating a p16/γ2 wound response. Distinguishing between these alternatives will be difficult, as it will require identifying and then microdissecting and PCR-analyzing the p16 locus of individual p16-positive and p16-negative cells at tumor margins.
Laminin 5 is a component of epithelial basement membranes. 50,58 Normal stratified squamous epithelia secrete and maintain an intact, continuous laminin 5 layer beneath them, on which they assemble stable, anchoring hemidesmosome structures. 59 Normally, the basal cell layer expresses very low levels of laminin 5 proteins, sufficient to maintain the basement membrane structure. In contrast, wounded epithelia 49,52-55 and invading regions of epidermal, oral, esophageal, and cervical SCC ( 35,36,47,48 and our present study) display a great increase in laminin 5 synthesis. The question of the molecular mechanisms underlying a migratory response versus a sessile response to a laminin 5 substratum remains unresolved. The protein has been reported to be migration-promoting and migration-inhibiting in culture in experiments having as variables different cell types, cell lines, culture conditions, and source and purity of the laminin 5 preparation used. 42,51,60-63 Laminin 5 is secreted from cells as a heterotrimeric precursor; the α3 subunit is subject to extracellular proteolytic cleavage by plasmin 42,64 and the γ2 subunit by MMP-2, 62 MT1-MMP, 63 and BMP-1. 65 Some of these proteases and their respective protease inhibitors are produced by the keratinocytes themselves, some are circulating, and some are produced by stromal cells. 66,67
Current evidence appears to best support a model of the wound response in which keratinocytes convert from an anchored state favored by α6β4 integrin adhesion to normal, mature laminin 5 to a motile state favored by α3β1 integrin adhesion to precursor laminin 5. 49,52 It has been proposed that further cleavage of the mature γ2 chain by tumor cell proteases produces a form of laminin associated with stromal invasive behavior in development, tissue remodeling, and cancer. 62,65 Our findings, and those reported previously, of induced γ2 expression in keratinocytes adjacent to a degraded or remodeling basement membrane, do not specifically support a hypothesis that γ2 production promotes invasion. The cells in regions of invasion may be responding to a lack of stable anchorage by increased synthesis and secretion of basement membrane protein in a futile attempt at stable cell-substratum adhesion. Our cell culture studies, however, revealed a close correlation between increased γ2 synthesis and increased directional motility, suggesting that the laminin 5 produced is a form permissive for motility.
Although the functional role of γ2 in neoplasia remains unknown, we found it to be expressed in epithelial regions that also expressed p16. Coordinate expression of p16 and γ2 in normal keratinocytes in culture was associated with growth arrest and enhanced directional motility. This occurred in two very different settings: in preconfluent cultures at senescence and in confluent cultures of early passage cells at the edge of an experimental wound. More detailed studies of p16/γ2 expression in cultured keratinocytes is in progress and will be presented elsewhere (E. Natarajan, J. Omobono, and J. Rheinwald, unpublished), but our results suggest to us that p16-enforced growth arrest as a suppressor of neoplastic progression in stratified squamous epithelia is triggered by activation of a mechanism that evolved originally to hasten wound closure in normal tissue. We speculate that sacrifice, by p16 induction, of the proliferative potential of cells confronting a wound edge facilitates the uninterrupted directional motility of these cells, which consequently can more efficiently pull their still-proliferative neighbors behind them across the wound surface.
It remains to be determined whether the same initial signal and pathway leading to activation of the p16/γ2 mechanism in early invasive carcinoma is the same as that in wounded normal epithelium, and how this may be related to keratinocyte replicative senescence in culture. It will be interesting to compare the patterns of p16 and γ2 expression in acute and chronic non-healing wounds in vivo, to determine whether the mechanism we have observed in culture goes awry in an understandable way in the latter situation. The p16-related growth arrest mechanism appears to block the proliferation of cells that are exposed to an abnormal epithelial-stromal interface, as occurs preceding and during invasion. Our data support a model in which p16 expression is activated in cells in response to basement membrane degradation/invasive behavior, whether of their own making or caused by neighboring cells. Successful SCCs would represent the progeny of rare cells arising within such lesions that retain migratory/invasive character while losing expression of p16, thereby simultaneously permitting cell division and invasion.
Table 4.
p16 and Laminin 5γ2 Staining of Oral Specimens, Set 2
| Specimen diagnosis | Region and p16 immunostaining | Lam 5γ2 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| NOR | HYP | MLD | MOD | SEV | CIS | EISC | SCC | ||
| Ridge keratosis | N | N | N | ||||||
| Ridge keratosis | N | N | N | ||||||
| Ridge keratosis | N | N | N | ||||||
| Ridge keratosis | N | N | N | ||||||
| Mod-Sev dysplasia | N | N | |||||||
| Mod-Sev dysplasia | N | N | |||||||
| Severe dysplasia | N | N | |||||||
| Carcinoma in situ* | N | P | PL | ||||||
| Early invasive SCC | V | V | P | ||||||
| Early invasive SCC | N | P | N | ||||||
| Early invasive SCC | N | P | V | P | |||||
| Early invasive SCC | N | N | V | P | |||||
| Early invasive SCC | P | N | P | ||||||
| Early invasive SCC | V | V | P | ||||||
| SCC | V | R | P | ||||||
| SCC | N | P | V | N | P | ||||
| SCC | N | V | N | P | |||||
| SCC | N | N | N | P | V | N | P | ||
| SCC | N | R | N | ||||||
NOR, normal epithelium; HYP, hyperplastic epithelium; MLD, mild dysplasia; MOD, moderate dysplasia; SEV, severe dysplasia; CIS, carcinoma in situ; EISC, microinvasion or superficial invasive margins of SCC; SCC, deeply invasive SCC; N, negative; V, variegated positive (>25% positive cells); P, all or most cells positive; R, rare positive cells; L, lack of complete colocalization with p16.
Underlined specimens were tested for HPV (all in this table were negative, except for specimen marked with asterisk
(*), which tested positive for HPV31).
Acknowledgments
We thank Denise Long-Woodward, Harvard Skin Disease Research Center, for preparing H&E-stained and unstained sections of tissue specimens. Jay Omobono, Sarah Browne, and Matt Ramsey provided expert technical assistance with immunocytochemical staining and cell culture. We thank Jim Koh (University of Vermont) and Jonathan Jones (Northwestern University) for generously providing p16 and Laminin 5 γ2 antibodies.
Footnotes
Address reprint requests to James G. Rheinwald, Harvard Institutes of Medicine, Room 664, 77 Avenue Louis Pasteur, Boston, MA 02115. E-mail: jrheinwald@rics.bwh.harvard.edu.
Supported by grant R01-DE13178 from the National Institute of Dental and Craniofacial Research (to J.G.R.) and by a Skin Disease Research Center grant (P30-AR42689) from the National Institute of Arthritis, Musculoskeletal and Skin Diseases.
References
- 1.Bouquot JE, Weiland LH, Kurland LT: Leukoplakia and carcinoma in situ synchronously associated with invasive oral/oropharyngeal carcinoma in Rochester, Minn, 1935–1984. Oral Surg Oral Med Oral Pathol 1988, 65:199-207 [DOI] [PubMed] [Google Scholar]
- 2.Mashberg A, Feldman LJ: Clinical criteria for identifying early oral and oropharyngeal carcinoma: erythroplasia revisited. Am J Surg 1988, 156:273-275 [DOI] [PubMed] [Google Scholar]
- 3.Lee JJ, Hong WK, Hittelman WN, Mao L, Lotan R, Shin DM, Benner SE, Xu XC, Lee JS, Papadimitrakopoulou VM, Geyer C, Perez C, Martin JW, El-Naggar AK, Lippman SM: Predicting cancer development in oral leukoplakia: ten years of translational research. Clin Cancer Res 2000, 6:1702-1710 [PubMed] [Google Scholar]
- 4.Kamb A, Gruis NA, Weaver-Feldhaus J, Liu Q, Harshman K, Tavtigian SV, Stockert E, Day RS, III, Johnson BE, Skolnick MH: A cell cycle regulator potentially involved in genesis of mamny tumor types. Science 1994, 264:436-440 [DOI] [PubMed] [Google Scholar]
- 5.Yeudall WA, Crawford RY, Ensley JF, Robbins KC: MTS1/CDK4I is altered in cell lines derived from primary and metastatic oral squamous cell carcinoma. Carcinogenesis 1994, 15:2683-2686 [DOI] [PubMed] [Google Scholar]
- 6.Merlo A, Herman JG, Mao L, Lee DJ, Gabrielson E, Burger PC, Baylin SB, Sidransky D: 5[prime] CpG island methylation is associated with transcriptional silencing of the tumour suppressor p16/CDKN2/MTS1 in human cancers. Nat Med 1995, 1:686-692 [DOI] [PubMed] [Google Scholar]
- 7.Shapiro GI, Park JE, Edwards CD, Mao L, Merlo A, Sidransky D, Ewen ME, Rollins BJ: Multiple mechanisms of p16INK4A inactivation in non-small cell lung cancer cell lines. Cancer Res 1995, 55:6200-6209 [PubMed] [Google Scholar]
- 8.Reed AL, Califano J, Cairns P, Westra WH, Jones RM, Koch W, Ahrendt S, Eby Y, Sewell D, Nawroz H, Bartek J, Sidransky D: High frequency of p16 (CDKN2/MTS-1/INK4A) inactivation in head and neck squamous cell carcinoma. Cancer Res 1996, 56:3630-3633 [PubMed] [Google Scholar]
- 9.Califano J, van der Riet P, Westra W, Nawroz H, Clayman G, Piantadosi S, Corio R, Lee D, Greenberg B, Koch W, Sidransky D: Genetic progression model for head and neck cancer: implications for field cancerization. Cancer Res 1996, 56:2488-2492 [PubMed] [Google Scholar]
- 10.Olshan AF, Weissler MC, Pei H, Conway K, Anderson S, Fried DB, Yarbrough WG: Alterations of the p16 gene in head and neck cancer: frequency and association with p53, PRAD-1 and HPV Oncogene 1997, 14:811-818 [DOI] [PubMed] [Google Scholar]
- 11.Papadimitrakopoulou V, Izzo J, Lippman SM, Lee JS, Fan YH, Clayman G, Ro JY, Hittelman WN, Lotan R, Hong WK, Mao L: Frequent inactivation of p16/INK4a in oral premalignant lesions. Oncogene 1997, 14:1799-1803 [DOI] [PubMed] [Google Scholar]
- 12.Soufir N, Daya-Grosjean L, de La Salmoniere P, Moles JP, Dubertret L, Sarasin A, Basset-Seguin N: Association between INK4a-ARF and p53 mutations in skin carcinomas of xeroderma pigmentosum patients. J Natl Cancer Inst 2000, 92:1841-1847 [DOI] [PubMed] [Google Scholar]
- 13.Poi MJ, Yen T, Li J, Song H, Lang JC, Schuller DE, Pearl DK, Casto BC, Tsai MD, Weghorst CM: Somatic INK4a-ARF locus mutations: a significant mechanism of gene inactivation in squamous cell carcinomas of the head and neck. Mol Carcinog 2001, 30:26-36 [DOI] [PubMed] [Google Scholar]
- 14.Rocco JW, Sidransky D: p16(MTS-1/CDKN2/INK4a) in cancer progression. Exp Cell Res 2001, 264:42-55 [DOI] [PubMed] [Google Scholar]
- 15.Sherr CJ: Cancer cell cycles. Science 1996, 274:1672-1677 [DOI] [PubMed] [Google Scholar]
- 16.Sano T, Oyama T, Kashiwabara K, Fukuda T, Nakajima T: Expression status of p16 protein is associated with human papillomavirus oncogenic potential in cervical and genital lesions. Am J Pathol 1998, 153:1741-1748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Keating JT, Cviko A, Riethdorf S, Riethdorf L, Quade BJ, Sun D, Duensing S, Sheets EE, Munger K, Crum CP: Ki-67, cyclin E, and p16INK4 are complimentary surrogate biomarkers for human papilloma virus-related cervical neoplasia. Am J Surg Pathol 2001, 25:884-891 [DOI] [PubMed] [Google Scholar]
- 18.Gillison ML, Koch WM, Capone RB, Spafford M, Westra WH, Wu L, Zahurak ML, Daniel RW, Viglione M, Symer DE, Shah KV, Sidransky D: Evidence for a causal association between human papillomavirus and a subset of head and neck cancers. J Natl Cancer Inst 2000, 92:709-720 [DOI] [PubMed] [Google Scholar]
- 19.Gruis NA, van der Velden PA, Sandkuijl LA, Prins DE, Weaver-Feldhaus J, Kamb A, Bergman W, Frants RR: Homozygotes for CDKN2 (p16) germline mutation in Dutch familial melanoma kindreds. Nat Genet 1995, 10:351-353 [DOI] [PubMed] [Google Scholar]
- 20.Sharpless NE, Bardeesy N, Lee KH, Carrasco D, Castrillon DH, Aguirre AJ, Wu EA, Horner JW, DePinho RA: Loss of p16/Ink4a with retention of p19Arf predisposes mice to tumorigenesis. Nature 2001, 413:86-91 [DOI] [PubMed] [Google Scholar]
- 21.Rane SG, Cosenza SC, Mettus RV, Reddy EP: Germ line transmission of the Cdk4(R24C) mutation facilitates tumorigenesis and escape from cellular senescence. Mol Cell Biol 2002, 22:644-656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nielsen GP, Stemmer-Rachamimov AO, Shaw J, Roy JE, Koh J, Louis DN: Immunohistochemical survey of p16INK4A expression in normal human adult and infant tissues. Lab Invest 1999, 79:1137-1143 [PubMed] [Google Scholar]
- 23.Klaes R, Friedrich T, Spitkovsky D, Ridder R, Rudy W, Petry U, Dallenbach-Hellweg G, Schmidt D, von Knebel Doeberitz M: Overexpression of p16(INK4A) as a specific marker for dysplastic and neoplastic epithelial cells of the cervix uteri. Int J Cancer 2001, 92:276-284 [DOI] [PubMed] [Google Scholar]
- 24.Lee G, Park BS, Han SE, Oh JE, You YO, Baek JH, Kim GS, Min BM: Concurrence of replicative senescence and elevated expression of p16(INK4A) with subculture-induced but not calcium-induced differentiation in normal human oral keratinocytes. Arch Oral Biol 2000, 45:809-818 [DOI] [PubMed] [Google Scholar]
- 25.Kiyono T, Foster SA, Koop JI, McDougall JK, Galloway DA, Klingelhutz AJ: Both Rb/p16INK4a inactivation and telomerase activity are required to immortalize human epithelial cells. Nature 1998, 396:84-88 [DOI] [PubMed] [Google Scholar]
- 26.Dickson MA, Hahn WC, Ino Y, Ronfard V, Wu JY, Weinberg RA, Louis DN, Li FP, Rheinwald JG: Human keratinocytes that express hTERT and also bypass a p16(INK4a)-enforced mechanism that limits life span become immortal yet retain normal growth and differentiation characteristics. Mol Cell Biol 2000, 20:1436-1447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Loughran O, Malliri A, Owens D, Gallimore PH, Stanley MA, Ozanne B, Frame MC, Parkinson EK: Association of CDKN2A/p16INK4A with human head and neck keratinocyte replicative senescence: relationship of dysfunction to immortality and neoplasia. Oncogene 1996, 13:561-568 [PubMed] [Google Scholar]
- 28.Munro J, Stott FJ, Vousden KH, Peters G, Parkinson EK: Role of the alternative INK4A proteins in human keratinocyte senescence: evidence for the specific inactivation of p16INK4A upon immortalization. Cancer Res 1999, 59:2516-2521 [PubMed] [Google Scholar]
- 29.Rheinwald JG, Hahn WC, Ramsey MR, Wu JY, Guo Z, Tsao H, De Luca M, Catricala C, O’Toole KM: A two-stage, p16(INK4A)- and p53-dependent keratinocyte senescence mechanism that limits replicative potential independent of telomere status. Mol Cell Biol 2002, 22:5157-5172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Farwell DG, Shera KA, Koop JI, Bonnet GA, Matthews CP, Reuther GW, Coltrera MD, McDougall JK, Klingelhutz AJ: Genetic and epigenetic changes in human epithelial cells immortalized by telomerase. Am J Pathol 2000, 156:1537-1547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ramirez RD, Morales CP, Herbert BS, Rohde JM, Passons C, Shay JW, Wright WE: Putative telomere-independent mechanisms of replicative aging reflect inadequate growth conditions. Genes Dev 2001, 15:398-403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bond JA, Wyllie FS, Wynford-Thomas D: Escape from senescence in human diploid fibroblasts induced directly by mutant p53. Oncogene 1994, 9:1885-1889 [PubMed] [Google Scholar]
- 33.Brown JP, Wei W, Sedivy JM: Bypass of senescence after disruption of p21CIP1/WAF1 gene in normal diploid human fibroblasts. Science 1997, 277:831-834 [DOI] [PubMed] [Google Scholar]
- 34.Stein GH, Drullinger LF, Soulard A, Dulic V: Differential roles for cyclin-dependent kinase inhibitors p21 and p16 in the mechanisms of senescence and differentiation in human fibroblasts. Mol Cell Biol 1999, 19:2109-2117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pyke C, Salo S, Ralfkiaer E, Romer J, Dano K, Tryggvason K: Laminin-5 is a marker of invading cancer cells in some human carcinomas and is coexpressed with the receptor for urokinase plasminogen activator in budding cancer cells in colon adenocarcinomas. Cancer Res 1995, 55:4132-4139 [PubMed] [Google Scholar]
- 36.Kainulainen T, Autio-Harmainen H, Oikarinen A, Salo S, Tryggvason K, Salo T: Altered distribution and synthesis of laminin-5 (kalinin) in oral lichen planus, epithelial dysplasias and squamous cell carcinomas. Br J Dermatol 1997, 136:331-336 [PubMed] [Google Scholar]
- 37.Lohi J: Laminin-5 in the progression of carcinomas. Int J Cancer 2001, 94:763-767 [DOI] [PubMed] [Google Scholar]
- 38.Pirisi L, Yasumoto S, Feller M, Doniger J, DiPaolo JA: Transformation of human fibroblasts and keratinocytes with human papillomavirus type 16 DNA. J Virol 1987, 61:1061-1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schön M, Rheinwald JG: A limited role for retinoic acid and retinoic acid receptors RAR alpha and RAR beta in regulating keratin 19 expression and keratinization in oral and epidermal keratinocytes. J Invest Dermatol 1996, 107:428-438 [DOI] [PubMed] [Google Scholar]
- 40.Hudson DL, Weiland KL, Dooley TP, Simon M, Watt FM: Characterisation of eight monoclonal antibodies to involucrin. Hybridoma 1992, 11:367-379 [DOI] [PubMed] [Google Scholar]
- 41.Mizushima H, Koshikawa N, Moriyama K, Takamura H, Nagashima Y, Hirahara F, Miyazaki K: Wide distribution of laminin-5 γ2 chain in basement membranes of various human tissues. Horm Res 1998, 50(Suppl 2):7-14 [DOI] [PubMed] [Google Scholar]
- 42.Goldfinger LE, Stack MS, Jones JC: Processing of laminin-5 and its functional consequences: role of plasmin and tissue-type plasminogen activator. J Cell Biol 1998, 141:255-265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Green H, Kehinde O, Thomas J: Growth of cultured human epidermal cells into multiple epithelia suitable for grafting. Proc Natl Acad Sci USA 1979, 76:5665-5668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fetsch PA, Abati A: Overview of the clinical immunohistochemistry laboratory: regulations and troubleshooting guidelines. Methods Mol Biol 1999, 115:405-414 [DOI] [PubMed] [Google Scholar]
- 45.Gravitt PE, Manos MM: Polymerase chain reaction-based methods for the detection of human papillomavirus DNA. IARC Sci Publ 1992, :121-133 [PubMed] [Google Scholar]
- 46.Lungu O, Wright TC, Jr, Silverstein S: Typing of human papillomaviruses by polymerase chain reaction amplification with L1 consensus primers and RFLP analysis. Mol Cell Probes 1992, 6:145-152 [DOI] [PubMed] [Google Scholar]
- 47.Ono Y, Nakanishi Y, Ino Y, Niki T, Yamada T, Yoshimura K, Saikawa M, Nakajima T, Hirohashi S: Clinocopathologic significance of laminin-5 γ2 chain expression in squamous cell carcinoma of the tongue: immunohistochemical analysis of 67 lesions. Cancer 1999, 85:2315-2321 [PubMed] [Google Scholar]
- 48.Yamamoto H, Itoh F, Iku S, Hosokawa M, Imai K: Expression of the γ2 chain of laminin-5 at the invasive front is associated with recurrence and poor prognosis in human esophageal squamous cell carcinoma. Clin Cancer Res 2001, 7:896-900 [PubMed] [Google Scholar]
- 49.Nguyen BP, Ryan MC, Gil SG, Carter WG: Deposition of laminin 5 in epidermal wounds regulates integrin signaling and adhesion. Curr Opin Cell Biol 2000, 12:554-562 [DOI] [PubMed] [Google Scholar]
- 50.Rousselle P, Lunstrum GP, Keene DR, Burgeson RE: Kalinin: an epithelium-specific basement membrane adhesion molecule that is a component of anchoring filaments. J Cell Biol 1991, 114:567-576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang K, Kramer RH: Laminin-5 deposition promotes keratinocyte motility. Exp Cell Res 1996, 227:309-322 [DOI] [PubMed] [Google Scholar]
- 52.Larjava H, Salo T, Haapasalmi K, Kramer RH, Heino J: Expression of integrins and basement membrane components by wound keratinocytes. J Clin Invest 1993, 92:1425-1435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kainulainen T, Hakkinen L, Hamidi S, Larjava K, Kallioinen M, Peltonen J, Salo T, Larjava H, Oikarinen A: Laminin-5 expression is independent of the injury and the microenvironment during reepithelialization of wounds. J Histochem Cytochem 1998, 46:353-360 [DOI] [PubMed] [Google Scholar]
- 54.Goldfinger LE, Hopkinson SB, deHart GW, Collawn S, Couchman JR, Jones JC: The α3 laminin subunit, α6β4 and α3β1 integrin coordinately regulate wound healing in cultured epithelial cells and in the skin. J Cell Sci 1999, 112:2615-2629 [DOI] [PubMed] [Google Scholar]
- 55.Ryan MC, Tizard R, VanDevanter DR, Carter WG: Cloning of the LamA3 gene encoding the α3 chain of the adhesive ligand epiligrin: expression in wound repair. J Biol Chem 1994, 269:22779-22787 [PubMed] [Google Scholar]
- 56.Anderson PJ, Zuk JA, Rao GS, Berry RB: Squamous cell carcinoma arising within seborrheic keratosis. Plast Reconstr Surg 1998, 102:453-455discussion 456–458 [DOI] [PubMed] [Google Scholar]
- 57.Saito T, Nakajima T, Mogi K: Immunohistochemical analysis of cell cycle-associated proteins p16, pRb, p53, p27 and Ki-67 in oral cancer and precancer with special reference to verrucous carcinomas. J Oral Pathol Med 1999, 28:226-232 [DOI] [PubMed] [Google Scholar]
- 58.Carter WG, Ryan MC, Gahr PJ: Epiligrin, a new cell adhesion ligand for integrin α3β1 in epithelial basement membranes. Cell 1991, 65:599-610 [DOI] [PubMed] [Google Scholar]
- 59.Jones JC, Hopkinson SB, Goldfinger LE: Structure and assembly of hemidesmosomes. Bioessays 1998, 20:488-494 [DOI] [PubMed] [Google Scholar]
- 60.Baker SE, DiPasquale AP, Stock EL, Quaranta V, Fitchmun M, Jones JC: Morphogenetic effects of soluble laminin-5 on cultured epithelial cells and tissue explants. Exp Cell Res 1996, 228:262-270 [DOI] [PubMed] [Google Scholar]
- 61.O’Toole EA, Marinkovich MP, Hoeffler WK, Furthmayr H, Woodley DT: Laminin-5 inhibits human keratinocyte migration. Exp Cell Res 1997, 233:330-339 [DOI] [PubMed] [Google Scholar]
- 62.Giannelli G, Falk-Marzillier J, Schiraldi O, Stetler-Stevenson WG, Quaranta V: Induction of cell migration by matrix metalloprotease-2 cleavage of laminin-5. Science 1997, 277:225-228 [DOI] [PubMed] [Google Scholar]
- 63.Koshikawa N, Giannelli G, Cirulli V, Miyazaki K, Quaranta V: Role of cell surface metalloprotease MT1-MMP in epithelial cell migration over laminin-5. J Cell Biol 2000, 148:615-624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Marinkovich MP, Lunstrum GP, Burgeson RE: The anchoring filament protein kalinin is synthesized and secreted as a high molecular weight precursor. J Biol Chem 1992, 267:17900-17906 [PubMed] [Google Scholar]
- 65.Amano S, Scott IC, Takahara K, Koch M, Champliaud MF, Gerecke DR, Keene DR, Hudson DL, Nishiyama T, Lee S, Greenspan DS, Burgeson RE: Bone morphogenetic protein 1 is an extracellular processing enzyme of the laminin 5 γ2 chain. J Biol Chem 2000, 275:22728-22735 [DOI] [PubMed] [Google Scholar]
- 66.Lukashev ME, Werb Z: ECM signalling: orchestrating cell behaviour and misbehaviour. Trends Cell Biol 1998, 8:437-441 [DOI] [PubMed] [Google Scholar]
- 67.Seiki M: The cell surface: the stage for matrix metalloproteinase regulation of migration. Curr Opin Cell Biol 2002, 14:624-632 [DOI] [PubMed] [Google Scholar]