

Abstract
The fibrinolytic system is known to play an important role in the inflammatory response to bacterial infections. In the present study, relationships between protein components of the fibrinolytic system and infectivity by Mycobacterium avium were analyzed. Infections were initiated through noninvasive intratracheal administration of M. avium 724 in mice individually deficient for plasminogen, tissue-type plasminogen activator, urokinase-type plasminogen activator, and urokinase-type plasminogen activator receptor, along with wild-type control mice. There were no differences in lung colony counts among all mouse genotypes throughout a 10-week infection. However, in tissue-type plasminogen activator and plasminogen-deficient mice an earlier dissemination of M. avium to other organs was observed. Nevertheless, the M. avium growth rates in the liver, spleen, and lung did not differ between the various mouse populations throughout a 10-week infection. Histochemical and immunohistochemical analyses at 5 and 10 weeks after infection demonstrated that plasminogen-deficient mice, compared to wild-type mice, had enhanced fibrin and fibronectin deposition, as well as increased neutrophil infiltration within liver granulomas. These results suggest that plasmin(ogen) plays a role in the turnover of extracellular matrix proteins within granulomas and has a limited effect in the early dissemination of M. avium from lungs. Thus, plasmin(ogen) functions in limiting progressive fibrosis in the granuloma during a chronic mycobacterial infection.
Mycobacteria are the etiological agents of numerous human and animal diseases, including tuberculosis and Mycobacterium avium complex. The lung is considered a major entry point for many pathogenic mycobacteria. Control of the mycobacterial infection in the lung requires granuloma formation and a proper Th1 cell response. Evidence suggests that dissemination of mycobacteria to the draining lymph nodes is required for initiating an effective T cell response. 1 The mycobacteria can then disseminate to other organs, such as the liver and spleen. The manner in which mycobacteria gain access to the lymph nodes is unclear, but may involve migration of infected macrophages or dendritic cells through the lymphatic system. 2 Degradation of extracellular matrix (ECM) proteins may play a role in this dissemination process. Further, migration of macrophages and other leukocytes to the site of a mycobacteria infection, as well as formation of a granulomatous response, require degradation of ECM and basement membrane. 3 Deposition of a proper ECM may also be important in maintaining the correct structure of a granuloma and in retaining the recruited leukocytes within the granuloma.
One important class of proteins involved in ECM and basement membrane degradation and remodeling are the fibrinolytic proteins. These include the zymogen, plasminogen (Pg), which is activated to a serine protease, plasmin (Pm), an enzyme that degrades the fibrin clot. At least two distinct serine protease-related Pg activators exist in mammals, namely, tissue-type plasminogen activator (tPA) and urokinase-type plasminogen activator (uPA). A cellular uPA receptor (uPAR) stimulates Pg activation on the cell surface. Regulation of this system at the protein level exists through inhibition of Pg activators by the serpins, PAI-1 and PAI-2, and via inhibition of Pm by another serpin, α2-antiplasmin. 4 Pm not only catalyzes lysis of fibrin(ogen), but also functions in a variety of pathophysiological processes, including degradation of the ECM and basement membrane. 5
Various studies have shown that pathogenic bacteria, such as group A, C, and G streptococci and Staphylococcus aureus, can activate Pg, 4,6 and it has been proposed that cell surface-bound Pm provides a possible mechanism for the acquisition of protease activity to assist in bacterial invasion of the surrounding matrix. 7 Binding of Pg and Pm to a variety of cell types occurs, and principally involves interactions of the lysine binding sites of kringle domains of Pg/Pm with appropriate ligands. One obvious protein-based ligand is the carboxyl-terminal lysine residue that exists on several Pg-binding proteins, 8-10 but an internal psuedo-lysine arrangement was found to be responsible for the high-affinity binding between Pg and the M-like protein from group A streptococci. 11-13 Recent studies also indicate that M. tuberculosis can bind and activate Pg. 14 Unlike most of the infectious agents that possess Pg/Pm receptors, mycobacteria are intracellular pathogens. Therefore, the role of Pg activation in mycobacterial pathogenesis and dissemination is less clear.
The fibrinolytic system has also been found to be important in immune and inflammatory responses. Urokinase-deficient (UPA−/−) mice showed increased susceptibility to Pneumocystis carinii and decreased levels of lymphocytes and macrophages were found in the lungs. 15 A similar finding was observed in UPA−/− mice infected with Cryptococcus neoformans. 16 Further, uPAR expression by human monocytes is required for chemotaxis of these cells, 17 likely because of the effect of uPAR on β2-integrin mediated adhesion to ECM and endothelial cells. 18
Therefore, to determine whether the fibrinolytic system plays a role in granuloma formation, dissemination, and immune responses to mycobacterial infections, mice individually deficient in Pg, tPA, uPA, and uPAR were infected with M. avium 724, and characterized in this regard. The results of this study are summarized herein.
Materials and Methods
Animals
Wild-type (WT), UPA−/−, TPA−/−, and UPAR−/− mice in C57BL/6 strain were purchased from Jackson Laboratories (Bar Harbor, ME). PG−/− mice were generated as previously described, 19 and backcrossed at least to the F8 generation in strain C57BL/6. Experimental mice were 8 to 12 weeks of age and included both genders. All animal experiments were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee (IACUC).
Infections
M. avium 724 (provided by Andrea Cooper, The Trudeau Institute, Saranac Lake, NY) and M. bovis BCG (American Type Culture Collection no. 35734, Rockville, MD) were passaged, in vivo, in C57BL/6 mice and cultured in Middlebrook 7H9 (Difco, Detroit, MI) medium with oleic acid, albumin, dextrose, Tween 20, and NaCl (OADC) (Sigma, St. Louis, MO) at 37°C in shaker flasks for 1 week. Before infection, aliquots of the inoculum were tested to confirm CFU of M. avium 724 and M. bovis BCG being delivered.
Mice were anesthetized with an intraperitoneal administration of 0.075 mg ketamine/0.015 mg xylazine/0.0025 mg aceprozamine/g weight of animal. Intratracheal administration of 1 × 108 CFU of M. avium 724 or 5 × 107 CFU of M. bovis BCG in 50 μl of sterile phosphate-buffered saline (PBS) was performed by infusion through the vocal cords using a fiber optic light source for illuminating the entrance into the trachea. Mycobacterial infections were performed on a set of gene-inactivated and WT control mice, and were repeated on two separate experimental groups for each genotype. Control animals included PBS-treated and nontreated mice.
Measurement of the Total Colony Counts in Organ Homogenates
At 1, 5, and 10 weeks after infection, mice were sacrificed and the spleen, one lobe of liver, and both lungs were removed aseptically. After the liver sample was weighed, the organs were homogenized individually in a sterile glass grinder with 5 ml of sterile PBS/1% IGEPAL (Sigma). To determine the CFU present in each organ, serial 10-fold dilutions of whole organ homogenates were made and plated on Middlebrook 7H10/OADC agar plates. Mycobacterial colony numbers were determined after 14 days (M. avium 724) or 21 days (M. bovis BCG) of incubation at 37°C. The lowest limit of detection was 2 × 103 CFU per organ.
Histochemistry and Immunohistochemistry
Lungs were inflated to total lung capacity by intratracheal infusion with 4% paraformaldehyde-PBS and then ligated. Lungs and liver were fixed in 4% paraformaldehyde-PBS for 2 hours, set in paraffin blocks, and serially sectioned at 3 to 4 μm thickness. Tissue sections were deparaffinated, rehydrated, and stained with hematoxylin and eosin (H&E), Masson’s Trichrome, and Ziel-Neelsen stain (acid-fast bacilli staining).
To identify macrophages and neutrophils, tissue sections were deparaffinated, and placed in Peroxoblock (Zymed Laboratories, South San Francisco, CA), followed by incubation in trypsin (macrophages) or heated in citrate buffer, pH 6.0 (neutrophils). The slides were placed in rat anti-mouse F4/80 IgG (Serotec, Raleigh, NC) as the primary antibody for macrophages or rat anti-mouse neutrophil IgG2a (Serotec). After this, the slides were incubated in horseradish peroxidase-conjugated STAR 72 goat anti-rat IgG (Serotec) as the secondary antibody. Diaminobenzidine chromogen was applied for positive staining. Slides were counterstained with hematoxylin.
For anti-fibrin(ogen) staining, all slides were placed in Peroxoblock (Zymed Laboratories), followed by a heat step for antigen retrieval. The slides were then incubated with a goat anti-mouse fibrin(ogen) IgG (Nordic Immunological Laboratories, Tilburg, The Netherlands), followed by rabbit-anti-goat IgG (DAKO, Carpinteria, CA) in 10% normal mouse serum. Afterward, a diaminobenzidine chromogen was applied and then the slides were counterstained.
For anti-fibronectin and anti-Factor XIII staining, all slides were heat treated for antigen retrieval and blocked for internal peroxidase activity. The slides were incubated with a rabbit anti-fibronectin AB-10 polyclonal antibody (LabVision, Fremont, CA), or a rabbit polyclonal antibody against the Factor XIII subunit A (BioGenex, San Ramon, CA), followed by biotinylated swine-anti-rabbit IgG F(ab′)2 (DAKO). After incubation with horseradish peroxidase-conjugated streptavidin (BioGenex), the slides were stained with 3′-aminoethyl carbazole (AEC) as a chromogen, and counterstained.
Immunohistochemical analyses were performed on sets of samples from both WT and gene-inactivated mice at the same time. For histopathological evaluation the slides were examined by independent observers in a randomized manner.
Cytokine Measurements
Enzyme-linked immunosorbent assay measurements of tumor necrosis factor (TNF)-α and interferon (IFN)-γ levels in liver and lung homogenates were performed using appropriate kits (Pharmingen, San Diego, CA).
Statistical Analysis
Values were expressed as mean ± SD. Comparisons were made using Student’s t-test or Mann-Whitney’s U-test between two groups or, when more than two values were compared, analysis of variance. P values <0.05 were considered significant.
Results
Course of M. avium 724 Infection in WT, UPA−/−, TPA−/−, and PG−/− Mice
The colony counts of M. avium 724 in the spleens, livers, and lungs of WT and gene-inactivated mice at 1 week after infection are shown in Figure 1 ▶ ; a to c. Some dissemination of M. avium 724 infection from the lungs to the spleens and livers was seen at this time point. However, ∼37% of the WT mice did not show detectable levels of mycobacteria in livers. The number of livers with undetectable levels of mycobacteria were lower in UPA−/−, TPA−/−, and PG−/− mice than WT mice (22%, 17%, and 11.1%, respectively). Further, it was found that TPA−/− mice had significantly higher total colony counts than WT mice in spleens, and PG−/− mice had significantly higher total colony counts than WT mice in spleens and livers at 1 week after infection. Together, these data indicate that the inactivation of the PG gene, and, to a lesser extent, the TPA gene, resulted in increased dissemination. In the lungs, there were no differences among all genotypes tested at 1 week after infection.
Figure 1.

The course of M. avium 724 infection in WT, UPA−/−, TPA−/−, and PG−/− mice. Mice were intratracheally infected with 1 × 108 CFU of M. avium 724. Total colony counts were determined in the spleens (CFU/organ) (a), livers (CFU/g weight) (b), and lungs (CFU/organ) (c) at 1 week after infection. d–f: Plots of the colony counts at 1, 5, and 10 weeks after infection in each genotype. The data represent the mean ± SD of CFU in the organs of gene-inactivated mice and WT mice. *, P < 0.05.
The initial rates of M. avium 724 growth from 1 to 10 weeks after infection were similar for each group (Figure 1 ▶ ; d to f). The total colony counts were increased more than 10,000-fold in the spleens and livers, and ∼100-fold in the lungs during this period. Almost all mice in each group were still alive at the end of this protocol, and there were no deaths because of mycobacterial infections. Although PG−/− mice showed more M. avium dissemination at 1 week after infection, there were no differences in the total colony counts of the spleens and livers at the late phase of M. avium 724 infection.
Histological Findings in the Livers and Lungs from WT and PG−/− Mice at 1 and 5 Weeks after Infection with M. avium 724
Histological analysis was performed on the lungs at 1 week after infection (Figure 2) ▶ . WT and PG−/− mouse lungs showed patchy densely stained areas near the arteries and the bronchioles, with normal alveolar spaces (Figure 2, b and c) ▶ . One week after PBS intratracheal administrations, WT mouse lungs displayed normal alveolar structure and no cell infiltration (Figure 2a) ▶ . In both infected groups of mice, each lesion was composed of collections of macrophages and lymphocytes in alveolar spaces around bronchioles and arteries (Figure 2, d and g) ▶ . These data demonstrate that the intratracheal method of administrating M. avium results in the terminal bronchiole as the initial site of entry. The macrophages and lymphocytes showed dense distribution throughout the lung. There were also accumulations of lymphocytes around the small blood vessels. Immunohistochemical staining for F4/80 antigen was used to display the distribution of alveolar macrophages in lung tissue. Both WT and PG−/− mouse lungs showed similar distributions of macrophages at 1 week after infection (Figure 2, e and h) ▶ . These cells were tightly packed and aggregated with lymphocytes and some neutrophils, and most of the macrophages contained acid-fast bacilli (Figure 2, f and i) ▶ . UPA−/− and TPA−/− mouse lungs showed similar histological findings to lungs from infected WT and PG−/− mice (data not shown). At 1 week after infection, all mice showed no significant liver pathologies (data not shown).
Figure 2.
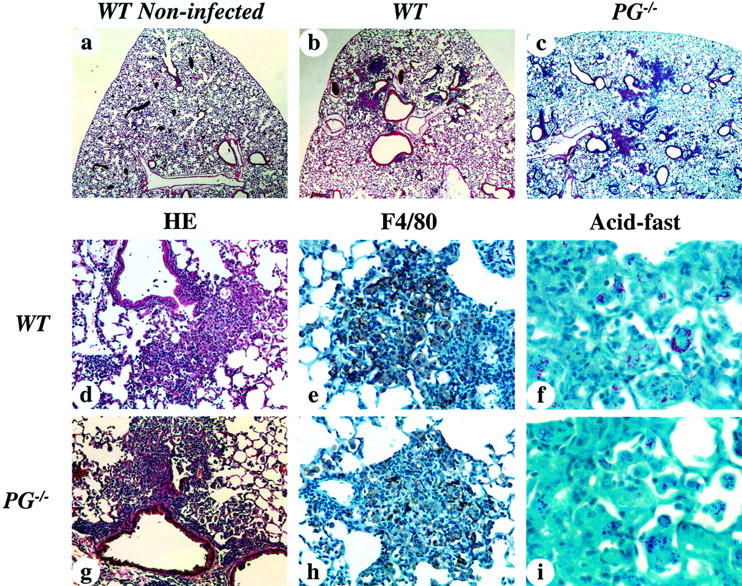
Photomicrographs of representative livers and lungs from WT and PG−/− mice 1 week after infection with 1 × 108 CFU of M. avium 724. a to d and g were stained with H&E, e and h were stained with anti-mouse F4/80 antibody, and f and i were stained for acid-fast bacilli. a: PBS-treated WT mouse lung showing normal alveolar structure and no cell infiltration. b and c: WT and PG−/− mouse lungs showing patchy distributions of lesions with normal alveolar space. d: WT mouse lung displaying focal cell infiltration of macrophages and lymphocytes in the alveolar space around bronchioles and arteries. e: In the focal cell-infiltrated area of the WT mouse lung, macrophages are tightly packed and aggregated with lymphocytes. f: WT mouse lung showing acid-fast bacilli in the cytoplasma of macrophages. g and h: A PG−/− mouse lung showing similar findings to those observed in WT mice. i: In a PG−/− mouse lung, macrophages with acid-fast bacilli were similar to WT mice. Original magnifications: ×40 (a–c); ×200 (d, g); ×400 (e, h); ×1000 (f, i).
Histological findings in the livers and lungs 5 weeks after infection are shown in Figure 3 ▶ . WT and PG−/− mouse livers possessed a large number of granulomas of varying sizes (Figure 3, a and c) ▶ . The granulomas showed peripheral lymphocyte infiltration and some granulomas appeared to have expanded and fused together. No differences were observed in the size, number, and distribution of granulomas between WT and PG−/− mouse livers, indicating that Pg was not required for granuloma formation. The macrophages in the liver granuloma displayed acid-fast bacilli (Figure 3e) ▶ . In the lungs, both WT and PG−/− mice had similar histological characteristics after the 5-week infection (Figure 3, b and d) ▶ . At this time, a significantly greater inflammatory cell infiltration was observed compared to 1 week after infection, as well as a decrease in the alveolar spaces and thickening of alveolar walls in both groups of mice. The macrophages within the lung showed a diffuse distribution and many were parasitized with acid-fast bacilli (Figure 3f) ▶ . UPA−/− and TPA−/− murine livers and lungs showed similar histological findings to PG−/− mice (data not shown).
Figure 3.
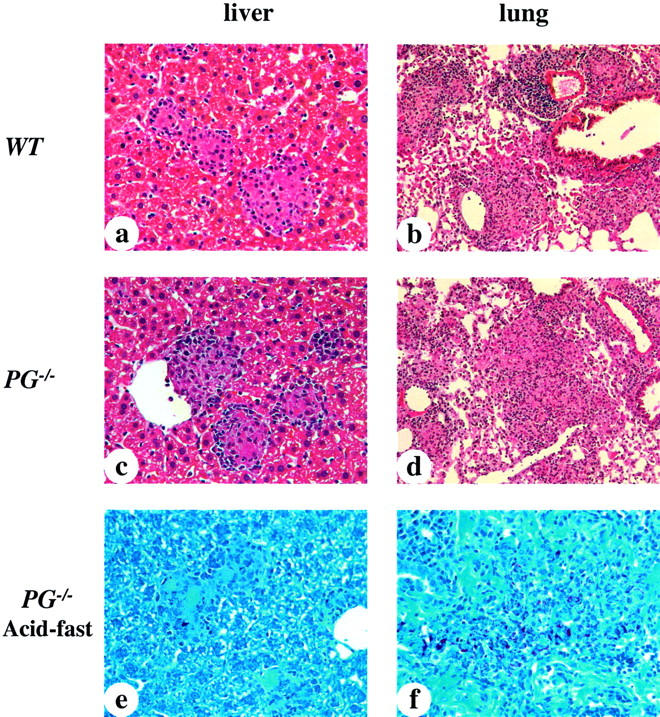
Photomicrographs of representative livers and lungs from WT and PG−/− mice 5 weeks after infection with 1 × 108 CFU of M. avium 724. a to d were stained with H&E and e and f were stained for acid-fast bacilli. a and c: WT and PG−/− mouse livers showing small and mature granulomas. The sizes and distributions of granuloma formations were similar in both groups. b and d: WT and PG−/− mouse lungs showing much more cell infiltration than that observed at 1 week after infection, as well as a decrease of alveolar space, and thickening of alveolar walls. e and f: PG−/− murine liver and lungs showing large macrophages parasitized by acid-fast bacilli. These findings were similar to those of WT mice. Original magnifications: ×400 (a, c, e, f); ×200 (b, d).
Histological Findings in the Livers and Lungs from WT and PG−/− Mice at 10 Weeks after Infection with M. avium 724
Histological analyses were performed on the livers and lungs at 10 weeks after infection in WT and PG−/− mice (Figure 4) ▶ . WT murine livers contained large numbers of mature, well-organized, and fused granulomas in the intact structure of hepatic lobe (Figure 4a) ▶ . Some mature granulomas had giant cell formations and a small amount of necrosis. PG−/− livers presented with granulomas of similar size, number, and distribution compared to those of WT mice (Figure 4b) ▶ . The macrophages in the granulomas were heavily infiltrated with acid-fast bacilli in both WT and PG−/− livers (Figure 4, e and f) ▶ .
Figure 4.
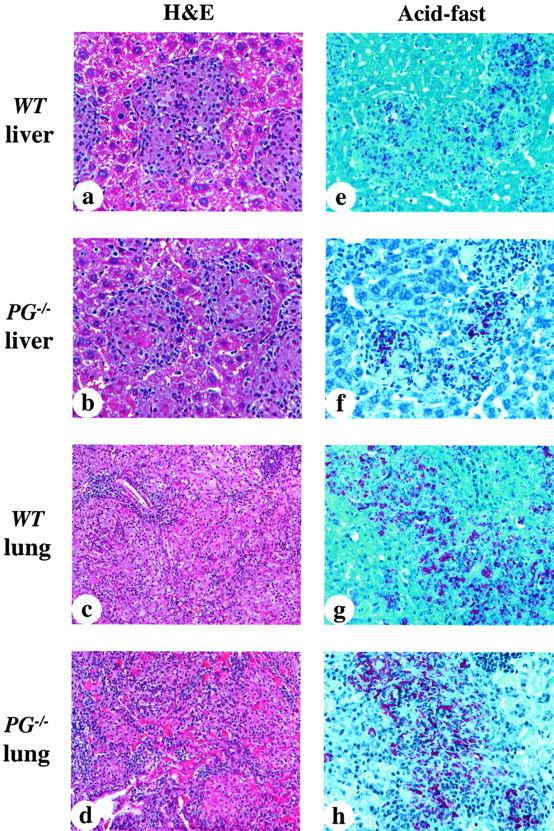
Photomicrographs of representative liver and lungs from WT and PG−/− mice at 10 weeks after infection. a to d were stained with H&E; e to h were stained for acid-fast bacilli. a: WT mouse liver showing mature and fused granulomas. The numbers and sizes of the granulomas were increased as compared to those at 5 weeks after infection. b: PG−/− mouse liver showing similar granuloma formations to those of WT livers, containing some fibrin deposition in granulomas. c: WT mouse lung showing extensive cell infiltration occupying most of the alveolar space. d: PG−/− murine lung showing similar findings to those of WT lungs, and more extensive fibrin deposition. e and f: WT and PG−/− mouse livers showing heavily parasitized macrophages within granulomas. g and h: WT and PG−/− mouse lungs showing diffuse distribution of heavily parasitized macrophages. Original magnifications: ×400 (a, b, e–h); ×200 (c, d).
WT mouse lungs displayed extensive cell infiltration occupying almost all of the alveolar area (Figure 4c) ▶ . A large number of lymphocytes, macrophages, and neutrophils infiltrated into the lungs and there were diffuse and extensive fibrin depositions in the alveolar space. PG−/− mouse lungs had similar histological findings to those of WT mouse (Figure 4d) ▶ . WT and PG−/− mouse lungs displayed no differences in the pattern and severity of inflammatory cell infiltration. PG−/− mouse lungs showed denser and more extensive fibrin depositions in the alveolar space. The macrophages, heavily parasitized with acid-fast bacilli, were diffusely distributed in the alveolar space in both WT and PG−/− mice (Figure 4, g and h) ▶ .
Fibrin and Fibronectin Deposition in the Livers and Lungs of WT and Gene-Inactivated Mice at 5 and 10 Weeks after Infection
Immunohistochemical stainings for fibrin(ogen) in the livers and lungs of WT and gene-inactivated mice at 5 weeks after infection are shown in Figure 5 ▶ . Liver granulomas from WT mice showed only limited fibrin deposition (Figure 5a) ▶ whereas the PG−/− livers showed dense fibrin deposition within the granulomas (Figure 5b) ▶ . Little fibrin deposition was observed outside of the granulomas in both WT and PG−/− livers. In the lungs, WT mice showed small levels of diffuse fibrin deposition in the area of inflammatory cell infiltration (Figure 5c) ▶ . In contrast, PG−/− lungs displayed an extensive deposition of fibrin (Figure 5d) ▶ . UPA−/− and TPA−/− mice had similar fibrin staining patterns compared to WT mice in both livers and lungs after the 5-week infection (data not shown).
Figure 5.

Anti-fibrin(ogen) staining in liver and lungs from infected WT and PG−/− mice 5 weeks after infection. a: WT mouse liver showing no fibrin deposition in granulomas. b: PG−/− mouse liver showing dense fibrin deposition in granulomas. c: WT murine lung showing a small amount of diffuse fibrin deposition. d: PG−/− mouse lung showing dense and highly extensive fibrin deposition. Original magnifications, ×400.
Fibrin deposition in the livers and lungs were also analyzed at 10 weeks after infection in WT and gene-inactivated mice (Figure 6) ▶ . WT, UPA−/−, and TPA−/− mouse livers showed no fibrin deposition in the granulomas (Figure 6 ▶ ; a, b, and c, respectively). In contrast, liver granulomas from infected PG−/− mice showed significant fibrin deposition, which was considerably higher than that observed in PG−/− mice after a 5-week infection (Figure 6d) ▶ . Little fibrin staining was observed in the intact hepatic structure in PG−/− mice. In the lungs, WT mice showed small amounts of diffuse fibrin deposition in the area of inflammatory cell infiltrations (Figure 6e) ▶ and these findings were similar to those of WT mice at 5 weeks after infection. The lungs of both infected UPA−/− and TPA−/− mice displayed increased fibrin deposition, as compared to WT mouse lungs (Figure 6, f and g) ▶ . After a 10-week infection, the lung lesions of PG−/− mice displayed a more dense and extensive fibrin deposition, compared to WT, UPA−/−, and TPA−/− mice (Figure 6h) ▶ . Overall, PG−/− mice had significant fibrin deposition within the liver granulomas and in areas of inflammatory cell infiltration in the lungs.
Figure 6.
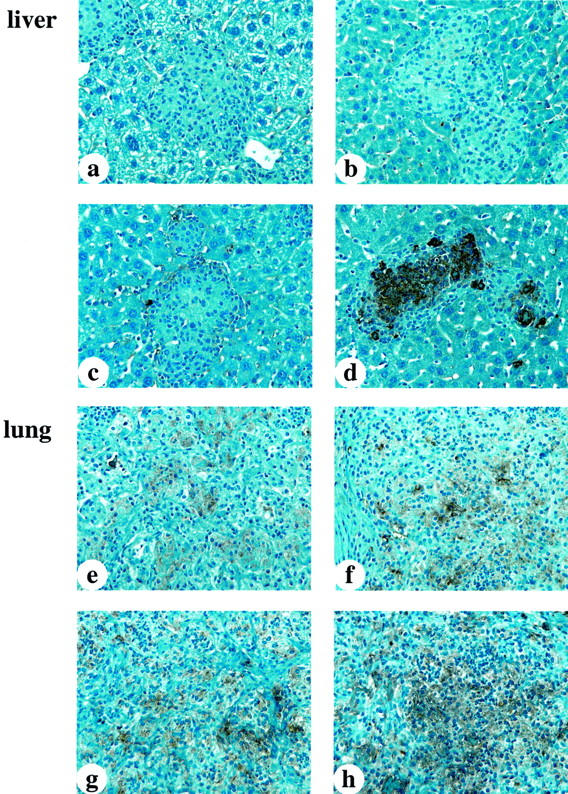
Anti-fibrin(ogen) staining in liver and lungs from infected WT and gene-inactivated mice 10 weeks after infection. a–c: WT, UPA−/−, and TPA−/− mouse livers, respectively, showing no fibrin deposition in granulomas. d: PG−/− mouse liver showing denser and more extensive fibrin deposition in granulomas, as compared with genotypically similar mice at 5 weeks after infection. e: WT mouse lung showing a small amount of diffuse fibrin deposition. f and g: Murine UPA−/− and TPA−/− lungs displaying increased fibrin deposition as compared to WT murine lungs. h: PG−/− mouse lung showing dense and more extensive fibrin deposition.
There were also significant differences in the deposition of fibronectin within the granulomas of WT and PG−/− mouse livers after 10 weeks (Figure 7, a and b) ▶ . Granulomas in the PG−/− mice showed a dense and extensive deposition of fibronectin, with the distribution being similar to that of fibrin in infected PG−/− mice. These findings suggest a general increase in the deposition of ECM proteins within the granulomas of infected PG−/− mice. Both WT and PG−/− mice demonstrated positive reactions in the hepatic sinuses, but the intensities were quite similar between the two groups, likely indicating a simple background reaction. Staining with Factor XIII was performed to determine whether this protein co-localized with fibrin(ogen), thus possibly cross-linking fibronectin and fibrin(ogen). Small amounts of this protein were found in both WT and PG−/− mice, but there were no differences between the two groups (Figure 7, c and d) ▶ .
Figure 7.
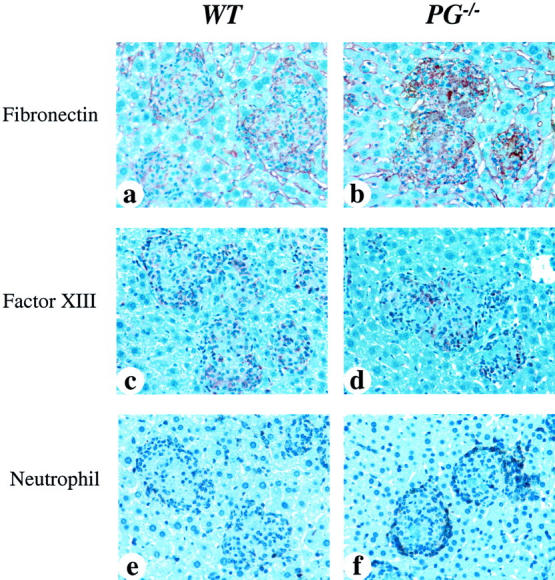
Immunohistochemical analyses for fibronectin, Factor XIII, and neutrophils in livers from WT and PG−/− mice at 10 weeks after infection. a and b were stained with anti-fibronectin antibody, c and d were stained with anti-Factor XIII subunit-A antibody, and e and f were stained with anti-neutrophil antibody. a: WT mouse liver showing a small amount of diffuse fibronectin deposition in granulomas. b: PG−/− mouse liver showing a more dense and extensive fibronectin deposition in granulomas, as compared with WT mice. c and d: WT and PG−/− mouse livers, respectively, showing a small amount of Factor XIII deposition in granulomas. e: WT mouse had a few neutrophils at the peripheral areas of the granulomas. f: PG−/− mouse had much stronger neutrophil infiltration around the granulomas. The distribution of fibronectin deposition was similar to fibrin in granulomas. Original magnifications, ×400.
Neutrophil Infiltration in WT and PG−/− Mouse Livers at 10 Weeks after Infection
Immunohistochemical analyses for neutrophils were performed on the livers of WT and PG−/− mice at 10 weeks after infection (Figure 7) ▶ . WT mice showed very limited neutrophil infiltration in the livers (Figure 7e) ▶ , whereas PG−/− mice presented a much larger neutrophil infiltration centered around the granulomas (Figure 7f) ▶ . There was no infiltration of neutrophils in the normal hepatic structure in PG−/− mice. It is possible that the accumulated fibrin deposits resulted in increased neutrophil migration to the granuloma. In the lungs, both WT and PG−/− mice displayed diffuse neutrophil infiltration and accumulation of neutrophils in the necrotic area (data not shown).
Course of Infection of M. avium 724 in WT and UPAR−/− Mice
The time course of colony counts in the spleens, livers, and lungs of WT and UPAR−/− mice infected with M. avium 724 is shown in Figure 8 ▶ . In the spleens and livers, both WT and UPAR−/− mice had similar mycobacterial growths at 1 and 5 weeks after infection and the colony counts in the lungs of both WT and UPAR−/− mice were similar through 10 weeks. However, at 10 weeks after infection, UPAR−/− mice had twofold to threefold higher colony counts in the spleens and livers than those of WT mice.
Figure 8.
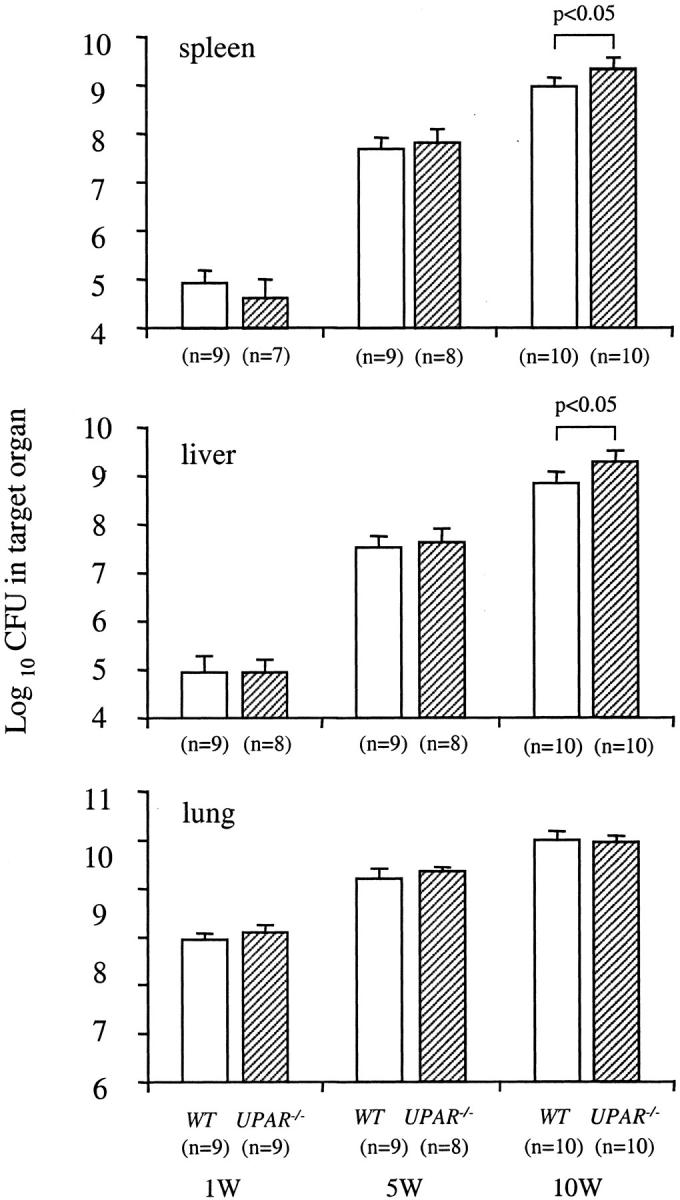
The time course of M. avium 724 infection in WT and UPAR−/− mice. Mice were infected with 1 × 108 CFU of M. avium 724. Total colony counts were determined in the spleens (CFU/organ), livers (CFU/g weight), and lungs (CFU/organ) of both genotypes at 1, 5, and 10 weeks after infection. The data represent the mean ± SD of CFU in the organs of WT and UPAR−/− mice. *, P < 0.05 versus WT mice.
Histological Findings in Liver and Lung from UPAR−/− Mice at 1 and 10 Weeks after Infection
At 1 week after infection, UPAR−/− mice lungs showed similar findings to those of WT mice (Figure 9, a and b) ▶ . Focal cell infiltrations of macrophages and lymphocytes in the alveolar spaces around the bronchioles and arteries were found in UPAR−/− mice. In immunohistochemical analyses for the F4/80 antigen, macrophages were aggregated with lymphocytes both in WT and UPAR−/− mouse lungs, and there were no discernable differences in the number or distribution of inflammatory cells between WT and UPAR−/− lungs. At 10 weeks after infection, the livers from UPAR−/− mice showed mature, well-organized, and fused granulomas (Figure 9c) ▶ , and these granulomas contained a small amount of fibrin deposition, similar to the UPA−/− and TPA−/− mice (Figure 9e) ▶ . In the 10-week infected lungs, an extensive cell infiltration was found in UPAR−/− mice, which occupied most of the alveolar space, and showed limited levels of diffuse fibrin deposition (Figure 9, d and f) ▶ .
Figure 9.
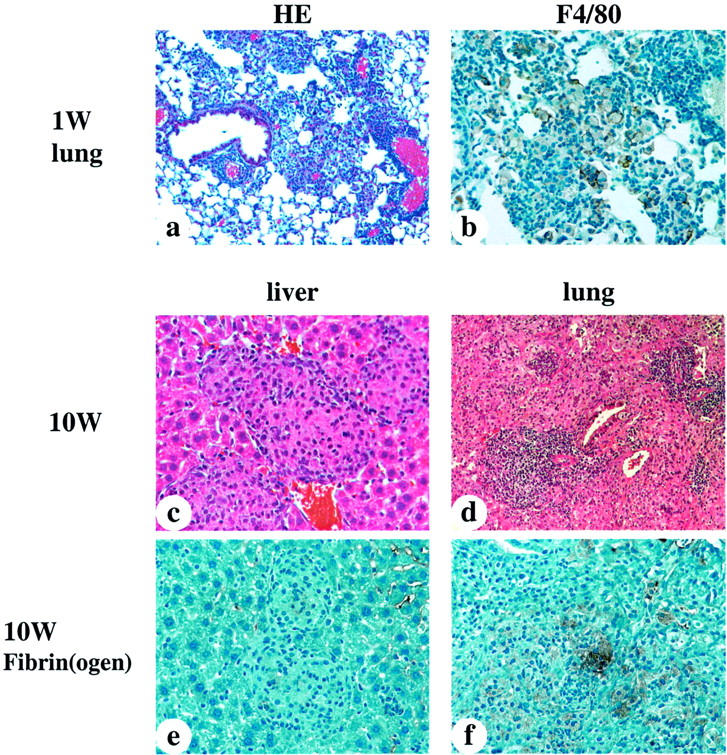
Photomicrographs of representative livers and lungs from UPAR−/− mice at 1 and 10 weeks after infection. a, c, and d were stained with H&E, b was stained with anti-mouse F4/80 antibody, and e and f were stained with anti-fibrin(ogen) antibody. a: UPAR−/− mouse lung at 1 week after infection showing focal cell infiltration of macrophages and lymphocytes in the alveolar space around bronchioles and arteries. These findings were similar to those of WT lung (Figure 2, a and b) ▶ . b: Anti-F4/80 antibody staining showing macrophages aggregated with lymphocytes in UPAR−/− mouse lung at 1 week after infection. These findings were similar to those of infected WT lungs (Figure 2, a to e) ▶ . c: UPAR−/− mouse liver at 10 weeks after infection showing mature and fused granulomas. d: UPAR−/− mouse lung at 10 weeks after infection displaying extensive cell infiltration that occupies most of the alveolar space. e and f: UPAR−/− mouse liver and lung showing a minimal amount of fibrin deposition. Original magnifications: ×200 (a); ×400 (b–f).
Cytokine Production in Liver and Lung Homogenates of M. avium 724-Infected WT and Gene-Inactivated Mice
The roles of TNF-α and IFN-γ in initiating an anti-mycobacterial immune reaction are well documented. 20-22 To determine whether the levels of TNF-α and IFN-γ varied between WT and the fibrinolytic-deficient mice at different stages of an M. avium infection, cytokine levels were measured by enzyme-linked immunosorbent assay in the organ homogenates of both liver and lungs. The concentrations of IFN-γ showed no increase from 1 week to 10 weeks of infection within each genotype in both livers and lungs. These results indicate that both WT and gene-inactivated mice infected with M. avium 724 did not initiate a strong IFN-γ response to control M. avium 724 infection. Liver and lung homogenates showed increased TNF-α production during the course of infection, although there were no consistent differences among the groups. These results suggest that the fibrinolytic proteins do not play major roles in the cytokine production during the chronic phase of a M. avium infection.
M. bovis BCG Infection in WT and PG−/− Mice at 5 Weeks after Infection
To determine whether results were specific to M. avium 724, WT and PG−/− mice were infected with M. bovis BCG, an attenuated strain of M. bovis, which shows limited pathogenicity in C57BL/6 mice. 23 The colony counts of M. bovis BCG in the spleens and lungs of WT and PG−/− mice at 5 weeks after infection are shown in Figure 10 ▶ . Unlike the M. avium 724 infection, the WT C57BL/6 mice were able to control the M. bovis BCG infection. In the spleens and lungs, both WT and PG−/− mice had similar total colony counts at 5 weeks after infection. These data suggest that Pg is not playing a significant role in controlling a mycobacterial infection during the chronic phase.
Figure 10.

M. bovis BCG infection in WT and PG−/− mice at 5 weeks after infection. Mice were intratracheally infected with 5 × 107 CFU of M. bovis BCG. Total colony counts (CFU/organ) were determined in the spleens (A) and lungs (B) at 5 weeks after infection. Total colony counts in livers were undetectable in both WT and PG−/− mice. The data represent the mean ± SD of five mice per group.
Discussion
A basic understanding of the steps involved in a pulmonary mycobacterial infection has been developed and involves ingestion by resident alveolar macrophages, migration of leukocytes to the site of an infection, granuloma formation, and mycobacterial dissemination. Yet a number of important questions remain. These include elaboration of the roles of host proteases in both granuloma formation and dissemination of mycobacteria, definition of the extracellular components that comprise a granuloma, and the manner in which these proteases are involved in the process of granuloma formation. Another important set of issues involves the nature of the proteases that function to promote leukocyte migration during a mycobacterial infection. To address these questions, studies were initiated to examine the role of the fibrinolytic proteins in controlling a mycobacterial infection. The importance of this system is emphasized by the fact that the serine protease, Pm, not only catalyzes limited digestion of the fibrin clot, but is the major plasma extracellular protease with a range of substrates. Its ability to degrade the ECM directly through its activity toward proteoglycans, 24 fibronectin, 25 laminin, 26 and type IV collagen, 27 and indirectly via its activation of prometalloproteases, such as stromelysin and procollagenase, 28,29 provides a means for Pm to facilitate cell migration for tissue remodeling. This enzyme also functions in the conversion of latent cell-associated transforming growth factor-β1 to its active form, and via this route may be involved in regulation of the inflammatory response. 30 Pm can function to activate the classic 31 and alternate 32 complement pathways, and in bradykinin generation from kininogen. 33 These activities implicate the fibrinolytic system in a wide range of pathophysiological processes that could affect the response to infection.
M. avium 724, a highly pathogenic M. avium strain in mice, was selected for the infection studies. An intratracheal route of infection was also chosen so that the involvement of fibrinolytic proteins in a primary infection, as well as in dissemination of the mycobacteria to secondary sites with establishment of a systemic infection, could be studied. Several reports indicate that a pulmonary infection with M. avium 724 can result in a systemic disseminated infection in C57BL/6 mice. 34,35 In the present studies, it was found that both WT and gene-inactivated mice were not able to control the M. avium 724 infection, leading to continuous mycobacterial growth and progressive tissue destruction in several organs. There were no differences in this effect between WT, PG−/−, UPA−/−, and TPA−/− mice after 5-week and 10-week infections. This result suggests that the fibrinolytic proteins do not play a major role in the immune responses during the chronic phase of a M. avium infection. Several studies have indicated a role for Pg and uPA in T cell activation during an immune response against various bacterial pathogens. 15,16,36 Although we did not directly evaluate the T cell response in the Pg-sufficient and -deficient mice after the M. avium and M. bovis BCG infections, our CFU results suggest that the T cell responses are similar between the WT and PG−/− mice.
The immune response to mycobacteria depends on the proper cytokine secretion profile. Both IFN-γ and TNF-α are key cytokines in controlling a mycobacterial infection. 20-22,37,38 uPA has been shown to have an important role in the production of IFN-γ and interleukin (IL)-12 after a pulmonary Cryptococcus neoformans infection. 39 Therefore, the levels of IFN-γ and TNF-α in liver and lung after a M. avium 724 infection were analyzed. The results showed similar cytokine production in these organs from WT, PG−/−, UPA−/−, and TPA−/− mice after a 5-week and 10-week M. avium infection, again indicating a limited role for the fibrinolytic proteins in dictating an immune response against mycobacterial infection.
Several studies have reported a role for uPAR in the immune response during the control of various bacterial pathogens. 40,41 Stimulation by the bacterial infection induces an up-regulation of uPAR expression on monocytes and neutrophils. 42-44 This receptor is involved in cellular movement by generating cell-surface Pm activity, via uPA binding, and by uPAR binding to β2-integrins, particularly CD11b/CD18 (Mac-1). 41 It is likely that the increased CFU in UPAR−/− mice is caused by Pm-independent mechanisms, because there were no differences in CFU after a 10-week infection among UPA−/−, PG−/−, and WT mice. In M. tuberculosis infection, it has been reported that absence of CD11b had a small effect on the control of infection. 45 Our data suggests a limited role for uPAR during the chronic phase of a mycobacterial infection.
Previous studies using thioglycollate to induce a peritoneal inflammatory reaction indicated that a reduction in Pm activity suppresses monocyte and lymphocyte recruitment. 46 Macrophages, as well as other leukocytes, can bind Pm and thus lead to increased proteolytic activity at the cell surface with consequent enhancement of migration of these cells by direct or indirect degradation of ECM proteins. 47 The histological results obtained in this study showed a similar number and distribution of lung macrophages in both WT and PG−/− mice 1 week after infection and similar mycobacterial growth after 5- and 10-week infections, suggesting that a deficiency in Pg is not affecting the recruitment of macrophages after a M. avium pulmonary infection.
The murine intratracheal infection model used in this study has the advantage of allowing both a pulmonary and systemic mycobacterial infection to be followed. This investigation showed increased CFU in the spleens and livers from PG−/− and TPA−/− mice after a 1-week M. avium infection, compared to infected WT mice, suggesting an earlier dissemination from lungs in these gene-inactivated mice. It is proposed that the CFU differences are because of changes in dissemination versus control of the M. avium infection, because at later time points the infection levels were similar in spleen and liver among the different gene-altered mice. Several lines of evidence support the concept that the extrapulmonary dissemination involves both phagocytic and nonphagocytic cells. 48 Alveolar macrophages and dendritic cells are thought to convey the engulfed bacteria from the lungs to the draining lymph nodes and to other organs. 2,49 Another pathway of dissemination, which has been shown for M. tuberculosis, is a direct interaction and subsequent migration of the mycobacteria through alveolar epithelial cells. 48 One of the possible mechanisms of increased dissemination in PG−/− mice may be because of differences in extra-pulmonary dissemination through the nonphagocytic cell. Pathogenic mycobacteria express on their surface a heparin-binding hemagglutinin adhesin glycoprotein, HBHA. 50,51 HBHA is involved in attachment of mycobacteria to pulmonary epithelial cells and plays an important role in extra-pulmonary dissemination. 52 The COOH-terminal region of HBHA contains two different lysine-rich repeats and a terminal lysine residue. 53 The previously described Pg-binding protein on mycobacteria could be HBHA because proteins that contain COOH-terminal lysine residues are known to bind Pg. This is supported by the similar molecular weight (ie, 30 kd) between one of the described Pg-binding proteins from M. tuberculosis and HBHA. 14 The ability of mycobacteria to bind Pg, likely through HBHA, may limit the ability of the mycobacterium to interact with the epithelial cells and, thus, its transmigration and dissemination. Therefore, a lack of Pg may favor mycobacterial interaction with alveolar epithelial cells through HBHA. At later stages of infection, with increased numbers of infected macrophages and granulomas, dissemination may be mediated almost exclusively through macrophages and dendritic cells.
Histological analysis of the infected mice showed progressive granuloma formation and fibrosis, which is a hallmark of a chronic mycobacterial infection. After 5-week and 10-week infections, both WT and gene-inactivated mice had well-developed granulomas in the liver. These results indicate that fibrinolytic proteins are not necessary for granuloma formation after a mycobacterial infection. However, the livers of PG−/− mice had denser and more extensive fibrin and fibronectin deposition in granulomas than those of WT and other gene-inactivated mice. Fibrin, and other ECM proteins, are believed to play a role in the formation and in maintenance of a granuloma, 54,55 and fibrin(ogen) reportedly stimulates TNF-α and IL-1β expression by macrophages/monocytes. 56,57 Our results suggest that fibrin is produced within a WT granuloma, but is rapidly degraded, because staining for fibrin in granulomas from WT-infected mice was limited, but strong fibrin staining was observed in PG−/− mice. This also suggests that fibrin turnover is regulated by Pm, perhaps through its binding to macrophages within the granuloma. Activated macrophages show increased binding to fibrin, 58 and also up-regulate the expression of uPAR, uPA, and PAI-1 at the cell surface. 59,60 Although, there was no apparent difference between WT and PG−/− mice at the gross histological level after a 10-week infection, a difference may become more apparent at later times after infection. This is suggested from the increased staining of fibrin and fibronectin in granulomas from the PG−/− mice.
In a mouse infection model, activated macrophages are thought to be responsible for ingesting and killing M. avium. The role of neutrophils in controlling a chronic mycobacterial infection is still unclear. However, it has been reported that neutrophils may also be involved in the defense against M. avium complex infection. 61 The histological analyses reported herein showed PG−/− mice to maintain a much larger neutrophil infiltration within liver granulomas than in WT mice. PG−/− mice showed increased neutrophil counts in the peripheral blood, but the kinetics of neutrophil recruitment into the peritoneal cavity in PG−/− mouse was similar to those for WT mouse after thioglycollate injection. 46 Increased neutrophil infiltration within the granuloma of PG−/− mice may be a consequence of both higher neutrophil counts in peripheral blood and increased concentrations of fibrin(ogen), fibronectin, and perhaps other ECM that can serve as chemotactic factors for neutrophils. 62
In summary, our study demonstrates that Pg and other fibrinolytic proteins do not play a significant role in initiating or maintaining the immune response against a M. avium 724 or M. bovis BCG infection. However, these fibrinolytic proteins may regulate the progression of fibrosis in the granuloma during the chronic phase of an infection.
Acknowledgments
We thank Ms. Stacey Raje and Ms. Angelik Anderson for maintenance of the mouse colonies and Ms. Mayra Sandoval-Cooper for assisting with the histology.
Footnotes
Address reprint requests to Francis J. Castellino, W. M. Keck Center for Transgene Research and the Department of Chemistry and Biochemistry, University of Notre Dame, Notre Dame, IN 46556. E-mail: fcastell@nd.edu.
Supported by the National Institutes of Health (grants HL-13423 to F. J. C. and HL-63682 to V. A. P.), the Kleiderer-Pezold Family Endowed Professorship (to F. J. C.), and the American Heart Association (grant 0030134N to J. S.).
References
- 1.Chackerian AA, Alt JM, Perera TV, Dascher CC, Behar SM: Dissemination of Mycobacterium tuberculosis is influenced by host factors and precedes the initiation of T-cell immunity. Infect Immun 2002, 70:4501-4509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Saunders BM, Cooper AM: Restraining mycobacteria: role of granulomas in mycobacterial infections. Immunol Cell Biol 2000, 78:334-341 [DOI] [PubMed] [Google Scholar]
- 3.Ehlers S: Immunity to tuberculosis: a delicate balance between protection and pathology. FEMS Immunol Med Microbiol 1999, 23:49-58 [DOI] [PubMed] [Google Scholar]
- 4.Castellino FJ, Ploplis VA: Plasminogen and streptokinase Bachmann F eds. Handbook of Experimental Pharmacology, Fibrinolytics and Antifibrinolytics 2000, vol 146:pp 25-56 Springer-Verlag, New York [Google Scholar]
- 5.Lijnen HR: Plasmin and matrix metalloproteinases in vascular remodeling. Thromb Haemost 2001, 86:324-333 [PubMed] [Google Scholar]
- 6.Lahteenmaki K, Kuusela P, Korhonen TK: Bacterial plasminogen activators and receptors. FEMS Microbiol Rev 2001, 25:531-552 [DOI] [PubMed] [Google Scholar]
- 7.Berge A, Sjobring U: PAM, a novel plasminogen-binding protein from Streptococcus pyogenes. J Biol Chem 1993, 268:25417-25424 [PubMed] [Google Scholar]
- 8.Suenson E, Thorsen S: Secondary-site binding of glu-plasmin, lys-plasmin and miniplasmin to fibrin. Biochem J 1981, 197:619-628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hortin GL, Gibson BL, Fok KF: Alpha 2-antiplasmin’s carboxy-terminal lysine residue is a major site of interaction with plasmin. Biochem Biophys Res Commun 1988, 155:591-596 [DOI] [PubMed] [Google Scholar]
- 10.Miles LA, Dahlberg CM, Plescia J, Felez J, Kato K, Plow EF: Role of cell-surface lysines in plasminogen binding to cells: identification of a-enolase as a candidate plasminogen receptor. Biochemistry 1991, 30:1682-1691 [DOI] [PubMed] [Google Scholar]
- 11.Wistedt AC, Ringdahl U, Mulleresterl W, Sjobring U: Identification of a plasminogen-binding motif in PAM, a bacterial surface protein. Mol Microbiol 1995, 18:569-578 [DOI] [PubMed] [Google Scholar]
- 12.Wistedt AC, Kotarsky H, Marti D, Ringdahl U, Castellino FJ, Schaller J, Sjobring U: Kringle 2 mediates high affinity binding of plasminogen to an internal sequence in streptococcal surface protein PAM. J Biol Chem 1998, 273:24420-24424 [DOI] [PubMed] [Google Scholar]
- 13.Rios-Steiner JL, Schenone M, Mochalkin I, Tulinsky A, Castellino FJ: Structure and binding determinants of the recombinant kringle-2 domain of human plasminogen to an internal peptide from a group A Streptococcal surface protein. J Mol Biol 2001, 308:705-719 [DOI] [PubMed] [Google Scholar]
- 14.Monroy V, Amador A, Ruiz B, Espinoza-Cueto P, Xolalpa W, Mancilla R, Espitia C: Binding and activation of human plasminogen by Mycobacterium tuberculosis. Infect Immun 2000, 68:4327-4330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beck JM, Preston AM, Gyetko MR: Urokinase-type plasminogen activator in inflammatory cell recruitment and host defense against Pneumocystis carinii in mice. Infect Immun 1999, 67:879-884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gyetko MR, Chen GH, McDonald RA, Goodman R, Huffnagle GB, Wilkinson CC, Fuller JA, Toews GB: Urokinase is required for the pulmonary inflammatory response to Cryptococcus neoformans. A murine transgenic model. J Clin Invest 1996, 97:1818-1826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gyetko MR, Todd RF, Wilkinson CC, Sitrin RG: The urokinase receptor is required for human monocyte chemotaxis in vitro. J Clin Invest 1994, 93:1380-1387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.May AE, Kanse SM, Lund LR, Gisler RH, Imhof BA, Preissner KT: Urokinase receptor (CD87) regulates leukocyte recruitment via beta 2 integrins in vivo. J Exp Med 1998, 188:1029-1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ploplis VA, Carmeliet P, Vazirzadeh S, Van Vlaenderen I, Moons L, Plow EF, Collen D: Effects of disruption of the plasminogen gene on thrombosis, growth, and health in mice. Circulation 1995, 92:2585-2593 [DOI] [PubMed] [Google Scholar]
- 20.Zerlauth G, Eibl MM, Mannhalter JW: Induction of anti-mycobacterial and anti-listerial activity of human monocytes requires different activation signals. Clin Exp Immunol 1991, 85:90-97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Murray PJ, Wang L, Onufryk C, Tepper RI, Young RA: T cell-derived IL-10 antagonizes macrophage function in mycobacterial infection. J Immunol 1997, 158:315-321 [PubMed] [Google Scholar]
- 22.Ottenhoff TH, de Boer T, Verhagen CE, Verreck FA, van Dissel JT: Human deficiencies in type 1 cytokine receptors reveal the essential role of type 1 cytokines in immunity to intracellular bacteria. Microbes Infect 2000, 2:1559-1566 [DOI] [PubMed] [Google Scholar]
- 23.Dunn PL, North RJ: Virulence ranking of some Mycobacterium tuberculosis and Mycobacterium bovis strains according to their ability to multiply in the lungs, induce lung pathology, and cause mortality in mice. Infect Immun 1995, 63:3428-3437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Edmonds-Alt X, Quisquater E, Vaes G: Proteoglycan- and fibrin-degrading neutral proteinase activities of Lewis lung carcinoma cells. Eur J Cancer 1980, 16:1257-1261 [DOI] [PubMed] [Google Scholar]
- 25.Jilek F: Cold-insoluble globulin III. Cyanogen bromide and plasminolysis fragments containing a label introduced by transamidation. Hoppe-Seyler’s Z Physiol Chem 1977, 358:1165-1168 [PubMed] [Google Scholar]
- 26.Schlechte W, Murano G, Boyd D: Examination of the role of the urokinase receptor in human colon cancer mediated laminin degradation. Cancer Res 1989, 49:6064-6069 [PubMed] [Google Scholar]
- 27.MacKay AR, Corbitt RH, Hartzler JL, Thorgeirsson UP: Basement membrane type IV collagen degradation: evidence for the involvement of a proteolytic cascade independent of metalloproteinases. Cancer Res 1990, 50:5997-6001 [PubMed] [Google Scholar]
- 28.Stricklin GP, Bauer EA, Jeffrey JJ, Eisen A: Human skin collagenase: isolation of precursor and active forms from both fibroblast and organ cultures. Biochemistry 1977, 16:1607-1615 [DOI] [PubMed] [Google Scholar]
- 29.He C, Wilhelm SM, Pentland AP, Marmer BL, Grant GA, Eisen AZ, Goldberg GI: Tissue cooperation in a proteolytic cascade activating human interstitial collagenase. Proc Natl Acad Sci USA 1989, 86:2632-2636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Khalil N, Corne S, Whitman C, Yacyshyn H: Plasmin regulates the activation of cell-associated latent TGF-beta(1) secreted by rat alveolar macrophages after in vivo bleomycin injury. Am J Respir Cell Mol Biol 1996, 15:252-259 [DOI] [PubMed] [Google Scholar]
- 31.Agostoni A, Gardinali M, Frangi D, Cafaro C, Conciato L, Sponzilli C, Salvioni A, Cugno M, Cicardi M: Activation of complement and kinin systems after thrombolytic therapy in patients with acute myocardial infarction: a comparison between streptokinase and recombinant tissue-type plasminogen activator. Circulation 1994, 90:2666-2670 [DOI] [PubMed] [Google Scholar]
- 32.Brade V, Nicholson A, Bitter-Suermann D, Hadding V: Formation of the C-3 cleaving properdin enzyme on zymosen. J Immunol 1974, 113:1735-1743 [PubMed] [Google Scholar]
- 33.Habal FM, Burrowes CE, Movat HZ: Generation of kinin by plasma kallikrein and plasmin and the effect of α1-antitrypsin and antithrombin III on the kininogenases. Adv Exp Med Biol 1976, 70:23-36 [DOI] [PubMed] [Google Scholar]
- 34.Hubbard RD, Flory CM, Collins FM: Memory T cell-mediated resistance to Mycobacterium tuberculosis infection in innately susceptible and resistant mice. Infect Immun 1991, 59:2012-2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Appelberg R, Sarmento AM: The role of macrophage activation and of Bcg-encoded macrophage function(s) in the control of Mycobacterium avium infection in mice. Clin Exp Immunol 1990, 80:324-331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goguen JD, Bugge T, Degen JL: Role of the pleiotropic effects of plasminogen deficiency in infection experiments with plasminogen-deficient mice. Methods 2000, 21:179-183 [DOI] [PubMed] [Google Scholar]
- 37.Cooper AM, Dalton DK, Stewart TA, Griffin JP, Russell DG, Orme IM: Disseminated tuberculosis in interferon gamma gene-disrupted mice. J Exp Med 1993, 178:2243-2248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bean AG, Roach DR, Briscoe H, France MP, Korner H, Sedgwick JD, Britton WJ: Structural deficiencies in granuloma formation in TNF gene-targeted mice underlie the heightened susceptibility to aerosol Mycobacterium tuberculosis infection, which is not compensated for by lymphotoxin. J Immunol 1999, 162:3504-3511 [PubMed] [Google Scholar]
- 39.Gyetko MR, Sud S, Chen GH, Fuller JA, Chensue SW, Toews GB: Urokinase-type plasminogen activator is required for the generation of a type 1 immune response to pulmonary Cryptococcus neoformans infection. J Immunol 2002, 168:801-809 [DOI] [PubMed] [Google Scholar]
- 40.Gyetko MR, Sud S, Kendall T, Fuller JA, Newstead MW, Standiford TJ: Urokinase receptor-deficient mice have impaired neutrophil recruitment in response to pulmonary Pseudomonas aeruginosa infection. J Immunol 2000, 165:1513-1519 [DOI] [PubMed] [Google Scholar]
- 41.Rijneveld AW, Levi M, Florquin S, Speelman P, Carmeliet P, van Der Poll T: Urokinase receptor is necessary for adequate host defense against pneumococcal pneumonia. J Immunol 2002, 168:3507-3511 [DOI] [PubMed] [Google Scholar]
- 42.Juffermans NP, Dekkers PE, Verbon A, Speelman P, van Deventer SJ, van der Poll T: Concurrent upregulation of urokinase plasminogen activator receptor and CD11b during tuberculosis and experimental endotoxemia. Infect Immunol 2001, 69:5182-5185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dekkers PE, ten Hove T, te Velde AA, van Deventer SJ, van Der Poll T: Upregulation of monocyte urokinase plasminogen activator receptor during human endotoxemia. Infect Immun 2000, 68:2156-2160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Coleman JL, Gebbia JA, Benach JL: Borrelia burgdorferi and other bacterial products induce expression and release of the urokinase receptor (CD87). J Immunol 2001, 166:473-480 [DOI] [PubMed] [Google Scholar]
- 45.Melo MD, Catchpole IR, Haggar G, Stokes RW: Utilization of CD11b knockout mice to characterize the role of complement receptor 3 (CR3, CD11b/CD18) in the growth of Mycobacterium tuberculosis in macrophages. Cell Immunol 2000, 205:13-23 [DOI] [PubMed] [Google Scholar]
- 46.Ploplis VA, French EL, Carmeliet P, Collen D, Plow EF: Plasminogen deficiency differentially affects recruitment of inflammatory cell populations in mice. Blood 1998, 91:2005-2009 [PubMed] [Google Scholar]
- 47.Plow EF, Herren T, Redlitz A, Miles LA, Hoover-Plow JL: The cell biology of the plasminogen system. EMBO J 1995, 9:939-945 [DOI] [PubMed] [Google Scholar]
- 48.Teitelbaum R, Cammer M, Maitland ML, Freitag NE, Condeelis J, Bloom BR: Mycobacterial infection of macrophages results in membrane-permeable phagosomes. Proc Natl Acad Sci USA 1999, 96:15190-15195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bodnar KA, Serbina NV, Flynn JL: Fate of Mycobacterium tuberculosis within murine dendritic cells. Infect Immun 2001, 69:800-809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Menozzi FD, Rouse JH, Alavi M, Laude-Sharp M, Muller J, Bischoff R, Brennan MJ, Locht C: Identification of a heparin-binding hemagglutinin present in mycobacteria. J Exp Med 1996, 184:993-1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reddy VM, Kumar B: Interaction of Mycobacterium avium complex with human respiratory epithelial cells. J Infect Dis 2000, 181:1189-1193 [DOI] [PubMed] [Google Scholar]
- 52.Pethe K, Alonso S, Biet F, Delogu G, Brennan MJ, Locht C, Menozzi FD: The heparin-binding haemagglutinin of M. tuberculosis is required for extrapulmonary dissemination. Nature 2001, 412:190-194 [DOI] [PubMed] [Google Scholar]
- 53.Menozzi FD, Bischoff R, Fort E, Brennan MJ, Locht C: Molecular characterization of the mycobacterial heparin-binding hemagglutinin, a mycobacterial adhesin. Proc Natl Acad Sci USA 1998, 95:12625-12630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Izaki S, Goldstein SM, Fukuyama K, Epstein WL: Fibrin deposition and clearance in chronic granulomatous inflammation: correlation with T-cell function and proteinase inhibitor activity in tissue. J Invest Dermatol 1979, 73:561-565 [DOI] [PubMed] [Google Scholar]
- 55.Marshall BG, Wangoo A, Cook HT, Shaw RJ: Increased inflammatory cytokines and new collagen formation in cutaneous tuberculosis and sarcoidosis. Thorax 1996, 51:1253-1261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kajikawa T, Oshima H, Yamazaki M, Mizuno D: Induction by heterologous fibrinogen of release of TNF-like cytotoxic factor from murine macrophages. J Biol Response Mod 1986, 5:283-287 [PubMed] [Google Scholar]
- 57.Perez RL, Roman J: Fibrin enhances the expression of IL-1 beta by human peripheral blood mononuclear cells. Implications in pulmonary inflammation. J Immunol 1995, 154:1879-1887 [PubMed] [Google Scholar]
- 58.Shetty S, Kumar A, Pueblitz S, Emri S, Gungen Y, Johnson AR, Idell S: Fibrinogen promotes adhesion of monocytic to human mesothelioma cells. Thromb Haemost 1996, 75:782-790 [PubMed] [Google Scholar]
- 59.Khan KM, Falcone DJ: Role of laminin in matrix induction of macrophage urokinase-type plasminogen activator and 92-kDa metalloproteinase expression. J Biol Chem 1997, 272:8270-8275 [DOI] [PubMed] [Google Scholar]
- 60.Beschorner R, Schluesener HJ, Nguyen TD, Magdolen V, Luther T, Pedal I, Mattern R, Meyermann R, Schwab JM: Lesion-associated accumulation of uPAR/CD87-expressing infiltrating granulocytes, activated microglial cells/macrophages and upregulation by endothelial cells following TBI and FCI in humans. Neuropathol Appl Neurobiol 2000, 26:522-527 [DOI] [PubMed] [Google Scholar]
- 61.Hartmann P, Becker R, Franzen C, Schell-Frederick E, Romer J, Jacobs M, Fatkenheuer G, Plum G: Phagocytosis and killing of Mycobacterium avium complex by human neutrophils. J Leukoc Biol 2001, 69:397-404 [PubMed] [Google Scholar]
- 62.Kuhns DB, Nelson EL, Alvord WG, Gallin JI: Fibrinogen induces IL-8 synthesis in human neutrophils stimulated with formyl-methionyl-leucyl-phenylalanine or leukotriene B(4). J Immunol 2001, 167:2869-2878 [DOI] [PubMed] [Google Scholar]