

Abstract
The platelet-derived growth factors are implicated in development of fibrotic reactions and disease in several organs. We have overexpressed platelet-derived growth factor-C in the heart using the α-myosin heavy chain promoter and created a transgenic mouse that exhibits cardiac fibrosis followed by hypertrophy with sex-dependent phenotypes. The transgenic mice developed several pathological changes including cardiac fibroblast proliferation and deposition of collagen, hypertrophy, vascular defects, and the presence of Anitschkow cells in the adult myocardium. Male mice developed a hypertrophic phenotype, whereas female mice were more severely affected and developed dilated cardiomyopathy, leading to heart failure and sudden death. The vascular defects initially included dilation of microvessels and vascular leakage. Subsequently, a marked loss of microvessels, formation of large vascular sac-like structures, and an increased density of smooth muscle-coated vessels were observed in the myocardium. In part, the observed vascular changes may be because of an up-regulation of vascular endothelial growth factor in cardiac fibroblasts of the transgenic hearts. This unique animal model reveals that a potent mitogen for cardiac fibroblasts result in an expansion of the interstitium that induce a secondary sex-dependent hypertrophic response in the cardiomyocytes.
Myocardial disease is the most common cause of death in humans. The reasons underlying cardiac malfunction are diverse but often relate to thromboembolic events, arrhythmias, or cardiomyopathies. The cardiomyopathies can be classified in several subgroups ranging from hypertrophic cardiomyopathy characterized by left ventricular hypertrophy to dilated cardiomyopathy with a thin left ventricle wall and limited contractile function. Cardiac hypertrophy is an adaptive response to compensate for a decreased cardiac output and may occur as a result of a variety of stimuli including myocardial infarction, hypertension, endocrine disorders, and mutations in proteins affecting contractile function. The cardiomyocytes respond to the decreased cardiac output by increasing in size by cellular hypertrophy rather than undergoing cell division. Although the hypertrophic response initially will increase the cardiac output it can ultimately lead to dilated cardiomyopathy and heart failure. As part of the hypertrophic response, interstitial cells, ie, cardiac fibroblasts, become activated and the expansion of the interstitium often results in a replacement fibrotic reaction, because the hypertrophic cardiomyocytes are unable to regenerate damaged tissue. 1-5
Several types of growth factors are able to induce cardiac hypertrophy and fibrosis, among them tumor necrosis factor (TNF)-α and transforming growth factor-β. 6,7 Platelet-derived growth factors (PDGFs) are potent mitogens for many cell types of mesenchymal origin, including cardiac fibroblasts. 8,9 The PDGF family consists of four different members; PDGF-A, PDGF-B, PDGF-C, and PDGF-D. 9,10 The PDGF isoforms bind to and activate two receptor tyrosine kinases, PDGFR-α and PDGFR-β, that are differentially expressed in target tissues. PDGF-A, -B, and -C chains bind to PDGFR-α with high affinity, whereas only PDGF-B and -D chains bind PDGFR-β. The two novel PDGFs, PDGF-C and PDGF-D, differ from PDGF-A and -B in that they are secreted as latent factors that require proteolytic activation by removal of the N-terminal CUB domain for activity. 11,12
In cardiovascular medicine, transgenic animals with induced myocardial diseases mimicking those found in humans are of great interest, and may provide unique opportunities to explore diverse aspects of the diseases, including drug development. Despite the well known roles of the PDGFs as potent mitogens for fibroblasts, potentially being directly involved in fibrotic reactions in a variety of organs, 9 only a few attempts to overexpress PDGFs in vivo using transgenic techniques have been performed. 13-16 In this work we have targeted transgenic overexpression of PDGF-C to the heart using the α-myosin heavy chain (α-MHC) promoter and have created a mouse model in which a potent fibroblast mitogen is overexpressed in the heart, leading to a progressive fibrosis and hypertrophy. Female mice were shown to develop a lethal dilated cardiomyopathy, whereas the males develop a more progressive hypertrophic phenotype. As a control, transgenic mice overexpressing the CUB domain only were also generated. These mice appeared primarily normal suggesting that the activated PDGF-C core domain is responsible for the pathological changes seen in the mice expressing full-length PDGF-C.
Materials and Methods
Generation of Transgenic Mice
Transgenic mice overexpressing full-length PDGF-C in the heart were generated as earlier described using CBA/C57BL/6 mice. 11 Transgenic mice were maintained by backcrossing onto C57BL/6 animals the first generation and then by brother-sister matings. The transgenic expression and the observed phenotypes were stable for at least five generations. All experiments were performed using heterozygous and wild-type animals.
To generate transgenic mice overexpressing the CUB domain of PDGF-C, mouse PDGF-C cDNA was subjected to polymerase chain reaction (PCR) mutagenesis using the following primers: 5′ CGC GGT CGA CGC CCC AGT CAG CCA A (forward) including a flanking SalI site and 5′ CGC GAA GCT TTT ACA AGT CTT CTT CAG AAA TGA GCT TTT GTT CTG TGA CTT GTG GCA (reverse) including the human c-Myc epitope and a flanking HindIII site. The PCR generated a 572-bp fragment that was digested with SalI/HindIII and cloned into the α-MHC vector 17 (kindly provided by K. Alitalo, Helsinki University, Helsinki, Finland). The linearized and purified transgene fragment was injected into male pronuclei of fertilized mouse oocytes (Mouse Camp, Karolinska Institutet, Stockholm, Sweden). Tail lysate prepared from the resulting pups was screened for the presence of the transgene by PCR using the CUB domain-specific primer used in the cloning 5′ CGC GGT CGA CGC CCC AGT CAG CCA A (forward) and the c-Myc/CUB-specific primer 5′ ATG AGC TTT TGT TCT GTG AC (reverse). A 542-bp fragment was amplified in the transgenic founders. The animal experiments in this study were approved by the local committee of the Swedish National Board for Laboratory Animals (CFN).
Histology and Immunohistochemistry
Wild-type and heterozygous littermates of different ages were sacrificed and weighed. The hearts were dissected and weighed or frozen on dry ice for protein and RNA extraction, or fixed in 4% paraformaldehyde in phosphate-buffered saline overnight at + 4°C and processed for paraffin-embedding using standard protocols. A few hearts were collected from females that died from heart failure. Microphotographs of whole hearts were taken with a Nikon Digital Camera finePix S1 pro. The paraffin-embedded hearts were sectioned (6 μm) and the sections stained with hematoxylin and eosin (H&E), or with Masson’s trichrome, using standard procedures. Visualization of myofibrils was performed using a phosphotungstic acid hematoxylin staining kit (Bio Optica, Milano, Italy). Immunolocalization of PDGF-C was performed using TSA indirect (NEN Life Science, Boston, MA, USA) with an affinity-purified rabbit Ig fraction to human PDGF-C core domain protein (1 μg Ig/ml). 11 Endothelial-specific staining was performed as above using rat anti-mouse CD31/platelet-endothelial cell adhesion molecule (PECAM) antibody (Pharmingen, San Diego, CA, USA) as earlier described. 18 α-Smooth muscle actin (SMA) staining was performed using a monoclonal anti-human α-SMA antibody 1A4 (DAKO, Glostrup, Denmark). Antigen retrieval was obtained by heating the tissue slides in 0.01 mol/L of citrate buffer, pH 6.0, at 94°C for 20 minutes. Vascular endothelial growth factor (VEGF) staining was performed using a rabbit polyclonal anti-VEGF antibody A 20 (Santa Cruz, Biotechnology, Santa Cruz, CA, USA). Antigen retrieval was obtained as above. Detection of proliferating cells was performed using a mouse monoclonal anti-proliferating cell nuclear antigen antibody (Chemicon, Temecula, CA, USA). For SMA, VEGF, and proliferating cell nuclear antigen stainings, Elite ABC Vectastain (Vector Laboratories, Burlingame, CA, USA) was used.
RNase Protection Assay
Total RNA extracted from hearts was analyzed by RNase protection assay using [32P]UTP-labeled riboprobes as earlier described. 19 A β-actin probe was used as an internal control. The results were visualized and quantified using a phosphorimager (Fuji Bas 1500).
Immunoblot Analysis
Proteins were extracted from heart tissue in lysis buffer including protease inhibitor mix (Complete; Roche, Mannheim, Germany) according to standard protocols. Aliquots were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (12%) under reducing conditions. Immunoblotting was performed using the affinity-purified rabbit Ig fraction to human PDGF-C core domain PDGF-C (see above, 1 μg Ig/ml) or a rabbit polyclonal Ig against human c-Myc for detection of transgenic CUB domain (1/500, Santa Cruz). Bound antibodies were visualized using ECL+ (Amersham Biosciences, Buckinghamshire, England). Expression levels were compared using a Luminiscent Image Analyzer (Fuji Film, Japan). As loading control, the ER protein calnexin was visualized using a rabbit antiserum against human calnexin (kindly provided by Jonas Fuxe, Ludwig Institute for Cancer Research Stockholm Branch, Stockholm, Sweden).
Northern Blot Analysis
Total RNA was extracted from heart tissue by sonicating frozen tissue in lysis buffer according to the guanidinum thiocyanate/acid phenol method. 20 Twenty μg of total RNA from each sample was subsequently subjected to agarose gel electrophoresis and blotted onto nylon filters. The hybridizations were performed in 50% formamide containing solutions at 42°C overnight and the filters were subsequently washed at high stringency. The results were visualized and quantified using a phosphoimager (Fuji Bas 1500). The probes were radiolabeled to high specific activity with [32P]dCTP (Amersham) using random priming as recommended by the supplier (Megaprime; Amersham). The probes were: GAPDH cDNA (200-bp PCR product generated using primers 5′ ATGCCAGTGAGCTTCCCGTTCAGC (forward) and 5′ TGGTATCGTGGAAGGACTCATGAC (reverse)), brain natriuretic peptide (BNP) DNA (500-bp PCR product generated using primer 5′ GATCTCCTGAAGGTGCTGTCC (forward) and 5′ ATCCGGTCTATCTTGTGCCCA (reverse)), and isolated inserts from IMAGE cDNA clones (Incyte Genomics, St. Louis, MO, USA) encoding atrial natriuretic peptide (ANP) (GenBank AI324503), and myosin light chain (MLC)-2 (GenBank AA434871).
PDGFR-α Analysis
Hearts were homogenized in lysis buffer containing 50 mmol/L Tris-HCl, pH 8.0, 150 mmol/L NaCl, 1% Triton X-100, 0.5% deoxycholate, 0.1% SDS, 0.5 mmol/L vanadate, and protease inhibitors (Complete; Roche). Supernatants were collected and an equal amount of protein from each lysate was immunoprecipitated using PDGFR-α rabbit antiserum (R7) on ice followed by incubation with protein A-Sepharose. Beads were washed and boiled in 2× SDS loading buffer and subjected to 7% SDS-PAGE under reducing conditions. Immunoblotting was performed using rabbit serum against C-terminal PDGFR (CED), or a monoclonal anti-phosphotyrosine antibody (Py99, 1 μg/ml). Bound antibodies were visualized using ECL+ (Amersham).
Echocardiography
Mice were anesthetized using isoflurane (Forene, Baxter, Deerfield, IL, USA) in 40% oxygen. The analysis was performed using echocardiography (Philips ATL HTI5000E equipped with a 15 MHz probe, CL 15-7). Posterior wall thickness, end systolic and end diastolic left ventricle diameters, and ECGs were recorded simultaneously. Fractional shortening was calculated.
Results
Cardiac Expression and Processing of PDGF-C in Transgenic Mice
Transgenic mice expressing full-length PDGF-C under the control of the α-MHC promoter were generated as earlier described. 11 Newborn transgenic pups appeared normal and transgenic mice were born with the expected frequency in each litter. Immunoblotting of heart lysates under reducing conditions from 1- or 3-month-old animals showed that transgenic PDGF-C appeared as species of 45 kd (full length) and 35 kd (partially processed) (Figure 1a) ▶ . To investigate whether there was a difference in the expression level or processing of transgenic PDGF-C between females and males, protein extracts from four different animals of each sex at the age of 1 month were compared. No differences were observed. In wild-type animals the expression of endogenous PDGF-C was below the level of detection. As a control, transgenic mice overexpressing the CUB domain only were generated and analyzed (see below).
Figure 1.
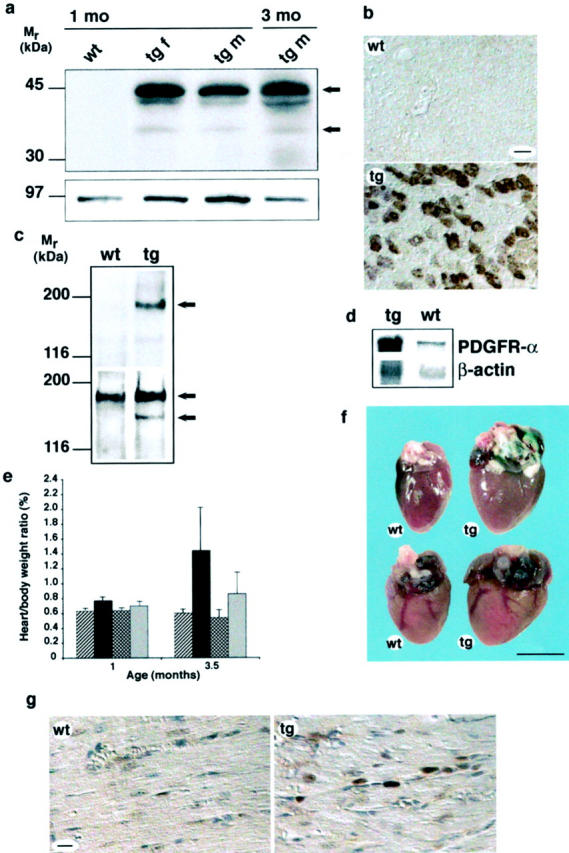
Transgenic expression of PDGF-C protein and gross phenotype of transgenic hearts. a: SDS-PAGE and immunoblot analysis of heart tissue lysates under reducing conditions. Expression of PDGF-C was detected in transgenic hearts (tg) at 1 month of age and the expression was persistent in hearts from 3-month-old animals. Both full-length PDGF-C (45 kd, top arrow) and processed species of ∼35 kd (bottom arrow) are visualized (top). The expression level and processing of transgenic PDGF-C were similar in females and males. Calnexin served as a loading control (bottom). b: Immunohistochemical staining of heart sections from 1-month-old mice. In wild-type hearts (wt) no expression was detected, whereas in the transgenic hearts (tg) PDGF-C was detected in the cytoplasm of a subset of cardiomyocytes at 1 month. Older mice expressed the transgene in the majority of cardiomyocytes. Scale, 20 μm. c: PDGFR-α is activated in PDGF-C transgenic mice. Heart lysates from 2-month-old transgenic animals were subjected to immunoprecipitation using a rabbit antiserum to PDGFR-α and the precipitates were analyzed by SDS-PAGE and immunoblotted using a monoclonal anti-phosphotyrosine antibody (top), or using an antiserum to the C-terminal of PDGFRs (bottom). Activated PDGFR-α is seen only in the transgenic heart (arrow, top). Mature (170 kd), and not fully processed (140 kd) PDGFR-α were found in the transgenic heart, whereas only mature (170 kd) PDGFR-α was found in wild-type hearts (arrows, bottom). d: RNase protection assay on total RNA isolated from transgenic (tg) and wild-type (wt) hearts using a PDGFR-α-specific riboprobe. Five transgenic and three wild-type animals were analyzed. PDGFR-α mRNA was up-regulated approximately threefold in transgenic hearts. A β-actin riboprobe was used as an internal control. e: PDGF-C transgenic mice have increased heart:body weight ratios. Comparison between mouse heart and body weights in wild-type (striped and checked bars) and transgenic animals (black and gray bars) of different ages (n = 4 to 8 animals in each group). The heart:body weight ratios in female transgenics (black bars) is dramatically increased compared to wild-type females (striped bars, P = 0.0013 for 1-month-old animals, and P = 0.013 for 3.5-month-old animals). Transgenic males (gray bars) develop a less abnormal ratio compared to the wild-type males (checked bars, P = 0.057 for 1-month-old animals, and P = 0.049 for 3.5-month-old animals). f: Cardiac hypertrophy in PDGF-C transgenic animals. Hearts from 3.5-month-old female wild-type (wt) and transgenic (tg) littermates are shown (top). Hearts from 6-month-old male wild-type (wt) and transgenic (tg) littermates (bottom). Scale, 5 mm. g: Fibroblasts proliferate in the transgenic heart. Immunohistochemical staining visualizing proliferating cell nuclear antigen in fibroblasts of transgenic (tg) heart at age 1 month compared with the wild-type (wt).
Immunohistochemical staining on tissue sections showed the presence of transgenic PDGF-C in the cardiomyocytes (Figure 1b) ▶ . During cardiac development, the major myosin isoform expressed is β-MHC and α-MHC is only expressed at low levels. After birth there is a switch in isoform expression and α-MHC becomes the major isoform. 21,22 In the newborn transgenic mouse PDGF-C protein could only be detected in a small portion of cells. Gradually the expression expanded and in the adult mouse (3 months) the majority of cardiomyocytes expressed PDGF-C. Female and male mice showed a similar expression pattern. Because the overexpression of PDGF-C was driven by the α-MHC promoter the observed expression pattern was expected. These results show expression and in vivo processing of transgenic PDGF-C and support earlier findings. 11
PDGFR-α Is Up-Regulated and Activated in PDGF-C Transgenic Heart
To investigate whether PDGFR-α was activated in vivo, tissue lysates from wild-type and transgenic littermates were subjected to immunoprecipitation using an antiserum to PDGFR-α, the precipitates were resolved by SDS-PAGE, and analyzed by immunoblotting using anti-phosphotyrosine antibodies, or an antiserum against PDGF receptors. In the transgenic hearts, PDGFR-α was strongly tyrosine phosphorylated, in contrast to the wild-type tissue (Figure 1c) ▶ . Control immunoblottings, using the antiserum to PDGF receptors, visualized a 170-kd band corresponding to mature PDGFR-α in both wild-type and transgenic hearts. An additional faster migrating species of ∼140 kd was detected only in transgenic hearts. This species may correspond to immature and not fully processed PDGFR-α, suggesting enhanced synthesis of the receptor in transgenic tissue. Both female and male mice were used in the analysis and no sex difference was observed regarding receptor activation or expression. Analysis by RNase protection assay showed an almost three-fold up-regulation of PDGFR-α mRNA in transgenic hearts compared with the wild type, confirming the previous suggestion (Figure 1d) ▶ . Immunostaining of tissue sections using an antibody against the proliferating cell nuclear antigen showed a significant increase of proliferating fibroblasts in the transgenic heart (Figure 1g ▶ and Table 1 ▶ ). These results suggest that transgenic PDGF-C becomes activated and is able to stimulate PDGFR-α expressed primarily by interstitial fibroblasts. 8 The up-regulation of the PDGFR-α mRNA probably reflects the increased number of this cell type in the transgenic tissue (see below).
Table 1.
Proliferating Fibroblast in Transgenic Hearts Compared with Wild-Type Hearts
| Age, months | Genotype | Positive nuclei* | P† |
|---|---|---|---|
| 1 | wt‡ | 6 ± 2 | |
| 1 | tg§ | 31 ± 12 | 0.0004 |
* Means ± SD.
† Student’s t-test comparing the two genotypes.
‡ Six animals (two males, four females).
§ Six animals (three males, three females). The numbers of PCNA-stained fibroblast nuclei/visual field were counted at × 40 magnification. The means from 11 visual fields were calculated for each mouse and used in the t-test.
PDGF-C Transgenic Mice Develop Cardiac Fibrosis, Hypertrophy, and Dilated Cardiomyopathy
PDGF-C transgenic mice appeared normal during embryonic development, and at birth as compared to wild-type littermates. At 2 weeks, mild fibrosis but no hypertrophy was observed, and the animals appeared normal. At 1 month, heart to body weight ratios in age-matched animals were significantly higher in transgenic females compared to wild-type animals (Figure 1e) ▶ . As the mice grew older, the difference increased dramatically, especially in females. The increase in heart:body weight ratios resulted from both larger hearts and a general weight loss probably caused by the dysfunctional heart. Starting from the age of 3 months, transgenic females died from heart failure. Abdominal swelling, because of edema, was sometimes observed and was probably a consequence of failing heart functions (data not shown). Male transgenic mice did not die spontaneously before sacrifice at 7 months. Generation of homozygous transgenic mice resulted in heart failure at younger age suggesting a dose-dependent phenotype, and these mice were not further analyzed in detail.
PDGF-C transgenic hearts developed a progressive hypertrophy (Figure 1f) ▶ . In H&E-stained tissue sections several pathological changes were observed (Figure 2, a to h, o and p) ▶ . The most striking feature was the increased number of interstitial fibroblasts in the transgenic hearts leading to a disorganized myocardial architecture, and increased thickness of the ventricular walls. Masson’s trichrome staining of tissue sections from transgenic mice of increasing age showed progressive collagen depositions in the transgenic hearts, suggesting that transgenic animals developed a progressive cardiac fibrosis (Figure 2, i to n) ▶ .
Figure 2.
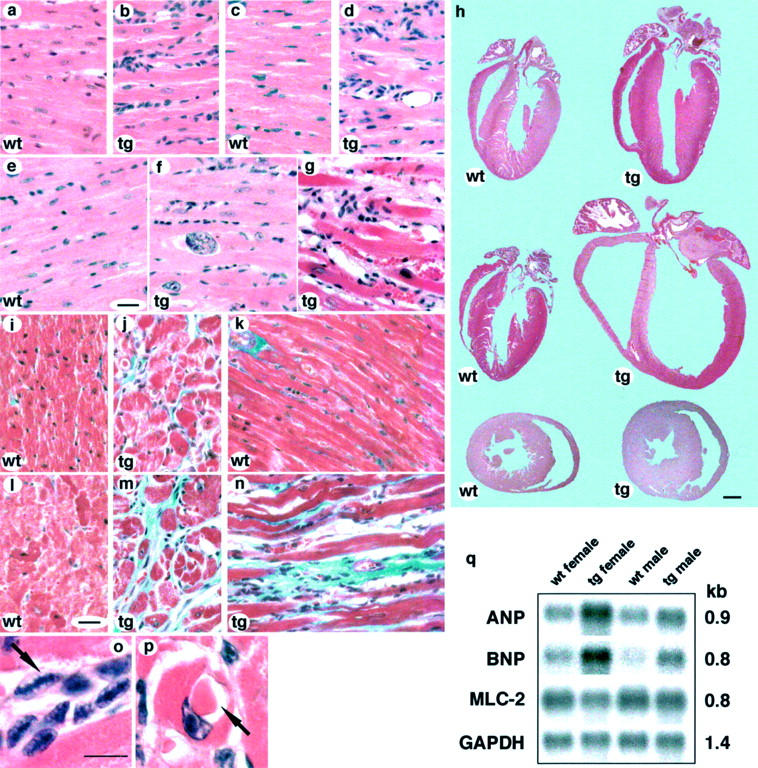
Histology of PDGF-C transgenic hearts. a to g: H&E stainings on tissue sections from the left ventricle of the heart from a: a 1-month-old wild-type female; b: a 1-month-old transgenic male showing proliferation of interstitial fibroblasts; c: a 3-month-old wild-type female, d: a 3-month-old transgenic female; e: a 6-month-old wild-type male; f: a 6-month-old transgenic male with enlarged myocyte nuclei; g: a transgenic female with heart failure exhibiting a disorganized heart tissue architecture because of extensive interstitial fibrosis. h: Comparisons of ventricle size and wall thickness on H&E-stained heart tissue sections from males and females. Top: A comparison between 3.5-month-old wild-type (wt) and transgenic (tg) males. Middle: A comparison between 3.5-month-old wild-type (wt) and transgenic (tg) females. Bottom: A comparison of heart cross-sections from 6-month-old wild-type (wt) and transgenic (tg) males. In the transgenic female the size of ventricles is enlarged and ventricle walls are dilated. The transgenic males develop a different phenotype with a thickening of left ventricle wall leading to a smaller ventricle volume. Scale, 1 mm. i to n: Masson’s trichrome staining on heart tissue sections from the left ventricle. In the Masson’s trichrome staining, collagen is stained in blue; the nucleus in black; and cytoplasm, keratin, muscle fibers, and fibrin in red. The presence of collagenous extracellular matrix is indicated by the blue staining. Scale, 20 μm. i: Normal appearance of myocardium in 3-month-old wild-type female; j: 3-month-old transgenic male with thickened myofibers and deposition of extracellular matrix; k: 3-month-old wild-type female; l: 6-month-old wild-type male; m: a 6-month-old transgenic male showing extensive fibrosis and variable myofiber diameter; n: a section from a 5.5-month-old transgenic female that died from heart failure; o and p: H&E stainings of transgenic tissue sections showing pathological findings. Anitschkow cells with caterpillar-like chromatin patterns o: and presence of hyaline drops as evidence of hyaline degeneration p: Scale, 10 μm. q: Northern blot analysis on total heart RNA from 3- to 4-month-old wild-type (wt) (two females, two males) and transgenic (tg) (two females, two males) mice. Expression of ANP and BNP was highly up-regulated in the transgenic mice, whereas the expression of MLC-2 showed variations. The differences were more prominent in the females; ANP was up-regulated twofold to sixfold and BNP was up-regulated twofold to ninefold (compared to sex-matched wild types). MLC-2 was slightly down-regulated in females, whereas in the younger male MLC-2 was up-regulated twofold and in the older male MLC-2 was slightly down-regulated (compared to sex-matched wild types). The amount of RNA in each lane was normalized by analyzing the expression level of GAPDH.
Transgenic heterozygous mice overexpressing the CUB domain appeared normal and were fertile. Immunoblotting of heart lysates under reducing conditions showed that transgenic CUB domain appeared as a species of ∼24 kd (Figure 3a) ▶ . In H&E-stained sections of the heart no morphological abnormalities could be detected (Figure 3b) ▶ .
Figure 3.

Transgenic mice with targeted overexpression of the CUB domain of PDGF-C in the heart a: SDS-PAGE and immunoblot analysis of heart tissue lysates under reducing conditions shows expression of 24-kd transgenic CUB. b: H&E stainings on tissue sections from the left ventricle of the heart from transgenic (tg) and wild-type (wt) littermates. Scale, 20 μm.
Detailed pathological examination of the PDGF-C transgenic hearts showed the presence of both hypertrophic and atrophic cardiac myocytes. Degenerating myofibers displayed granulation and loss of striation, abnormally shaped and misplaced nuclei, sometimes with misplaced chromatin, hyaline degeneration seen as hyaline drops (Figure 2p) ▶ , and possible intracellular and extracellular edema. Hypertrophic myofibers frequently displayed enlarged nuclei (Figure 2f) ▶ . Interestingly, Anitschkow cells, or cells with Anitschkow chromatin pattern of the nuclei, were frequently observed in the transgenic hearts at 3 months of age (Figure 2o) ▶ . The blood vessels were dilated and leaky, and in the failing hearts, small hemorrhages may have occurred (see below).
The effect of PDGF-C overexpression was partially sex-dependent (Figure 2h) ▶ . Male and female mice displayed similar phenotypes up to 1 month of age, including the proliferation of interstitial cells, and some pathological changes in the myofibers. However, at the age of 3 months, prominent differences between the sexes had developed. Transgenic males developed the progressive cardiac hypertrophy, including a thickened left ventricle wall and reduced ventricle volume, whereas transgenic females developed dilated cardiomyopathy. To visualize muscle myofibrils heart sections were stained with phosphotungstic acid hematoxylin. In the transgenic male, muscle fibers were thickened (data not shown). We were unable to detect a similar hypertrophic response in the transgenic female myocardium before the dilatation. Atrial thrombosis was frequently observed in females with a failing heart and female transgenic mice were very seldom were alive at 6 months of age. There was a variability in the severity of the phenotypes between the male transgenic mice, probably as a result of nonhomogenous genetic background. Mild dilation of male hearts was observed in some cases.
Up-Regulation of Hypertrophic Markers in PDGF-C Transgenic Mice
Cardiac hypertrophy and fibrosis is frequently associated with re-expression of genes expressed during fetal and perinatal development, and up-regulation of some cardiac proteins (eg, ANP, BNP, α-SMA, and MLC-2). 23,24 Northern blot analysis of RNA from wild-type and transgenic mice showed that ANP and BNP were up-regulated in the transgenic hearts compared to the wild-type. MLC-2 was slightly down-regulated in the females but was differentially regulated in males, which most likely reflects the variability in the severity of the phenotypes between the analyzed male transgenic mice (Figure 2q) ▶ . The differences in expression levels were more prominent in females, supporting the observation that females develop a more severe dilated phenotype. By immunohistochemistry, expression of α-SMA was shown to be up-regulated in cardiomyocytes of transgenic hearts (Figure 4, i and j) ▶ . In the CUB transgenic heart, SMA was also found to be up-regulated in the cardiomyocytes (data not shown).
Figure 4.
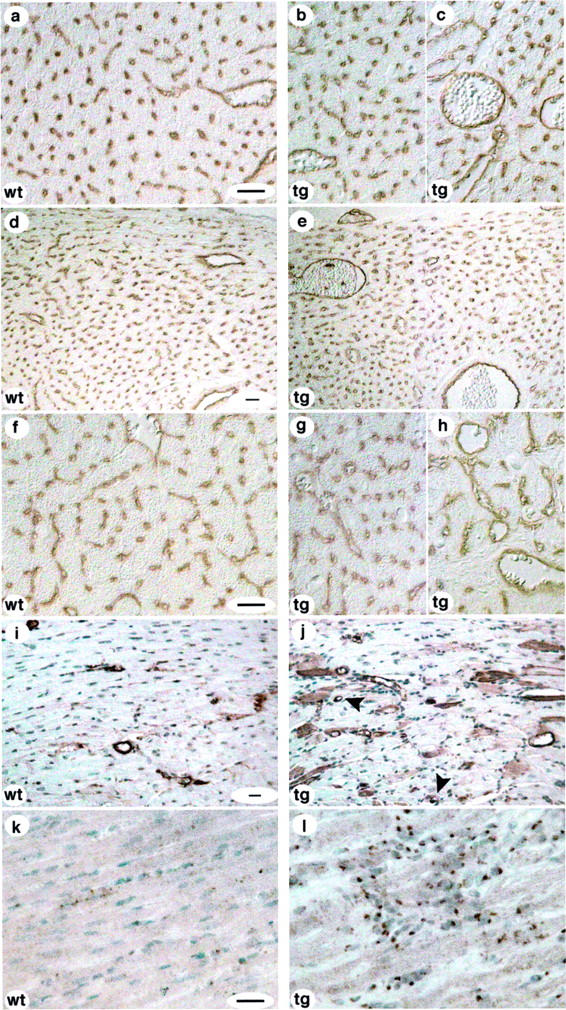
Vascular changes in PDGF-C transgenic mice. Endothelial cell-specific staining using antibodies to PECAM (a–h), SMA-specific staining using antibodies to SMA (i, j), and VEGF-specific staining (k, l) of heart tissue sections from the left ventricle. a: A 3-month-old wild-type female showing a normal appearance of microvascular network. b and c: Variable degrees of vascular proliferation in a PDGF-C transgenic 3-month-old female. d and e: Wild-type female age 3 months compared (d) with transgenic male at lower magnification (e), showing the presence of larger vessels in the transgenic ventricle wall. f: Wild-type male age 6 months. g and h: Variable degrees of vascular proliferation in the same transgenic male at age 6 months. h: Clearly illustrated is a loss of capillaries in the transgenic heart. i: Normal distribution of smooth muscle-coated vessels in a 3-month-old wild-type female. j: Increased number of smooth muscle-coated vessels (arrows) and up-regulation of SMA in cardiomyocytes in a female transgenic littermate. Scale, 20 μm. k: VEGF staining of wild-type myocardium. l: VEGF staining of transgenic myocardial fibroblasts at 1 month of age. Scale, 20 μm.
The Vasculature Is Altered in PDGF-C Transgenic Hearts
To investigate the potential angiogenic activity of PDGF-C and its impact on the vasculature, blood vessels were identified by immunostaining of tissue sections for PECAM/CD31 expression. No differences in the structure of the vasculature were observed between wild-type and transgenic mice at 1 month of age (data not shown). However, as the mice grew older significant differences were observed (ages 3 to 4 and 6 to 7 months). The blood vessel densities, mainly capillaries, were generally lower in older transgenic hearts compared to wild-type (Figure 4, a to h ▶ , and Table 2 ▶ ). In transgenic hearts, the normal capillary network was gradually replaced by dilated capillaries, and sac-like structures. Also, normally larger vessels were enlarged in transgenic hearts. For quantification, corresponding areas of left ventricle were selected in sections from both transgenic and wild-type animals.
Table 2.
Comparison of Blood Vessel Densities in Wild-Type and PDGF-C Transgenic Mouse Hearts
| Age, months | Genotype | Vessel counts, means ± SD | P* |
|---|---|---|---|
| 3–4 | wt† | 66 ± 9 | |
| 3–4 | tg‡ | 48 ± 10 | 0.0033 |
| 6–7 | wt§ | 58 ± 9 | |
| 6–7 | tg¶ | 44 ± 4 | 0.009 |
* Student’s t-test comparing the two genotypes of the same age.
† Seven animals (four females, three males).
‡ Seven animals (four females, three males).
§,¶ Five animals each (males). The numbers of PECAM-stained vessels/visual field (left ventricles) were counted at × 200 magnification. The means from seven visual fields were calculated for each mouse and used in the t-test.
To quantify smooth muscle-coated vessels, sections were stained for SMA and (Figure 4, i and j ▶ , and Table 3 ▶ ). The densities of SMA-positive vessels were higher in transgenic hearts compared to wild-type age-matched controls. Mainly arterioles accounted for the increased densities. The dilated microvessels and sac-like vascular structures observed with PECAM-staining were all negative for SMA. There was some variability in the densities of the arterioles throughout the transgenic heart. At some locations, no or very little differences were observed compared with the wild-type, whereas other areas contained significantly more of these vessels (Table 3) ▶ . For quantification, corresponding areas of left ventricle were selected in sections from both transgenic and wild-type animals.
Table 3.
Comparison of SMA-Positive Vessel Density in Wild-Type and PDGF-C Transgenic Mouse Hearts
| Age, months | Genotype | SMA-positive vessels, means ± SD | P* |
|---|---|---|---|
| 3–4 | wt† | 11 ± 3 | |
| 3–4 | tg‡ | 12 ± 2 | 0.4141 |
| 6–7 | wt§ | 13 ± 1 | |
| 6–7 | tg¶ | 21 ± 3 | 0.0002 |
* Student’s t-test comparing the two genotypes of the same age.
† Six animals (three females, three males).
‡ Six animals (three females, three males).
§,¶ Five animals each (males). The numbers of SMA-positive vessels/visual field (left ventricles) were counted at × 40 magnification. The means from seven visual fields were calculated for each mouse and used in the t-test.
In the CUB transgenic hearts we could not detect any vascular abnormalities. SMA staining of tissue sections showed a normal distribution and appearance of smooth muscle-coated vessels (data not shown).
Immunohistochemical staining revealed an up-regulation of VEGF in interstitial fibroblasts of PDGF-C transgenic hearts (Figure 4, k and l) ▶ . This up-regulation was observed already at the age of 2 weeks at the same time as proliferation of interstitial fibroblasts was first observed, before changes in the vasculature. These data show that the vasculature is significantly altered in the PDGF-C transgenic animals. The up-regulation of VEGF may at least in part explain the observed alterations.
Echocardiography Analysis of PDGF-C Transgenic Hearts
Transgenic animals and littermate controls (3.5 month of age) were analyzed by echocardiography and electrocardiography (ECG) (Figure 5) ▶ . Posterior left ventricle wall thickness, end systolic and end diastolic left ventricle diameters, and ECG were recorded simultaneously. The contractile function was also estimated by calculating the fractional shortening (Figure 5 ▶ and Table 4 ▶ ). The results essentially confirm the histological findings showing that PDGF-C overexpression leads to cardiac hypertrophy with decreased contractile function measured as fractional shortening, and that females develop a severe dilated phenotype. Expectedly, the conduction properties in the transgenic hearts were also severely affected with prolonged PQ interval, broadened QRS complex, and changes in the ST area. There was some variability in the degree of hypertrophy between transgenic males, probably because of a nonhomologous genetic background.
Figure 5.
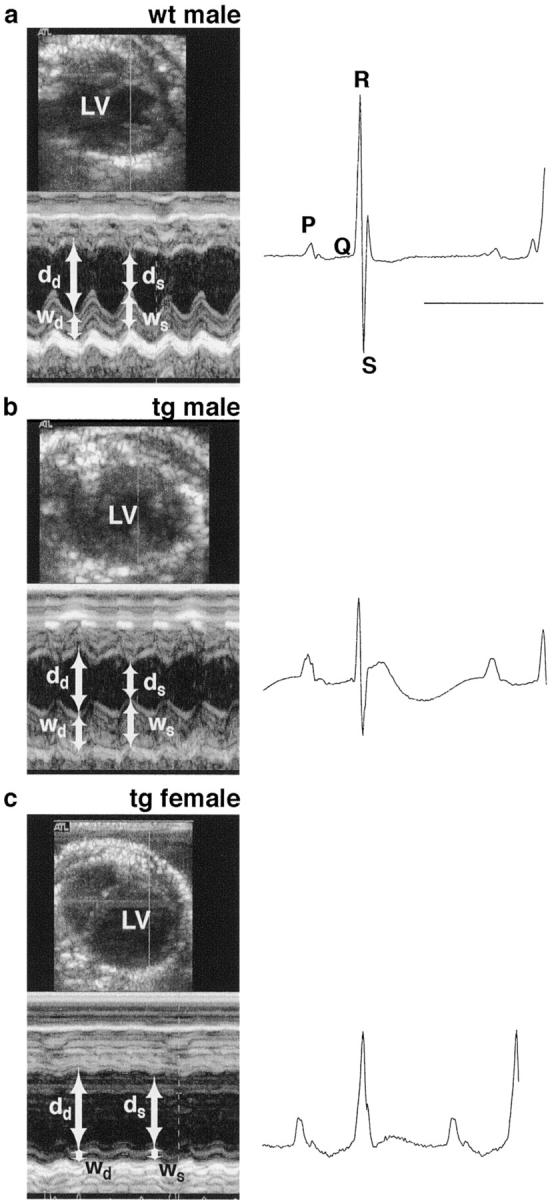
Representative transthoracic echocardiograms on wild-type and PDGF-C transgenic littermates at 3.5 months of age. End-diastolic left ventricle diameter (LV), and end-systolic and end-diastolic left ventricle wall thickness (ws and wd, respectively), and diameters (ds and dd, respectively) were measured (left). The corresponding electrocardiograms are shown at the right (bar, 100 ms). Fractional shortening, defined as (dd − ds)/dd, was impaired in the PDGF-C transgenic mice. a: A wild-type male with a normal appearance of the heart. b: A transgenic male with a thicker ventricle wall, a resulting smaller left ventricle volume, and decreased contractile function. c: A transgenic female with a dramatically enlarged left ventricle, a dilated ventricle wall, and an almost absent contractile function.
Table 4.
Echocardiography Data of Representative PDGF-C Transgenic and Wild-Type Mice
| Genotype* | Diameter† | Wall thickness‡ | FS§ | ||
|---|---|---|---|---|---|
| Diastole | Systole | Diastole | Systole | ||
| wt male | 3.7 | 2.1 | 0.7 | 1.4 | 43% |
| tg male | 2.5 | 1.7 | 1.6 | 1.7 | 32% |
| tg female | 5.5 | 5.0 | 0.7 | 0.9 | 9% |
* n = 3, wild-type male; n = 2 transgenic male; n = 1 transgenic female, three representative littermates at 3.5 months of age are viewed in Figure 5 ▶ .
† Left ventricle diameter in mm.
‡ Left ventricle wall thickness in mm.
§ Fraction shortening in %.
Discussion
To find out a role in vivo for the novel PDGF-C and possible action in cardiac tissue we generated transgenic mice overexpressing PDGF-C in the heart. The mice developed cardiac fibrosis and cardiomyopathy with phenotypes similar to those described for human hypertrophy and dilated cardiomyopathy. 1,4,5 The transgenic PDGF-C expression primarily induced fibroblast proliferation and deposition of extracellular matrix including collagen, via activation of PDGFR-α. The hypertrophic induction of the cardiomyocytes in this model is interesting because it is generally believed that the hypertrophic response of the cardiomyocytes, or tissue damage is proceeding the expansion of the interstitium and subsequent fibrosis, and not vice versa. It should be noted that the increase in heart mass probably reflects an excessive number of fibroblasts as well as hypertrophic cardiomyocytes.
Interestingly, α-MHC promoter-driven overexpression of PDGF-A in the mouse heart leads to a phenotype similar to that observed in PDGF-C transgenic animals with the difference that the hypertrophic phenotype is seen already in postnatal animals (Christer Betsholtz, personal communication). The temporal differences may reflect the fact that PDGF-C is a latent growth factor 11 and that proteolytic processing and activation is rate limiting in the PDGF-C transgenic animals. Only a portion of transgenic PDGF-C protein was processed, which may reflect the fact that PDGF-C is normally expressed at low levels in the adult mouse heart in contrast to human hearts where it is highly expressed. 11,25 Therefore the necessary protease(s) may not be expressed at sufficient levels to be able to quantitatively process transgenic PDGF-C.
The general function of CUB domains is unclear. 26 We have however shown that the CUB domain of PDGF-C has no pathological effect in the mouse heart because transgenic mice overexpressing this domain in the heart appear normal. The study also excluded that the α-MHC promoter itself caused any pathological changes. We suggest that the activated PDGF-C core domain is responsible for the phenotype in transgenic mice expressing full-length PDGF-C. It has been suggested that the CUB domain of PDGF-C expressed in Escherichia coli is able to stimulate proliferation of human artery smooth muscle cells. 27 We could not confirm this result because we could not detect any obvious vascular abnormalities or proliferation of smooth muscle cells in the CUB transgenic mice.
One of the hallmarks of cardiac hypertrophy is an altered profile of gene expression. Genes normally expressed during fetal development are re-expressed during hypertrophy, eg, α-SMA, ANP, and BNP. 28-32 The expression of certain cardiac genes expressed in adult myocardium is also up-regulated, such as MLC-2. 23,24 In the hearts of adult PDGF-C transgenic mice, ANP and BNP mRNAs and SMA protein expression were up-regulated as expected, whereas MLC-2 mRNA was differentially regulated, which probably reflects the degree of hypertrophy. It is possible that up-regulation of MLC-2 is correlated with cardiomyopathies in which hypertrophy is the primary pathological change, whereas in our model, the primary histopathological change is the proliferation of fibroblasts.
We observed sex-dependent phenotypes with the transgenic males developing the progressive hypertrophy and the females developing a lethal dilated cardiomyopathy. The different phenotypes were also reflected in the regulation of the molecular markers in which the females displayed larger changes in expression levels compared to the males. The most striking feature was the up-regulation of BNP in females, supporting the suggestion that female transgenic mice suffered from heart failure. 30 In humans, the risk of developing cardiovascular disease is lower in premenopausal women than in men, whereas postmenopausal women are more susceptible. The differences in susceptibility are believed to be related to estrogen status. 33 Several animal models of cardiomyopathies are available, most of which affect males more seriously. However, atrial thrombosis, a common consequence in dilated cardiomyopathy occurs earlier in female hamsters than in males. 1 A similar complication was observed in the PDGF-C transgenic females. The reason why females develop a more severe phenotype in our transgenic model is not clear (see below).
In a similar study in which TNF-α was overexpressed in mouse heart the animals developed opposite sex differences, with males having the dilated phenotype. 6 Further analysis showed that the expression levels of the transgenic TNF-α, as well as several cytokines regulated by TNF-α signaling, were equal in both females and males. However, the expression of TNFR-1 and -2 as well as the TNF-α-responsive second messenger molecule, ceramide, was higher in males than in females. In the present study the expression level of transgenic PDGF-C is equal between the two sexes. Expression and activation of PDGFR-α does not seem to differ between the sexes. It is possible that the sex difference observed arises from the downstream targets of PDGF-C signaling or reflects a secondary response in the sensitivity to the induced fibrosis.
Characteristic vascular abnormalities developed in the PDGF-C transgenic hearts. The microvessels gradually became dilated and sometimes developed into large sac-like structures, similar to the large vessels and vascular pools seen in muscle tissue transplanted with myoblasts engineered to overexpress VEGF. 34 In support of a role of VEGF in this phenotype is the observation that VEGF is up-regulated in cardiac fibroblasts in the transgenic hearts. The up-regulation of VEGF may be directly linked to PDGF-C overexpression and PDGF receptor-mediated signaling, or an indirect consequence of tissue damage, increased deposition of extracellular matrix, or tissue hypoxia. At least in part the increase in VEGF production may be due to PDGF-C overexpression because we observed abundant VEGF expression in the cardiac fibroblasts already in 2-week-old animals, at the early onset of fibroblast proliferation and before any detectable hypertrophy or any other myocardial abnormalities. In vitro studies have also shown that overexpression of PDGF-C in NIH/3T3 cells causes up-regulation of VEGF expression. 35 In the cornea pocket assay, PDGF-C is a potent inducer of angiogenesis, but the underlying mechanism is unclear. 36 It is possible that the role of PDGF-C in angiogenesis is indirect via induction of VEGF in connective tissue cells or that the activated fibroblasts produce a permissive extracellular matrix for blood vessels. However, regardless of mechanism, the outcome of PDGF-mediated stimulation of the vasculature appears site-specific because the PDGF-C transgenic mice initially developed a phenotype associated with the angiogenic properties of VEGF, but in later stages lose microvessels. Another feature of the vasculature in the transgenic animals is the increased number of smooth muscle-coated vessels. PDGF-C is a potent mitogen for vascular smooth muscle cells 37 and the remodeling of the vascular tree is likely to be a direct consequence of increased numbers of these cells. This observation may have bearings on therapeutic angiogenesis using endothelial cell mitogens, like VEGF, that generate immature and fragile vessels in need of a stabilizing coat of perivascular cells. A combination therapy using VEGF and a mitogen for vascular smooth muscle cells may generate more stabile vessels, and PDGF-C is an attractive candidate for such a mitogen.
A striking feature of the transgenic adult myocardium is the presence of Anitschkow cells. It is currently believed that the caterpillar-like appearance of chromatin in Anitschkow cells indicates cellular immaturity and undifferentiation, rather than any specific cell type, and it has been shown to be a characteristic of human fetal cardiac myocytes. The presence of Anitschkow cells in the myocardium and cardiac valves is also known to be a hallmark of rheumatic fever, and so-called Aschoff nodules are reportedly composed of Anitschkow cells. 38 Cells with Anitschkow-like chromatin have been shown to participate in response to trauma and foreign materials. 39,40 It is unknown whether the Anitschkow cells in the PDGF-C transgenic myocardium are present as a consequence of hypertrophy and fibrosis, or whether these cells are induced to proliferate, or home to the myocardium by PDGF-mediated signaling. Further characterization of the Anitschkow cells is clearly warranted.
The data presented in this work suggests that overexpression of PDGFs induces fibrosis and a unique secondary sex-dependent hypertrophic response. It remains to be established whether PDGFs might also have a role in cardiac fibrosis and fibrous replacement reactions in humans.
Acknowledgments
We thank B. Åkerblom for technical assistance and Dr. Ricardo E. Feinstein at the National Veterinary Institute and Björn Rozell at Huddinge Hospital for excellent histological analysis.
Footnotes
Address reprint requests to Dr. Ulf Eriksson, Ludwig Institute for Cancer Research, Stockholm Branch, Box 240, S-171 77 Stockholm, Sweden. E-mail: ueri@licr.ki.se.
Supported by grants from the Swedish Research Council, the Novo Nordisk Foundation, and the Karolinska Institutet.
Present address of K. A.: Department of Oncology, Karolinska Hospital, CCK, Karolinska Institutet, S-171 76 Stockholm, Sweden.
References
- 1.Van Vleet JF, Ferrans VJ: Myocardial diseases of animals. Am J Pathol 1986, 124:98-178 [PMC free article] [PubMed] [Google Scholar]
- 2.Sabbah HN: Apoptotic cell death in heart failure. Cardiovasc Res 2000, 45:704-712 [DOI] [PubMed] [Google Scholar]
- 3.Fukuda K: Development of regenerative cardiomyocytes from mesenchymal stem cells for cardiovascular tissue engineering. Artif Organs 2001, 25:187-193 [DOI] [PubMed] [Google Scholar]
- 4.Marian AJ, Roberts R: The molecular genetic basis for hypertrophic cardiomyopathy. J Mol Cell Cardiol 2001, 33:655-670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schönberger J, Seidman CE: Many roads lead to a broken heart: the genetics of dilated cardiomyopathy. Am J Hum Genet 2001, 69:249-260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kadokami T, McTiernan CF, Kubota T, Frye CS, Feldman AM: Sex-related survival differences in murine cardiomyopathy are associated with differences in TNF-receptor expression. J Clin Invest 2000, 106:589-597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nakajima H, Nakajima HO, Salcher O, Dittie AS, Dembowsky K, Jing S, Field LJ: Atrial but not ventricular fibrosis in mice expressing a mutant transforming growth factor-beta(1) transgene in the heart. Circ Res 2000, 86:571-579 [DOI] [PubMed] [Google Scholar]
- 8.Simm A, Nestler M, Hoppe V: Mitogenic effect of PDGF-AA on cardiac fibroblasts. Basic Res Cardiol 1998, 93:40-43 [DOI] [PubMed] [Google Scholar]
- 9.Heldin CH, Westermark B: Mechanism of action and in vivo role of platelet-derived growth factor. Physiol Rev 1999, 79:1283-1316 [DOI] [PubMed] [Google Scholar]
- 10.Heldin CH, Eriksson U, Östman A: New members of the platelet-derived growth factor family of mitogens. Arch Biochem Biophys 2002, 398:284-290 [DOI] [PubMed] [Google Scholar]
- 11.Li X, Pontén A, Aase K, Karlsson L, Abramsson A, Uutela M, Bäckström G, Hellström M, Boström H, Li H, Soriano P, Betsholtz C, Heldin CH, Alitalo K, Östman A, Eriksson U: PDGF-C is a new protease-activated ligand for the PDGF alpha-receptor. Nat Cell Biol 2000, 2:302-309 [DOI] [PubMed] [Google Scholar]
- 12.Bergsten E, Uutela M, Li X, Pietras K, Östman A, Heldin CH, Alitalo K, Eriksson U: PDGF-D is a specific, protease-activated ligand for the PDGF beta-receptor. Nat Cell Biol 2001, 3:512-516 [DOI] [PubMed] [Google Scholar]
- 13.Li J, Hoyle GW: Overexpression of PDGF-A in the lung epithelium of transgenic mice produces a lethal phenotype associated with hyperplasia of mesenchymal cells. Dev Biol 2001, 239:338-349 [DOI] [PubMed] [Google Scholar]
- 14.Hoyle GW, Li J, Finkelstein JB, Eisenberg T, Liu JY, Lasky JA, Athas G, Morris GF, Brody AR: Emphysematous lesions, inflammation, and fibrosis in the lungs of transgenic mice overexpressing platelet-derived growth factor. Am J Pathol 1999, 154:1763-1775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reneker LW, Overbeek PA: Lens-specific expression of PDGF-A in transgenic mice results in retinal astrocytic hamartomas. Invest Ophthalmol Vis Sci 1996, 37:2455-2466 [PubMed] [Google Scholar]
- 16.Reneker LW, Overbeek PA: Lens-specific expression of PDGF-A alters lens growth and development. Dev Biol 1996, 180:554-565 [DOI] [PubMed] [Google Scholar]
- 17.Subramaniam A, Jones WK, Gulick J, Wert S, Neumann J, Robbins J: Tissue-specific regulation of the alpha-myosin heavy chain gene promoter in transgenic mice. J Biol Chem 1991, 266:24613-24620 [PubMed] [Google Scholar]
- 18.Dumont DJ, Jussila L, Taipale J, Lymboussaki A, Mustonen T, Pajusola K, Breitman M, Alitalo K: Cardiovascular failure in mouse embryos deficient in VEGF receptor-3. Science 1998, 282:946-949 [DOI] [PubMed] [Google Scholar]
- 19.Aase K, von Euler G, Li X, Pontén A, Thorén P, Cao R, Cao Y, Olofsson B, Gebre-Medhin S, Pekny M, Alitalo K, Betsholtz C, Eriksson U: Vascular endothelial growth factor-B-deficient mice display an atrial conduction defect. Circulation 2001, 104:358-364 [DOI] [PubMed] [Google Scholar]
- 20.Chomczynski P, Sacchi N: Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 1987, 162:156-159 [DOI] [PubMed] [Google Scholar]
- 21.Lyons GE, Schiaffino S, Sassoon D, Barton P, Buckingham M: Developmental regulation of myosin gene expression in mouse cardiac muscle. J Cell Biol 1990, 111:2427-2436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ng WA, Grupp IL, Subramaniam A, Robbins J: Cardiac myosin heavy chain mRNA expression and myocardial function in the mouse heart. Circ Res 1991, 68:1742-1750 [DOI] [PubMed] [Google Scholar]
- 23.Vikstrom KL, Bohlmeyer T, Factor SM, Leinwand LA: Hypertrophy, pathology, and molecular markers of cardiac pathogenesis. Circ Res 1998, 82:773-778 [DOI] [PubMed] [Google Scholar]
- 24.Wagner M, Mascareno E, Siddiqui MA: Cardiac hypertrophy: signal transduction, transcriptional adaptation, and altered growth control. Ann NY Acad Sci 1999, 874:1-10 [DOI] [PubMed] [Google Scholar]
- 25.Aase K, Abramsson A, Karlsson L, Betsholtz C, Eriksson U: Expression analysis of PDGF-C in adult and developing mouse tissues. Mech Dev 2002, 110:187-191 [DOI] [PubMed] [Google Scholar]
- 26.Bork P, Beckmann G: The CUB domain. A widespread module in developmentally regulated proteins. J Mol Biol 1993, 231:539-545 [DOI] [PubMed] [Google Scholar]
- 27.Dijkmans J, Xu J, Masure S, Dhanaraj S, Gosiewska A, Geesin J, Sprengel J, Harris S, Verhasselt P, Gordon R, Yon J: Characterization of platelet-derived growth factor-C (PDGF-C): expression in normal and tumor cells, biological activity and chromosomal localization. Int J Biochem Cell Biol 2002, 34:414-426 [DOI] [PubMed] [Google Scholar]
- 28.Chien KR, Knowlton KU, Zhu H, Chien S: Regulation of cardiac gene expression during myocardial growth and hypertrophy: molecular studies of an adaptive physiologic response. FASEB J 1991, 5:3037-3046 [DOI] [PubMed] [Google Scholar]
- 29.Rubattu S, Volpe M: The atrial natriuretic peptide: a changing view. J Hypertens 2001, 19:1923-1931 [DOI] [PubMed] [Google Scholar]
- 30.Sakata Y, Yamamoto K, Masuyama T, Mano T, Nishikawa N, Kuzuya T, Miwa T, Hori M: Ventricular production of natriuretic peptides and ventricular structural remodeling in hypertensive heart failure. J Hypertens 2001, 19:1905-1912 [DOI] [PubMed] [Google Scholar]
- 31.Scheuermann-Freestone M, Freestone NS, Langenickel T, Hohnel K, Dietz R, Willenbrock R: A new model of congestive heart failure in the mouse due to chronic volume overload. Eur J Heart Fail 2001, 3:535-543 [DOI] [PubMed] [Google Scholar]
- 32.McConnell BK, Fatkin D, Semsarian C, Jones KA, Georgakopoulos D, Maguire CT, Healey MJ, Mudd JO, Moskowitz IP, Conner DA, Giewat M, Wakimoto H, Berul CI, Schoen FJ, Kass DA, Seidman CE, Seidman JG: Comparison of two murine models of familial hypertrophic cardiomyopathy. Circ Res 2001, 88:383-389 [DOI] [PubMed] [Google Scholar]
- 33.Sugden PH, Clerk A: Akt like a woman: gender differences in susceptibility to cardiovascular disease. Circ Res 2001, 88:975-977 [DOI] [PubMed] [Google Scholar]
- 34.Springer ML, Chen AS, Kraft PE, Bednarski M, Blau HM: VEGF gene delivery to muscle: potential role for vasculogenesis in adults. Mol Cell 1998, 2:549-558 [DOI] [PubMed] [Google Scholar]
- 35.Li H, Fredriksson L, Li X, Eriksson U: PDGF-D is a potent transforming and angiogenic growth factor. Oncogene 2003, 22:1501-1510 [DOI] [PubMed] [Google Scholar]
- 36.Cao R, Bråkenhielm E, Li X, Pietras K, Widenfalk J, Östman A, Eriksson U, Cao Y: Angiogenesis stimulated by PDGF-CC, a novel member in the PDGF family, involves activation of PDGFR-αα and -αβ receptors. FASEB J 2002, 16:1575-1583 [DOI] [PubMed] [Google Scholar]
- 37.Uutela M, Lauren J, Bergsten E, Li X, Horelli-Kuitunen N, Eriksson U, Alitalo K: Chromosomal location, exon structure, and vascular expression patterns of the human PDGFC and PDGFD genes. Circulation 2001, 103:2242-2247 [DOI] [PubMed] [Google Scholar]
- 38.Fraser WJ, Haffejee Z, Cooper K: Rheumatic Aschoff nodules revisited: an immunohistological reappraisal of the cellular component. Histopathology 1995, 27:457-461 [DOI] [PubMed] [Google Scholar]
- 39.Stehbens WE, Zuccollo JM: Anitschkow myocytes or cardiac histiocytes in human hearts. Pathology 1999, 31:98-101 [DOI] [PubMed] [Google Scholar]
- 40.Satoh F, Tsutsumi Y: Anitschkow cells in the human heart. Pathol Int 1999, 49:85-87 [DOI] [PubMed] [Google Scholar]