

Abstract
The ataxia telangiectasia mutated (ATM) protein plays a central role in the cellular response to DNA double-strand breaks (DSBs). Developmentally programmed DSBs are restricted to cellular subsets within lymphoid tissues and we asked whether ATM expression is differentially regulated during lymphoid differentiation. We showed that immature B cells in bone marrow and immature T cells of the thymic cortex were negative or weakly ATM-positive. T cells of thymic medulla and peripheral tissues strongly expressed ATM. High levels of ATM were present in the B lymphocytes of the mantle zone and in plasma cells, while the majority of germinal center B cells were negative or weakly labeled. Therefore, ATM expression appears to be down-regulated at those stages of lymphoid development where physiological DNA DSBs occur. In B-chronic lymphocytic leukemia and mantle cell lymphoma we observed two categories: ATM-negative tumors, most likely reflecting the presence of ATM mutation, and tumors with abundant ATM expression. Most follicular center-cell lymphomas and diffuse large B-cell lymphomas, which rarely show inactivation of the ATM gene, were negative or weakly ATM-positive. Tumor cells from most cases of Hodgkin’s disease were ATM-negative. Therefore, unless ATM inactivation occurs, ATM expression in lymphoid tumors is likely to reflect their cellular origin. As a result, immunostaining to identify lymphoid neoplasias with ATM inactivation might only be feasible for tumors derived from the stages where ATM is constitutively highly expressed.
Individuals with biallelic inactivation of the ataxia telangiectasia mutated (ATM) gene show a high predisposition to the development of lymphoid tumors of both B- and T-cell origin. While T-cell malignancies in A-T patients show a wide spectrum of phenotypes and include tumors of mature as well as immature T cells, B-cell tumors are derived mostly from the later stages of B-cell differentiation. 1
ATM is a 370-kd protein belonging to a family of PI-3 protein kinases with a role in DNA processing, regulation of the cell cycle, and control of telomere length. The principal function of the ATM protein is the integration of cellular responses to DNA double-strand breaks (DSBs). 2-4 In lymphoid tissues DNA DSBs can be created either during normal lymphoid development by the processes of V(D)J recombination, somatic hypermutation, and isotype switching, 5 or are caused by extrinsic factors such as ionizing radiation (IR). ATM-dependent cellular responses to DNA DSBs include the activation of DNA repair, cell-cycle checkpoints and apoptosis, and involve a complex network of protein-protein interactions that act to prevent propagation of DNA damage and the transmission of DNA errors that could be potentially tumorigenic for the cell. 4,6 Direct phosphorylation of p53 protein and activation of the p53 pathway is one of the crucial ATM-dependent responses and is important for the ATM-mediated activation of the G1/S checkpoint and the induction of apoptosis. 3
Human cells can process DSBs by either homology-directed or non-homologous repair pathways. Non-homologous end-joining (NHEJ) repair is based on error-prone ligation of broken ends and involves the proteins Ku, DNA-PKCS, Xrcc4, and DNA ligase IV. This pathway is also used during V(D)J recombination of the immune system genes. 5 In contrast, a high-fidelity mechanism of homologous recombination (HR) between sister chromatids is conducted by the proteins Rad51, Rad52, Rad54, and BRCA. The Nbs1/hMre11/Rad50 protein complex is implicated in both HR and NHEJ repair pathways. ATM directly phosphorylates repair proteins such as Nbs1 and BRCA1, and was shown to bind directly to the sites of DNA DSBs 7 as well as V(D)J intermediates. 8 Furthermore, both NHEJ and HEJ repair pathways are found to be defective in the absence of ATM. 9,10 The precise mechanism of DNA repair regulation by ATM, however, is yet to be elucidated.
Earlier reports suggested that ATM expression remains constant during cell-cycle progression and also after induction of DNA DSBs by γ irradiation. 11-13 In these circumstances ATM function appears to be determined by regulation of its kinase activity rather than by protein expression. 14 In contrast, a rapid change in protein expression has been reported under certain conditions. ATM was found to be up-regulated in the proliferative myoepithelium of breast ducts compared with the quiescent myoepithelial cells. 15 In addition, the level of ATM protein dramatically increases in quiescent lymphocytes in response to mitogenic agents. 16 Finally, both fibroblasts and lymphoid cells were found to down-regulate ATM in response to epidermal growth factor through a mechanism that involves alteration in DNA binding activity of the transcription factor Sp1. 17 Accordingly, several Sp1 consensus binding sites were previously identified within the sequence of the ATM/NPAT/E14 bi-directional promoter. 18
It has not been reported whether regulation of ATM during normal lymphoid differentiation involves variation in ATM protein expression. Although variations in expression of ATM transcripts between different tissues have been observed, with particularly high-level expression in tissues that are frequently exposed to DNA DSBs such as spleen and thymus, 19 ATM expression in specific cell types within lymphoid tissues has not been analyzed. To address this question, we studied a spectrum of human lymphoid tissues using an antibody directed against ATM. Our findings revealed a clear difference in ATM expression between different stages of lymphoid development. ATM was generally absent in both immature B and T cells of the bone marrow and the thymic cortex, respectively. In contrast, T lymphocytes of the thymic medulla and the peripheral tissues generally expressed high levels of ATM. During B-cell differentiation high-level expression was observed in pre- and postgerminal center B cells, but not in germinal center B cells. These findings suggest that down-regulation of ATM expression may be important during developmentally programmed genomic recombinations. Because of the variations in ATM expression observed during B-cell differentiation we extended our study to include an analysis of B-cell tumors derived from these different stages of B-cell development. Our results revealed that the majority of tumors derived from the germinal center stages did not express ATM. In tumors derived from the stages of B-cell differentiation where we had previously demonstrated high-level ATM expression we observed two distinct categories: ATM-negative tumors, presumably the result of the presence of inactivating ATM mutations, and tumors exhibiting strong ATM expression. Our results have important implications for the use of protein detection in the identification of tumors harboring inactivating ATM mutations.
Materials and Methods
Tissues
ATM expression was studied in paraffin-embedded normal lymphoid tissues (lymph node, thymus, spleen, bone marrow) as well as in frozen thymus and tonsil. Paraffin wax-embedded specimens of a variety of B-lymphoid tumors, including B-cell chronic lymphocytic leukemia (B-CLL), mantle cell lymphoma (MCL), follicular center cell lymphoma (FCCL), diffuse large B-cell lymphoma (DLBCL), and classic Hodgkin’s disease (cHD) were also investigated. Five-μm paraffin wax sections were cut to Vectabond-coated slides and left at 37°C for a minimum of 2 hours before being dewaxed and transferred to PBS buffer pH 7.4. Frozen sections were cut at 6 μm to coated slides, fixed in 10% formal saline for 20 minutes and washed in PBS. Lymphoblastoid cell lines (LCLs) prepared from A-T patients and from normal donors were used to confirm the specificity of the ATM antibody and subsequently as controls for the ATM staining.
Production of 11G12 ATM Monoclonal Antibody
A 474-bp fragment representing amino acids 992-1144 of the ATM cDNA sequence was cloned in frame with the hexa-histidine tag of the vector pQE-32 (Qiagen, Crawley, UK) to generate the clone, designated FP8. Bulk expression in E. coli and purification of the His-tagged ATM fusion protein was performed using protocols suggested by the manufacturer (Qiagen). Aliquots (50 μg) of ATM fusion protein in Freund’s adjuvant were injected into three mice at two-week intervals for a total of eight weeks, with the fourth and final injection in the absence of Freund’s adjuvant. Sera from the mice were tested by Western blotting for good antibody responses to the fusion protein, before proceeding to monoclonal antibody production. Spleen cells were fused to SP2 mouse myeloma cells, plated out in 96-well plates and hybridomas selected in medium containing HAT (Sigma Ltd., Poole, Dorset, UK). Supernatants were tested by ELISA toidentify hybridoma clones producing antibodies to ATM fusion protein. Positive clones were expanded andsupernatants tested by Western blotting of protein cell extracts from normal and A-T individuals, to identify those hybridomas producing monoclonal antibodies specific to the ∼370-kd ATM protein (hereafter referred to as 11G12 antibody).
Immunohistochemistry
The 11G12 ATM antibody was used at a dilution of 1:10 in immunohistochemical assays and was detected by the standard peroxidase-based Avidin Biotin Duet system (catalog number K492; Dako Corp., Ely, UK) or the Chemicon IHC Select (Chemicon, Temecula, CA) followed by the demonstration of peroxidase activity using the DAB reaction. Tissue sections were microwaved in 0.01 mol/L-citrate buffer (pH 6.0) for a minimum of 45 minutes. In the case of the lymphoma specimens tumors were classed as ATM-negative if fewer than 10% of tumor cells were labeled. Tissues were also analyzed by sequential double-labeling to demonstrate ATM and a range of hematopoietic differentiation markers, including CD3 (for T cells), CD20, CD21, and CD22 (for B cells), CD34 (for early hematopoietic cells), CD45 (leukocyte progenitors in bone marrow), CD42b (for megakaryocytes), CD68 (for macrophages), CD79a (for mature B cells), CD138 (for post-GC B cells, including plasma cells), CD235a (glycophorin A, for cells of the erythroid series), and myeloperoxidase (for cells of the myeloid series).
Bound antibodies were detected using Chemicon IHC Select kit and a combination of DAB (brown) with either Vector VIP (purple) (Vector Laboratories, Burlingame, CA) or Vector SG (gray/black). DAB was generally but not always used in the first labeling reaction. The antigen which required the shorter retrieval time was labeled in the first reaction. Controls were used to ensure non-cross-reactivity between first and second labeling steps and these consisted of a series of consecutive sections in which individual antibody steps were omitted. Immunophenotyping of FCCL, DLBCL, and cHD was performed using antibodies to CD10 and CD138. ATM expression in HRS cells of cHD was assessed by dual-labeling with anti-CD30 antibodies. The presence of DSBs was demonstrated using antibodies against phosphorylated histone H2AX (γ-H2AX). 20
Results
ATM Antibody 11G12
The specificity of the monoclonal 11G12 antibody for ATM was confirmed by Western blotting and by immunohistochemistry using LCLs derived from three classical A-T patients (A-TLCL118–3, A-TLCL77–3, A-TLCL142), each carrying two ATM truncating mutations, and from three normal donors. Figure 1A ▶ shows the detection of a 370-kd band corresponding to the ATM protein in the LCLs from normal donors but not in the LCLs from the A-T patients. In cytospins and paraffin-embedded cell pellets, strong labeling of nuclei was observed in normal donor LCLs, whereas LCLs from A-T patients were not labeled with the ATM antibody (Figure 1B) ▶ .
Figure 1.
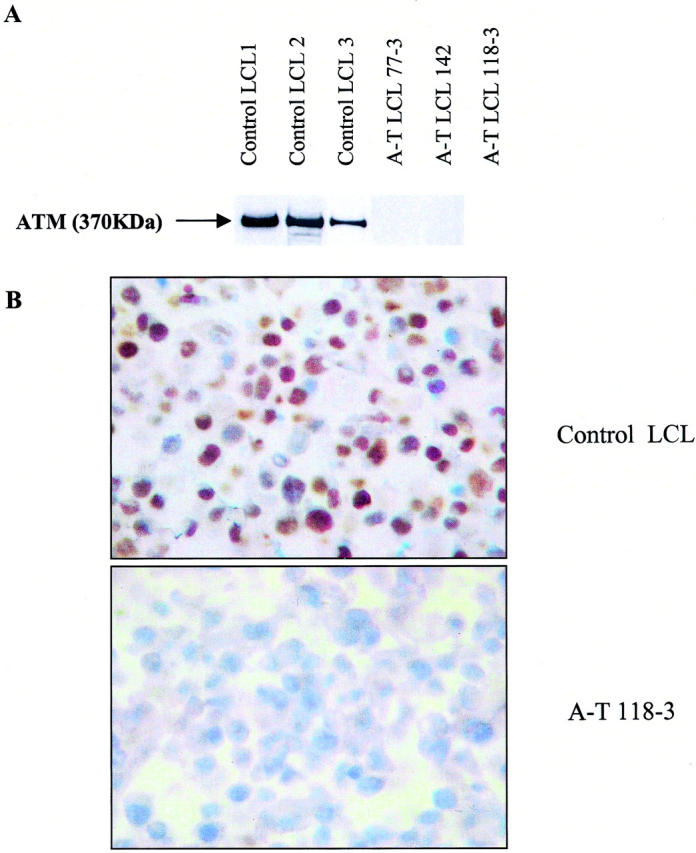
Specificity of ATM monoclonal antibody 11G12. Western blotting (A) shows expression of a 370-kd band in 3 LCLs from normal donors and absence of the same band in the LCLs from three A-T donors with truncating ATM mutations of both alleles. Immunohistochemistry using the 11G12 antibody produced strong nuclear staining in cytospin preparations of the normal donor LCLs, whereas the LCLs from the A-T donors were negative (B).
ATM Protein Expression in Normal Lymphoid Tissues
Our studies revealed that ATM-positivity in the bone marrow was restricted to a minor subpopulation of mostly small cells, comprising 5% to 10% of the total cellular compartment (Figure 2) ▶ . CD34-positive cells (stem cells/pro-B cells) were mostly ATM-negative, although occasional cells were labeled. More mature B cells, as evidenced by their expression of CD45 (early pro-B cells, mature B cells), CD20, CD21, or CD79a (all mature B cells), were more frequently ATM-positive (mean numbers of ATM-positive cells across 3 separate normal bone-marrow specimens that co-expressed B-cell markers were for CD45, 72.8%; CD20, 65.3%; CD21, 35.6%; CD79a, 49.4%). Triple-labeling experiments revealed that the majority of ATM-positive cells not expressing B-cell markers (CD20, CD21, CD79a) included plasma cells (Figure 2c ▶ , inset), macrophages, and T-lymphocytes. Occasional CD20-, CD79a-, and CD21-expressing cells appeared to lack ATM expression although these were generally rare. Remarkably, developing cells of the erythroid (as detected by expression of CD235a) and myeloid lineages (by expression of myeloperoxidase) were almost exclusively ATM-negative. Cells of the platelet series, including megakaryocytes, expressed moderate levels of ATM.
Figure 2.
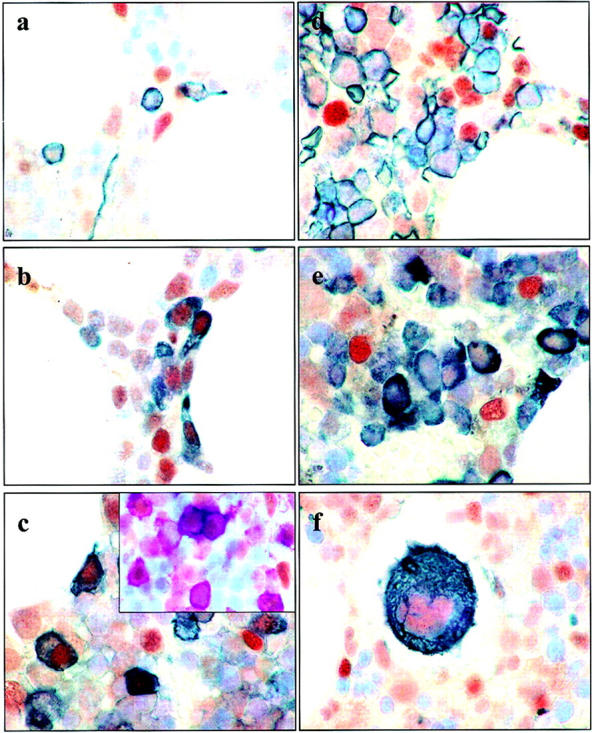
ATM expression in bone marrow. ATM-positive cells of the bone marrow were small and comprised 5% to 10% of the total cellular compartment. Dual-labeling was performed for ATM (brown) and a range of hematopoietic differentiation markers (dark gray/black). a: Dual-labeling with an antibody specific for CD34; the majority of CD34-expressing cells were ATM-negative. ATM expression was apparent in more mature lymphoid cells, including those that expressed CD45 and CD20 (b and c, respectively). However, a proportion of ATM-positive cells were not labeled with either CD20 or CD45. Triple-labeling with ATM, CD20, and either CD138, CD68, or CD3 revealed that these cells were mostly either plasma cells, macrophages, or T-lymphocytes. Inset of c shows triple-labeling for ATM (brown), CD20 (purple), and CD138 (gray/black). Cells of the erythroid series (shown in d by staining with antibodies to glycophorin A) and those of the myeloid series (e, by staining with antibodies to myeloperoxidase) were consistently ATM-negative. Megakaryocytes (f, stained for CD42b) showed weak to moderate staining with the ATM antibody.
In the thymic cortex <5% of cells expressed detectable ATM; however, these larger cells were mostly either macrophages or epithelial cells as evidenced by dual expression with cytokeratin and only occasional small cells showed dual-positivity for ATM and CD3. In the medulla, however, >75% of cells were stained positively for ATM (Figure 3) ▶ ; most of these were T cells as detected by dual-staining (Figure 3) ▶ . Epithelial cells of the thymic medulla were also labeled with the ATM antibody (Figure 3) ▶ . In contrast to the clear differences in T-lymphocyte expression of ATM at the medulla and cortex, there were no differences in the level of ATM expression in epithelial cells at these locations.
Figure 3.
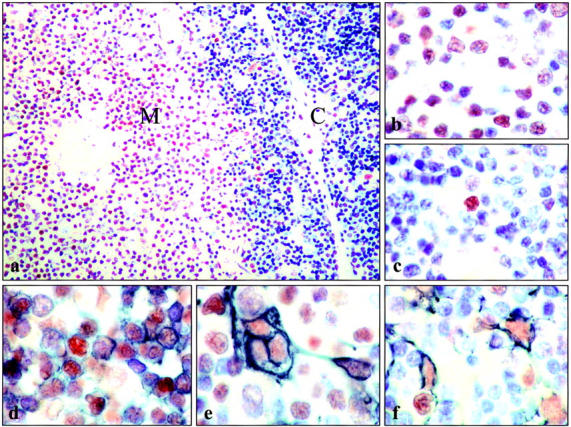
ATM expression in thymus. a: Immunostaining of thymus revealed strong expression of ATM in cells of the thymic medulla, but not the cortex. b: Higher magnification of thymic medulla showing that the majority of cells with typical lymphocyte morphology are ATM-positive, whereas in cortex (c) such cells were rarely ATM-positive. Double-labeling of ATM (brown) and CD3 (gray/black; d) confirms the majority of ATM-positive cells in the medulla are T-lymphocytes. Epithelial cells of the thymic medulla were also ATM-positive as evidenced by their co-expression of ATM (brown) and cytokeratin (gray/black; e). f: Epithelial cells of the cortex were also ATM-positive.
We observed a marked variation of ATM expression in lymph nodes and tonsils. In the case of B-lymphocytes, the level of expression varied dramatically with the stage of B-cell development. Mantle-zone B cells were all strongly labeled, while germinal center B cells were only weakly stained or negative, and post-GC CD138-positive B cells, including plasma cells, were strongly ATM-positive (Figure 4) ▶ . Thus, in B-lymphocytes ATM appears to be down-regulated at those sites where DNA DSBs occur. The presence of DSBs in ATM-low germinal-center B lymphocytes was confirmed in consecutive sections using antibodies against γ-H2AX, which is associated with this type of DNA damage (Figure 4) ▶ . Cells in interfollicular areas were strongly labeled mainly because of the generally strong expression of ATM by T cells in these areas (Figure 4) ▶ . In contrast to germinal-center B lymphocytes, macrophages at this location were strongly ATM-positive (Figure 5) ▶ . Moderate staining of basal and parabasal cells of the tonsillar squamous epithelium was also noted, whereas the more superficial cells were negative.
Figure 4.
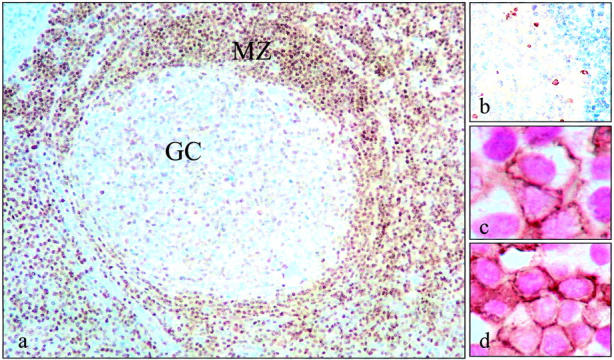
Variation in ATM expression among different cellular subsets in secondary lymphoid organs. In tonsils and lymph nodes ATM expression was remarkably variable between different cellular subsets. a: Mantle zone (MZ) but not germinal center (GC) B-lymphocytes were strongly labeled, suggesting that ATM is down-regulated at those sites undergoing DSBs. We confirmed the presence of DSBs in ATM-low cells in the germinal centers by staining consecutive sections for γ-H2AX (b). c: Plasma cells expressing CD138 (brown membrane staining) were also strongly ATM-positive (shown here as purple nuclear reactivity). d: In interfollicular areas, CD3-positive T cells (brown membrane staining) were mostly also ATM-positive (shown here as purple).
Figure 5.
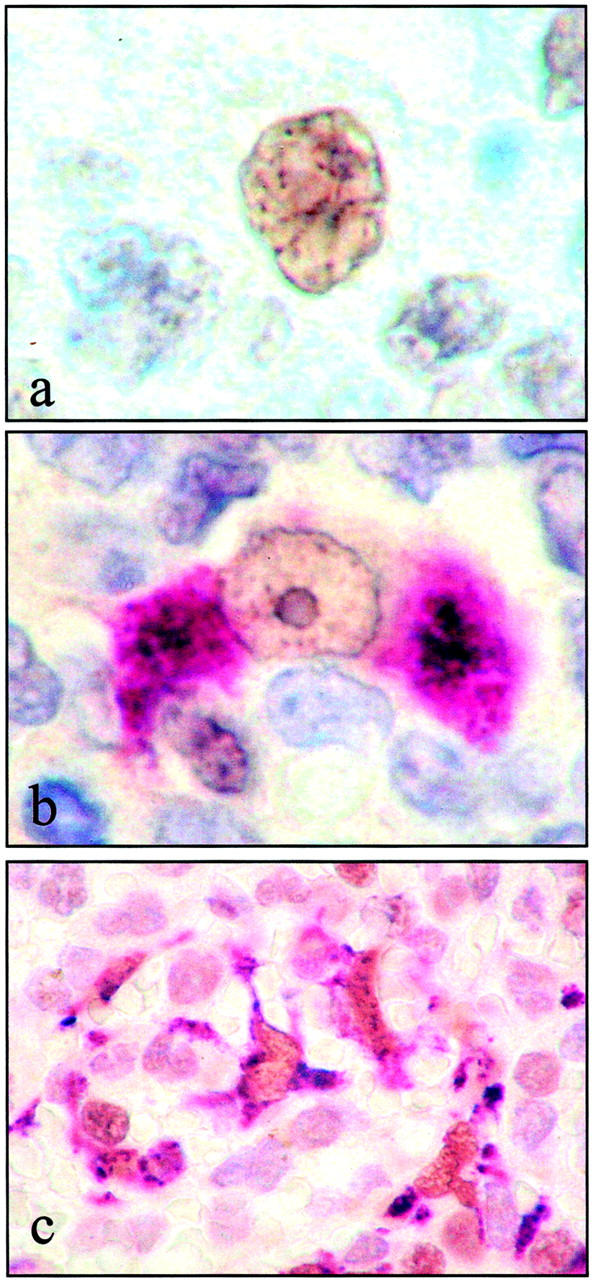
Strong expression of ATM by macrophages. a: In contrast to the weak labeling of GC B lymphocytes, macrophages in the GC were strongly labeled for ATM (brown) as identified by typical morphology, and by dual-labeling with CD68 (b; ATM brown, CD68 purple). Macrophages of the red pulp of spleen were also ATM-positive, shown here in c by dual-labeling for ATM (brown) and CD68 (purple).
A similar pattern of staining was observed in spleen with strong ATM staining in periarteriolar T lymphocytes and in B cells of primary follicles but not in germinal centers where the pattern was identical to that seen in lymph nodes and tonsil. Macrophages of the red pulp and germinal centers of spleen also strongly expressed ATM (Figure 5) ▶ .
ATM Protein Expression in B-Lymphoid Tumors (Table 1) ▶
Table 1.
ATM Expression in B-Lymphoid Tumors
| Tumor | No. ATM-positive cases | |
|---|---|---|
| Diffuse large B-cell lymphoma | 11/47 | |
| Follicular lymphoma | 19/59 | |
| CD10-positive | 7/41* | |
| CD10-negative | 11/14 | |
| Mantle cell lymphoma | 7/15 | |
| Chronic lymphocytic leukemia† | 32/42 | |
| Wild-type ATM | 15/15 | |
| Mutant ATM | 0/2 | B-CLL1 1058delGT; 5464G→A(E1822Q) |
| B-CLL2 G1048T(A350T); T1055C(I352T) | ||
| 29/31 | ||
| Hodgkin’s disease (classic) | 5/25 | |
| CD138-positive | 5/6 | |
| CD138-negative | 0/19 | |
| EBV status‡ | ||
| (−) | 1/10 | |
| (+) | 3/14 | |
| Subtype | ||
| MC | 2/12 | |
| NS | 2/11 | |
| Lymphocyte-rich classic HD | 1/2 | |
| Age | ||
| <15 years | 1/6 | |
| 15 and over | 4/19 |
*CD10 status not determinable in four cases.
†ATM status had previously been determined for these CLL tumors.
‡EBV status not available for one ATM-positive case.
The majority of B-cell tumors derived from the germinal-center stages of B-cell differentiation where ATM is normally down-regulated (ie, DLBCL, FCCL), and in which prior studies have not generally identified inactivating ATM mutations, were either negative or weakly labeled with the ATM antibody (Figure 6) ▶ . However, some DLBCL and FCCL strongly expressed ATM and interestingly, in the case of FCCL but not DLBCL we observed phenotypic differences between the ATM-positive and ATM-negative variants. Thus, the majority of ATM-negative FCCLs displayed a typical GC phenotype characterized by expression of CD10, whereas the rarer ATM-positive tumors were mostly CD10-negative.
Figure 6.
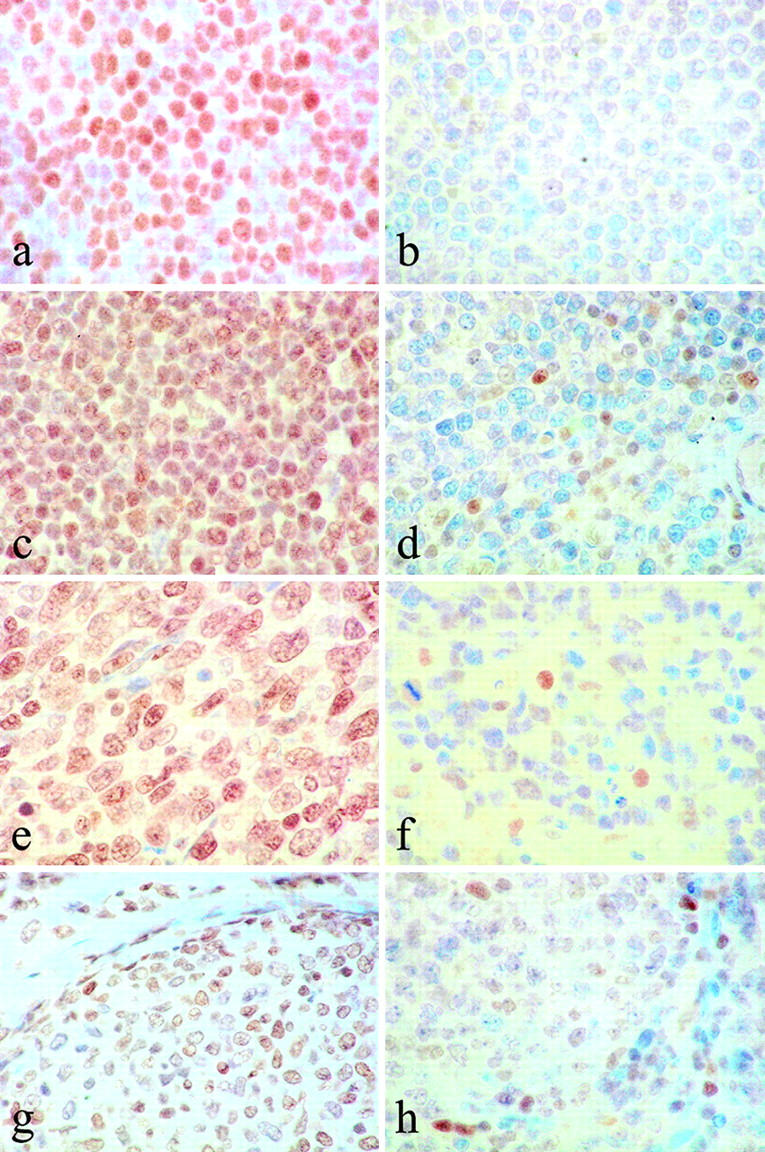
Variation of ATM expression in subsets of B-cell neoplasias. B-cell tumors derived from the different stages of B-cell differentiation showed variation in ATM expression. a: A B-CLL tumor where previous analysis had revealed wild-type ATM status, showing strong ATM expression. In contrast, B-CLL1 tumor (b) carrying an inactivating mutation in the ATM gene did not express detectable levels of ATM protein. The other tumor where ATM mutations are frequent is mantle cell lymphoma (MCL) and two subgroups of tumor were recognizable in our study; a group showing strong ATM expression, an example of which is shown in c, and a group of ATM-negative tumors where we presume ATM is mutant (d). In diffuse large B-cell lymphomas (DLBCL) both ATM-positive (e) and ATM-negative tumors (f) were observed. Likewise, ATM-positive and ATM-negative groups of FCCL tumors were seen (g and h, respectively). In DLBCL and FCCL tumors variation in ATM expression is most likely due to variations in the differentiation status of the malignant B cells.
In tumors derived from the B-cell stages where we demonstrated high levels of ATM expression, and in which ATM mutations have been frequently detected (ie, MCL, B-CLL), we observed two distinct patterns (Figure 6) ▶ : 1) ATM-negative tumors, most likely the result of the presence of inactivating ATM mutations, and 2) tumors showing abundant ATM expression. In each of the two B-CLL cases with a loss of ATM expression we previously identified two ATM mutations. In B-CLL1 we identified a truncating mutation 1058delGT and the missense mutation 5464G->A, predicted to lead to the substitution of glutamic acid by glutamine at position 1822. In B-CLL 2 we identified two missense mutations: 1048G->T predicted to lead to the substitution of alanine by threonine at position 350, and 1055T->C predicted to cause the substitution of isoleucine by threonine at position 352. 21 Both tumors showed a defect in damage-induced activation of the p53 pathway, 21 suggesting the pathogenic nature of the identified mutations. All CLL cases with wild-type ATM showed high-level expression of ATM. Notably, the majority of multiple myelomas was strongly ATM-positive. In the majority of the B-cell lymphomas examined, irrespective of ATM status, we could identify small, scattered ATM-positive non-tumor cells. Double-labeling revealed that these were mostly T lymphocytes (data not shown).
The majority of cases of cHD showed no detectable ATM expression in the malignant Hodgkin/Reed-Sternberg (HRS) cells despite strong staining of surrounding reactive cells (Figure 7) ▶ . However, there was no relationship between ATM expression and histological subtype, EBV status and whether the tumors were from adults or children. In contrast to HD cases harboring ATM-negative HRS cells, which were with one exception either negative or showed only rare expression of CD138 (<5% HRS cells stained), in all ATM-positive tumors HRS cells strongly expressed CD138, suggesting that the majority of tumor cells in these cases had the phenotype of post-GC B-cells.
Figure 7.
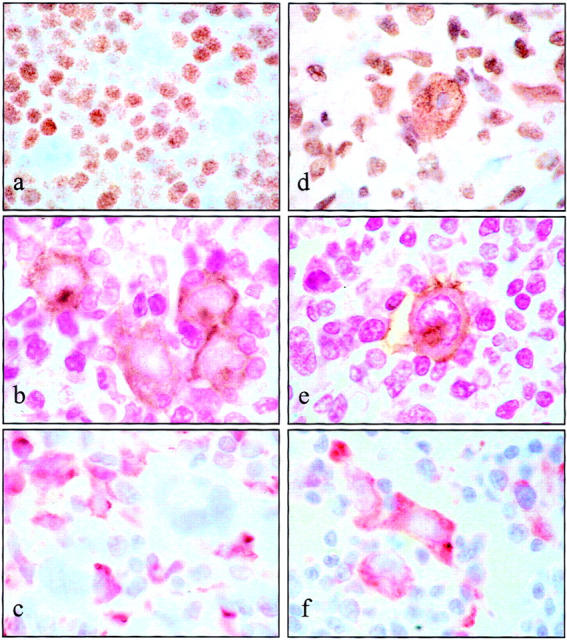
Expression of ATM in the malignant cells of Hodgkin’s disease. HRS cells from the majority of cases lacked detectable ATM expression. a: A representative ATM-negative HD tumor. b: Dual-labeling of ATM (purple) with CD30 (brown), confirming the absence of ATM expression in the malignant population in this case. d: Strong expression of ATM in the HRS cell of a tumor classified as ATM-positive. e: Expression of ATM (purple) in CD30-positive (brown) malignant cells from this case. We also observed differences in the phenotype of the malignant cells between ATM-positive and ATM-negative HD cases; thus with one exception, ATM-negative tumors either did not express CD138 or only expressed this marker at low levels in malignant cells (c), whereas ATM-positive HRS cells strongly expressed CD138 (f).
Discussion
In this study we have demonstrated significant variations in ATM expression within different B- and T-cell subpopulations in human lymphoid organs that are likely to reflect differences in the requirement for ATM at different stages of B- and T-cell development.
B- and T-lymphocyte development is characterized by progressive differentiation within distinct tissue compartments. In the case of B lymphocytes, early antigen-independent development occurs in the bone marrow and is characterized by stepwise gene rearrangement, involving first the heavy chain D-J segments (pro-B cell stage), followed by heavy chain and then light chain V(D)J recombination. Mature B cells exported to the periphery then undergo a second round of proliferation if they encounter antigen. This occurs in the germinal center and leads to the production of high-affinity antibody producing plasma cells and memory B cells. Class switching and somatic hypermutation also occur in the activated B cells of the germinal center. In contrast, T-cell development occurs in the thymus. Pre-T cells of the thymic cortex rearrange their TCR genes and undergo positive selection for self-MHC restriction. T cells that survive this process migrate to the medulla from where they travel to the peripheral tissues and localize to T-cell areas (interfollicular regions of lymph nodes and periarteriolar cuffs in spleen). Unlike B cells there is no formation of an equivalent to the germinal center during T-cell differentiation. Our results show that ATM is expressed at low levels at sites where physiological DNA DSBs are required and where cells are undergoing rapid proliferation, ie, in germinal center B cells, in immature B cells and T cells of the bone marrow and thymic cortex, respectively. Interestingly, we observed high-level ATM expression at both pre-germinal and postgerminal stages of B-cell differentiation, as well as in the thymic medulla at the locations of terminal T-cell differentiation.
One possible explanation for the variations in ATM expression we have observed in lymphoid cells might be the requirement for developmentally regulated cellular responses to DNA DSBs. The alteration in ATM expression might be required to prevent apoptosis in immature lymphocytes undergoing immune system gene rearrangements. Indeed, Bhandoola and colleagues 22 recently reported that immature thymocytes are able to tolerate DNA DSBs induced by DNA intercalating agents, whereas mature T cells are signaled to die by an ATM-dependent death mechanism. Here, we demonstrate a clear decrease in ATM expression at the locations of immune system gene rearrangements in lymphoid organs. We suggest that reduced ATM expression may represent a mechanism for the down-regulation of responses to DNA DSBs. Indeed, we have previously shown that decreased levels of ATM expression can result in significantly reduced ATM function and impaired responses to DNA DSBs. 23 Furthermore, it has been reported that during lymphoid development, activation of the p53 pathway plays an important role in induction of apoptosis and elimination of lymphocytes with the accumulation of V(D)J intermediates. 24 Down-regulation of ATM in immature human B and T lymphocytes observed in this study would therefore be consistent with protection from excessive apoptosis during lymphoid development. In contrast, high-level expression of ATM in pre- as well as in postgerminal B cells and in mature T lymphocytes may ensure that cells with disadvantageous V(D)J recombinations, or potentially tumorigenic DNA damage are eliminated by apoptosis.
During the processing of developmentally programmed DSBs, ATM co-operates with other repair proteins. Expression of the DSB repair protein, Nbs1, which is involved in V(D)J recombination, is also low at the sites of developmentally programmed DNA DSBs, both in the spleen and thymus. 25 However, DNA PKcs and Ku 80 show low expression in mantle zone and high expression in germinal centers, the opposite of that observed for ATM. 26 The differences in expression of various repair proteins throughout lymphoid development may reflect their different roles in response to developmentally regulated DNA damage.
The mechanism by which differential ATM expression is regulated throughout lymphoid development is currently unknown. The separation of lymphoid subsets from different stages of B- and T-cell differentiation and quantitation at both the protein and transcriptional level could potentially provide such an answer but these separation procedures are difficult to perform. Interestingly, our previous studies conducted on a spectrum of T- and B-cell lines of different maturity revealed no correlation between endogenous ATM protein levels and ATM promoter activity determined by analysis of transfected luciferase reporter gene constructs (unpublished data). There is a possibility, therefore, that throughout lymphoid development ATM protein expression is regulated at a posttranscriptional level.
Our results revealed a striking difference in ATM expression between different B-cell malignancies. Differences in ATM expression could be the result of variation in the frequency of ATM inactivation within B-cell tumors. Alternatively, patterns of ATM expression might reflect the different stages of B-cell differentiation from which individual tumors are derived.
The ATM gene is inactivated in sporadic lymphoid tumors of both mature B- and T-cell phenotype. 27 The frequency of ATM inactivation varies between the different histological subentities. Vorechovsky and colleagues 28 reported the presence of ATM mutations in 17 of 37 analyzed sporadic T-PLLs. The majority of these 17 tumors had lost the second ATM allele, suggesting a complete inactivation of the ATM gene. Similar results were reported in two other studies where the remaining ATM allele was found to be mutated in up to 70% of sporadic T-prolymphocytic leukemia (T-PLL) with loss of heterozygosity (LOH) across the ATM gene. 29,30 Forty percent of MCL with LOH across 11q22–23 also show mutation of the second allele. 31,32 In addition, we and others have reported inactivation of the ATM gene in some 20% of B-CLL. 21,33-36 In contrast, ATM is rarely mutated in FCCL, DLBCL, or T-NHL. 37
MCL is a tumor of resting B lymphocytes lacking VH somatic hypermutation and derived from the mantle zone. Judging by the variable heavy (VH) somatic mutation status, B-CLL tumors could be of either pre-germinal or postgerminal origin. 38,39 Interestingly, we previously observed that ATM mutant B-CLL tumors uniformly lack VH somatic hypermutation, suggesting their common pre-germinal origin. 21 Therefore, both sporadic B-celltumors with frequent inactivation of the ATM gene, B-CLL and MCL, are derived from cells that normally display high-level expression of ATM protein. Absence of ATM in either MCL or B-CLL is likely to represent the presence of inactivating ATM mutations. Indeed, we were able to demonstrate that in the case of B-CLL, tumors that were wild-type for ATM uniformly expressed high levels of ATM protein, whereas two B-CLL tumors carrying ATM mutations did not. Despite the fact that the region against which our ATM antibody was raised (encompassing amino acids 992 to 1144) did not coincide with the sites of amino acid substitutions of the mutant alleles, we could not identify these mutant proteins by immunostaining. This is not surprising bearing in mind that the missense mutant ATM alleles tend to show variable expression at the protein level, regardless of the position of mutation. Failure to detect expression of the mutant missense alleles might also reflect the impact of the amino acid substitution on the conformation of the mutant protein and its ability to bind the ATM antibody. Furthermore, truncating ATM mutant alleles are not expressed at the protein level due to their instability 27 and therefore the lack of the expression of the truncating 1058delGT allele was also predictable. 27
In tumors previously shown to only infrequently harbor an ATM mutation, such as FCCL and DLBCL, the most likely explanation for the lack of ATM expression is their origin from germinal center cells where ATM is not normally highly expressed. Intriguingly, however, ATM was highly expressed in a subset of both FCCL and DLBCL (30 of 106 analyzed tumors), which might suggest their origin from another point in the B-cell differentiation pathway. This interpretation was supported, at least in the case of FCCL, by our immunophenotypic analysis where ATM-positive tumors were more frequently CD10-negative, while ATM-negative tumors were more frequently CD10-positive, although the significance of CD10 expression by FCCL in relation to cell of origin has yet to be established. Surprisingly we found no relationship between ATM and CD10 expression in DLBCL where CD10 has been useful in making the distinction between the GC-derived and activated forms of DLBCL. An alternative explanation is that DLBCL and FCCL with high ATM expression might have involvement of genetic factors that deregulate ATM. We conclude that unless there is evidence of ATM inactivation, ATM expression in sporadic lymphoid tumors broadly reflects their cellular origin.
In Hodgkin’s disease, where a role for ATM inactivation is suggested by the increased incidence of this tumor in AT patients, low levels of ATM protein expression might indicate the presence of inactivating mutations in the ATM gene. Alternatively, it might merely reflect the origin of HRS cells from a germinal center stage of B-cell differentiation. Carbone et al 40 recently described variation in the extent to which tumor cells from individual cHD cases express a post-GC phenotype as assessed by their expression of CD138. We used immunohistochemistry to examine CD138 expression in tumor cells from our series. Our finding that all of the ATM-positive tumors strongly expressed CD138, whereas almost all of the ATM-negative tumors were either negative or showed only low CD138 expression favors the stage of differentiation as a determinant of ATM expression in HRS cells. However, we did find a single tumor that strongly expressed CD138 but lacked ATM expression; this might represent a post-GC subgroup that carry inactivating ATM mutations. Further studies are required to confirm this possibility.
The observations from our study have practical implications for the analysis of ATM in different sporadic lymphoid tumors. We have demonstrated remarkable variations in ATM expression among the lymphoid subsets of different maturity, indicating that not only the presence of ATM mutations, but also the stage of differentiation, can influence ATM expression in lymphoid cells. Consequently, it may be difficult to detect a decrease in ATM expression caused by ATM inactivation in tumors derived from cells with constitutively low levels of ATM expression. In neoplasias derived from stages with high ATM expression, however, immunocytochemistry could provide an ideal screening procedure for the detection of tumors harboring mutant ATM.
Acknowledgments
We thank Marie Smith and Pamela Edwards for help in the collection of the pathology samples.
Footnotes
Address reprint requests to Dr. P.G. Murray, Department of Pathology, Division of Cancer Studies, The Medical School, University of Birmingham, Edgbaston, Birmingham, B15 2TT, UK. E-mail: p.g.murray@bham.ac.uk.
Supported by the Leukemia Research Fund, Cancer Research UK and the Kendall (Kay) Leukemia Fund.
References
- 1.Taylor AMR, Metcalfe JA, Thick J, Mak Y-F: Leukaemia and lymphoma in ataxia telangiectasia. Blood 1996, 87:423-438 [PubMed] [Google Scholar]
- 2.Shiloh Y: Ataxia-telangiectasia and the Nijmegen breakage syndrome: related disorders but genes apart. Ann Rev Genet 1997, 31:635-662 [DOI] [PubMed] [Google Scholar]
- 3.Shiloh Y: ATM and ATR: networking cellular responses to DNA damage. Curr Opin Genet Dev 2001, 11:71-77 [DOI] [PubMed] [Google Scholar]
- 4.Kastan MB, Lin D: The many substrates and functions of ATM. Nat Rev 2000, 1:179-186 [DOI] [PubMed] [Google Scholar]
- 5.Vanasse GJ, Concannon P, Willerford DM: Regulated genomic instability and neoplasia in the lymphoid lineage. Blood 1999, 94:3997-4010 [PubMed] [Google Scholar]
- 6.Khanna KK, Jackson SP: DNA double strand breaks: signalling, repair and cancer connection. Nat Genet 2001, 27:247-254 [DOI] [PubMed] [Google Scholar]
- 7.Suzuki K, Kodama S, Watanabe M: Recruitment of ATM protein to double strand DNA irradiated with ionizing radiation. J Biol Chem 1999, 274:25571-25575 [DOI] [PubMed] [Google Scholar]
- 8.Perkins EJ, Nair A, Cowley DO, Van Dyke T, Chang Y, Ramsden DA: Sensing of intermediates in V(D)J recombination by ATM. Genes Dev 2002, 16:159-164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Luo CM, Tang W, Mekeel KL, DeFrank JS, Anne PR, Powell SN: High frequency and error-prone DNA recombination in ataxia telangiectasia cell lines. J Biol Chem 1996, 271:4497-4503 [DOI] [PubMed] [Google Scholar]
- 10.Morrison C, Sonoda E, Takao N, Shinohara A, Yamamoto K, Takeda S: The controlling role of ATM in homologous recombinational repair of DNA damage. EMBO J 2000, 19:463-471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lakin ND, Weber P, Stankovic T, Rottinghaus ST, Taylor AMR, Jackson P: Analysis of the ATM protein in wild-type and ataxia telangiectasia cells. Oncogene 1996, 13:2707-2716 [PubMed] [Google Scholar]
- 12.Watters D, Khanna KK, Beamish H, Birrell G, Spring K, Kedar P, Gatei M, Stenzel D, Hobson K, Kozlov S, Zhang N, Farrell A, Ramsay J, Gatti R, Lavin M: Cellular localisation of the ataxia-telangiectasia (ATM) gene product and discrimination between mutated and normal forms. Oncogene 1997, 14:1911-1921 [DOI] [PubMed] [Google Scholar]
- 13.Brown KD, Ziv Y, Sadanandan SN, Chessa L, Collins FS, Shiloh Y, Tagle DA: The ataxia-telangiectasia gene product, a constitutively expressed nuclear protein that is not up-regulated following genome damage. Proc Natl Acad Sci USA 1997, 94:1840-1845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pandita TK, Lieberman HB, Lim DS, Dhar S, Zheng W, Taya Y, Kastan MB: Ionizing radiation activates the ATM kinase throughout the cell cycle. Oncogene 2000, 19:1386-1391 [DOI] [PubMed] [Google Scholar]
- 15.Clarke RA, Kairouz R, Watters D, Lavin MF, Kearsley JH, Lee CS: Upregulation of ATM in sclerosing adenosis of the breast. Mol Pathol 1998, 51:224-226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fukao T, Kaneko H, Birrell G, Gatei M, Tashita H, Yoshida T, Cross S, Kedar P, Watters D, Khana KK, Misko I, Kondo N, Lavin MF: ATM is upregulated during the mitogenic response in peripheral blood mononuclear cells. Blood 1999, 94:1998-2006 [PubMed] [Google Scholar]
- 17.Gueven N, Keating KE, Chen P, Fukao T, Khanna KK, Watters D, Rodemann PH, Lavin MF: Epidermal growth factor sensitizes cells to ionizing radiation by down-regulating protein mutated in ataxia-telangiectasia. J Biol Chem 2001, 276:8884-8891 [DOI] [PubMed] [Google Scholar]
- 18.Byrd PJ, Cooper PR, Stankovic T, Kullar HS, Watts GDJ, Taylor AMR: A gene transcribed from the bidirectional ATM promoter coding for a serine rich protein: amino acid sequence, structure and expression studies. Hum Mol Genet 1996, 5:1785-1791 [DOI] [PubMed] [Google Scholar]
- 19.Savitsky K, Bar-Shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L, Tagle DA, Smith S, Uziel T, Sfez S: A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science 1995, 268:1749-1753 [DOI] [PubMed] [Google Scholar]
- 20.Tomilin NV, Solovjeva LV, Svetlova MP, Pleskach NM, Zalenskaya IA, Yau PM, Bradbury EM: Visualization of focal nuclear sites of DNA repair synthesis induced by bleomycin in human cells. Radiat Res 2001, 156:347-354 [DOI] [PubMed] [Google Scholar]
- 21.Stankovic T, Stewart G, Fegan C, Biggs P, Last J, Byrd, Moss PAH, Taylor AMR: ATM deficient B-CLL occurs in pre-germinal center cells and results in defective damage response and unrepaired chromosome damage. Blood 2002, 99:300-309 [DOI] [PubMed] [Google Scholar]
- 22.Bhandoola A, Dolnick B, Fayad N, Nussenzweig A, Singer A: Immature thymocytes undergoing receptor rearrangements are resistant to an ATM-dependent death pathway activated in mature T cells by double-stranded DNA breaks. J Exp Med 2000, 192:891-897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stewart GS, Last JIK, Stankovic T, Haites N, Byrd PJ, Taylor AMR: Presence of residual ATM function in cells derived from A-T patients with a less severe clinical and cellular phenotype. J Biol Chem 2001, 276:30133-30141 [DOI] [PubMed] [Google Scholar]
- 24.Guidos CJ, Williams CJ, Grandal I, Knowles G, Huang MT, Danska JS: V(D)J recombination activates a p53-dependent DNA damage checkpoint in SCID lymphocyte precursors. Genes Dev 1996, 10:2038-2054 [DOI] [PubMed] [Google Scholar]
- 25.Wilda M, Demuth I, Concannon P, Sperling K, Hameister H: Expression pattern of the Nijmegen breakage syndrome gene, Nbs1, during murine development. Hum Mol Genet 2000, 22:1739-1744 [DOI] [PubMed] [Google Scholar]
- 26.Moll U, Lau R, Sypes MA, Gupta MM, Anderson CW: DNA-PK, the DNA-activated protein kinase, is differentially expressed in normal and malignant human tissues. Oncogene 1999, 18:3114-3126 [DOI] [PubMed] [Google Scholar]
- 27.Stankovic T, Kidd AM, Sutcliffe A, McGuire GM, Robinson P, Weber P, Bedenham T, Bradwell AR, Easton DF, Lennox GG, Haites N, Byrd PJ, Taylor AMR: ATM mutations and phenotypes in ataxia-telangiectasia families in the Br Isles: expression of mutant ATM and the risk of leukemia, lymphoma, and breast cancer. Am J Hum Genet 1998, 2:334-345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vorechovsky I, Luo L, Dyer MJ, Catovsky D, Amlot PL, Yaxley JC, Foroni L, Hammarstrom L, Webster AD, Yuille MA: Clustering of missense mutations in the ataxia telangiectasia gene in a sporadic T cell leukaemia. Nat Genet 1997, 17:96-99 [DOI] [PubMed] [Google Scholar]
- 29.Stilgenbauer S, Schaffner C, Litterst A, Liebisch P, Gilad S, Bar-Shira A, James MR, Lichter P, Dohner H: Biallelic mutations in the ATM gene in T-prolymphocytic leukaemia. Nat Med 1997, 3:1155-1159 [DOI] [PubMed] [Google Scholar]
- 30.Stoppa-Lyonnet D, Soulier J, Lauge A, Dastot H, Garand R, Sigaux F, Stern MH: Inactivation of the ATM gene in T-cell prolymphocytic leukaemia. Blood 1998, 91:3920-3926 [PubMed] [Google Scholar]
- 31.Schaffner C, Idler I, Stilgenbaue S, Dohner H, Lichter P: Mantle cell lymphoma is characterized by inactivation of the ATM gene. Proc Natl Acad Sci USA 2000, 97:2773-2778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Camacho E, Hernandez L, Hernadez S, Tort F, Bellosillo B, Bea S, Bosch F, Montserrat E, Cardesa A, Fernandez PL, Campo E: ATM gene inactivation in mantle cell lymphoma mainly occurs by truncating mutations and missense mutations involving the phosphatidylinositol-3 kinase domain and is associated with increasing numbers of chromosomal imbalances. Blood 2002, 99:238-244 [DOI] [PubMed] [Google Scholar]
- 33.Stankovic T, Weber P, Stewart G, Bedenham T, Murray J, Byrd PJ, Moss PA, Taylor AM: (1999) Inactivation of ataxia telangiectasia mutated gene in B-cell chronic lymphocytic leukaemia. Lancet 1999, 353:26-29 [DOI] [PubMed] [Google Scholar]
- 34.Bullrich F, Rasio D, Kitada S, Starostik P, Kipps T, Keating M, Albitar M, Reed JC, Croce CM: ATM mutations in B-cell chronic lymphocytic leukemia. Cancer Res 1999, 59:24-27 [PubMed] [Google Scholar]
- 35.Schaffner C, Stilgenbauer S, Rappold GA, Dohner H, Lichter P: Somatic ATM mutations indicate a pathogenic role of ATM in B-cell chronic lymphocytic leukemia. Blood 1999, 94:748-753 [PubMed] [Google Scholar]
- 36.Pettitt AR, Sherrington PD, Stewart G, Cawley JC, Taylor AM, Stankovic T: p53 dysfunction in B-cell chronic lymphocytic leukemia: inactivation of ATM as an alternative to TP53 mutation. Blood 2001, 98:814-822 [DOI] [PubMed] [Google Scholar]
- 37.Gronbaek K, Worm J, Ralfkiaer E, Ahrenkiel V, Hokland P, Guldberg P: ATM mutations are associated with inactivation of the ARF-TP53 tumor suppressor pathway in diffuse large B-cell lymphoma. Blood 2002, 100:1430-1437 [DOI] [PubMed] [Google Scholar]
- 38.Hamblin TJ, Davis Z, Gardiner A, Oscier DG, Stevenson FK: Unmutated Ig VH genes are associated with a more aggressive form of chronic lymphocytic leukaemia. Blood 1999, 94:1848-1854 [PubMed] [Google Scholar]
- 39.Damle RN, Wasil T, Fais F, Ghiotto F, Valetto A, Allen SL, Buchbinder A, Budman D, Dittmar K, Kolitz J, Lichtman SM, Schulman P, Vinciguerra VP, Rai KR, Ferrarini M, Chiorazzi N: Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukaemia. Blood 1999, 94:1840-1847 [PubMed] [Google Scholar]
- 40.Carbone A, Gloghini A, Gaidano G, Franceschi S, Capello D, Drexler HG, Falini B, Dalla-Favera R: Expression status of BCL-6 and syndecan-1 identifies distinct histogenetic subtypes of Hodgkin’s disease. Blood 1998, 92:2220-2228 [PubMed] [Google Scholar]