

Abstract
The molecular and cellular mechanisms that maintain proper collagen homeostasis in healthy human skin and are responsible for the dysregulated collagen synthesis in scleroderma remain primarily unknown. This study demonstrates that Fli1 is a physiological negative regulator of collagen gene expression in dermal fibroblasts in vitro and in human skin in vivo. This conclusion is supported by the analyses of mouse embryonic fibroblasts from Fli1−/−, Fli1+/−, and Fli1+/+ mice. In cultured human and mouse fibroblasts Fli1 expression levels are inversely correlated with the collagen type I expression levels. These in vitro observations were validated in vivo. In healthy human skin Fli1 protein is expressed in fibroblasts and endothelial cells. Significantly, absence of Fli1 expression in individual fibroblasts correlates with elevated collagen synthesis. In contrast to healthy skin, Fli1 protein is consistently absent from fibroblasts and significantly reduced in endothelial cells in clinically involved scleroderma skin, which correlates with enhanced collagen synthesis in systemic sclerosis skin. This study supports the role of Fli1 as a suppressor of collagen transcription in human skin in vivo. Persistent down-regulation of Fli1 in scleroderma fibroblasts in vivo may directly contribute to uncontrolled matrix deposition in scleroderma skin.
Systemic sclerosis (SSc) is a complex disease characterized by activation of the immune system, vascular injury, and tissue fibrosis. 1 Evidence supports a role for activation of fibroblasts as a central mechanism responsible for tissue fibrosis and disease progression. 2,3 Fibroblasts in SSc skin lesions, particularly those in deep dermis, produce increased amounts of extracellular matrix proteins, including collagen type I, 4 type III, 5 type VI, 6 and type VII. 7 In contrast, in healthy adult skin, only a small proportion of fibroblasts actively produce collagen at any given time. 8 The molecular and cellular mechanisms that maintain proper collagen homeostasis in healthy skin in vivo and are responsible for the dysregulated collagen synthesis in SSc skin remain to be determined.
Current knowledge regarding collagen biosynthesis is based on studies with cultured cells, which are activated by adherence to plastic and propagation in the presence of serum. These extensive in vitro studies indicate that regulation at the transcriptional level plays a central role in both physiological and pathological collagen turnover. 9,10 A number of transcription factors have been shown to regulate collagen synthesis at the basal level and in response to cytokines and stress. 9-11 There is also an increasing evidence that elevated collagen production by SSc fibroblasts in vitro, is in part because of the alterations of the specific transcriptional complexes involved in regulation of the collagen type I gene. For example, the activity of transfected COL1A2 and COL1A1 promoter/reporter constructs is elevated in SSc fibroblasts compared to healthy skin fibroblasts. 12,13 It has been also observed that nuclear extracts from SSc fibroblasts exhibit increased binding of the CBF/NF-Y complex to the proximal CCAAT box present in the COL1A1 promoter. 14 CBF/NF-Y and Sp1 are potent activators of collagen gene transcription in human fibroblasts. 15,16 Increased phosphorylation of Sp1 was also observed in SSc cells. 17 Although the role of Smads in SSc phenotype is currently controversial, 18 down-regulation of Smad 7 may play a role in a subset of SSc patients. 19 Collectively, these studies suggest that alterations of specific transcriptional complexes contribute to the up-regulation of collagen gene expression in SSc cells in vitro. The role of the specific transcription factors in the abnormal collagen deposition in SSc skin in vivo has not been validated.
We have recently characterized Fli1, a transcription factor that inhibits collagen gene transcription via an Sp1-dependent pathway. 20 Fli1, a member of the Ets family of transcription factors, has been shown to play roles in hematopoiesis, embryonic development, and vasculogenesis. 21-24 Collective evidence indicates that Ets transcription factors are the important mediators of cellular programs involved in extracellular matrix degradation, 25 and are frequently dysregulated in diseases characterized by abnormal matrix turnover, including invasive tumors and arthritis. 26 In contrast, the specific role(s) of the Ets factors in the maintenance of collagen level homeostasis in healthy skin and their possible role in fibroproliferative disorders, including SSc, has not yet been adequately assessed.
The goal of this study was to determine the specificity of Fli1 as an inhibitor of collagen gene expression in dermal fibroblasts in vitro and in human skin in vivo. We also wished to determine whether Fli1 plays a role in the abnormal collagen deposition characteristic of SSc skin. The findings from this study further strengthen the role of Fli1 as a repressor of collagen biosynthesis in human dermal fibroblasts in vitro. In agreement with these findings, we observed a significantly increased collagen type I mRNA and protein levels in mouse embryonic fibroblasts (MEFs) obtained from Fli1−/− mice. Furthermore, this study demonstrates for the first time that Fli1 is expressed by dermal fibroblasts in human skin in vivo and supports the role for Fli1 as a physiological negative regulator of collagen gene expression in healthy skin. We also observed consistent down-regulation of Fli1 expression levels in cultured SSc fibroblasts and in SSc skin in vivo. Together, our observations suggest that dysregulation of Fli1 may directly contribute to the development of fibrosis in SSc.
Materials and Methods
Patients
The study group consisted of 25 patients with diffuse cutaneous SSc (dcSSc) and 21 healthy volunteers. All patients fulfilled the criteria of the American College of Rheumatology for dcSSc. On informed consent and in compliance with the Institutional Review Board for Human Studies, skin biopsies were obtained from the affected areas (dorsal forearm), and in three patients from clinically unaffected area. Skin biopsies were either embedded in paraffin and used for immunohistochemistry and in situ hybridization (see below) or were used to establish in vitro cell cultures (see below).
Immunohistochemistry
Skin biopsies obtained from 12 patients with dcSSc and 8 healthy volunteers were used for immunohistochemistry. Clinical features of the patients used for this study are described in Table 1 ▶ . The modified Rodnan method was used to determine skin score. 27 Skin biopsy specimens were fixed in neutral buffered formalin, embedded in paraffin, stained with hematoxylin and eosin, and used for in situ hybridization and immunohistochemistry. Immunohistochemical staining of Fli1 was performed using a Vectastain ABC kit (Vector, Burlingame, CA) according to the manufacturer’s recommendations. Five-μm-thick sections were mounted on APES-coated slides, deparaffinized with xylene, and rehydrated through a graded series of ethyl alcohol and phosphate-buffered saline (PBS). The sections were then incubated with antibodies against Fli1 (C-19) (Santa Cruz Biotechnology, Santa Cruz, CA) diluted 1:200 in PBS overnight at 4°C, followed by the incubation with biotinylated anti-rabbit secondary antibody. The immunoreactivity was visualized with diaminobenzidine and the sections were counterstained with hematoxylin. Independent scoring was performed blindly by ES and openly by MK and JC-L.
Table 1.
Clinical Features of Patients with SSc
| Subject | Sex/age | SSc type | Duration | Skin score | Presence of skin infiltrates | |
|---|---|---|---|---|---|---|
| Involved | Uninvolved | |||||
| SSc 1 | M | Diffuse | 2 years | 15 | +++ | + |
| SSc 2 | M | Diffuse | 1 year | 28 | + | − |
| SSc 3 | F | Diffuse | 2 years | 38 | ++ | − |
| SSc 4 | F/53 | Diffuse | 3 months | 17 | ++ | N/A |
| SSc 5 | F/62 | Diffuse | 2 years | 24 | + | N/A |
| SSc 7 | M/57 | Diffuse | 18 years | 25 | + | N/A |
| SSc 8 | F/47 | Diffuse | 1 year | 15 | + | N/A |
| SSc 10 | F/55 | Diffuse | 8 years | 16 | −/+ | N/A |
| SSc 11 | F/71 | Diffuse | 1 year | 34 | + | N/A |
| SSc 12 | F/47 | Diffuse | 8 years | 15 | + | N/A |
| SSc 13 | F/42 | Diffuse | 8 years | 18 | −/+ | N/A |
| SSc 14 | F/73 | Diffuse | 1 year | 38 | ++ | N/A |
Collagen mRNA Measurements by in Situ Hybridization
A nonradioactive in situ hybridization technique using digoxigenin (DIG)-labeled RNA probes was used as described previously 28 with some modifications. Briefly, paraffin-embedded sections were cut to a thickness of 5 μm, mounted on silane-coated slides, and deparaffinized. The sections were treated with 0.2 mol/L HCl for 15 minutes, followed by 1.5 μg/ml proteinase K (Sigma, St. Louis, MO) digestion for 15 minutes at 37°C. The sections were postfixed with 4% paraformaldehyde in PBS for 30 minutes and treated with PBS containing 2 mg/ml glycine twice, for 15 minutes each time. After rinsing with PBS, the samples were soaked twice in standard saline-standard saline citrate (SSC) buffer with 50% formamide and subjected to hybridization. A 650-bp fragment of COL1A1 cDNA (kindly provided by Dr. Vuorio, Turku, Finland) was subcloned into the Bluescript SK II phagemid (Stratagene, La Jolla, CA). The sense probes and anti-sense probes for COL1A1 were labeled with DIG-11-UTP using a DIG RNA-labeling kit (Roche, Indianapolis, IN). The labeled RNA probes (final concentration, 1 ng/μl) in a mixture containing 50% formamide, 10% dextran sulfate, 1× Denhardt’s solution, 100 μg/ml transfer RNA, 5× SSC, 0.25% sodium dodecyl sulfate, 1 mmol/L ethylenediaminetetraacetic acid, and 50 mmol/L NaH2PO4 were placed on the slides and overlaid with a coverslip. Hybridization was performed in a humidified chamber for 18 hours at 45°C, after which the specimens were washed in 2× SSC with 50% formamide at 50°C. Unhybridized probes were digested in 2.5 μg/ml RNase A, 500 mmol/L NaCl, 1 mmol/L ethylenediaminetetraacetic acid, and 10 mmol/L Tris-HCl (pH 8.0) for 15 minutes at 37°C. The slides were then washed for 15 minutes in 2× SSC and for 15 minutes in 0.2× SSC, twice at 50°C. After posthybridization washing, DIG-labeled probes were visualized as described in the DIG nucleic acid detection kit protocol (Roche). The slides were weakly counterstained with hematoxylin.
Cell Culture
Human dermal fibroblast cultures were established from skin biopsies taken from the dorsal forearm in compliance with the Institutional Review Board for Human Studies. Tissue was dissociated enzymatically by 0.25% collagenase type I (Sigma) and 0.05% DNase I (Sigma) in Dulbecco’s modified Eagle’s medium (DMEM) with 20% fetal bovine serum (HyClone, Logan, UT). Cells were grown at 37°C in a 5% CO2 atmosphere in DMEM supplemented with 10% fetal calf serum and 50 μg/ml gentamicin (Sigma). All studies used cells from passage number 3 to 6. Before stimulation with cytokines and infection with adenoviruses, fibroblasts were incubated in serum-free medium (DMEM/0.1% bovine serum albumin) for 48 hours.
Primary murine embryo fibroblasts (MEF) were prepared from wild-type, heterozygous, and homozygous mutant Fli1embryos (+/+, +/− and −/−). MEFs were dispersed from each embryo using 200 μl of 0.25% trypsin solution (containing 0.53 mmol/L EDTA, collagenase, and DNAse) and were cultured in DMEM supplemented with 10% FBS, l-glutamine and penicillin/streptomycin (Invitrogen, Carlsbad, CA) at 37°C in a 5% CO2 atmosphere.
Adenoviral Vectors
Replication-deficient recombinant adenoviruses were generated by homologous recombination using a bacterial system based on the method previously described. 29 Construction of Ets1 adenovirus was previously described. 30 We also constructed a Fli1 adenovirus. Briefly, human Fli1 cDNA from pSG5-Fli1 31 was polymerase chain reaction-amplified using Pfu polymerase with Fli1-specific primers designed to incorporate BclI and NotI restriction enzyme recognition sites, digested with BclI and NotI and subcloned into a modified pcDNA3 vector between BamHI and NotI. This vector (pFcDNA3, provided by Dr. C. Hauser, La Jolla Cancer Research Center, The Burnham Institute, La Jolla, CA) allowed incorporation of a Flag epitope tag at the amino terminus to monitor protein expression. The sequence of this polymerase chain reaction-generated insert was verified, released by HindIII and recloned into the shuttle plasmid, pAdTrackCMV at the unique HindIII site. For homologous recombination, PmeI digested shuttle plasmid bearing the kanamycin resistance gene was co-electroporated with the circular adenoviral genome plasmid pAdEasy-1 (E1A deleted) into competent BJ5183 bacterial cells (Stratagene) and plated on kanamycin media. Recombinant adenoviral DNA was purified, linearized with PacI, and transfected into the 293A cells using FuGene6 (Roche). Recombinant adenoviruses were plaque-purified and screened for the expression of the Fli11 construct by Western blot using M5 Flag antibody (Sigma). After large-scale preparation, adenoviruses were purified by cesium chloride gradient centrifugation, dialyzed, and titered using the TCID50 method.
Procollagen Analysis by [3H]Proline Incorporation, Sodium Dodecyl Sulfate (SDS)-Polyacrylamide Gel Electrophoresis, and Autoradiography
Analysis of proline incorporation into secreted protein was performed as previously described. 30 Fibroblasts were grown to visual confluence in 12-well plates. The medium was changed to serum-free medium (DMEM) supplemented with 50 μg/ml ascorbic acid for the duration of the experiment and cells were transduced with Fli1 (or Ets1)/GFP-adenovirus or control GFP-adenovirus (multiplicity of infection, 100). This condition allowed >95% of cells to be transduced with virus as visualized by expression of GFP. Eighteen hours after infection, fibroblasts were stimulated with 2 ng/ml of human recombinant transforming growth factor (TGF)-β1 (R&D Systems, Minneapolis, MN), and 6 hours later, 20 μCi/ml of [3H]proline (Amersham, Piscataway, NJ) was added to the medium for 24 hours. Medium was harvested from each well and cells were trypsinized and counted. Medium was dehydrated in a SpeedVac (Savant, Farmingdale, NY) and resuspended in 2× SDS/dithiothreitol sample buffer. Aliquots of conditioned media normalized for cell number were denatured and loaded on a 6% SDS-polyacrylamide gel. After electrophoresis the gel was enhanced by Fluoro-Hance (Research Products Int. Corp., Mt. Prospect, IL) and visualized by autoradiography. The intensity of bands of collagenous protein was quantitated using NIH-Image densitometry software, version 1.55.
Western Blot
Western blot was performed using total cell lysates [5 μg for protein disulfide isomerase (PDI)], 30 μg for prolyl 4-hydroxylase and heat-shock protein 47 (HSP 47), and 100 μg for Fli1). Anti-PDI antibody was obtained from Calbiochem (San Diego, CA), anti-prolyl 4-hydroxylase antibody was from Chemicon (Temecula, CA) and anti-HSP 47 antibody was from Santa Cruz. Anti-Fli1 antibody (C-19) from Santa Cruz was also used for immunostaining. The specificity of this antibody (lack of cross-reactivity with Erg1 and Ets1) was confirmed using respective antigens. To determine the levels of Fli1, nuclear extracts (25 μg) were used for Western blot analysis with polyclonal anti-Fli1 Ab (C-19, Santa Cruz). Ets1 levels were determined in whole cell lysates (100 μg) with polyclonal Ets1 Ab (C-20, Santa Cruz) or monoclonal Ets1 Ab (Transduction Laboratories). β-actin antibody was from Sigma.
Analyses of MEFs
MEF cultures from Fli1 wild-type (+/+), heterozygous (+/−), and knockout (−/−) mouse embryos were propagated in DMEM supplemented with 10% fetal bovine serum. Total RNA was extracted and analyzed by Northern blot. Filters were hybridized sequentially with radioactive probes for Fli1 and collagen α1(I). For Western blot analyses, 25 μg of total cell lysates were used. Fli1 protein levels were determined with polyclonal anti-Fli1 Ab (C-19, Santa Cruz) and collagen type I levels were determined with goat anti-type I collagen Ab (Southern Biotechnology, Birmingham, AL).
Results
Fli1 Inhibits Basal and TGF-β-Induced Synthesis of Collagenous Proteins and Down-Regulates Synthesis of Prolyl 4-Hydroxylase in Dermal Fibroblasts
Fli1 has been previously implicated in collagen gene regulation using stable and transiently transfected human fibroblasts. 20 To gain further insights into the Fli1 function, an adenoviral vector expressing Fli1 was generated to ensure efficient gene delivery into dermal fibroblasts. To assess the effect of Fli1 on newly synthesized collagenous proteins at the basal level and in response to TGF-β, conditioned media from cells metabolically labeled with 3H-proline were analyzed by SDS-polyacrylamide gel electrophoresis and autoradiography. Consistent with our previous observations, Fli1 reduced secretion of collagenous proteins at the basal level including procollagens type I and III and fibronectin (Figure 1) ▶ . In addition, Fli1 inhibited TGF-β induction of collagen. Under similar experimental conditions, Ets1 abrogated TGF-β stimulation of collagen, but did not affect basal production of collagenous proteins consistent with our previous study. 30 Together, these data suggest that either Fli1 or Ets1 is capable of inhibiting profibrotic effects of TGF-β; however inhibition of basal collagen expression is a distinctive property of Fli1.
Figure 1.

Fli1 inhibits basal and TGF-β-induced synthesis of collagenous proteins. A: Newly synthesized collagenous proteins in control (GFP) and either Fli1- or Ets1-infected fibroblasts were measured in a [3H]proline incorporation assay after 30 hours of TGF-β stimulation. Aliquots of conditioned media normalized for cell number were analyzed for collagenous protein content via SDS-polyacrylamide gel electrophoresis and fluorography. B: Fli1 and Ets1 levels in GFP- and either Fli1 or Ets1 adenovirus-transduced fibroblasts (multiplicity of infection = 100) before and after TGF-β treatment. Blots were reprobed with β-actin antibody. C: Fli1 down-regulates synthesis of prolyl 4-hydroxylase α in dermal fibroblasts. Protein levels of prolyl 4-hydroxylase α (P4Hα), PDI, and HSP 47 in control and Fli1-infected fibroblasts (multiplicity of infection = 100) were determined using specific antibodies. The blots were probed with anti-p4Hα antibody, anti-PDI antibody, anti-HSP 47 antibody, and anti-β-actin antibody.
We also examined whether Fli1 affects synthesis of selected enzymes involved in collagen production. Prolyl 4-hydroxylases catalyze the formation of 4-hydroxyproline in collagens. Prolyl 4-hydroxylase is an α2β2 tetramer consisting of either α(I)2 or α(II)2 subunits and a common β subunit. The α(I) subunit is a major form. The β subunit of prolyl 4-hydroxylase is identical to the PDI, a multifunctional, ubiquitously expressed peptide. 32 HSP 47 is a collagen-binding protein whose expression correlates with that of collagen under both physiological and pathological conditions. It most likely acts as a collagen chaperon, but its specific functions are not fully understood. 33 The protein levels of prolyl 4-hydroxylase α, PDI, and HSP 47 were measured by Western blot. The protein levels of prolyl 4-hydroxylase were significantly reduced in cells overexpressing Fli1, whereas the levels of PDI and HSP47 remained unchanged (Figure 1) ▶ .
Fli1 Protein Levels Are Modulated by TGF-β and Tumor Necrosis Factor (TNF)-α in Dermal Fibroblasts
Our previous studies, 20 as well as experiments using adenoviral expression of Fli1, strongly suggest that Fli1 is a potent inhibitor of collagen biosynthesis. We therefore asked whether the signaling pathways known to modulate collagen gene expression control expression levels of Fli1. We selected TGF-β, a primary inducer of collagen biosynthesis, and TNF-α, a potent inhibitor of TGF-β-induced collagen synthesis. 34 Expression levels of Fli1 protein were determined in dermal fibroblasts stimulated for 24 and 48 hours with either TGF-β or with the combination of TGF-β and TNF-α. The Fli1 protein levels were determined by Western blot. As shown in Figure 2 ▶ , TGF-β transiently inhibited Fli1 levels at 24 hours after stimulation, but at 48 hours, Fli1 levels returned to the levels expressed by unstimulated fibroblasts. TNF-α stimulated Fli1 protein levels at both 24 and 48 hours (data not shown) and completely abrogated the inhibitory effect of TGF-β. These data suggest that Fli1 protein levels are modulated by the signaling pathways involved in collagen regulation, in a manner consistent with its role as a collagen gene repressor.
Figure 2.

Fli1 protein level is modulated by cytokines. Serum-starved, confluent fibroblasts were treated with TGF-β (2 ng/ml) alone or a combination of TGF-β and TNF-α (10 ng/ml) added simultaneously for indicated periods of time. Fli1 protein was determined in 100 μg of total cell extract by Western blot. The blots were reprobed with anti-β actin antibody.
Collagen Type I Gene Is Overexpressed in Fli−/− Embryonic Fibroblasts
To further investigate the relationship between Fli1 expression levels and collagen production, we used MEFs obtained from Fli1+/+, Fli1+/−, and Fli1−/− embryos. 22 Collagen type I mRNA and protein levels versus Fli1 levels were examined in Fli1−/−, Fli1+/−, and Fli1+/+ MEFs. As shown in Figure 3, A and B ▶ , COL1A1 mRNA and collagen type I protein levels are inversely proportional to Fli1 expression levels. Absence of Fli1 results in a large increase of collagen type I levels as compared to the wild-type fibroblasts, while Fli1+/− MEFs exhibit intermediate phenotype.
Figure 3.
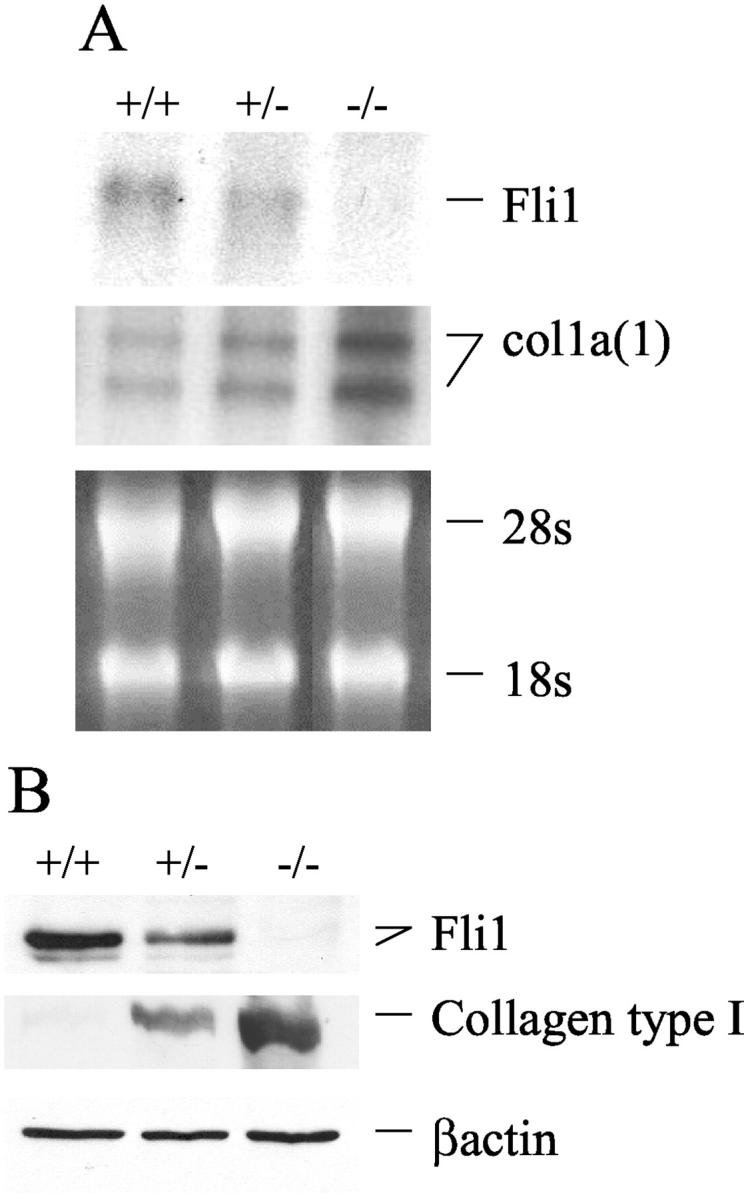
Fli1 expression levels inversely correlate with collagen type I synthesis in Fli1+/+, Fli1+/−, and Fli1−/− MEFs. A: Representative Northern blot of Fli1 and collagen α1(I) mRNAs. B: Representative Western blot of Fli1 and collagen type I proteins. β-actin was used as loading control.
Fli1 Protein Is Down-Regulated in Dermal Fibroblasts Synthesizing Collagen in the Skin in Vivo
The above experiments indicated an inverse relationship between expression of Fli1 and collagen in cultured human dermal fibroblasts. We next examined whether these findings are also applicable to the fibroblasts in human skin in vivo. In Figure 4, A to F ▶ , representative photomicrographs of serial sections of skin are presented, demonstrating immunostaining for Fli1 protein and for α1(I) collagen mRNA. Fli1 protein was detected in dermal fibroblasts located in the upper, medium, and lower dermis. Furthermore, we consistently observed inverse correlation between Fli1 and collagen expression in dermal fibroblasts in vivo. For example, an area shown in Figure 4A-a ▶ contains numerous Fli1-positive fibroblasts with very little collagen synthesis present (Figure 4B-c ▶ ). In contrast, the areas found to contain mainly Fli1-negative fibroblasts (eg, Figure 4A-b ▶ ) correspond to those containing many cells actively synthesizing collagen (Figure 4B-d ▶ ). Compare also Figure 4F-a ▶ with Figure 4F-b ▶ , which shows Fli1 and collagen presence in closely matched fibroblasts from serial sections. Fli1 was also detected in endothelial cells. In both fibroblasts and endothelial cells, Fli1 exhibited nuclear staining.
Figure 4.
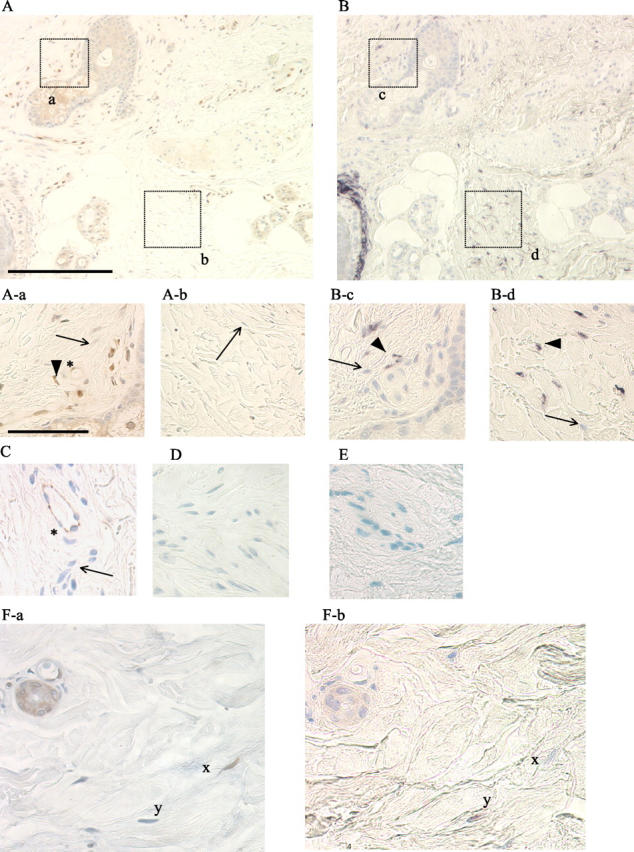
Fli1 and collagen expression in healthy human skin in vivo. Serial skin sections were used for Fli1 immunodetection (A) and COL1A1 in situ hybridization (B) (original magnifications, ×100; scale bar, 200 μm). Note: Fli1-positive fibroblasts represented by brown nuclear staining (thick arrow) and Fli-1-negative fibroblasts (blue counterstain, thin arrow) in A-a and A-b (original magnifications, ×400; scale bar, 50 μm). B-c and B-d represent collagen mRNA expression from the corresponding fields. Note: COL1A1-positive fibroblasts (thick arrowhead) and Fli1-negative fibroblasts (thin arrow). Endothelial origin of cells expressing Fli1 (marked by asterisk) in A-a was confirmed using anti-CD31 antibody (C). In D (negative control), primary antibody was omitted. In E, sense (negative control) COL1A1 probe was used. F-a and F-b (original magnifications, ×400) show serial sections of skin in which corresponding fibroblasts are marked x and y. Note that Fli1-positive fibroblast (x) does not express COL1A1 mRNA, while Fli1-negative fibroblast (y) is positive for collagen mRNA.
Fli1 Protein Is Underexpressed in a Subset of Cultured SSc Fibroblasts
Because both Fli1 and Ets1 are implicated in collagen gene regulation, 20,30 we next examined expression levels ofFli1 and Ets1 in cultured dermal SSc and healthy control fibroblasts. SSc and closely matched healthy control pairs were always propagated and used for experiments at the same time to avoid variations of culture conditions. Different pairs were processed at different times and depend on the availability of biopsies obtained from patients with SSc. Expression levels of Fli1 were determined in 13 pairs of SSc and healthy dermal fibroblasts by Western blot with polyclonal anti-Fli1 antibody. Fli1 protein expression by SSc fibroblasts was consistently reduced, albeit to a various degree, in nine SSc/NS pairs (Figure 5A) ▶ and was not changed in four additional pairs (data not shown). Expression of Ets1 was determined in seven pairs of SSc and healthy dermal control fibroblasts. There was no difference in Ets1 expression levels in all seven SSc/NS pairs tested (Figure 5B) ▶ .
Figure 5.

Fli1 protein is underexpressed in a subset of SSc fibroblasts. Dermal SSc and healthy control fibroblasts (NS) were analyzed for Fli1 and Ets1 protein expression levels. Nuclear extracts were used for Western blot with polyclonal Fli1 antibody or with polyclonal Ets1 antibody. Blots were reprobed with β-actin antibody. A: Representative data of Fli1 protein levels in SSc and NS fibroblasts B: Representative data of Ets1 protein levels in SSc and NS fibroblasts.
Fli1 Is Underexpressed in SSc Skin in Vivo
To examine expression pattern of Fli1 in SSc skin, we focused our analysis on deep reticular dermis because previous reports demonstrated that pathological differences are especially pronounced in this skin layer. 35,36 In healthy skin, the majority of the cells with fibroblastic morphology, as well as endothelial cells in deep dermis, express Fli1 (Figure 6, a and b) ▶ . In contrast, in clinically involved skin of SSc patients with active disease, Fli1-positive fibroblasts were either absent or only occasionally seen. (Figure 6; c, e, g) ▶ . Fli1-positive fibroblasts were counted in deep dermis of 11 patients with SSc (with <8 years duration) and 8 matched healthy controls (Figure 6k) ▶ . This analysis revealed significant differences between SSc and healthy skin with respect to a number of Fli1-positive fibroblasts. In healthy skin, the percentage of Fli1-positive fibroblasts equals 66 ± 14 (mean ± SD) versus 8 ± 8 (mean ± SD) in SSc skin (P < 0.001). Because previous reports indicated that pathological changes also occur in clinically uninvolved skin, 37 we examined Fli1 expression in skin biopsies from uninvolved skin obtained from SSc patients. Fli1 staining was also mainly absent from the majority of fibroblasts in the uninvolved skin from two patients (SSc 1 and SSc 3), whereas moderate expression of Fli1 was observed in uninvolved skin of one patient (SSc 2) (Figure 6; d, f, h ▶ , and Table 2 ▶ ). Thus, uninvolved skin appears to represent an intermediate stage between healthy and lesional SSc skin with regard to Fli1 expression in fibroblasts.
Figure 6.

Immunodetection of Fli1 in skin from healthy individuals and uninvolved and involved skin from patients with SSc. Positively stained fibroblasts and endothelial cells are present in healthy skin (a, b) (original magnifications, ×400; scale bar, 100 μm). Thick arrow points to Fli1-positive fibroblasts, an asterisk indicates endothelial cell; c and d, e and f, and g and h involved and uninvolved skin sections from patients with SSc. Note absence of Fli1 in fibroblasts (thin arrow points to Fli1-negative fibroblast) and in some endothelial cells (asterisk) in SSc skin. k: Percentage of Fli-1-positive fibroblasts in SSc and healthy skin. At least 100 fibroblasts were counted for each specimen.
Table 2.
Fli1 and Collagen Staining in Endothelial Cells (EC) and Fibroblasts (FB) in Skin Sections of Patients with SSc Compared with Healthy Skin
| Patient | Fli1/EC | Fli1/FB | COL1A1 mRNA | ||
|---|---|---|---|---|---|
| Involved | Uninvolved | Involved | Uninvolved | ||
| SSc 1 | − | +/− | − | +/− | N/D |
| SSc 2 | + | ++ | + | ++ | N/D |
| SSc 3 | − | +/++ | +/− | + | N/D |
| SSc 4 | +/− | +/− | ++ | ||
| SSc 5 | +/++ | +/++ | ++ | ||
| SSc 7 | +++ | +++ | + | ||
| SSc 8 | +/++ | + | +++ | ||
| SSc 10 | ++ | +/++ | +++ | ||
| SSc 11 | +/− | −/+ | +++ | ||
| SSc 12 | ++ | +/++ | +/++ | ||
| SSc 13 | +/− | +/++ | ++ | ||
| SSc 14 | +/− | −/+ | ++ | ||
| NS 1 | +++ | ++/+++ | + | ||
| NS 2 | ++ | ++ | + | ||
| NS 3 | ++/+++ | ++ | + | ||
| NS 4 | +++ | ++ | +/++ | ||
| NS 5 | +++ | ++ | + | ||
All cells with fibroblast morphology (>100) were counted in each specimen. −, negative; +, occasionally positive cell in the field—weak; ++, 50% cells positive—moderate; +++, more than 80% cells positive—strong.
N/D, not done.
Significantly, altered Fli1 protein expression was also noted in endothelial cells. Intense uniform nuclear staining of Fli1 was present in endothelial cells throughout the healthy skin (Figure 7, a and b) ▶ . In contrast, in clinically uninvolved SSc skin most of the individual blood vessels consisted of heterogeneously stained endothelial cells. Within a blood vessel, some endothelial cells were positive for Fli1, whereas other cells displayed either decreased or undetectable Fli1 staining (Figure 7, c and d) ▶ . In addition, the number of the vessels was greatly reduced in clinically involved skin, and the remaining vessels appeared damaged. 38,39 Fli1-positive cells were detectable, but the structure of the vessel was destroyed (data not shown).
Figure 7.

Immunodetection of Fli1 in endothelial cells in healthy skin (a, b) and in uninvolved skin from patients with SSc (c, d) (original magnifications, ×400; scale bar, 100 μm). Thick arrow points to Fli1-positive endothelial cells, thin arrow points to Fli1-negative endothelial cells.
Because elevated collagen synthesis, especially by fibroblasts in deeper reticular dermis is a hallmark of SSc, we examined α1(I) collagen mRNA expression. Fibroblasts, positive for α1(I) collagen mRNA were present in skin sections from SSc patients, while such cells were almost absent in sections from healthy skin. Positive staining for collagen type I mRNA correlated with the absence of Fli1 in fibroblasts as observed in consecutive sections (Table 2) ▶ .
A summary of the in vivo data are presented in Table 2 ▶ . Our data demonstrate heterogeneous Fli1 expression in fibroblasts and endothelial cells in healthy and SSc skin. Significantly, however, healthy and SSc skin differ in a relative proportion of Fli1-positive cells. In healthy skin Fli1-positive cells predominate, whereas in SSc skin a majority of the cells do not express Fli1. Interestingly, in a patient with an 18-year disease duration (SSc 7), Fli1 staining in endothelial and fibroblastic cells was undistinguishable from healthy skin.
Discussion
Turnover of collagen type I, the main component of human dermis, is tightly regulated under normal physiological conditions. However, very little is currently known regarding regulatory mechanisms that control this process. The present study provides the evidence for the role of transcriptional repressor, Fli1, in regulation of collagen synthesis in cultured dermal fibroblasts and in human skin in vivo. This study extends our previous findings that established Fli1 as a transcriptional repressor of the collagen gene. 20 The role of Fli1 as a physiological negative regulator of extracellular matrix genes is further supported by the observations that Fli1 protein levels inversely correlate with production of collagen type I in fibroblast cultures generated from Fli1−/−, Fli1+/−, and Fli1+/+ embryos. Consistent with these findings we observed that Fli1 levels are positively regulated by TNF-α, a well-established inhibitor of collagen synthesis, and negatively regulated by TGF-β, a potent inducer of extracellular matrix synthesis. Importantly, TGF-β response is transient and at 48 hours after stimulation, Fli1 expression returns to control levels. This may be a part of a negative feedback regulatory loop that prevents uncontrolled matrix synthesis in response to this potent fibrogenic cytokine. As shown in this study, absence of Fli1 correlates with the pathological tissue fibrosis.
This study provides the first demonstration that Fli1 is expressed in human dermal fibroblasts in vivo. In the human skin, Fli1 expression is heterogenous because both positive and negative fibroblasts are present. Systematic examination of serial skin sections indicates that absence of Fli1 expression in fibroblasts correlates with collagen synthesis. Taken together, our data support a role for Fli1 as a specific negative regulator of collagen synthesis in dermal fibroblasts in healthy skin.
Analysis of skin biopsies from SSc patients showed remarkable down-regulation of Fli1 in SSc skin. In SSc skin obtained from patients with active disease, Fli1 was absent from fibroblasts present in all skin layers. Fibroblast staining for Fli1 in clinically uninvolved skin revealed an intermediate pattern between healthy and involved skin, most likely reflecting the initial phase of the fibrotic process. Down-regulation of Fli1 correlated with induction of collagen synthesis in patients with active disease. On the other hand, in patients with late disease, Fli1 was re-expressed in fibroblasts, concomitant with a reduction of collagen synthesis to levels comparable with healthy skin. Our data strongly suggest that the prolonged absence of Fli1 in SSc fibroblasts in vivo may directly contribute to the process of fibrosis. Down-regulation of Fli1 may also be responsible for other abnormally expressed genes in SSc skin, including integrins, growth factor receptors, and adhesion molecules; 40-43 however, at present the spectrum of Fli1-dependent genes in fibroblasts is unknown. Our future studies will be directed toward identification of genes regulated by Fli1 in cultured fibroblasts in vitro and to examine their expression in SSc skin in vivo. Down-regulation of Fli1 was also exhibited by SSc fibroblasts in vitro. These differences, however, were less pronounced than those observed in fibroblasts in vivo, suggesting partial loss of the phenotype during in vitro culturing.
Expression of Fli1 was also markedly decreased in endothelial cells in SSc lesions. Significantly, endothelial cells in the uninvolved skin exhibited decreased or negative staining for Fli1 in the majority of the vessels. Furthermore, vessels in the involved skin contained mainly Fli1-negative cells. Down-regulation of Fli1 in the endothelial cells may be particularly relevant to the pathogenesis of SSc. Vascular damage associated with endothelial cell apoptosis has been considered an initiating event in the disease process, followed by the perivascular and tissue infiltration of the inflammatory cells and subsequent development of fibrosis. 1,44 Fli1 is known to play an essential, although not fully defined, role in endothelial cell function. 22,23 Mice with a targeted mutation in Fli1 locus, die of hemorrhage at day 11.5 of embryogenesis because of, at least in part, the loss of vascular integrity. Interestingly, Fli1−/− embryos are able to form a functional network of blood vessels, but appear to fail to recruit pericytes or smooth muscle cells during late embryonic angiogenesis and vascular remodeling. Furthermore, it was demonstrated that Fli1−/− endothelial cells were not viable even in the chimeric embryos consisting of Fli1+/+ and Fli1−/− cells. 23 These previous studies demonstrate that Fli1 plays a critical role in endothelial cell function during embryogenesis. Presence of Fli1 in endothelial cells in healthy skin suggests that it may also have an important function in adult tissues. Although, further studies are needed to delineate a specific role of Fli1 in microvascular endothelial cells, it is tempting to speculate that the absence of Fli1 in these cells may be an additional manifestation of the endothelial cell dysfunction in SSc and may directly contribute to endothelial cell apoptosis in SSc.
In conclusion, this study provides evidence that Fli1 is a physiological inhibitor of collagen gene expression in healthy human skin in vivo. The importance of Fli1 in the collagen homeostasis is further underscored by the observation that Fli1 is absent in SSc patient skin that is characterized by fibrosis. This study strongly supports the notion that dysregulation at the level of transcription as manifested by the persistent down-regulation of Fli1 in SSc skin underlies the mechanism of the uncontrolled collagen deposition in SSc. Further studies are needed to determine the nature of the pathways that regulate Fli1 expression in healthy human skin in vivo as well as the mechanisms responsible for down-regulation of Fli1 in skin of patients with SSc.
Acknowledgments
We thank Dr. Monika Gooz for determining the specificity of the anti-Fli1 antibody, and Dr. Demetri Spyropoulos for providing expertise in establishing MEF primary cultures.
Footnotes
Address reprint requests to Dr. Maria Trojanowska, Division of Rheumatology and Immunology, Medical University of South Carolina, 96 Jonathan Lucas St., Suite 912, P.O. Box 250637, Charleston, SC 29425. E-mail: trojanme@musc.edu.
Supported by the National Institutes of Health (grants AR 42334, AR 44883, NCI-PO1 CA78582) and the Scleroderma Foundation. J. C.-L. was a recipient of a postdoctoral fellowship award from the Arthritis Foundation.
M. K. and J. C.-L. contributed equally to this study.
References
- 1.LeRoy EC: The connective tissue in scleroderma. Coll Relat Res 1981, 1:301-308 [DOI] [PubMed] [Google Scholar]
- 2.Trojanowska M, LeRoy EC, Eckes B, Krieg T: Pathogenesis of fibrosis: type 1 collagen and the skin. J Mol Med 1998, 76:266-274 [DOI] [PubMed] [Google Scholar]
- 3.Kahari VM: Activation of dermal connective tissue in scleroderma. Ann Med 1993, 25:511-518 [PubMed] [Google Scholar]
- 4.Fleischmajer R, Gay S, Meigel WN, Perlish JS: Collagen in the cellular and fibrotic stages of scleroderma. Arthritis Rheum 1978, 21:418-428 [DOI] [PubMed] [Google Scholar]
- 5.Ohtsuka T, Koibuchi N, Sakai H, Yamakage A, Yamazaki S: Quantitative analysis of alpha 1(I) and alpha 1(III) procollagen mRNA expression in systemic sclerosis skin tissue—an in situ hybridization study. Arch Dermatol Res 1999, 291:575-582 [DOI] [PubMed] [Google Scholar]
- 6.Peltonen J, Kahari L, Uitto J, Jimenez SA: Increased expression of type VI collagen genes in systemic sclerosis. Arthritis Rheum 1990, 33:1829-1835 [DOI] [PubMed] [Google Scholar]
- 7.Rudnicka L, Varga J, Christiano AM, Iozzo RV, Jimenez SA, Uitto J: Elevated expression of type VII collagen in the skin of patients with systemic sclerosis. Regulation by transforming growth factor-beta. J Clin Invest 1994, 93:1709-1715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fisher GJ, Datta S, Wang Z, Li XY, Quan T, Chung JH, Kang S, Voorhees JJ: c-Jun-dependent inhibition of cutaneous procollagen transcription following ultraviolet irradiation is reversed by all-trans retinoic acid. J Clin Invest 2000, 106:663-670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Widom RL: Regulation of matrix biosynthesis and degradation in systemic sclerosis. Curr Opin Rheumatol 2000, 12:534-539 [DOI] [PubMed] [Google Scholar]
- 10.Trojanowska M: Molecular aspects of scleroderma. Front Biosci 2002, 7:D608-D618 [DOI] [PubMed] [Google Scholar]
- 11.Ghosh AK: Factors involved in the regulation of type I collagen gene expression: implication in fibrosis. Exp Biol Med (Maywood) 2002, 227:301-314 [DOI] [PubMed] [Google Scholar]
- 12.Kikuchi K, Hartl CW, Smith EA, LeRoy EC, Trojanowska M: Direct demonstration of transcriptional activation of collagen gene expression in systemic sclerosis fibroblasts: insensitivity to TGF beta 1 stimulation. Biochem Biophys Res Commun 1992, 187:45-50 [DOI] [PubMed] [Google Scholar]
- 13.Hitraya EG, Varga J, Artlett CM, Jimenez SA: Identification of elements in the promoter region of the alpha1(I) procollagen gene involved in its up-regulated expression in systemic sclerosis. Arthritis Rheum 1998, 41:2048-2058 [DOI] [PubMed] [Google Scholar]
- 14.Saitta B, Gaidarova S, Cicchillitti L, Jimenez SA: CCAAT binding transcription factor binds and regulates human COL1A1 promoter activity in human dermal fibroblasts: demonstration of increased binding in systemic sclerosis fibroblasts. Arthritis Rheum 2000, 43:2219-2229 [DOI] [PubMed] [Google Scholar]
- 15.Tamaki T, Ohnishi K, Hartl C, LeRoy EC, Trojanowska M: Characterization of a GC-rich region containing Sp1 binding site(s) as a constitutive responsive element of the alpha 2(I) collagen gene in human fibroblasts. J Biol Chem 1995, 270:4299-4304 [DOI] [PubMed] [Google Scholar]
- 16.Ihn H, Ohnishi K, Tamaki T, LeRoy EC, Trojanowska M: Transcriptional regulation of the human alpha2(I) collagen gene. Combined action of upstream stimulatory and inhibitory cis-acting elements. J Biol Chem 1996, 271:26717-26723 [DOI] [PubMed] [Google Scholar]
- 17.Ihn H, Tamaki K: Increased phosphorylation of transcription factor Sp1 in scleroderma fibroblasts: association with increased expression of the type I collagen gene. Arthritis Rheum 2000, 43:2240-2247 [DOI] [PubMed] [Google Scholar]
- 18.Varga J: Scleroderma and Smads: dysfunctional Smad family dynamics culminating in fibrosis. Arthritis Rheum 2002, 46:1703-1713 [DOI] [PubMed] [Google Scholar]
- 19.Dong C, Zhu S, Wang T, Yoon W, Li Z, Alvarez RJ, ten Dijke P, White B, Wigley FM, Goldschmidt-Clermont PJ: Deficient Smad7 expression: a putative molecular defect in scleroderma. Proc Natl Acad Sci USA 2002, 99:3908-3913 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 20.Czuwara-Ladykowska J, Shirasaki F, Jackers P, Watson DK, Trojanowska M: Fli-1 inhibits collagen type I production in dermal fibroblasts via an Sp1-dependent pathway. J Biol Chem 2001, 276:20839-20848 [DOI] [PubMed] [Google Scholar]
- 21.Truong AH, Ben-David Y: The role of Fli-1 in normal cell function and malignant transformation. Oncogene 2000, 19:6482-6489 [DOI] [PubMed] [Google Scholar]
- 22.Spyropoulos DD, Pharr PN, Lavenburg KR, Jackers P, Papas TS, Ogawa M, Watson DK: Hemorrhage, impaired hematopoiesis, and lethality in mouse embryos carrying a targeted disruption of the Fli1 transcription factor. Mol Cell Biol 2000, 20:5643-5652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hart A, Melet F, Grossfeld P, Chien K, Jones C, Tunnacliffe A, Favier R, Bernstein A: Fli-1 is required for murine vascular and megakaryocytic development and is hemizygously deleted in patients with thrombocytopenia. Immunity 2000, 13:167-177 [DOI] [PubMed] [Google Scholar]
- 24.Brown LA, Rodaway AR, Schilling TF, Jowett T, Ingham PW, Patient RK, Sharrocks AD: Insights into early vasculogenesis revealed by expression of the ETS-domain transcription factor Fli-1 in wild-type and mutant zebrafish embryos. Mech Dev 2000, 90:237-252 [DOI] [PubMed] [Google Scholar]
- 25.Wasylyk B, Hagman J, Gutierrez-Hartmann A: Ets transcription factors: nuclear effectors of the Ras-MAP-kinase signaling pathway. Trends Biochem Sci 1998, 23:213-216 [DOI] [PubMed] [Google Scholar]
- 26.Trojanowska M: Ets factors and regulation of the extracellular matrix. Oncogene 2000, 19:6464-6471 [DOI] [PubMed] [Google Scholar]
- 27.Clements PJ, Lachenbruch PA, Seibold JR, Zee B, Steen VD, Brennan P, Silman AJ, Allegar N, Varga J, Massa M, Wigley FM, Ingenito F, Weisman M, White B, Martin RW, McCloskey D, Moreland LW, Mayes M, Lally EV, Unanue M, Collier DH, Weiner S, Weinstein A, Medsger TA, Jr, Andrews B, Dixon M, Furst DE: Skin thickness score in systemic sclerosis: an assessment of interobserver variability in 3 independent studies. J Rheumatol 1993, 20:1892-1896 [PubMed] [Google Scholar]
- 28.Kubo M, Ihn H, Yamane K, Tamaki K: Up-regulated expression of transforming growth factor beta receptors in dermal fibroblasts in skin sections from patients with localized scleroderma. Arthritis Rheum 2001, 44:731-734 [DOI] [PubMed] [Google Scholar]
- 29.He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B: A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci USA 1998, 95:2509-2514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Czuwara-Ladykowska J, Sementchenko VI, Watson DK, Trojanowska M: Ets1 is an effector of the transforming growth factor Beta (tgf-beta) signaling pathway and an antagonist of the profibrotic effects of tgf-beta. J Biol Chem 2002, 277:20399-20408 [DOI] [PubMed] [Google Scholar]
- 31.Shirasaki F, Makhluf HA, LeRoy C, Watson DK, Trojanowska M: Ets transcription factors cooperate with Sp1 to activate the human tenascin-C promoter. Oncogene 1999, 18:7755-7764 [DOI] [PubMed] [Google Scholar]
- 32.Kivirikko KI, Myllyharju J: Prolyl 4-hydroxylases and their protein disulfide isomerase subunit. Matrix Biol 1998, 16:357-368 [DOI] [PubMed] [Google Scholar]
- 33.Nagata K: Expression and function of heat shock protein 47: a collagen-specific molecular chaperone in the endoplasmic reticulum. Matrix Biol 1998, 16:379-386 [DOI] [PubMed] [Google Scholar]
- 34.Kouba DJ, Chung KY, Nishiyama T, Vindevoghel L, Kon A, Klement JF, Uitto J, Mauviel A: Nuclear factor-kappa B mediates TNF-alpha inhibitory effect on alpha 2(I) collagen (COL1A2) gene transcription in human dermal fibroblasts. J Immunol 1999, 162:4226-4234 [PubMed] [Google Scholar]
- 35.Strehlow D, Jelaska A, Strehlow K, Korn JH: A potential role for protease nexin 1 overexpression in the pathogenesis of scleroderma. J Clin Invest 1999, 103:1179-1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Scharffetter K, Lankat-Buttgereit B, Krieg T: Localization of collagen mRNA in normal and scleroderma skin by in-situ hybridization. Eur J Clin Invest 1988, 18:9-17 [DOI] [PubMed] [Google Scholar]
- 37.Claman HN, Giorno RC, Seibold JR: Endothelial and fibroblastic activation in scleroderma. The myth of the “uninvolved skin.” Arthritis Rheum 1991, 34:1495-1501 [DOI] [PubMed] [Google Scholar]
- 38.Prescott RJ, Freemont AJ, Jones CJ, Hoyland J, Fielding P: Sequential dermal microvascular and perivascular changes in the development of scleroderma. J Pathol 1992, 166:255-263 [DOI] [PubMed] [Google Scholar]
- 39.Freemont AJ, Hoyland J, Fielding P, Hodson N, Jayson MI: Studies of the microvascular endothelium in uninvolved skin of patients with systemic sclerosis: direct evidence for a generalized microangiopathy. Br J Dermatol 1992, 126:561-568 [DOI] [PubMed] [Google Scholar]
- 40.Postiglione L, Montagnani S, Riccio A, Montuori N, Sciorio S, Ladogana P, Spigna GD, Castaldo C, Rossi G: Enhanced expression of the receptor for granulocyte macrophage colony stimulating factor on dermal fibroblasts from scleroderma patients. J Rheumatol 2002, 29:94-101 [PubMed] [Google Scholar]
- 41.Majewski S, Hunzelmann N, Johnson JP, Jung C, Mauch C, Ziegler-Heitbrock HW, Riethmuller G, Krieg T: Expression of intercellular adhesion molecule-1 (ICAM-1) in the skin of patients with systemic scleroderma. J Invest Dermatol 1991, 97:667-671 [DOI] [PubMed] [Google Scholar]
- 42.Koch AE, Kronfeld-Harrington LB, Szekanecz Z, Cho MM, Haines GK, Harlow LA, Strieter RM, Kunkel SL, Massa MC, Barr WG, Jimenez HA: In situ expression of cytokines and cellular adhesion molecules in the skin of patients with systemic sclerosis. Their role in early and late disease. Pathobiology 1993, 61:239-246 [DOI] [PubMed] [Google Scholar]
- 43.Gruschwitz MS, Vieth G: Up-regulation of class II major histocompatibility complex and intercellular adhesion molecule 1 expression on scleroderma fibroblasts and endothelial cells by interferon-gamma and tumor necrosis factor alpha in the early disease stage. Arthritis Rheum 1997, 40:540-550 [DOI] [PubMed] [Google Scholar]
- 44.Jimenez SA: Alterations in the regulation of expression of the a1(I) collagen gene (COL1A1) in systemic sclerosis (scleroderma). Springer Semin Immunopathol 2000, 21:397-414 [DOI] [PubMed] [Google Scholar]