

Abstract
Activation of the insulin-like growth factor-I receptor (IGF-IR) was recently shown to modulate angiogenesis by up-regulating the expression of vascular endothelial growth factor (VEGF). We hypothesized that inhibiting IGF-IR function would inhibit angiogenesis and growth of pancreatic cancer in vivo and sought to identify major signaling pathways regulated by IGF-IR in pancreatic cancer cells. Human pancreatic cancer cells (L3.6pl) were stably transfected with a dominant-negative form of IGF-IR (IGF-IR DN) or an empty vector (pcDNA). In vitro, IGF-IR DN cells exhibited a decrease in both constitutive and inducible phosphorylation of IGF-IR and Erk1/2. Constitutive expression of nuclear hypoxia-inducible factor-1α and secreted VEGF (P < 0.01) protein levels also were significantly lower in IGF-IR DN cells than in pcDNA cells. In vivo, IGF-IR inhibition led to decreases in pancreatic tumor volume and weight, vessel density, and tumor cell proliferation (P < 0.01 for all) and increases in tumor cell apoptosis (P < 0.02). Our results suggest that autocrine activation of the IGF-IR system significantly affects VEGF expression and angiogenesis in human pancreatic cancer. Thus, IGF-IR may be a valid target in the treatment of pancreatic cancer.
The insulin-like growth factor-I receptor (IGF-IR) is often overexpressed in a variety of human cancers, including pancreatic cancer, and has been associated with aggressive disease and formation of metastases. 1-8 IGF-IR is a heterotetrameric tyrosine kinase receptor that consists of two α- and two β-subunits. 9 The α-subunits are entirely extracellular and contain a cysteine-rich domain responsible for ligand binding. 9,10 The β-subunits are divided by a hydrophobic transmembrane domain into extracellular and intracellular domains, with the intracellular portion containing the tyrosine kinase domain. 9,10 The IGF-IR seems to be crucial for maintaining normal growth and development, 10,11 and IGF-IR activation has been implicated in the malignant progression of several types of human cancer. 12,13 In pancreatic cancer, several mechanisms regulate or render autocrine IGF-IR activation which may contribute to the aggressive nature of this disease. 14-18 Both IGF-IR and its ligands have been shown to be up-regulated in various pancreatic cancer cell lines and tissue specimens. 4,8,17,18 Increased Src kinase activity, activation of phosphatidylinositol 3′-kinase (PI-3K)/Akt, or both was shown to lead to IGF-IR overexpression in these cancer cells, 8,16 and its ligand IGF-I has been found to be overexpressed in human pancreatic cancer specimens relative to normal pancreatic tissue. 4
Activation of the IGF-IR system is important in mediating angiogenesis by means of up-regulating vascular endothelial growth factor (VEGF) expression in neoplastic 19-23 and nonneoplastic 24,25 tissues. VEGF (also termed VEGF-A) is a fundamental promoter of angiogenesis, a process essential for the outgrowth and metastatic dissemination of tumors. 26-28 VEGF is an important mediator of tumor growth and metastasis in experimental models of pancreatic cancer. 29-31 Blockade of VEGF activity in experimental models has been shown to significantly reduce tumor growth and metastasis. 30,31 Because VEGF expression in tumors is partially promoted by the tumor microenvironment, 29 means of inhibiting autocrine/paracrine mechanisms of VEGF regulation in cells may be a promising approach for enhancing the efficacy of anti-angiogenic therapies.
In the current study, we hypothesized that inhibition of IGF-IR function in human pancreatic cancer cells would decrease VEGF expression and angiogenesis and in turn inhibit tumor growth in vivo. We further sought to elucidate the role of the IGF-I receptor/ligand system in the constitutive activation of intracellular pathways that mediate VEGF expression in pancreatic cancer cells. We found that constitutive activation of IGF-IR-mediated VEGF expression in pancreatic cancer cells occurred in part through increasing the nuclear translocation of hypoxia-inducible factor-1α (HIF-1α). Moreover, inhibition of IGF-IR function inhibited angiogenesis and growth of pancreatic tumors, suggesting that the IGF-IR system is a valid molecular target in the treatment of human pancreatic cancer.
Materials and Methods
Cell Culture and Stable Transfection
The human pancreatic cancer cell line L3.6pl 32 was kindly provided by I. J. Fidler, Ph.D., D.V.M. (The University of Texas M. D. Anderson Cancer Center, Houston, TX). Cells were cultured and maintained in minimal essential medium (MEM) supplemented with 10% fetal bovine serum (FBS), 2 U/ml of a penicillin-streptomycin mixture (Flow Laboratories, Rockville, MD), vitamins (Life Technologies, Inc., Grand Island, NY), 1 mmol/L sodium pyruvate, 2 mmol/L l-glutamine, and nonessential amino acids and incubated in 5% CO2-95% air at 37°C. To clarify the influence of IGF-IR inhibition on VEGF regulation and angiogenesis, we stably transfected L3.6pl cells with a mutated dominant-negative (DN) form of IGF-IR (truncated at position 952 in the β-subunit transmembrane region), which was the generous gift of D. Prager M.D. (Cedars-Sinai Medical Center-UCLA School of Medicine, Los Angeles, CA), 33,34 or an empty vector (pcDNA3) as a control. Cell were transfected by using FuGENE 6 transfection reagent (Roche Diagnostics Corporation, Indianapolis, IN) according to the manufacturer’s protocol as described elsewhere. 19,20 Cells were then grown and expanded in selective medium containing neomycin (G418, Life Technologies). Successful transfection was verified by changes in insulin receptor substrate-1 (IRS-1) phosphorylation on stimulation with recombinant human IGF-I (rhIGF-I) (R&D Systems Inc., Minneapolis, MN), which was reduced in IGF-IR DN cells. 19,20
Immunohistochemical Analysis of IGF-IR and Activated IGF-R in Human Pancreatic Cancer Specimens
Paraffin-embedded tissues from primary human pancreatic adenocarcinomas (n = 11) [confirmed by a pathologist on hematoxylin and eosin (H&E)-stained slides] or normal pancreas (n = 10) were stained for IGF-IR expression and IGF-IR activation by using antibodies to IGF-IR-α (Santa Cruz Biotechnology, Santa Cruz, CA) or phosphorylated IGF-IR (Biosource International, Camarillo, CA), respectively. Patients did not receive neoadjuvant therapy before surgery. Specimens were collected under a protocol approved by the Institutional Review Board of the M.D. Anderson Cancer Center. Tissue sections were deparaffinized in xylene, followed by treatment with a graded series of alcohol washes [100%, 95%, 80% ethanol/ddH2O (v/v)], rehydration in phosphate-buffered saline (PBS) (pH 7.5), and microwaving for 10 minutes in 100 mmol/L of citrate solution (pH 6.0) for antigen retrieval. Slides were then washed in PBS and incubated for 20 minutes in a protein-blocking solution consisting of PBS supplemented with 1% normal goat serum and 5% normal horse serum. Primary antibody was diluted in protein-blocking solution (1:200 for anti-IGF-IR-α, 1:50 for anti-phospho-IGF-IR) and applied for 18 hours at 4°C. Antibodies were then washed off, protein-block applied for 20 minutes, followed by 1 hour of incubation with Alexa594 conjugated goat anti-rabbit secondary antibody (1:600) (Jackson ImmunoResearch Laboratories, West Grove, PA). Tissues were then counterstained with Hoechst dye (1:2000). Tumor and normal tissue regions were identified by parallel comparison to H&E-stained slides and images were obtained at ×100 magnification.
IGF-I/IGF-II Secretion by Cells and Signaling Pathway Activation
Western blot analysis was done to detect secreted IGF-II protein in conditioned medium from parental L3.6pl cells. 35 IGF-I protein in conditioned medium was measured by using an enzyme-linked immunosorbent assay (ELISA) kit for human IGF-I (R&D Systems Inc.). Briefly, L3.6pl pancreatic cancer cells were incubated in 10% FBS-MEM for 48 hours and conditioned medium was harvested, centrifuged (360 × g, 5 minutes) and filtered through a 0.22-μm filter (Corning Inc., Corning, NY). For IGF-II detection, proteins in samples of conditioned media (100 μg/sample) were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis on a denaturing 15% gel under reducing conditions. Western blotting was performed with a monoclonal primary antibody against human IGF-II (1:3000 in 5% milk) (Upstate Biotechnology, Lake Placid, NY) and a secondary antibody for chemiluminescent analysis (ECL; Amersham Biosciences, Piscataway, NJ) as described previously. 19 IGF-IR phosphorylation was investigated using a rabbit anti-phospho-IGF-IR antibody at 1:1000 dilution (Biosource International) for Western blot analysis.
Signaling pathways activated by IGF-I in IGF-IR DN- or pcDNA-transfected cells were investigated by Western blotting. 20,36 Briefly, cells were grown to 50 to 60% cell confluence, incubated overnight in 1% FBS-MEM, and treated thereafter for various times with rhIGF-I (100 ng/ml) in 1% FBS-MEM. To investigate constitutive phosphorylation levels of signaling intermediates in IGF-IR DN- and pcDNA-transfected cells, cells were incubated in 10% FBS-MEM for 48 hours and harvested in PBS. Protein was extracted from cell lysates with RIPA buffer, and 50-μg protein samples were subjected to Western blot analysis by sodium dodecyl sulfate-polyacrylamide gel electrophoresis on a denaturing 10% gel. Activated signaling pathways were identified by using the following antibodies: anti-extracellular signal-regulated kinase (Erk)-1/2 (p42/44) (Oncogene Research Products, San Diego, CA); and anti-phosphospecific Erk-1/2 (p42/44), anti-Akt, anti-phosphospecific Akt, anti-phosphospecific c-jun amino-terminal kinase (JNK), anti-P38, and anti-phosphospecific P38 from Cell Signaling Technologies (Beverly, MA).
Regulation of VEGF Expression by the IGF-I Receptor System
For Northern blot analyses, total RNA was extracted from cells by using Tri Reagent (Molecular Research Center, Cincinnati, OH) according to the manufacturer’s instructions. Northern blot analysis was performed as described previously. 19,37 Briefly, probes for VEGF (human VEGF-specific 204-bp cDNA probe) 19 or glyceraldehyde-3-phosphate dehydrogenase (American Type Culture Collection, Manassas, VA) were radiolabeled by the random primer technique with a commercially available kit (Rediprime II, Amersham Biosciences), and nylon membranes (Hybond-N+, Amersham Biosciences) were subsequently hybridized overnight (Rapid-hyb Solution, Amersham Biosciences) at 65°C. Autoradiography was performed thereafter in the linear range of the film (Hyperfilm MP, Amersham Biosciences). To investigate VEGF induction by IGF-I, parental and transfected cells were grown to 50 to 60% confluence in standard medium as described above and then incubated in 5% FBS-containing medium overnight. Cells were then incubated in the presence or absence of rhIGF-I (100 ng/ml) in 1% FBS-MEM for 24 hours because induction of VEGF mRNA expression in this cell line was maximal by this time. Total RNA was extracted, and VEGF mRNA expression was determined by Northern blot analysis.
VEGF protein concentrations in conditioned media from IGF-IR DN- or pcDNA- transfected L3.6pl cells were measured by using an ELISA kit for human VEGF (Biosource International). Supernatant (3 ml) from the transfected cells was collected after 48 hours of incubation in 10% FBS-MEM. Cells were rinsed with cold PBS, trypsinized, and counted. VEGF ELISA was performed according to the manufacturer’s protocol, and VEGF levels (in pg) were calculated per 1 × 106 cells.
Experiments using the cyclooxygenase-2 (Cox-2) inhibitor celecoxib were performed by dissolving 200-mg celecoxib capsules (obtained from the Pharmacy at M. D. Anderson Cancer Center) in dimethyl sulfoxide (100 mmol/L stock solution). Preliminary experiments confirmed that dimethyl sulfoxide, at a final concentration of 0.01% in cell culture medium, did not induce VEGF expression.
Regulation of Nuclear HIF-1α Translocation by the IGF-IR System
Western blot analyses for HIF-1α were performed from nuclear extracts from parental and transfected cells as described elsewhere. 38,39 Briefly, IGF-IR DN- and pcDNA-transfected L3.6pl cells were incubated for 48 hours in 10% FBS-MEM, washed twice with ice-cold PBS, and harvested into cold PBS (1 ml). After centrifugation (400 × g for 5 minutes at 4°C), cell pellets were incubated with ice-cold lysis buffer (buffer A: 10 mmol/L HEPES, 1.5 mmol/L MgCl2, 10 mmol/L KCl, 0.5 mmol/L dithiothreitol, 0.2 mmol/L phenylmethyl sulfonyl fluoride, and 1× protein inhibitor) for 30 minutes on ice, centrifuged again (7150 × g for 3 minutes at 4°C), and the remaining cell pellets were washed twice with ice-cold buffer A. For nuclear protein extraction, washed pellets were incubated in ice-cold buffer C (20 mmol/L HEPES, 25% glycerol, 450 mmol/L NaCl, 1.5 mmol/L MgCl2, 0.2 mmol/L ethylenediaminetetraacetic acid, 0.5 mmol/L dithiothreitol, 0.5 mmol/L phenylmethyl sulfonyl fluoride, and 1× protein inhibitor) for 30 minutes on ice, and supernatant was collected after centrifugation (22,000 × g for 15 minutes at 4°C). Protein concentrations were then determined 19 and 50-μg aliquots subjected to Western blot analysis on a 6% sodium dodecyl sulfate-polyacrylamide gel electrophoresis gel. Membranes were probed with an anti-HIF-1α monoclonal antibody (BD Biosciences Pharmingen, San Diego, CA) at 1:1000 dilution in 5% milk overnight at 4°C, followed by incubation with a secondary antibody for chemiluminescent analysis (ECL, Amersham Biosciences).
In Vivo Studies
Eight-week-old male nude mice (Animal Production Area of the National Cancer Institute-Frederick Cancer Research and Development Center, Frederick, MD) were acclimated for 1 to 2 weeks while caged in groups of five. Mice were housed under laminar flow conditions and fed a diet of animal chow and water ad libitum throughout the experiment. All experiments were approved by the Institutional Animal Care and Use Committee of M. D. Anderson Cancer Center.
Orthotopic Model of Pancreatic Cancer
Cells for in vivo experiments were grown to 70% cell confluence and prepared for orthotopic implantation in mice as follows. Cells were rinsed briefly with PBS, trypsinized for 3 minutes in 0.25% trypsin with 0.02% ethylenediaminetetraacetic acid, washed in 10% FBS-MEM, and counted. Cell viability was assessed by trypan blue exclusion; cell viability was >90% in both transfected cell lines. Cells were then centrifuged and resuspended in Hanks’ balanced salt solution for injection into mice. For the development of pancreatic tumors, 250,000 cells of pcDNA (control)- or IGF-IR DN-transfected L3.6pl cells were injected (in a volume of 50 μl) into the pancreas of mice with a 30-gauge needle after anesthesia had been induced with pentobarbital (Nembutal, 50 mg/kg). Mice were observed daily, and the experiment was terminated when three mice in any one group showed decreased mobility or discomfort. Mice were killed by cervical dislocation after being anesthetized with Nembutal. Body weights and pancreatic tumor diameters were measured, and tumors were subsequently excised and weighed. Tumor volumes were calculated as width 2 × length × 0.5. Tumor tissue was then snap-frozen in optimum cutting temperature (OCT) solution (Miles Inc., Elkhart, IN) in preparation for subsequent immunohistochemical analyses.
Immunohistochemical Analysis of Microvessel Density and Tumor Cell Proliferation
Antibodies for immunohistochemical analyses were obtained as follows: rat anti-mouse CD31/PECAM-1 antibody from PharMingen (San Diego, CA), mouse anti-proliferating cell nuclear antigen (PCNA) PC10 (IgG2a) from DAKO (Carpinteria, CA), rat anti-mouse IgG2a-horseradish peroxidase (HRP) conjugate from Serotec Inc. (Raleigh, NC), and peroxidase-conjugated goat anti-rat IgG from Jackson Research Laboratories (West Grove, PA). Tumors that had been frozen in OCT were sectioned in 8-μm slices, mounted on positively charged slides, air-dried for 30 minutes, fixed in cold acetone followed by 1:1 acetone/chloroform and acetone, and then washed with PBS. Specimens were then incubated with 3% H2O2 in methanol for 12 minutes at room temperature to block endogenous peroxidases, washed three times with PBS (pH 7.5), and incubated for 20 minutes at room temperature in a protein-blocking solution, as described above. For PCNA staining, sections were treated (with intermittent washing steps) with 0.2 N HCl (10 minutes), then 0.07 N NaOH (in 70% ethanol) for 5 minutes and incubated in 1% Triton X-100 (5 minutes) before being subjected to protein-blocking solution. In protein-blocking solution, diluted primary antibodies directed against CD31 (1:800) or PCNA (1:100) were then applied to the sections, which were incubated overnight at 4°C. Sections were then rinsed in PBS and incubated for 10 minutes in protein-blocking solution before the addition of peroxidase-conjugated secondary antibody. The secondary antibodies used for CD31 (peroxidase-conjugated goat anti-rat IgG) or PCNA (rat anti-mouse IgG2a HRP) staining were diluted 1:200 in protein-blocking solution. After being incubated with the secondary antibody for 1 hour at room temperature, the samples were washed and incubated with stable diaminobenzidine (Research Genetics, Huntsville, AL) substrate. Staining was monitored under a bright-field microscope, and the reaction was stopped by washing with distilled water. Sections were counterstained with Gill’s No. 3 hematoxylin (Sigma Chemical Co., St. Louis, MO) and mounted with Universal Mount (Research Genetics). CD31-stained vessels were counted (at ×50 magnification) in four different quadrants of each tumor (2 mm inside the tumor-normal tissue interface) and means were calculated. Slides were also stained with H&E to study overall tissue structure. The number of tumor cells that stained positively or negatively for PCNA was determined in four random fields per tumor (at ×100 magnification), and the percentage of PCNA-positive cells was then calculated. For all immunohistochemical studies, the primary antibody was omitted as a negative control. 40
Immunofluorescent Staining for Analysis of Tumor Cell Apoptosis
For immunofluorescent terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) staining, frozen tissue was fixed in cold acetone and chloroform as described above. The TUNEL assay was performed with a commercial kit (Promega, Madison, WI) according to the manufacturer’s protocol. Tissue sections were also counterstained with Hoechst dye (1:2000) to allow identification of tumor cells. Immunofluorescence microscopy was done on an epifluorescence microscope equipped with narrow bandpass excitation filters (Chroma Technology Corp., Brattleboro, VT). Images were captured with a C5810 Hamamatsu camera (Hamamatsu Photonics K.K., Japan) mounted on a Zeiss Axioplan microscope (Carl Zeiss Inc.) with Optimas image analysis software (Media Cybernetics, Silver Spring, MD). Images were further processed with Adobe Photoshop software (Adobe Systems, Mountain View, CA). TUNEL-positive cells were counted at ×100 magnification in four different quadrants of each tumor according to the guidelines noted above for vessel counts.
Immunohistochemical Analysis of VEGF Expression in Pancreatic Tumors
Tumor sections were fixed as described above and treated for immunohistochemical staining as described for CD31, except that rabbit anti-VEGF antibody (A20, Santa Cruz Biotechnology) was added in a 1:500 dilution (protein-block) for overnight incubation at 4°C. Slides were then washed, incubated for 20 minutes in protein-blocking solution, and goat anti-rabbit IgG biotinylated secondary antibody (Biocare Medical, Walnut Creek, CA) was added for 30 minutes. After being washed in PBS, streptavidin complex (DAKO, 1:300) was then applied for 30 minutes. Slides were washed and incubated with stable diaminobenzidine (Research Genetics) substrate. Staining was monitored under a bright-field microscope, and the reaction was stopped by washing with distilled water. Counterstaining was omitted. VEGF expression in tumor sections was quantified by optical densitometry analysis at ×50 magnification with Optimas Imaging software (Media Cybernetics, Carlsbad, CA).
Densitometric Quantification and Statistical Analyses
Densitometric analysis of autoradiographs was performed with NIH Image Analysis software (V1.62) from the National Institutes of Health (Bethesda, MD) to quantify the results of Northern and Western blot analyses. All statistical analyses were done with InStat Statistical Software (V2.03; GraphPad Software, San Diego, CA), with P values of less than 0.05 considered to be statistically significant. Tumor-associated variables were tested for statistical significance with the two-tailed Student’s t-test or the Mann-Whitney U-test (for nonparametric data) as specified in the figure legends.
Results
Expression and Activation of IGF-I Receptor in Human Pancreatic Adenocarcinomas
The presence of IGF-IR expression and activation in human pancreatic cancer was investigated by immunohistochemical analysis of primary pancreatic adenocarcinoma specimens from patients that underwent surgical resection without having received neoadjuvant therapy. The expression of IGF-IR was detectable in all (11 of 11) pancreatic adenocarcinomas and in the epithelium of normal pancreatic ducts (10 of 10) (Figure 1) ▶ . However, activated (phosphorylated) IGF-IR was only detectable in pancreatic adenocarcinoma specimens (11 of 11), whereas nonmalignant ductal epithelium lacked IGF-IR activation. Activated IGF-IR was heterogeneously distributed within the tumor. Relatively high levels of IGF-IR activation were detectable in 7 of 11 (63%) malignant specimens and weak activity was noted in 4 of 11 (37%) cancers. In addition, stromal cells in pancreatic cancer tissues also demonstrated some evidence of activation of IGF-IR.
Figure 1.
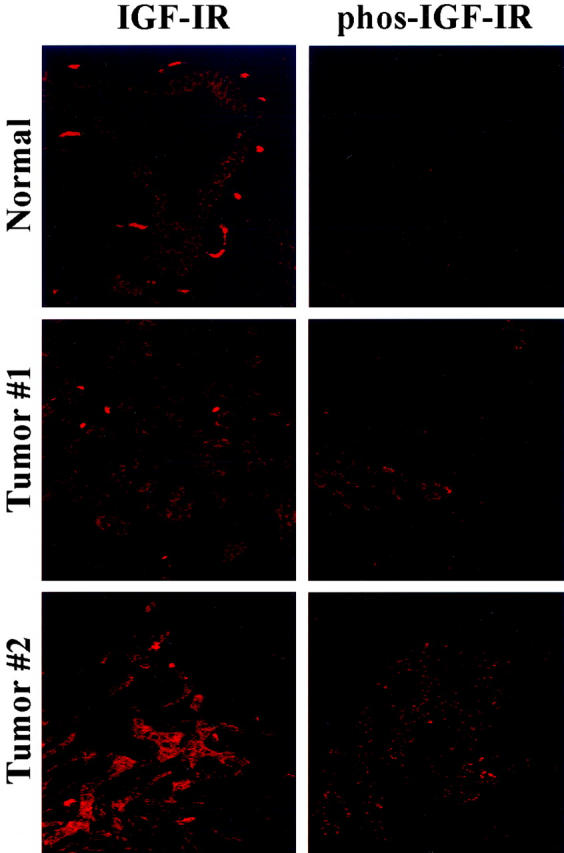
Immunohistochemical staining for IGF-IR and activated IGF-IR in human pancreatic cancers and nonmalignant pancreatic tissues. Paraffin-embedded specimens from human pancreatic cancers and nonmalignant tissues were immunofluorescently stained for IGF-IR and phosphorylated IGF-IR. IGF-IR was expressed in normal ductal epithelium as well as tumor epithelium. However, only tumor epithelium stained positive for phosphorylated IGF-IR.
Expression of IGF-I Receptor and Its Ligands by Pancreatic Cancer Cells
To verify that IGF-IR is expressed in human pancreatic cancer cells, we screened several pancreatic cancer cells lines by immunoprecipitation for IGF-IR expression. Eight of nine cell lines tested expressed IGF-IR at levels that were markedly higher than those in colon cancer cell lines (positive control) (data not shown). 19,20 The highly aggressive L3.6pl cell line, previously used as an orthotopic pancreatic cancer model, 30,32,40,41 demonstrated high IGF-IR expression levels and thus were selected for use in this study. Western blotting and ELISA of conditioned media from L3.6pl cells confirmed that these cells secrete both IGF-I and IGF-II into the culture medium. By ELISA, L3.6pl cells produced detectable IGF-I protein levels [9.2 ± 2.4 ng/ml/106cells (mean ± SEM)] in conditioned media. IGF-II expression was detected by Western blotting of conditioned media from parental and transfected cells, and showed no differences in IGF-II protein levels among those cells (not shown). Autocrine activation of the IGF-I receptor was investigated by incubating L3.6pl cells for 48 hours in 10% FBS-MEM, and either leaving the medium unchanged during this time (conditioned medium), or replacing the media with fresh 10% FBS-MEM 8 hours before cell lysis (control) (to lower cumulative secreted levels of IGF-I and IGF-II). By Western blot analysis, cells grown in conditioned medium for 48 hours demonstrated basal phosphorylation of the IGF-I receptor, in contrast to the absence of IGF-IR phosphorylation in the controls where media was replaced (Figure 2A) ▶ .
Figure 2.

A: Autocrine IGF-IR activation in parental L3.6pl pancreatic cancer cells. Cells were grown for 48 hours and medium was either replaced with fresh 10% FBS-MEM (lane 1) 8 hours before harvesting the cells (control), or remained unchanged for 48 hours (conditioned medium) (lane 2). Cell lysates were subjected to Western blot analysis of IGF-IR phosphorylation. Equal loading was verified by probing for β-actin. When cells are allowed to grow in their own conditioned medium, the IGF-IR is phosphorylated. In contrast, when the medium is changed to prevent the cells from conditioning their own medium, there is no receptor phosphorylation. B: Effect of IGF-IR inhibition on constitutive and inducible activation of signaling pathways. IGF-IR DN- and pcDNA-transfected L3.6pl cells were treated for the indicated times with rhIGF-I (100 ng/ml) under serum-reduced conditions (1% FBS-MEM). Protein extracts were examined by Western blot analysis for IGF-IR phosphorylation and activated signaling pathways. Equal loading was verified by probing for β-actin. No differences in Akt phosphorylation were observed among transfected cell lines. P38 and JNK phosphorylation levels were not affected by rhIGF-I (not shown).
Effect of IGF-I Receptor Inhibition on Intracellular Signaling Pathway Activation and VEGF Expression in Pancreatic Cancer Cells
We next investigated the role of the IGF-IR system in regulation of VEGF expression in pancreatic cancer cells by stably transfecting a dominant-negative IGF-IR construct (IGF-IR DN) or an empty vector (pcDNA) into L3.6pl cancer cells. Western blotting revealed that lysates from IGF-IR DN-transfected cells (harvested from subconfluent cultures) demonstrated lower constitutive phosphorylation levels of the mitogen-activated protein kinases Erk1/2 than did pcDNA-transfected cells. To identify time-dependent phosphorylation of IGF-IR and signaling intermediates, this experiment was repeated after reducing constitutive phosphorylation levels by overnight incubation in 1% FBS-MEM and then adding rhIGF-I. Again, constitutive Erk1/2 phosphorylation levels were significantly reduced in IGF-IR DN cells (0 minute time point, Figure 2B ▶ ). IGF-IR phosphorylation was diminished in IGF-IR DN cells after stimulation with rhIGF-I, whereas IGF-IR phosphorylation in pcDNA cells was detectable for up to 30 minutes. However, rhIGF-I treatment activated Erk1/2, JNK and P38 phosphorylation in IGF-IR DN cells, but to a markedly lesser degree than controls (Figure 2B) ▶ , indicating that impaired IGF-IR function inhibited IGF-I activation of these signaling intermediates. The PI-3K/Akt pathway was not significantly activated by rhIGF-I in these cells.
In preliminary experiments with parental L3.6pl cells in vitro, we determined that the extent of induction of VEGF mRNA expression by rhIGF-I stimulation peaked at approximately two times the original value at 24 hours (data not shown). This finding was unexpected, as IGF-I treatment of colon cancer cells leads to significantly greater VEGF induction. 19,20 pcDNA-transfected cells exhibited relatively high constitutive VEGF mRNA expression that could be only modestly increased (by 1.3 times) by the addition of rhIGF-I at 24 hours. However, the inhibition of IGF-IR function led to a significant reduction (by 50%) of constitutive VEGF mRNA expression in IGF-IR DN cells (Figure 3A) ▶ . Moreover, VEGF induction in response to rhIGF-I was blunted in those cells. To verify these results, we used ELISA to assess VEGF protein levels in conditioned medium with or without the addition of rhIGF-I (Figure 3B) ▶ . Constitutive VEGF protein levels were relatively high in conditioned medium from pcDNA-transfected cells and, consistent with the findings from Northern blot analysis, could not be further increased by the addition of rhIGF-I. VEGF concentrations in medium from IGF-IR DN cells were significantly lower (40 to 50%) than that in pcDNA-control cells and remained unchanged on addition of IGF-I.
Figure 3.

Effect of IGF-IR inhibition on constitutive and inducible VEGF expression. A: Northern blot analysis of VEGF expression in pcDNA- and IGF-IR DN-transfected L3.6pl cells after a 24-hour incubation in serum-reduced conditions (1% FBS-MEM) with or without rhIGF-I (100 ng/ml). Relative changes in VEGF mRNA expression with respect to controls (unstimulated pcDNA cells) are shown beneath each lane. B: ELISA of VEGF protein concentration in conditioned medium of transfected L3.6pl cells. Medium was collected after a 48-hour incubation period in 10% FBS-MEM in the presence or absence of rhIGF-I (100 ng/ml). *, P < 0.01 versus treated and untreated pcDNA cells, Student’s t-test. Data are presented as means ± SEM.
Effect of IGF-I on HIF-1α Activation in Parental and Transfected Cells
In light of a recent report that IGF-I led to increased HIF-1α activation and in turn up-regulation of VEGF expression in colon cancer cells, 39 we hypothesized that an IGF-I/IGF-IR autocrine loop would contribute via HIF-1α to the induction of VEGF in pancreatic cancer cells. After overnight serum starvation, parental cells were stimulated for various times with rhIGF-I and nuclear extracts were analyzed for nuclear translocation of HIF-1α. Throughout the course of 8 hours, the addition of IGF-I increased nuclear HIF-1α protein levels over those in untreated controls (Figure 4A) ▶ . However, constitutive HIF-1α levels in the control cells increased slightly in a time-dependent manner, suggesting the possibility of autocrine activation. This mechanism of HIF-1α activation was then further investigated by incubating pcDNA- and IGF-IR DN-transfected cells for 48 hours to allow sufficient IGF-I/IGF-II protein to accumulate in the cell culture medium. Western blot analysis for HIF-1α expression of nuclear extracts showed an ∼50% reduction in HIF-1α nuclear translocation in IGF-IR DN cells as compared with pcDNA cells (Figure 4B) ▶ .
Figure 4.

Effect of IGF-IR function on constitutive and inducible HIF-1α expression. A: Parental L3.6pl cells were serum-deprived in 1% FBS-MEM overnight, treated for the indicated times with rhIGF-I (100 ng/ml), and subjected to Western blot analysis of HIF-1α protein in nuclear extracts. B: Western blot analysis of constitutive HIF-1α nuclear translocation in pcDNA- and IGF-IR DN-transfected L3.6pl cells. Cells were incubated for 48 hours in 10% FBS-MEM to evaluate the possible effects of an IGF-I autocrine loop on HIF-1α activation. Nuclear protein extracts from each cell line were analyzed by Western blot analysis. By densitometry, IGF-IR DN-transfected cells showed approximately half the nuclear translocation of HIF-1α as that of control (pcDNA-transfected) cells.
Effect of Cox-2 Inhibition on VEGF Expression in Pancreatic Cancer Cells
In preliminary experiments, we showed by Western blot analysis that Cox-2 was highly expressed in L3.6pl cells and could be up-regulated further by the addition of rhIGF-I (data not shown). To determine whether IGF-IR regulation of VEGF occurred through a Cox-2-dependent pathway in human pancreatic cancer cells, we incubated parental L3.6pl cells for 24 hours with 10 μmol/L of celecoxib (a Cox-2 inhibitor) or vehicle alone (0.01% dimethyl sulfoxide), added rhIGF-I, and subjected the cells to Northern blotting 24 hours later. Celecoxib did not block rhIGF-I induction of VEGF in L3.6pl cells (Figure 5) ▶ , but did lead to a compensatory increase in Cox-2 mRNA expression (data not shown).
Figure 5.

Effect of Cox-2 inhibition on VEGF expression in L3.6pl pancreatic cancer cells by Northern blot analysis. After a 24-hour incubation in 5% FBS-MEM with either vehicle (0.01% dimethyl sulfoxide) or 10 μmol/L of celecoxib, L3.6pl cells were incubated for another 24 hours with or without rhIGF-I (100 ng/ml). Relative changes in VEGF mRNA expression associated with rhIGF-I stimulation relative to those in unstimulated cells are shown beneath each lane.
Effect of IGF-I Receptor Inhibition on Pancreatic Tumor Growth in Mice
In vivo tumor growth of IGF-IR DN- and pcDNA-transfected cells was investigated in an orthotopic pancreatic cancer model in which tumor cells were injected directly into the pancreatic tails of nude mice. After 29 days of tumor growth, 10 of 10 mice in the pcDNA group and 9 of 10 mice in the IGF-IR DN group had detectable pancreatic tumors. Inhibition of IGF-IR function significantly reduced the tumor burden (P < 0.01 for tumor weight and tumor volume) relative to tumors in mice injected with pcDNA-transfected cells (Figure 6, A and B) ▶ . Representative images of pancreatic tumors in each group are shown in Figure 6C ▶ . These results were similar to those of previous experiments in which a different IGF-IR DN clone was used in a subcutaneous xenograft model. In this previous study, IGF-IR DN tumors demonstrated a dramatic reduction in growth rate and tumor volumes after 14 days of tumor growth [133 ± 39.5 mm3 (mean ± SEM)], compared to control tumors (732 ± 119 mm3) (P < 0.05).
Figure 6.

Effect of impaired IGF-I receptor function on pancreatic tumor growth in vivo. In an orthotopic tumor model, pcDNA- or IGF-IR DN-transfected L3.6pl cells (250,000 cells in 50 μl) were injected into the pancreas of nude mice, and 29 days later the mice were killed and the pancreatic tumors excised. A: Tumors weighed less in the IGF-IR DN group versus in the pcDNA group (*, P < 0.01). B: Inhibition of IGF-IR function significantly reduced pancreatic tumor volumes (width2 × length × 0.5) (*, P < 0.01; Mann-Whitney U-test). Data are presented as means ± SEM. C: Representative pancreatic tumors (arrows) from each group.
Effect of Impaired IGF-I Receptor Function on Vessel Density, Tumor Cell Proliferation, Apoptosis, and VEGF Expression in Pancreatic Tumors
Tumor vessel counts (by CD31 staining) were lower in the IGF-IR DN tumors than in the pcDNA tumors (P < 0.01) (Figure 7A) ▶ . The proportion of proliferating tumor cells, evaluated by immunohistochemical staining for PCNA, was also lower in the IGF-IR DN tumors than in pcDNA tumors (P < 0.01) (Figure 7B) ▶ . The number of apoptotic tumor cells, detected by TUNEL analysis, was higher in the IGF-IR DN tumors, despite the smaller tumor volumes, than in the pcDNA tumors (P < 0.02) (Figure 7C) ▶ . Further, VEGF expression in the IGF-IR DN tumors was significantly less than that in control tumors (P < 0.01) (Figure 8) ▶ . Representative images of immunohistochemical analysis of pcDNA and IGF-IR DN tumors are shown in Figure 9 ▶ .
Figure 7.
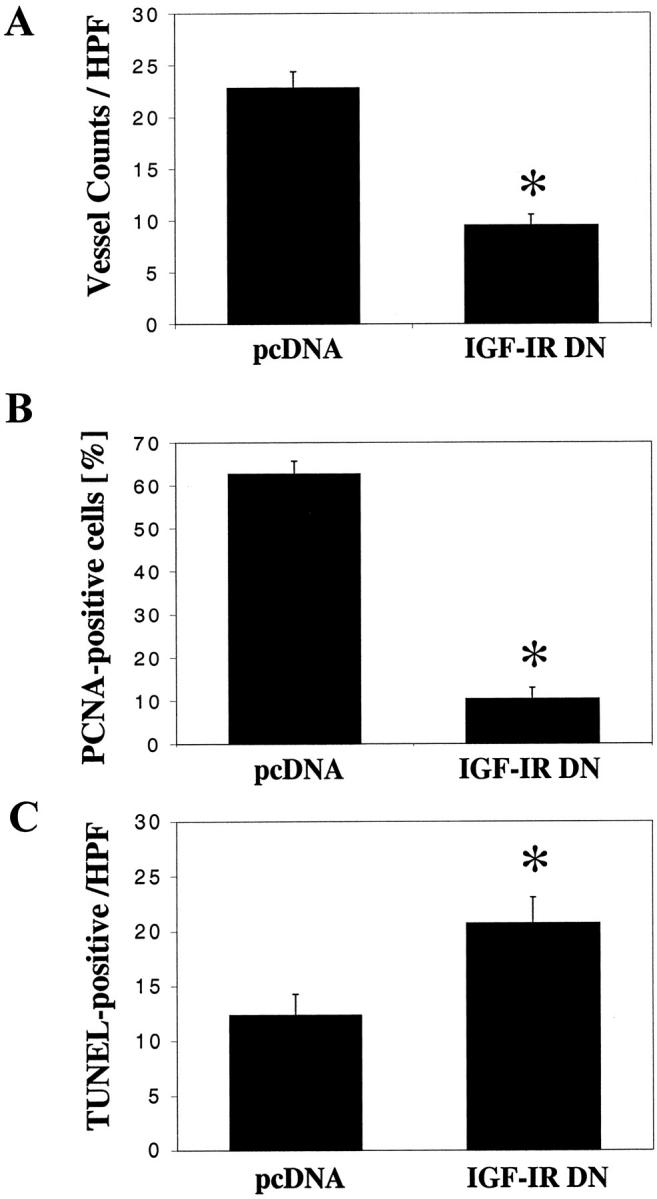
Effect of IGF-IR inhibition on vessel counts, tumor cell proliferation, and tumor cell apoptosis. Tumor sections were stained for CD31 (vessels), PCNA (cell proliferation), and TUNEL (apoptosis) as described in Materials and Methods. A: Tumor vessels were counted at four different quadrants in each tumor (1 to 2 mm inside the tumor/normal interface) and the mean numbers of vessels calculated. IGF-IR DN tumors had significantly fewer vessels than pcDNA tumors (*, P < 0.01; Student’s t-test). B: Inhibition of IGF-I receptor function in IGF-IR DN cells led to significant reductions in numbers of proliferating tumor cells with respect to controls (*, P < 0.001; Student’s t-test). C: Tumor cell apoptosis was detected by TUNEL and nuclear counterstaining. Tumors in the IGF-IR DN group showed higher numbers of apoptotic cells than pcDNA tumors (*, P < 0.02; Student’s t-test). Data are presented as means ± SEM.
Figure 8.

Effect of IGF-I receptor inhibition on VEGF expression in pancreatic tumors. Tumor sections were stained with anti-VEGF antibody and quantified in terms of optical densities. IGF-IR DN tumors expressed significantly less VEGF than pcDNA tumors (*, P < 0.001; Student’s t-test). Data are presented as means ± SEM.
Figure 9.
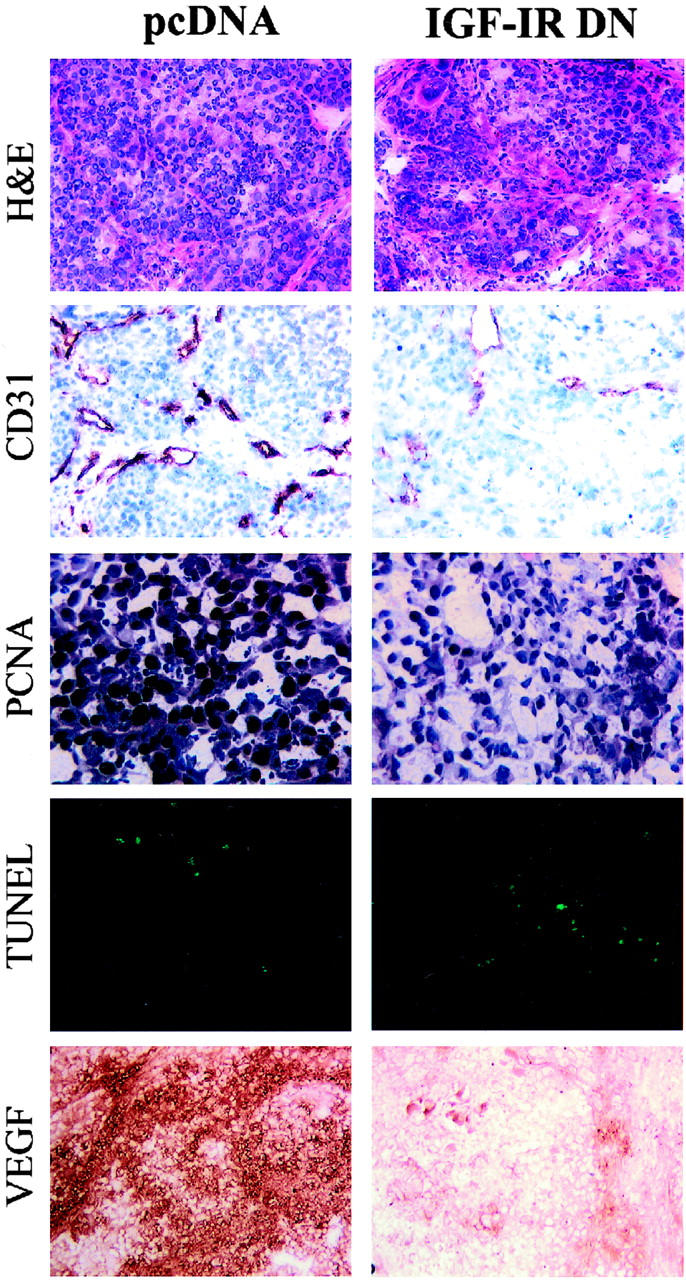
Immunohistochemical analysis of pancreatic tumors. Tumor sections were stained with H&E, anti-CD31 antibody, anti-PCNA antibody, TUNEL, and anti-VEGF antibody as described in Materials and Methods. Original magnifications: ×50 (H&E, CD31, VEGF); ×100 (PCNA, TUNEL).
Discussion
In the current study, impairing IGF-IR function by stable transfection with a dominant-negative form of IGF-IR significantly reduced constitutive VEGF expression in pancreatic tumor cells in vitro and drastically inhibited angiogenesis and orthotopic tumor growth in vivo. VEGF expression by L3.6pl cells was partially mediated via constitutive activation (nuclear translocation) of HIF-1α, which itself was regulated by an activated IGF-IR system. This mechanism could contribute to the high VEGF expression levels in the pancreatic cancer microenvironment. In contrast to IGF-IR inhibition, the blockade of Cox-2 function by treating cells with celecoxib did not reduce constitutive VEGF expression in these cancer cells, as suggested by other studies. 42
Although the IGF-I receptor/ligand system has been implicated in cell proliferation and cancer progression, 43 its role in tumor growth still remains to be elucidated. We focused here on mechanisms underlying the regulation of VEGF expression and angiogenesis mediated by the IGF-IR system in pancreatic cancer, which had not been investigated. Only a few studies have addressed the role of IGF-I in angiogenesis (ie, regulation of VEGF expression) in benign or neoplastic disorders. Smith and colleagues 25 recently demonstrated that interactions of IGF-I with IGF-IR increased retinal neovascularization by up-regulating VEGF expression in retinal endothelial cells by means of activating the MAPK signaling pathway. IGF-IR activation also seems to be necessary for VEGF to have the maximal effect in ocular angiogenesis. 25 Similar results were also described by Reinmuth and colleagues 20 in their investigations of an IGF-IR DN-transfected colon cancer cell line (HT29).
IGF-I induced regulation of VEGF expression in cancer cells in vitro has been reported for breast cancer cells, 44 endometrial adenocarcinoma cells, 45 and colorectal cancer cells. 21,22,39 However, the angiogenic effects of the IGF-IR system in malignant tumors in vivo have only recently been described for colorectal carcinomas. 19 In those studies, we showed that inhibition of IGF-IR function reduced VEGF expression in vitro in two human colon carcinoma cell lines (HT29, KM12L4), which resulted in a significant reduction of angiogenesis and hepatic colorectal tumor growth in two different in vivo models. 20 Tumors from IGF-IR DN-transfected cell lines in those studies also expressed greatly less VEGF than tumors from control (pcDNA) cells. 19,20 In light of the anti-angiogenic effects of an IGF-IR blockade in these colon cancer models, we wanted to investigate whether IGF-IR could also be a target in a more aggressive disease such as pancreatic cancer. We found that the anti-angiogenic and growth-inhibitory effects of inhibiting IGF-IR function were even more pronounced in the pancreatic cancer model than in the colon cancer model. To rule out a clonal effect for the growth inhibition of IGF-IR DN-transfected L3.6pl cells in vivo, we tested another IGF-IR DN-transfected clone in a subcutaneous xenograft model and obtained similar results, suggesting that our orthotopic findings were not because of clonal selection. We also tested whether the functionality of the epidermal growth factor receptor would be affected by impaired IGF-IR function, as has been described in other studies of the interactions between IGF-IR and ErbB2 receptor systems. 46 To do so, we stimulated IGF-IR DN- and pcDNA-transfected L3.6pl cells with recombinant epidermal growth factor in vitro; at 48 hours, MTT assays showed increased numbers of pcDNA-transfected control cells but no change in number of IGF-IR DN cells (data not shown). Targeting IGF-IR in pancreatic cancer may therefore be valuable, as it also affects the functionality of the epidermal growth factor receptor system. 47
One important factor in the regulation of cellular VEGF expression and angiogenesis is the transcription factor HIF-1α, which is frequently overexpressed or activated in various human cancers. 48-50 Fukuda and associates 39 recently demonstrated that activation of IGF-IR by IGF-I or IGF-II induced HIF-1α and VEGF expression in colon cancer cells and that this effect was mediated by both MAPK (Erk1/2) and PI-3K (Akt) signaling pathways. 39 Similarly, we found that recombinant IGF-I mediated the nuclear translocation of HIF-1α in L3.6pl pancreatic cancer cells in vitro. In contrast to parental and pcDNA cells, IGF-IR DN-transfected cells showed not only reduced IGF-IR and Erk1/2 phosphorylation levels but also a 50% reduction in constitutive nuclear HIF-1α protein, suggesting that an autocrine pathway involving IGF-IR and HIF-1α activation may contribute to high VEGF levels in these tumors. Results from recent studies investigating the role of HIF-1α in human pancreatic cancer suggest that the reduction of constitutive HIF-1α activation/expression, as in our IGF-IR DN cells, may potentially account for the pronounced growth inhibitory and anti-angiogenic effect seen in this tumor system. 51,52 We also investigated if other angiogenesis related genes such as bFGF or thrombospondin-1 would be regulated by the IGF-IR system. We did not observe increased bFGF levels or decreased thrombospondin-1 expression, respectively, on IGF-I treatment. However, it is entirely possible that IGF-I may regulate other angiogenic molecules; however, the large number of angiogenic mediators makes it difficult to characterize the complete angiogenic profile of IGF-I-stimulated cells.
Further, the activation of HIF-1α may up-regulate not only VEGF expression but also expression of the IGF-IR ligand, IGF-II. 53 This mechanism could participate in an autocrine IGF-IR activation/VEGF expression loop in human pancreatic cancer. IGF-II has been implicated in angiogenesis of other tumors as well. 54,55 In one study, IGF-II induced VEGF expression in human hepatoma cells, an action that may contribute to hepatocarcinogenesis. 54 Ritter and colleagues 55 recently used microarray technology and reverse transcriptase-polymerase chain reaction to show that IGF-II was highly expressed during the proliferative phase of human hemangiomas and that it was substantially decreased during hemangioma involution.
Autocrine loops of IGF-I receptor activation in human pancreatic cancer seem to play an important role in the regulation and maintenance of high VEGF expression levels. Targeting IGF-IR function may therefore be a promising approach for anti-angiogenic treatment of human pancreatic cancer. However, the role of Cox-2 inhibitors as anti-angiogenic agents requires further study. We found that Cox-2 inhibition did not reduce VEGF expression in tumor cells in vitro; others’ findings that Cox-2 inhibitors may inhibit angiogenesis by down-regulation of cytokine-induced VEGF expression may reflect differences in the cell lines used. 56 Interestingly, treating cells with celecoxib resulted in a compensatory increase of Cox-2 mRNA expression, suggesting that a negative feedback loop exists.
In summary, we found that IGF-IR is expressed and activated in all human pancreatic adenocarcinomas examined in this study. In an experimental model, blocking IGF-IR function inhibits angiogenesis and growth of pancreatic tumors, suggesting that the IGF-IR system is a valid molecular target for the treatment of human pancreatic cancer.
Acknowledgments
We thank Christine Wogan from the Department of Scientific Publications and Rita Hernandez from the Department of Surgical Oncology at M. D. Anderson for editorial assistance; and Donna Reynolds and Carol Oborn, both from the Department of Cancer Biology at M. D. Anderson, and Guido Sclabas, M.D., Department of Surgical Oncology, for technical assistance.
Footnotes
Address reprint requests to Lee M. Ellis, M.D., Department of Surgical Oncology, Box 444, The University of Texas M.D. Anderson Cancer Center, 1515 Holcombe Blvd., Houston, TX 77030-4009. E-mail: lellis@mdanderson.org.
Supported by the National Institutes of Health (grants T-32 09599 to A. A. P. and CA74821 and core grant CCSG CA16672 to L. M. E.); the Dr. Mildred Scheel Stiftung für Krebsforschung, Deutsche Krebshilfe (to N. R.); and the Lockton Foundation for Pancreatic Cancer Research (to L. M. E.).
References
- 1.Xie Y, Skytting B, Nilsson G, Brodin B, Larsson O: Expression of insulin-like growth factor-1 receptor in synovial sarcoma: association with an aggressive phenotype Cancer Res 1999, 59:3588-3591 [PubMed] [Google Scholar]
- 2.Hakam A, Yeatman TJ, Lu L, Mora L, Marcet G, Nicosia SV, Karl RC, Coppola D: Expression of insulin-like growth factor-1 receptor in human colorectal cancer Hum Pathol 1999, 30:1128-1133 [DOI] [PubMed] [Google Scholar]
- 3.Long L, Rubin R, Brodt P: Enhanced invasion and liver colonization by lung carcinoma cells overexpressing the type 1 insulin-like growth factor receptor Exp Cell Res 1998, 238:116-121 [DOI] [PubMed] [Google Scholar]
- 4.Bergmann U, Funatomi H, Yokoyama M, Beger HG, Korc M: Insulin-like growth factor I overexpression in human pancreatic cancer: evidence for autocrine and paracrine roles Cancer Res 1995, 55:2007-2011 [PubMed] [Google Scholar]
- 5.Basso D, Plebani M, Fogar P, Panozzo MP, Meggiato T, De Paoli M, Del Favero G: Insulin-like growth factor-I, interleukin-1 alpha and beta in pancreatic cancer: role in tumor invasiveness and associated diabetes Int J Clin Lab Res 1995, 25:40-43 [DOI] [PubMed] [Google Scholar]
- 6.Parisot JP, Hu XF, DeLuise M, Zalcberg JR: Altered expression of the IGF-1 receptor in a tamoxifen-resistant human breast cancer cell line Br J Cancer 1999, 79:693-700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burtscher I, Compagni A, Lamm GM, Christofori G: An insulin-like growth factor-mediated, phosphatidylinositol 3′ kinase-independent survival signaling pathway in beta tumor cells Cancer Res 1999, 59:3923-3926 [PubMed] [Google Scholar]
- 8.Tanno S, Mitsuuchi Y, Altomare DA, Xiao GH, Testa JR: AKT activation up-regulates insulin-like growth factor I receptor expression and promotes invasiveness of human pancreatic cancer cells Cancer Res 2001, 61:589-593 [PubMed] [Google Scholar]
- 9.Baserga R, Hongo A, Rubini M, Prisco M, Valentinis B: The IGF-I receptor in cell growth, transformation and apoptosis Biochim Biophys Acta 1997, 1332:F105-F126 [DOI] [PubMed] [Google Scholar]
- 10.Werner H, Le Roith D: The insulin-like growth factor-I receptor signaling pathways are important for tumorigenesis and inhibition of apoptosis Crit Rev Oncog 1997, 8:71-92 [DOI] [PubMed] [Google Scholar]
- 11.Liu ZZ, Wada J, Alvares K, Kumar A, Wallner EI, Kanwar YS: Distribution and relevance of insulin-like growth factor-I receptor in metanephric development Kidney Int 1993, 44:1242-1250 [DOI] [PubMed] [Google Scholar]
- 12.Steller MA, Delgado CH, Bartels CJ, Woodworth CD, Zou Z: Overexpression of the insulin-like growth factor-1 receptor and autocrine stimulation in human cervical cancer cells Cancer Res 1996, 56:1761-1765 [PubMed] [Google Scholar]
- 13.Cullen KJ, Yee D, Sly WS, Perdue J, Hampton B, Lippman ME, Rosen N: Insulin-like growth factor receptor expression and function in human breast cancer Cancer Res 1990, 50:48-53 [PubMed] [Google Scholar]
- 14.Ohmura E, Okada M, Onoda N, Kamiya Y, Murakami H, Tsushima T, Shizume K: Insulin-like growth factor I and transforming growth factor alpha as autocrine growth factors in human pancreatic cancer cell growth Cancer Res 1990, 50:103-107 [PubMed] [Google Scholar]
- 15.Freeman JW, Mattingly CA, Strodel WE: Increased tumorigenicity in the human pancreatic cell line MIA PaCa-2 is associated with an aberrant regulation of an IGF-1 autocrine loop and lack of expression of the TGF-beta type RII receptor J Cell Physiol 1995, 165:155-163 [DOI] [PubMed] [Google Scholar]
- 16.Flossmann-Kast BB, Jehle PM, Hoeflich A, Adler G, Lutz MP: Src stimulates insulin-like growth factor I (IGF-I)-dependent cell proliferation by increasing IGF-I receptor number in human pancreatic carcinoma cells Cancer Res 1998, 58:3551-3554 [PubMed] [Google Scholar]
- 17.Nair PN, De Armond DT, Adamo ML, Strodel WE, Freeman JW: Aberrant expression and activation of insulin-like growth factor-1 receptor (IGF-1R) are mediated by an induction of IGF-1R promoter activity and stabilization of IGF-1R mRNA and contributes to growth factor independence and increased survival of the pancreatic cancer cell line MIA PaCa-2 Oncogene 2001, 20:8203-8214 [DOI] [PubMed] [Google Scholar]
- 18.Murphy LO, Abdel-Wahab YH, Wang QJ, Knezetic JA, Permnert J, Larsson J, Hollingsworth AM, Adrian TE: Receptors and ligands for autocrine growth pathways are up-regulated when pancreatic cancer cells are adapted to serum-free culture Pancreas 2001, 22:293-298 [DOI] [PubMed] [Google Scholar]
- 19.Reinmuth N, Fan F, Liu W, Parikh AA, Stoeltzing O, Jung YD, Bucana CD, Radinsky R, Gallick GE, Ellis LM: Impact of insulin-like growth factor receptor-I function on angiogenesis, growth, and metastasis of colon cancer Lab Invest 2002, 82:1377-1389 [DOI] [PubMed] [Google Scholar]
- 20.Reinmuth N, Liu W, Fan F, Jung YD, Ahmad SA, Stoeltzing O, Bucana CD, Radinsky R, Ellis LM: Blockade of insulin-like growth factor I receptor function inhibits growth and angiogenesis of colon cancer Clin Cancer Res 2002, 8:3259-3269 [PubMed] [Google Scholar]
- 21.Akagi Y, Liu W, Zebrowski B, Xie K, Ellis LM: Regulation of vascular endothelial growth factor expression in human colon cancer by insulin-like growth factor-I Cancer Res 1998, 58:4008-4014 [PubMed] [Google Scholar]
- 22.Warren RS, Yuan H, Matli MR, Ferrara N, Donner DB: Induction of vascular endothelial growth factor by insulin-like growth factor 1 in colorectal carcinoma J Biol Chem 1996, 271:29483-29488 [DOI] [PubMed] [Google Scholar]
- 23.Wu Y, Yakar S, Zhao L, Hennighausen L, LeRoith D: Circulating insulin-like growth factor-I levels regulate colon cancer growth and metastasis Cancer Res 2002, 62:1030-1035 [PubMed] [Google Scholar]
- 24.Grant MB, Mames RN, Fitzgerald C, Ellis EA, Aboufriekha M, Guy J: Insulin-like growth factor I acts as an angiogenic agent in rabbit cornea and retina: comparative studies with basic fibroblast growth factor Diabetologia 1993, 36:282-291 [DOI] [PubMed] [Google Scholar]
- 25.Smith LE, Shen W, Perruzzi C, Soker S, Kinose F, Xu X, Robinson G, Driver S, Bischoff J, Zhang B, Schaeffer JM, Senger DR: Regulation of vascular endothelial growth factor-dependent retinal neovascularization by insulin-like growth factor-1 receptor Nat Med 1999, 5:1390-1395 [DOI] [PubMed] [Google Scholar]
- 26.Ellis LM, Fidler IJ: Angiogenesis and metastasis Eur J Cancer 1996, 32A:2451-2460 [DOI] [PubMed] [Google Scholar]
- 27.Folkman J: The role of angiogenesis in tumor growth Semin Cancer Biol 1992, 3:65-71 [PubMed] [Google Scholar]
- 28.Fidler IJ, Ellis LM: The implications of angiogenesis for the biology and therapy of cancer metastasis Cell 1994, 79:185-188 [DOI] [PubMed] [Google Scholar]
- 29.Tsuzuki Y, Mouta Carreira C, Bockhorn M, Xu L, Jain RK, Fukumura D: Pancreas microenvironment promotes VEGF expression and tumor growth: novel window models for pancreatic tumor angiogenesis and microcirculation Lab Invest 2001, 81:1439-1451 [DOI] [PubMed] [Google Scholar]
- 30.Bruns CJ, Shrader M, Harbison MT, Portera C, Solorzano CC, Jauch KW, Hicklin DJ, Radinsky R, Ellis LM: Effect of the vascular endothelial growth factor receptor-2 antibody DC101 plus gemcitabine on growth, metastasis and angiogenesis of human pancreatic cancer growing orthotopically in nude mice Int J Cancer 2002, 102:101-108 [DOI] [PubMed] [Google Scholar]
- 31.Solorzano CC, Baker CH, Bruns CJ, Killion JJ, Ellis LM, Wood J, Fidler IJ: Inhibition of growth and metastasis of human pancreatic cancer growing in nude mice by PTK 787/ZK222584, an inhibitor of the vascular endothelial growth factor receptor tyrosine kinases Cancer Biother Radiopharm 2001, 16:359-370 [DOI] [PubMed] [Google Scholar]
- 32.Bruns CJ, Harbison MT, Kuniyasu H, Eue I, Fidler IJ: In vivo selection and characterization of metastatic variants from human pancreatic adenocarcinoma by using orthotopic implantation in nude mice Neoplasia 1999, 1:50-62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Prager D, Yamasaki H, Weber MM, Gebremedhin S, Melmed S: Human insulin-like growth factor I receptor function in pituitary cells is suppressed by a dominant negative mutant J Clin Invest 1992, 90:2117-2122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Prager D, Li HL, Asa S, Melmed S: Dominant negative inhibition of tumorigenesis in vivo by human insulin-like growth factor I receptor mutant Proc Natl Acad Sci USA 1994, 91:2181-2185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Quinn KA, Treston AM, Unsworth EJ, Miller MJ, Vos M, Grimley C, Battey J, Mulshine JL, Cuttitta F: Insulin-like growth factor expression in human cancer cell lines J Biol Chem 1996, 271:11477-11483 [DOI] [PubMed] [Google Scholar]
- 36.Jung YD, Liu W, Reinmuth N, Ahmad SA, Fan F, Gallick GE, Ellis LM: Vascular endothelial growth factor is upregulated by interleukin-1 beta in human vascular smooth muscle cells via the P38 mitogen-activated protein kinase pathway Angiogenesis 2001, 4:155-162 [DOI] [PubMed] [Google Scholar]
- 37.Ahmad SA, Liu W, Jung YD, Fan F, Wilson M, Reinmuth N, Shaheen RM, Bucana CD, Ellis LM: The effects of angiopoietin-1 and -2 on tumor growth and angiogenesis in human colon cancer Cancer Res 2001, 61:1255-1259 [PubMed] [Google Scholar]
- 38.Jung YD, Fan F, McConkey DJ, Jean ME, Liu W, Reinmuth N, Stoeltzing O, Ahmad SA, Parikh AA, Mukaida N, Ellis LM: Role of P38 MAPK, AP-1, and NF-kappaB in interleukin-1beta-induced IL-8 expression in human vascular smooth muscle cells Cytokine 2002, 18:206-213 [DOI] [PubMed] [Google Scholar]
- 39.Fukuda R, Hirota K, Fan F, Jung YD, Ellis LM, Semenza GL: Insulin-like growth factor 1 induces hypoxia-inducible factor 1-mediated vascular endothelial growth factor expression, which is dependent on MAP kinase and phosphatidylinositol 3-kinase signaling in colon cancer cells J Biol Chem 2002, 277:38205-38211 [DOI] [PubMed] [Google Scholar]
- 40.Baker CH, Solorzano CC, Fidler IJ: Blockade of vascular endothelial growth factor receptor and epidermal growth factor receptor signaling for therapy of metastatic human pancreatic cancer Cancer Res 2002, 62:1996-2003 [PubMed] [Google Scholar]
- 41.Bruns CJ, Solorzano CC, Harbison MT, Ozawa S, Tsan R, Fan D, Abbruzzese J, Traxler P, Buchdunger E, Radinsky R, Fidler IJ: Blockade of the epidermal growth factor receptor signaling by a novel tyrosine kinase inhibitor leads to apoptosis of endothelial cells and therapy of human pancreatic carcinoma Cancer Res 2000, 60:2926-2935 [PubMed] [Google Scholar]
- 42.Fujiwaki R, Iida K, Kanasaki H, Ozaki T, Hata K, Miyazaki K: Cyclooxygenase-2 expression in endometrial cancer: correlation with microvessel count and expression of vascular endothelial growth factor and thymidine phosphorylase Hum Pathol 2002, 33:213-219 [DOI] [PubMed] [Google Scholar]
- 43.Rubin R, Baserga R: Insulin-like growth factor-I receptor. Its role in cell proliferation, apoptosis, and tumorigenicity Lab Invest 1995, 73:311-331 [PubMed] [Google Scholar]
- 44.Oh JS, Kucab JE, Bushel PR, Martin K, Bennett L, Collins J, DiAugustine RP, Barrett JC, Afshari CA, Dunn SE: Insulin-like growth factor-1 inscribes a gene expression profile for angiogenic factors and cancer progression in breast epithelial cells Neoplasia 2002, 4:204-217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bermont L, Lamielle F, Fauconnet S, Esumi H, Weisz A, Adessi GL: Regulation of vascular endothelial growth factor expression by insulin-like growth factor-I in endometrial adenocarcinoma cells Int J Cancer 2000, 85:117-123 [DOI] [PubMed] [Google Scholar]
- 46.Coppola D, Ferber A, Miura M, Sell C, D’Ambrosio C, Rubin R, Baserga R: A functional insulin-like growth factor I receptor is required for the mitogenic and transforming activities of the epidermal growth factor receptor Mol Cell Biol 1994, 14:4588-4595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bruns CJ, Harbison MT, Davis DW, Portera CA, Tsan R, McConkey DJ, Evans DB, Abbruzzese JL, Hicklin DJ, Radinsky R: Epidermal growth factor receptor blockade with C225 plus gemcitabine results in regression of human pancreatic carcinoma growing orthotopically in nude mice by antiangiogenic mechanisms Clin Cancer Res 2000, 6:1936-1948 [PubMed] [Google Scholar]
- 48.Zhong H, De Marzo AM, Laughner E, Lim M, Hilton DA, Zagzag D, Buechler P, Isaacs WB, Semenza GL, Simons JW: Overexpression of hypoxia-inducible factor 1alpha in common human cancers and their metastases Cancer Res 1999, 59:5830-5835 [PubMed] [Google Scholar]
- 49.Birner P, Gatterbauer B, Oberhuber G, Schindl M, Rossler K, Prodinger A, Budka H, Hainfellner JA: Expression of hypoxia-inducible factor-1 alpha in oligodendrogliomas: its impact on prognosis and on neoangiogenesis Cancer 2001, 92:165-171 [DOI] [PubMed] [Google Scholar]
- 50.Carmeliet P, Dor Y, Herbert JM, Fukumura D, Brusselmans K, Dewerchin M, Neeman M, Bono F, Abramovitch R, Maxwell P, Koch CJ, Ratcliffe P, Moons L, Jain RK, Collen D, Keshert E, Keshet E: Role of HIF-1alpha in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis Nature 1998, 394:485-490 [DOI] [PubMed] [Google Scholar]
- 51.Buchler P, Reber HA, Buchler M, Shrinkante S, Buchler MW, Friess H, Semenza GL, Hines OJ: Hypoxia-inducible factor 1 regulates vascular endothelial growth factor expression in human pancreatic cancer Pancreas 2003, 26:56-64 [DOI] [PubMed] [Google Scholar]
- 52.Akakura N, Kobayashi M, Horiuchi I, Suzuki A, Wang J, Chen J, Niizeki H, Kawamura K, Hosokawa M, Asaka M: Constitutive expression of hypoxia-inducible factor-1alpha renders pancreatic cancer cells resistant to apoptosis induced by hypoxia and nutrient deprivation Cancer Res 2001, 61:6548-6554 [PubMed] [Google Scholar]
- 53.Feldser D, Agani F, Iyer NV, Pak B, Ferreira G, Semenza GL: Reciprocal positive regulation of hypoxia-inducible factor 1alpha and insulin-like growth factor 2 Cancer Res 1999, 59:3915-3918 [PubMed] [Google Scholar]
- 54.Kim KW, Bae SK, Lee OH, Bae MH, Lee MJ, Park BC: Insulin-like growth factor II induced by hypoxia may contribute to angiogenesis of human hepatocellular carcinoma Cancer Res 1998, 58:348-351 [PubMed] [Google Scholar]
- 55.Ritter MR, Dorrell MI, Edmonds J, Friedlander SF, Friedlander M: Insulin-like growth factor 2 and potential regulators of hemangioma growth and involution identified by large-scale expression analysis Proc Natl Acad Sci USA 2002, 99:7455-7460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gallo O, Franchi A, Magnelli L, Sardi I, Vannacci A, Boddi V, Chiarugi V, Masini E: Cyclooxygenase-2 pathway correlates with VEGF expression in head and neck cancer. Implications for tumor angiogenesis and metastasis Neoplasia 2001, 3:53-61 [DOI] [PMC free article] [PubMed] [Google Scholar]