

Abstract
Recessively inherited mutations in the base excision repair gene MYH have recently been associated with predisposition to colorectal adenomas and cancer in materials selected for occurrence of multiple adenomas. In particular, variants Y165C and G382D have been shown to play a role in Caucasian patients. To evaluate the contribution of MYH mutations to colorectal cancer burden on the population level, and to examine the MYH-associated phenotype in an unselected series of colorectal cancer patients, we determined the frequencies of Y165C and G382D MYH mutations in a population-based series of 1042 Finnish colorectal cancer patients. Four (0.4%) patients had both MYH alleles mutated. Although all these patients had multiple adenomatous polyps, the phenotypes tended to be less extreme than in previous studies on selected cases. The lowest number of colorectal adenomas at the time of cancer diagnosis was five. Cases with one mutant MYH allele were subjected to sequencing of all exons to detect possible Finnish founder mutations, but no additional changes were detected. The Y165C and G382D variants were not present in 424 Finnish cancer-free controls showing that MYH mutations are not enriched in the population. As evaluated against national Finnish Polyposis Registry data MYH-associated colorectal cancer appears to be as common as colorectal cancer associated with familial adenomatous polyposis.
Up to 35% of all colorectal cancer (CRC) cases are because of genetic predisposition. 1-3 Some conditions are associated with a particularly high CRC risk, and these are typically autosomal-dominant traits, often with well-characterized molecular background. Hereditary nonpolyposis colorectal cancer is caused by mutations in the DNA mismatch repair genes, 4-9 and appears to cause up to 3% of the total CRC burden. The most common polyposis syndrome familial adenomatous poly-posis (FAP) is mainly caused by mutations in the adenomatous polyposis coli (APC) gene. 10,11 No more than 1% of CRCs is caused by FAP: 12,13 in populations with an efficient screening program this proportion is less than 0.5%. For example in Finland in the late 1980s the proportion was 0.14%. 13 In the very rare juvenile polyposis and Peutz-Jeghers syndrome, hamartomatous polyps develop in the gastrointestinal tract, mostly in the small intestine and colorectum. The genes underlying juvenile polyposis and Peutz-Jeghers syndrome are SMAD4 and ALK3, 14,15 and LKB1, 16 respectively.
Recently susceptibility to multiple adenomatous polyps and carcinoma of the colon has been found as an autosomal-recessive trait. The base excision repair gene human homolog of mutY (MYH) has been proposed as a new CRC predisposing gene. 17 In base excision repair an adenine-specific DNA glycosylase MYH removes adenines mispaired with guanines or damaged DNA base 8-oxo-dG. 18,19 Human homolog of mutM (OGG1) and human homolog of mutT (MTH1) remove the oxidized base from 8-oxo-G:C bp and prevent the incorporation of 8-oxo-dGMP into DNA. 20,21
Al-Tassan and colleagues 17 linked a somatic APC mutation pattern biased toward G to A transversions in adenomas derived from three siblings with multiple lesions to a base excision repair defect caused by compound germline heterozygosity for Y165C and G382D MYH variants. This elegant work, including functional analysis of the variants in a bacterial homologue, showed that biallelic inactivation of MYH predisposes to colorectal neoplasia. Al-Tassan and colleagues 17 found both Y165C and G382D once in 100 British controls demonstrating that heterozygosity on population level is not rare. Jones and colleagues 22 and Sieber and colleagues 23 studied patients with multiple colorectal adenomas and reported several additional patients with MYH germline mutations. Importantly, some patients had displayed more than 100 colorectal adenomas fulfilling the classical clinical criteria of FAP. Sieber and colleagues 23 showed that 9% of 157 multiple adenoma patients had germline MYH mutations. Five percent had double mutations. Twenty-nine percent of 28 patients with 15 to 100 adenomas were double-mutation carriers and the CRC frequency differed significantly from the general population.
Earlier studies assessing contribution of MYH to colorectal neoplasia susceptibility have been performed in series of patients with multiple colorectal adenomas. MYH mutation frequencies have not, to our knowledge, yet been determined in unselected and population-based series of CRC patients. We examined the contribution of MYH in such a series of 1042 patients, collected from Finland May 1994 to July 1998. 24,25 We screened germline missense variants Y165C and G382D of the MYH gene by solid-phase minisequencing, 26,27 to detect cases associated with biallelic loss of MYH function. DNA samples with only one mutant allele were subjected to sequencing of the whole gene, to detect possible Finnish founder mutations. DNAs from 424 anonymous blood donors were used as controls. The occurrence of MYH-associated CRC was compared with the occurrence of FAP-associated CRC in the studied population in the study period, using data from the Finnish Polyposis Registry.
Materials and Methods
DNA
Normal and tumor DNA samples were collected at nine Finnish regional central hospitals between May 1994 and July 1998. Samples were derived from 1042 patients diagnosed with CRC. This is approximately two-thirds of the total number of patients treated in the units within the time frame. One third was missed but no bias toward any particular phenotype such as young age could be observed, and this series of 1042 cases is expected to represent well the examined population. 24,25 BAT26 mononucleotide marker was used to determine the MSI status of the tumors. MSI cases were sequenced for MLH1 and MSH2 germline mutations, and 29 of 130 MSI patients had an MLH1 or MSH2 defect (unpublished data). 24,25
Eighteen DNA samples from the series of 1042, representing a sampling of patients with three or more polyps, were earlier screened for MYH mutations by Sieber and colleagues. 23 One MYH double-mutation carrier, C585, was detected in this selected group of samples. 23 For the purpose of this study, all samples with sufficient DNA amounts, 1003 samples, entered the analysis. In addition to the CRC patient DNAs, DNAs from 424 anonymous cancer-free blood donors were used to examine occurrence of Y165C and G382D in the Finnish population.
DNA Pooling Strategy, Polymerase Chain Reaction (PCR), and Solid-Phase Minisequencing
DNA samples (n = 16 to 22) were mixed in one pool. Fifty-three master pools representing 1003 DNA samples were amplified by PCR for screening known mutations in exons 7 and 13. PCR reactions were performed in 50-μl reaction volume containing 125 ng of pooled genomic DNA, 10× PCR buffer (Applied Biosystems, Branchburg, NJ), 500 μmol/L of each dNTP (Finnzymes, Espoo, Finland), 10 pmol of biotin-labeled forward primer, and 50 pmol of nonbiotinylated reverse primer (Table 1) ▶ , MgCl2 in 5 mmol/L (for exon 7 PCR) or in 3.5 mmol/L (for exon 13) concentrations, 5 U of AmpliTaqGOLD polymerase (AB). The following PCR cycles were used for amplification: 95°C for 10 minutes, 35 cycles of 95°C for 45 seconds denaturation, annealing temperature of 60°/58°C (exons 7 and 13) for 45 seconds, and 72°C for 30 seconds extension. Final extension was 72°C for 10 minutes.
Table 1.
PCR and Detection Primers for Screening Mutations in Exons 7 and 13 of MYH Gene
| Primer sequences | |
|---|---|
| 7Fb* | CGG GTG ATC TCT TTG ACC TC |
| 7R | GTT CCT ACC CTC CTG CCA TC |
| 7d† | GCC GCC GGC CAC GAG AAT AG |
| 13Fb* | AGG GAA TCG GCA GCT GAG |
| 13R | GGC TAT TCC GCT GCT CAC TT |
| 13d† | CCC ACA GTC CTG CCA GCA GA |
*Biotin-labeled primers.
†Detection primers.
Solid-phase minisequencing was performed as previously described. 26,27 Detection primers were designed to hybridize just adjacent to the nucleotide to be analyzed (Table 1) ▶ . If mutant alleles were detected, the PCR and solid-phase minisequencing were repeated. Double-positive master pools were split into minor pools, each containing 10 DNA samples. DNA samples from positive minor pools were amplified individually and their exact alleles were determined by solid-phase minisequencing. Pools with known positive and negative samples were included in the analyses as controls.
Patients with only one mutated allele in the germline were sequenced for all 16 exons from both normal and tumor DNA samples, to search for additional MYH defects. Primer sequences were obtained from Al-Tassan and colleagues 17 Big Dye 3 Terminator chemistry (AB) was used for direct sequencing. Cycle-sequencing products were run on ABI 3100 capillary sequencer (AB) according to the manufacturer’s instructions.
Somatic APC Mutation Analysis in MYH-Associated CRCs
Tumors from MYH mutation carriers were examined for mutations in the APC gene by fluorescent single-stranded nucleotide polymorphism analysis and by denaturing high performance liquid chromatography analysis. Exons 15A to 15L of APC were screened, which encompasses the mutation cluster region, codons 1286 to 1513. The majority of somatic APC mutations in MYH carriers have been found within this region. 22 For fluorescent single-stranded nucleotide polymorphism analysis, tumor DNA was isolated from fresh-frozen samples using standard methods. Primer pairs to amplify overlapping fragments of APC from exon 15A to 15L inclusive were used for the coding regions including exon-intron boundaries (sequences from the authors available on request).
Each 25-μl PCR reaction contained 1× PCR reaction buffer without MgCl2 (Promega, Madison, WI), 1.5mmol/L MgCl2, 200 μmol/L dNTPs, 200 nmol/L of each primers, 50 ng of genomic DNA, and 1 U of TaqDNA polymerase (Qiagen), and the PCR conditions consisted of 95°C for 5 minutes, followed by 35 cycles of 95°C for 1 minute, 55°C or 60°C for 1 minute, 72°C for 1 minute, and a final extension step at 72°C for 10 minutes. The resulting PCR products were screened for variants by being run at 18°C and 24°C on the ABI 3100 and analyzed using Genotyper 2.5 software (Perkin-Elmer Applied Biosystems). Fragments showing aberrant migration were reamplified, purified using Qiaquick columns (Qiagen), and then sequenced in both forward and reverse orientations using the ABI Big Dye Terminator kit (PE AB) in parallel with control samples.
Denaturing high performance liquid chromatography was done using the 3500HT WAVE nucleic acid fragment analysis system (Transgenomic, Crewe, UK). The same primer set was used as for fluorescent single stranded-nucleotide polymorphism. To enhance the formation of heteroduplexes before analysis, the PCR products were denatured at 94°C and reannealed by cooling to 50°C at a rate of 1°C per minute. Denaturing high performance liquid chromatography was performed at the melting temperature predicted by Wavemaker (version 4.0) software (Transgenomic) with a 12% acetonitrile gradient throughout 2.5 minutes. Samples displaying aberrant denaturing high performance liquid chromatography elution profiles were sequenced directly.
Patient Records
Patient records of MYH mutation carriers were studied for polyp number, sex, age at the time of carcinoma diagnosis, the degree of tumor differentiation, histology of tumor, Dukes stage, and tumor site (Table 2) ▶ . Family history of cancer in first-degree relatives had been examined through population and Cancer Registry 24,25 for all of the 1042 cases.
Table 2.
Clinical Features of CRC Patients with Germline MYH Mutations
| Patient | MYH mutation Y165D | MYH mutation G382D | Gender | Age at diagnosis | MSI status | Polyp number | Histology/grade/location/Dukes | Extracolonic features | Family history |
|---|---|---|---|---|---|---|---|---|---|
| C585 | X | X | M | 52 | − | ∼50–100 adenomas | Ac*/II/rectum/A | — | Father; cancer with unknown primary location (58 years) |
| Mother; melanoma (60 years) | |||||||||
| C697 | XX | F | 66 | − | 5 adenomas | Ac/II/rectosigma/D | Melanoma, 55 years | Mother; gastric cancer (59 years) | |
| Sister; CRC (61 years) | |||||||||
| C761 | X | X | F | 40 | − | Several | Ac/III/rectum/D | — | Mother; basal cell cancer (41 years) |
| Sister; astrocytoma (17 years) | |||||||||
| C965 | X | X | M | 55 | − | ∼80 adenomas | Ac/II/caecum/D | — | Mother; breast cancer (61 years) |
| C544 | X | M | 72 | − | > 6 adenomas | Ac/I/sigma/D | — | Brother 1; lung cancer (50 years) | |
| Brother 2; basal cell cancer (48 years), prostate cancer (70 years) | |||||||||
| Sister; basal cell cancer (75 years) | |||||||||
| C560 | X | F | 82 | + | — | Ac/II/caecum/B | — | Mother; gastric cancer (58 years) | |
| Sister; cerebral tumor (age unknown) | |||||||||
| Daughter; pancreatic cancer (age unknown) | |||||||||
| Son 1; renal cancer (50 years) | |||||||||
| Son 2; renal cancer (50 years) | |||||||||
| C595 | X | F | 58 | − | — | Ac/II/rectum/C | — | Mother; thyroid gland cancer (65 years) | |
| C691 | X | F | 77 | + | — | Acpm†/II/caecum/C | — | None | |
| C936 | X | F | 46 | − | 1 adenoma | Ac/II/rectum/B | Breast cancer, 44 years | Brother; urinary bladder cancer (34 years) |
*Ac adenocarcinoma.
†Acpm; adenocarcinoma, partially mucinous.
Biallelic MYH mutation patients in bold.
Statistical Methods
Chi-square test statistic was used for examining MYH mutation distribution between colorectal carcinoma patients and cancer-free controls. P < 0.05 was considered statistically significant.
Results and Discussion
Four patients (0.4%) with biallelic MYH mutations were detected among the 1042 CRC cases, including C585 previously reported in Sieber and colleagues. 23 Three patients were Y165C/G382C compound heterozygotes (Figure 1) ▶ . One patient was homozygous for the G382D mutation. The clinical features of the patients are depicted in Table 2 ▶ . Although the age at CRC diagnosis and number of polyps observed in the bowel appeared to give valuable clues as to the diagnosis of MYH-associated CRC, it became also clear that the phenotype could be quite subtle.
Figure 1.
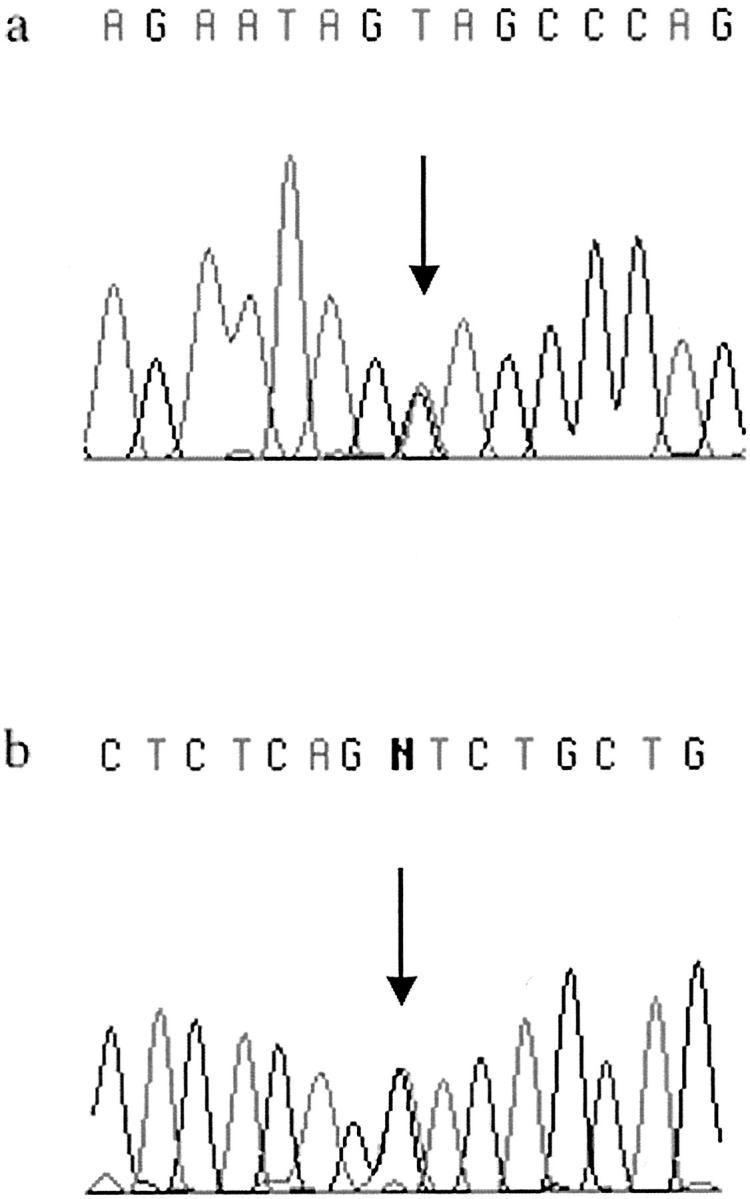
MYH germline mutations found in Finnish CRC cases. The missense changes Y165C in exon 7 (a) and G382D in exon 13 (b) are indicated by arrows.
Five patients were detected with only one mutation in either Y165C or G382D. No additional MYH defects were found in these individuals either in germline or cancer DNA (the latter analysis including loss of heterozygosity analysis in addition to sequencing), suggesting that MYH defect has had no significant role in the development of these lesions. Compatible with this notion, the patients had no specific clinical features; one patient had one polyp and one patient had more than five polyps (Table 2) ▶ .
Little is known about cancer in families segregating MYH defects. Table 2 ▶ lists all first-degree relatives of the nine CRC probands with biallelic or heterozygous MYH mutation, as well as possible other cancers diagnosed in the CRC proband. Although the data are compatible with the notion that no obvious excess of a particular type of malignancy is associated with heterozygous MYH defects, this aspect should be studied in a larger material because eight of nine probands had one to five first-degree relatives with cancer. Some of the cancers have occurred at relatively early age. Unfortunately, determination of the MYH genotype was possible only in the CRC probands.
In the series of CRC patient samples analyzed the allele frequency of Y165C was 0.002 (4 of 2006) and allele frequency of G382D was 0.004 (9 of 2006). Allele frequency of any of these two MYH mutations was 0.006 (13 of 2006). In a British control sample series of 100 DNAs both MYH mutant alleles Y165C and G382D were found once. 17 In the Finnish population examining a set of 424 blood donor samples did not reveal any mutations, and the difference between CRC patients and controls reached significance (P < 0.025). Furthermore, none of the five heterozygous Y165C and G382D carriers displayed another MYH defect elsewhere in the gene. Thus it appears that, similar to APC defects, 28 MYH mutations are not particularly enriched in the Finnish population. The low frequency of MYH mutations in blood donors supports the view that at present large-scale screening for biallelic MYH mutations is not justified. 29
The CRCs from the four patients with biallelic MYH germline mutations and five heterozygous carriers were analyzed for mutations in the APC mutation cluster region. One mutation, E1378X (G>T), was found in the MYH heterozygote C544 (Table 2) ▶ . The analysis was initially performed by single-stranded nucleotide polymorphism, and after the primarily negative results confirmed by denaturing high performance liquid chromatography. Because of the small number of cases analyzed little can be concluded from this effort.
Table 3 ▶ depicts the spectrum of predisposing gene defects observed in our population-based series of 1042 CRC cases in this and previous studies (unpublished). 24,25,28,30 At present altogether 35 patients (3.4%) have a molecular diagnosis of a hereditary CRC syndrome and this number is likely to increase in the future. According to the Finnish Polyposis Registry between May 1994 and July 1998 four FAP-associated CRCs were diagnosed in the target population. Table 3 ▶ depicts only one patient with molecular diagnosis of APC mutation. 28 One case was missed, in one case the tumor sample contained adenoma tissue only and the case was excluded from the series, and in one case a predisposing APC mutation has not been searched for. The comparison between occurrence of FAP- and MYH-associated disease is of interest. Four CRCs patients with biallelic MYH mutations in this population-based series represent an absolute minimum. That at the same time the national Polyposis registry recorded four cases of FAP-associated CRCs suggests that the contributions of APC and MYH germline variants are relatively equal, in a population with active clinical screening of FAP families. Thus the role of MYH as a cancer predisposition gene is not limited to extremely rare cases.
Table 3.
Number of HNPCC, FAP, JP, and Recessive Polyposis Cases with Molecular Diagnosis, in a Population-Based Series of 1042 Finnish CRC Patients
| Syndrome | Gene | Number of cases | % |
|---|---|---|---|
| HNPCC | MLH1 | 27 | 2.6 |
| HNPCC | MSH2 | 2 | 0.2 |
| FAP | APC | 1 | 0.1 |
| JP | ALK3 | 1 | 0.1 |
| Recessive polyposis | MYH | 4 | 0.4 |
| Total | 35 | 3.4% |
Identification of carriers of biallelic MYH mutations will be challenging as the number of polyps is far less than typically seen in FAP. Most MYH-associated CRC patients so far reported have displayed more than 100 adenomatous polyps. 22,23 In our unselected series the polyposis phenotype appears somewhat less aggressive (Table 2) ▶ . Two cases had the phenotype resembling attenuated FAP (50 to 100 adenomas). However, two others did not display polyposis. C761 was described to have a few or several polyps, whereas C697 had only five adenomas, in addition to CRC. From the clinical point of view the presence of tens of adenomas in the large bowel makes the choice of subtotal colectomy instead of segmental resection or hemicolectomy straight forward as recommended for classical FAP or even hereditary nonpolyposis colorectal cancer. 31 In the case of only few adenomas besides the carcinoma, the choice is more difficult, especially because family history is not a very powerful tool to detect MYH-associated cancer, and the molecular genetic diagnosis is available typically after the surgical treatment, if ever. In our opinion, undertaking subtotal colectomy is supported on light grounds in young CRC patients who have few additional adenomas or family history of colorectal neoplasia, or even without these factors because of the much easier follow-up with flexible sigmoidoscopy. This way undiagnosed MYH-associated CRC and hereditary nonpolyposis colorectal cancer patients will be protected from the increased risk of metachronous cancer. However, it is clear that more experience should be derived for a more profound understanding of the natural history of MYH-associated colorectal neoplasia. Number and histology of polyps are the cornerstones of detection of many CRC predisposition syndromes. Appropriate documentation of both is a prerequisite to accurate diagnosis and treatment of these conditions, and will also facilitate research on natural history of inherited forms of CRC.
Acknowledgments
We thank Sini Marttinen, Tuula Lehtinen, Laura Sonninen, and Kirsi Pylvänäinen for technical assistance.
Footnotes
Address reprint requests to Lauri A. Aaltonen, Department of Medical Genetics, Haartmaninkatu 3, Helsinki, FIN-00014, Finland. E-mail: lauri.aaltonen@helsinki.fi.
Supported by the European Commission (QLG2-CT-2001-01861), the Helsinki University Central Hospital, Biocentrum Helsinki, the Sigrid Juselius Foundation, the Paulo Foundation, the Finnish Cancer Society, and the Academy of Finland (44870, Finnish Center of Excellence Program 2000–2005).
References
- 1.Cannon-Albright LA, Skolnick MH, Bishop DT, Lee RG, Burt RW: Common inheritance of susceptibility to colonic adenomatous polyps and associated colorectal cancers. N Engl J Med 1988, 319:533-537 [DOI] [PubMed] [Google Scholar]
- 2.Houlston RS, Collins A, Slack J, Norton NE: Dominant genes for colorectal cancer are not rare. Ann Hum Genet 1992, 56:99-103 [DOI] [PubMed] [Google Scholar]
- 3.Lichtenstein P, Holm NV, Verkasalo PK, Iliadou A, Kaprio J, Koskenvuo M, Pukkala E, Skytthe A, Hemminki K: Environmental and heritable factors in the causation of cancer—analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med 2000, 343:78-85 [DOI] [PubMed] [Google Scholar]
- 4.Leach F, Nicolaides N, Papadopoulos N, Liu B, Jen J, Parsons R, Peltomäki P, Sistonen P, Aaltonen L, Nyström-Lahti M, Guan X-Y, Zhang J, Meltzer PS, Yu J-W, Kao F-T, Chen DJ, Cerosaletti KM, Fournier REK, Todd S, Lewis T, Leach RJ, Naylor SL, Weissenbach J, Mecklin J-P, Järvinen H, Petersen GM, Hamilton SR, Green J, Jass J, Watson P, Lynch HT, Trent JM, de la Chapelle A, Kinzler KW, Vogelstein B: Mutations of a mutS homolog in hereditary nonpolyposis colorectal cancer. Cell 1993, 75:1215-1225 [DOI] [PubMed] [Google Scholar]
- 5.Fishel R, Lescoe M, Rao M, Copeland N, Jenkins N, Garber J, Kane M, Kolodner R: The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell 1993, 75:1027-1038 [DOI] [PubMed] [Google Scholar]
- 6.Bronner C, Baker S, Morrison P, Warren G, Smith L, Lescoe M, Kane M, Earabino C, Lipford J, Lindblom A, Tannengård P, Bollag R, Godwin A, Ward D, Nordenskjöld M, Fishel R, Kolodner R, Liskay R: Mutation in the DNA mismatch repair gene homologue hMLH1 is associated with hereditary non-polyposis colon cancer. Nature 1994, 368:258-261 [DOI] [PubMed] [Google Scholar]
- 7.Papadopoulos N, Nicolaides N, Wei Y, Ruben S, Carter K, Rosen C, Haseltine W, Fleischmann R, Fraser C, Adams M, Venter J, Hamilton S, Petersen G, Watson P, Lynch H, Peltomäki P, Mecklin J-P, de la Chapelle A, Kinzler K, Vogelstein B: Mutation of mutL homolog in hereditary colon cancer. Science 1994, 263:1625-1629 [DOI] [PubMed] [Google Scholar]
- 8.Nicolaides N, Papadopoulos N, Liu B, Wei Y, Carter K, Ruben S, Rosen C, Haseltine W, Fleischmann R, Fraser C, Adams M, Venter J, Dunlop M, Hamilton S, Petersen G, de la Chapelle A, Vogelstein B, Kinzler K: Mutations of two PMS homologues in hereditary nonpolyposis colon cancer. Nature 1994, 371:75-80 [DOI] [PubMed] [Google Scholar]
- 9.Miyaki M, Konishi M, Tanaka K, Kikuchi-Yanoshita R, Muraoka M, Yasuno M, Igari T, Koike M, Chiba M, Mori T: Germline mutation of MSH6 as the cause of hereditary nonpolyposis colorectal cancer. Nat Genet 1997, 17:271-272 [DOI] [PubMed] [Google Scholar]
- 10.Groden J, Thiliveris A, Samowitz W, Carlson M, Gelbert L, Albertsen H, Joslyn G, Stevens J, Spirio L, Robertson M, Sargeant L, Krapcho K, Wolff E, Burt R, Hughes JP, Warrington J, McPherson J, Wasmuth J, LePaslier D, Abderrahim H, Cohen D, Leppert M, White R: Identification and characterization of the familial adenomatous polyposis coli gene. Cell 1991, 66:589-600 [DOI] [PubMed] [Google Scholar]
- 11.Nishisho I, Nakamura Y, Miyoshi Y, Miki Y, Ando H, Koyama K, Utsunomiya J, Baba S, Hedge P, Markham A, Krush AJ, Peterson G, Hamilton SR, Nilbert MC, Levy DB, Bryan TM, Preisinger AC, Smith KJ, Su LK, Kinzler KW, Vogelstein B: Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients. Science 1991, 253:665-669 [DOI] [PubMed] [Google Scholar]
- 12.Mulvihill JJ: The frequency of hereditary large bowel cancer. Ingall JR Mastromarino AJ eds. Prevention of Hereditary Large Bowel Cancer. 1983:pp 61-75 Alan R Liss New York [PubMed]
- 13.Järvinen HJ: Epidemiology of familial adenomatous polyposis in Finland: impact of family screening on the colorectal cancer rate and survival. Gut 1992, 33:357-360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Howe JR, Roth S, Ringold JC, Summers RW, Järvinen HJ, Sistonen P, Tomlinson IPM, Houlston RS, Bevan S, Mitros FA, Stone EM, Aaltonen LA: Mutations in the SMAD4/DPC4 gene in juvenile polyposis. Science 1998, 289:1086-1088 [DOI] [PubMed] [Google Scholar]
- 15.Howe JR, Bair JL, Sayed MG, Anderson ME, Mitros FA, Petersen GM, Velculescu VE, Traverso G, Vogelstein B: Germline mutations of the gene encoding bone morphogenetic protein receptor 1A in juvenile polyposis. Nat Genet 2001, 28:184-187 [DOI] [PubMed] [Google Scholar]
- 16.Hemminki A, Markie D, Tomlinson I, Avizienyte E, Roth S, Loukola A, Bignell G, Warren W, Aminoff M, Höglund P, Järvinen H, Kristo P, Pelin K, Ridanpää M, Salovaara R, Toro T, Bodmer W, Olschwang S, Olsen AS, Stratton MR, de la Chapelle A, Aaltonen LA: A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature 1998, 391:184-187 [DOI] [PubMed] [Google Scholar]
- 17.Al-Tassan N, Chmiel NH, Maynard J, Fleming N, Livingston AL, Williams GT, Hodges AK, Davies DR, David SS, Sampson JR, Cheadle JP: Inherited variants of MYH associated with somatic G: C→T:A mutations in colorectal tumors. Nat Genet 2002, 30:227-232 [DOI] [PubMed] [Google Scholar]
- 18.Slupska MM, Baikalov C, Luther WM, Chiang JH, Wei YF, Miller JH: Cloning and sequencing a human homolog (hMYH) of the Escherichia coli mutY gene whose function is required for the repair of oxidative DNA damage. J Bacteriol 1996, 178:3885-3892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Slupska MM, Luther WM, Chiang JH, Yang H, Miller JH: Functional expression of hMYH, a human homolog of the Escherichia coli MutY protein. J Bacteriol 1999, 181:6210-6213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roldan-Arjona T, Wei YF, Carter KC, Klungland A, Anselmino C, Wang RP, Augustus M, Lindahl T: Molecular cloning and functional expression of a human cDNA encoding the antimutator enzyme 8-hydroxygianine-DNA glycosylase. Proc Natl Acad Sci USA 1997, 94:8016-8020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sakumi K, Furuichi M, Tsuzuki T, Kakuma T, Kawabata S, Maki H, Sekiguchi M: Cloning and expression of cDNA for a human enzyme that hydrolyzes 8-oxo-dGTP, a mutagenic substrate for DNA-synthesis. J Biol Chem 1993, 268:23524-23530 [PubMed] [Google Scholar]
- 22.Jones S, Emmerson P, Maynard J, Best J, Jordan S, Williams G, Sampson J, Cheadle J: Biallelic germline mutations in MYH predispose to multiple colorectal adenoma and somatic G: C→T:A mutations. Hum Mol Genet 2002, 11:2961-2967 [DOI] [PubMed] [Google Scholar]
- 23.Sieber OM, Lipton L, Crabtree M, Heinimann K, Fidalgo P, Phillips RKS, Bisgaard M-L, Orntoft TF, Aaltonen LA, Hodgson SV, Huw DM, Thomas JW, Tomlinson PM: The multiple colorectal adenoma phenotype, familial adenomatous polyposis and germline mutations in. MYH. N Engl J Med 2003, 348:791-799 [DOI] [PubMed] [Google Scholar]
- 24.Aaltonen LA, Salovaara R, Kristo P, Canzian F, Hemminki A, Peltomäki P, Chadwick RB, Percesepe A, Kääriäinen H, Ahtola H, Eskelinen M, Härkönen N, Julkunen R, Kangas E, Ojala S, Tulikoura J, Valkamo E, Järvinen H, Mecklin J-P, de la Chapelle A: Incidence of hereditary nonpolyposis colorectal cancer, and molecular screening for the disease. N Engl J Med 1998, 338:1481-1487 [DOI] [PubMed] [Google Scholar]
- 25.Salovaara R, Loukola A, Kristo P, Kääriäinen H, Ahtola H, Eskelinen M, Härkönen N, Julkunen R, Kangas E, Ojala S, Tulikoura J, Valkamo E, Järvinen H, Mecklin J-P, Aaltonen LA, de la Chapelle A: Population-wide molecular detection of hereditary nonpolyposis colorectal cancer. J Clin Oncol 2000, 18:2193-2200 [DOI] [PubMed] [Google Scholar]
- 26.Syvänen A-C, Sajantila A, Lukka M: Identification of individuals by analysis of biallelic DNA markers, using PCR and solid-phase minisequencing. Am J Hum Genet 1993, 42:46-59 [PMC free article] [PubMed] [Google Scholar]
- 27.Suomalainen A, Syvänen A-C: Quantitative analysis of human DNA sequences by PCR and solid-phase minisequencing. Mol Biotechnol 2000, 15:123-131 [DOI] [PubMed] [Google Scholar]
- 28.Moisio A-L, Järvinen H, Peltomäki P: Genetic and clinical characterization of familial adenomatous polyposis: a population based study. Gut 2000, 50:845-850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marra G, Jiricny J: Multiple colorectal adenomas—is their number up? N Engl J Med 2003, 348:845-847 [DOI] [PubMed] [Google Scholar]
- 30.Zhou X-P, Woodford-Richens K, Lehtonen R, Kurose K, Aldred M, Hampel H, Launonen V, Virta S, Pilarski R, Salovaara R, Bodmer WF, Conrad BA, Dunlop M, Hodgson SV, Iwama T, Järvinen H, Kellokumpu I, Kim JC, Leggett B, Markie D, Mecklin J-P, Neale K, Phillips R, Piris J, Rozen P, Houlston RS, Aaltonen LA, Tomlinson IPM, Eng C: Germline mutation in BMPR1A/ALK3 cause a subset of cases of juvenile polyposis syndrome and of Cowden and Bannayan-Riley-Ruvalcaba syndromes. Am J Hum Genet 2001, 69:704-711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mecklin J-P, Järvinen HJ: Treatment and follow-up strategies in hereditary nonpolyposis colorectal carcinoma. Dis Colon Rectum 1993, 36:927-929 [DOI] [PubMed] [Google Scholar]