

Abstract
Tumor necrosis factor-α (TNF-α) production is a critical factor in the pathogenesis of alcoholic liver injury. Both oxidative stress and endotoxin have been implicated in the process of alcohol-induced TNF-α production. However, a cause-and-effect relationship between these factors has not been fully defined. The present study was undertaken to determine the mediators of acute alcohol-induced TNF-α production using a mouse model of acute alcohol hepatotoxicity. Alcohol administration via gavage at a dose of 6 g/kg to 129/Sv mice induced hepatic TNF-α production in Kupffer cells as demonstrated by measuring protein levels, immunohistochemical localization, and mRNA expression. Alcohol intoxication caused liver injury in association with increases in plasma endotoxin and hepatic lipid peroxidation. Treatment with an endotoxin neutralizing protein significantly suppressed alcohol-induced elevation of plasma endotoxin, hepatic lipid peroxidation, and inhibited TNF-α production. Treatment with antioxidants, N-acetyl-l-cysteine, or dimethylsulfoxide, failed to attenuate plasma endotoxin elevation, but significantly inhibited alcohol-induced hepatic lipid peroxidation, TNF-α production and steatosis. All treatments prevented alcohol-induced necrotic cell death in the liver. This study thus systemically dissected the relationship among plasma endotoxin elevation, hepatic oxidative stress, and TNF-α production following acute alcohol administration, and the results demonstrate that oxidative stress mediates endotoxin-induced hepatic TNF-α production in acute alcohol intoxication.
Alcohol consumption is a leading cause of illness and death from liver disease in the United States. 1 Although some agents have been evaluated for the prevention and treatment of alcoholic liver disease in experimental models or clinic trials, 2 there are currently no FDA-approved therapies available. Investigations on the pathogenesis of alcohol-induced cell injury would likely provide a basis for developing novel therapies. A number of hypotheses regarding the mechanisms by which alcohol causes cell injury have been suggested, with oxidative stress and proinflammatory cytokine production being leading putative etiological factors. 3-5
Three major pathways for alcohol metabolism exist in the liver, each located in a different subcellular compartment: alcohol dehydrogenase in the cytosol, microsomal ethanol oxidizing system in the endoplasmic reticulum, and aldehyde oxidase in the mitochondria. 6,7 All three result in the generation of reactive oxygen species (ROS), including superoxide, hydroxyl radical, and hydrogen peroxide. When the cellular antioxidant capacity is insufficient to cope with ROS accumulation, oxidative stress occurs in the liver. 3,4 Alcohol-induced hepatic oxidative stress has been repeatedly demonstrated by detecting ROS 8,9 or by measuring lipid peroxidation, 10-12 a marker for oxidative stress in both alcoholic patients and animal models. The accumulation of ROS in the liver was found to cause dysfunction of cellular membrane systems, protein and DNA oxidation, and eventually hepatocyte injury. 13-15
Proinflammatory cytokines such as TNF-α play a critical role in the initiation and development of alcoholic hepatitis. 5 Kupffer cells are the main source of TNF-α in the liver after alcohol exposure. It has been suggested that alcohol-induced hepatic TNF-α production is mediated by endotoxin (lipopolysaccharide, LPS). 5,16,17 Simultaneous increases in the plasma endotoxin level and hepatic TNF-α expression were repeatedly reported in alcohol-fed rats. 18-20 Mechanistic studies have demonstrated that endotoxin binds to the LPS CD14/toll-like receptor 4 complex on Kupffer cells causing NF-κB activation and TNF-α expression. 16,17,21
While the role of endotoxin is well studied, oxidative stress also plays an important role in alcohol-induced TNF-α expression. Liver perfusion studies have demonstrated that Kupffer cells are largely responsible for hepatic superoxide release during the early phase of acute alcohol intoxication and the recovery period. 22,23 Many reports are consistent with the hypothesis that alcohol-induced ROS not only act as toxic substances, but also stimulate signal transduction by activating redox-sensitive nuclear transcription factor, NF-κB, which in turn leads to TNF-α production. 24-27 However, there is accumulating evidence that TNF-α signaling in the hepatocyte causes an increase in mitochondrial ROS generation through ubiquinone cycling via the electron transport chain. 28,29 Thus, whether oxidative stress reflects a response to alcohol-induced TNF-α production or serves as a critical mediator for endotoxin-induced TNF-α production remains controversial. The present study was therefore undertaken to define the relationship among endotoxin, oxidative stress and TNF-α in a mouse model of acute alcoholic hepatotoxicity.
Materials and Methods
Animals
129/Sv mice were obtained from the Jackson Laboratories (Bar Harbor, ME) and housed in the animal quarters at the University of Louisville Research Resources Center. They were maintained at 22°C with a 12-hour light/dark cycle and had free access to rodent chow and tap water. The mice were used at 9 weeks of age, which has previously been a routine age selection for the same kind of studies. 24,30 The experimental procedures were approved by the Institutional Animal Care and Use Committee, which is certified by the American Association for Accreditation of Laboratory Animal Care.
Treatments
1) Alcohol administration: one dose of alcohol (Aldrich, Milwaukee, WI) at 6 g/kg was given by gavage. 2) Endotoxin neutralization: to neutralize alcohol-induced plasma endotoxin elevation, one dose of endotoxin-neutralizing protein (ENP) (Sigma Chemical Co., St. Louis, MO) at 10 mg/kg was injected intravenously after acute alcohol administration. 3) Inhibition of alcohol-induced oxidative stress: two antioxidants, N-acetyl-l-cysteine (NAC) and dimethylsulfoxide (DMSO) were used. Two doses of NAC (Calbiochem Corp., La Jolla, CA) were given at 150 mg/kg by i.p. injection at 12 hours and 30 minutes before alcohol treatment. One dose of DMSO was given at 2 g/kg by i.p. injection at 30 minutes before alcohol treatment. To perform a time-course study, plasma and liver samples were taken at 1.5, 3, 6, and 12 hours after alcohol administration. Mice were anesthetized with sodium pentobarbital (0.05 mg/g body weight). Blood was drawn with a heparinized syringe from the dorsal vena cava and plasma was obtained by centrifugation at 500 × g for 10 minutes at 4°C and stored at −80°C. The liver was perfused with saline and samples were processed for the measurements of TNF-α and lipid peroxidation, and for histopathological observation.
Alanine Aminotransferase Assay
Plasma alanine aminotransferase (ALT: EC2.6.1.2.) activity was colorimetrically measured using a Diagnostic kit (procedure number 505; Sigma) according to the instruction provided.
Histopathological Observation
Liver tissues were fixed with 10% neutral formalin and embedded in paraplast. Tissue sections of 5 μm were cut and stained by hematoxylin and eosin.
TNF-α Quantification in the Liver
Liver samples for TNF-α assay were prepared according to a previous report 31 with some minor modifications. Briefly, liver samples were disintegrated in 5 volumes of ice-cold Ripa buffer. 31 After incubation on ice for 30 minutes, samples were centrifuged twice at 20,000 × g for 15 minutes at 4°C. The resulting supernatants were used for assay. The TNF-α levels were detected by enzyme-linked immunosorbent assay (ELISA) using a murine kit (BioSource International, Inc., Camarillo, CA), and expressed as pg/g
Liver Total RNA Isolation and RT-PCR for TNF-α Expression
Total RNA from liver tissue was extracted by RNeasy Mini kit (QIAGEN, Valencia, CA) according to the manufacturer’s instructions. Two μg total RNA was used for cDNA synthesis using cDNA CYCLE kit (Invitrogen Corp., Carlsbad, CA) according to the manufacturer’s instructions. For polymerase chain reaction (PCR), 5 μl of cDNA from each preparation were added to 45 μl of Master Mix containing 5 μl of 10X PCR buffer, 0.1 μl of 100 mmol/L dNTP mix, 2 μl 50 mmol/L MgSO4, 0.2 μl (5 U/μl) TaqDNA polymerase (GIBCO-BRL, Grand Island, NY), 2 μl corresponding primers and 35.7 μl ddH2O. Primers for both murine TNF-α and β-actin were obtained from R&D Systems (Minneapolis, MN). PCR was initiated in a thermal cycler programmed at 94°C for 4 minutes followed by 96°C for 1 minute, 57°C for 4 minutes for 2 cycles, and then at 94°C for 1 minute, 59°C for 2 minutes, and 72°C for 4 minutes (33 cycles). After PCR, the content of each treatment was analyzed by Ready Gel Precast Gels (Bio-Rad, Hercules, CA). The bands were visualized with ethidium bromide.
Immunohistochemical Localization of TNF-α in the Liver
Cryostat liver sections were cut at 7 μm, air-dried, and fixed in acetone for 10 minutes at −20°C. Endogenous peroxidase activity was quenched by incubating sections in 3% H2O2. Non-specific binding sites were blocked by 10% normal goat sera for 30 minutes. Sections were incubated with polyclonal rabbit anti-TNF-α antibody (BioSource International, Inc.) overnight at 4°C, followed by incubation with horseradish peroxidase (HRP)-conjugated goat anti-rabbit antibody (Zymed Laboratories, Inc., San Francisco, CA) for 1 hour. The antibody binding sites were visualized by incubation with DAB-H2O2 solution.
Plasma Endotoxin Levels
A chromogenic endotoxin detection kit (QCL-1000, Whittaker Bioproducts Inc., Walkersville, MD) based on limulus amebocyte lysate assay was used for measuring plasma endotoxin levels following the manufacture’s instructions. Briefly, plasma samples were diluted 1:10 and heated to 75°C for 20 minutes to denature endotoxin inhibitors in the plasma. Samples were incubated with limulus amebocyte lysate for 10 minutes at 37°C, followed by incubation with the provided chromagen for 6 minutes. Absorbance at 405 nmol was measured, and the endotoxin concentrations were expressed as EU/ml.
Lipid Peroxidation Assay
Hepatic lipid peroxidation was quantified by measuring thiobarbituric acid-reactive substance (TBARS) as described previously. 32 The TBARS concentrations were expressed as nmol/g liver.
Statistics
All measurements are expressed as mean ± SD (n = 4–6). The data were analyzed by analysis of variance and Newman-Keuls’ multiple-comparison test. Differences between groups were considered significant at P < 0.05.
Results
Alcohol-Induced TNF-α Production and Liver Injury
TNF-α concentrations, gene expression, and localization in the liver were determined by ELISA, RT-PCR and immunohistochemistry, respectively. As shown in Figure 1 ▶ , hepatic TNF-α levels were significantly elevated as early as 1.5 hours after alcohol treatment. Alcohol-induced TNF-α production peaked at 6 hours, reaching 4.6-fold elevation. Although the TNF-α production decreased at 12 hours, the level was still higher than control values. RT-PCR analysis of TNF-α mRNA in the liver at 3 and 6 hours after alcohol administration demonstrated that TNF-α gene expression was enhanced by alcohol treatment (Figure 2) ▶ . By immunohistochemistry, TNF-α-positive staining was found on the Kupffer cells which localize on the sinusoid wall (Figure 3) ▶ .
Figure 1.

Time-course changes in hepatic TNF-α levels after acute alcohol administration. One gastric alcohol was given at 6 g/kg, and hepatic TNF-α levels at different time-points were measured by ELISA. The data were analyzed by analysis of variance and Newman-Keuls’ multiple-comparison test. Significant difference (P < 0.05) is identified by various superscript letters. Cont, control.
Figure 2.

Alcohol-induced TNF-α mRNA expression in the liver. Livers were removed at 3 or 6 hours after alcohol administration (6 g/kg), and TNF-α and housekeeping gene mRNA were determined by RT-PCR analysis. Alcohol administration increased TNF-α mRNA expression in the liver at both 3 and 6 hours after treatment. Cont, control.
Figure 3.

Immunohistochemical staining of TNF-α-positive cells in the liver. Livers were removed 6 hours after alcohol administration (6 g/kg) and 7-μm cryostat section were made. Sections were incubated with a rabbit polyclonal anti-mouse TNF-α antibody, followed by incubation with HRP-conjugated goat anti-rabbit IgG antibody. A: Control liver. B: Alcohol-treated liver. Arrowheads: TNF-α-positive cells. Arrows: Liver sinusoid. Magnification, ×260.
Alcohol-induced liver injury was estimated by measuring plasma ALT activities and histopathological changes. As shown in Figure 4 ▶ , alcohol induced a significant increase in plasma ALT activities at 6 and 12 hours after alcohol administration, though the value at 12 hours was significantly lower than 6 hours. Histopathological changes were observed as early as 3 hours after alcohol administration (Figure 5) ▶ . Minor microvesicular steatosis was found at 3 hours, while necrotic cell death also occasionally occurred as suggested by cell enlargement, vacuolization, and nuclear dissolution. However, both microvesicular steatosis and necrosis were prominent at 6 and 12 hours.
Figure 4.

Time-course changes in plasma ALT activities after acute alcohol administration. One gastric alcohol was given at 6 g/kg, and plasma ALT activity was measured by using a Sigma diagnostic kit. The data were analyzed by analysis of variance and Newman-Keuls’ multiple-comparison test. Significant difference (P < 0.05) is identified by different letter superscripts. Cont, control.
Figure 5.
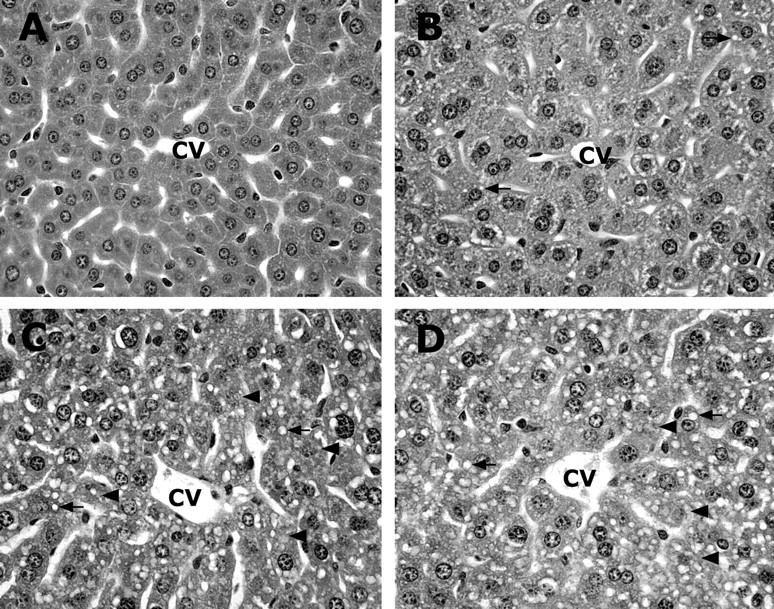
Alcohol-induced histopathological changes in the liver. One gastric alcohol was given at 6 g/kg. A: Control. B: Alcohol for 3 hours. C: Alcohol for 6 hours. D: Alcohol for 12 hours. Alcohol treatment induced prominent microvesicular steatosis (arrows) along with necrosis (arrowheads) in the liver. The necrotic hepatocytes are characterized by cell enlargement and nuclear dissolution. Hematoxylin and eosin staining; magnification, ×260.
Simultaneous Increases in Plasma Endotoxin and Hepatic Oxidative Stress in Association with Acute Alcohol Intoxication
Endotoxin is an important factor that has been implicated to trigger TNF-α production in alcohol-induced liver injury. To determine whether TNF-α production is associated with endotoxin elevation, plasma endotoxin levels were measured after ethanol administration. As shown in Figure 6 ▶ , plasma endotoxin levels were significantly increased at 1.5 and 3 hours after alcohol administration. The plasma endotoxin levels decreased to normal after 6 hours.
Figure 6.

Time-course changes in plasma endotoxin levels after acute alcohol administration. One gastric alcohol was given at 6 g/kg, and plasma endotoxin levels at different time-points were measured with a chromogenic detection kit based on limulus amebocyte lysate assay. The data were analyzed by analysis of variance and Newman-Keuls’ multiple-comparison test. Significant difference (P < 0.05) is identified by various superscript letters. Cont, control.
Because oxidative stress could be the cause or the result, or both, of TNF-α production, alcohol-induced oxidative stress in the liver was assessed by measuring lipid peroxidation (TBARS concentrations). As shown in Figure 7 ▶ , hepatic TBARS concentrations were elevated as early as 1.5 hours after alcohol administration, and time-dependent increases in TBARS concentrations were observed up to 12 hours, a pattern that was different from that of alcohol-induced hepatic TNF-α elevation.
Figure 7.

Time-course changes in hepatic lipid peroxidation after acute alcohol administration. One gastric alcohol was given at 6 g/kg, and hepatic lipid peroxidation at different time-points was evaluated by measuring TBARS concentrations. The data were analyzed by analysis of variance and Newman-Keuls’ multiple-comparison test.
Role of Endotoxin in Alcohol-Induced Hepatic TNF-α Production and Liver Injury
To determine the role of endotoxin in hepatic TNF-α production under acute alcohol exposure, endotoxin neutralization was performed by i.v. injection of ENP. As shown in Figure 8 ▶ , ENP injection significantly inhibited the increase in plasma endotoxin level at 1.5 hours after acute alcohol exposure. As shown in Figure 9 ▶ , endotoxin neutralization resulted in suppression of TNF-α production in the liver after acute alcohol administration. Endotoxin neutralization also partially inhibited acute alcohol-induced lipid peroxidation in the liver at 6 hours after acute alcohol administration (Figure 10) ▶ .
Figure 8.

Effects of endotoxin neutralization and antioxidants on alcohol-induced plasma endotoxin elevation. One gastric alcohol was given at 6 g/kg for 1.5 hours. ENP (10 mg/kg) was injected intravenously after alcohol administration. NAC (300 mg/kg) or DMSO (2 g/kg) was injected intravenously before alcohol treatment. Plasma endotoxin levels were measured with a chromogenic detection kit based on limulus amebocyte lysate assay. The data were analyzed by analysis of variance and Newman-Keuls’ multiple-comparison test. Significant difference (P < 0.05) is identified by various superscript letters.
Figure 9.

Effects of endotoxin neutralization and antioxidants on alcohol-induced hepatic TNF-α production. One gastric alcohol was given at 6 g/kg for 1.5 hours. ENP (10 mg/kg) was injected intravenously after alcohol administration. NAC (300 mg/kg) or DMSO (2 g/kg) was injected intravenously before alcohol treatment. Hepatic TNF-α levels were measured by ELISA. The data were analyzed by analysis of variance and Newman-Keuls’ multiple-comparison test. Significant difference (P < 0.05) is identified by various superscript letters.
Figure 10.

Effects of endotoxin neutralization and antioxidants on alcohol-induced hepatic lipid peroxidation. One gastric alcohol was given at 6 g/kg for 1.5 hours. ENP (10 mg/kg) was injected intravenously after alcohol administration. NAC (300 mg/kg) or DMSO (2 g/kg) was injected intravenously before alcohol treatment. Hepatic lipid peroxidation was evaluated by measuring TBARS concentrations. The data were analyzed by analysis of variance and Newman-Keuls’ multiple-comparison test. Significant difference (P < 0.05) is identified by various superscript letters.
The effects of endotoxin neutralization on alcohol-induced liver injury were assessed by measuring plasma ALT activity and histopathological changes in the liver. As shown in Figure 11 ▶ , alcohol-induced elevation of plasma ALT activity was abrogated by endotoxin neutralization. In concurrence, light microscopy demonstrated that endotoxin neutralization resulted in abrogation of alcohol-induced necrotic cell death (Figure 12c) ▶ . Furthermore, alcohol-induced microvesicular steatosis in the liver was partially inhibited by endotoxin neutralization.
Figure 11.

Effects of endotoxin neutralization and antioxidants on alcohol-induced plasma ALT elevation. One gastric alcohol was given at 6 g/kg for 1.5 hours. ENP (10 mg/kg) was injected intravenously after alcohol administration. NAC (300 mg/kg) or DMSO (2 g/kg) was injected intravenously before alcohol treatment. Plasma ALT activity was measured using a Sigma diagnostic kit. The data were analyzed by analysis of variance and Newman-Keuls’ multiple-comparison test. Significant difference (P < 0.05) is identified by various superscript letters.
Figure 12.
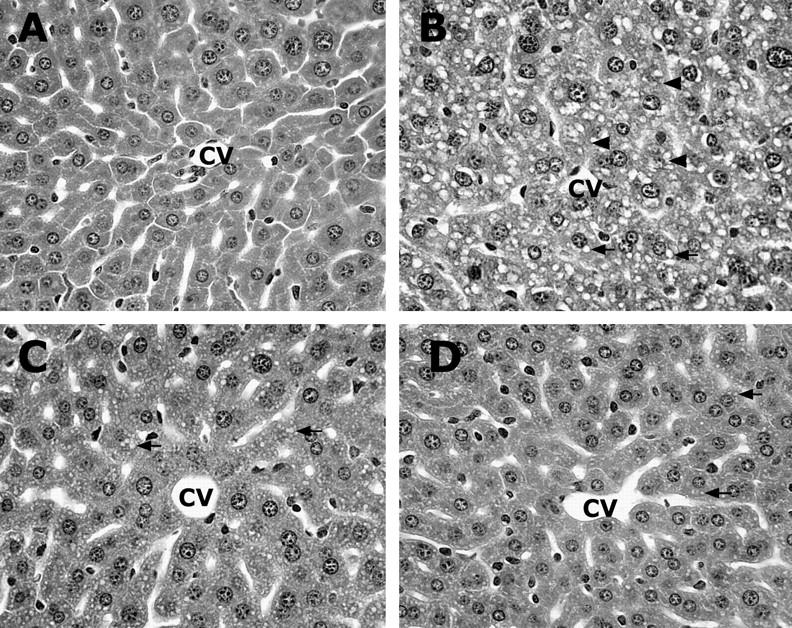
Effects of ENP and NAC on alcohol-induced histopathological changes in the liver. One gastric ethanol was given at 6 g/kg. ENP (10 mg/kg) was injected intravenously after alcohol administration. NAC (300 mg/kg) was injected intravenously before alcohol treatment. Livers were removed at 6 hours after alcohol administration. A: Control. B: Alcohol. C: ENP/alcohol. D: NAC/alcohol. Endotoxin neutralization by ENP resulted in abrogation of necrotic cell death and inhibition of lipid accumulation. NAC treatment attenuated alcohol-induced liver injury, with only minor microvesicular steatosis being found. Hematoxylin and eosin staining; magnification, ×260.
Role of Oxidative Stress in Alcohol-Induced Hepatic TNF-α Production and Liver Injury
To determine the role of oxidative stress in TNF-α production and liver injury, NAC and DMSO, two exogenous antioxidants, were used to attenuate oxidative stress after acute alcohol exposure. As shown in Figure 8 ▶ , treatment with either NAC or DMSO had no effect on the plasma endotoxin elevation at 1.5 hours after alcohol administration. However, the TNF-α production in the liver at 6 hours after alcohol administration was significantly inhibited by both NAC and DMSO (Figure 9) ▶ . Hepatic lipid peroxidation at 6 hours after alcohol administration was also attenuated by pretreatment with either NAC or DMSO (Figure 10) ▶ .
Alcohol-induced liver injury was attenuated by antioxidant treatment as indicated by measurements of plasma ALT activity and histopathological observation. As shown in Figure 11 ▶ , treatment with either NAC or DMSO abrogated alcohol-induced increase in plasma ALT activity (Figure 11) ▶ . Antioxidant (data not shown for DMSO) treatment not only prevented alcohol-induced necrotic cell death, but also inhibited microvesicular steatosis (Figure 12) ▶ .
Discussion
TNF-α production has been repeatedly demonstrated both in animal models and in patients with alcoholic hepatitis. 5 Many studies demonstrated that TNF-α plays an important role in the pathogenesis of alcoholic hepatitis. Neutralization of TNF-α by a specific antibody has been shown to attenuate hepatic necrosis and inflammation caused by chronic alcohol exposure. 33 Long-term alcohol feeding caused liver injury in wild-type mice but not in the TNF-α receptor-1 knockout mice. 26,34 The present study provided direct evidence that acute alcohol administration significantly increases intrahepatic TNF-α protein levels. While the role of TNF-α in alcoholic liver injury has been well defined, the mechanisms by which alcohol consumption leads to TNF-α overproduction have not been fully understood.
Endotoxemia has long been known to be associated with alcohol exposure in both animal models and patients with alcoholic liver disease. 18-20,35,36 Alcohol consumption deleteriously affects the anatomical and functional integrity of intestinal mucosa and increases intestinal permeability, thus allowing gut-derived endotoxin to escape into the blood. 37,38 In our experimental paradigm, the elevation of plasma endotoxin occurred before the increase in hepatic TNF-α production. Moreover, ENP significantly suppressed alcohol-induced elevation of plasma endotoxin levels and inhibited alcohol-induced hepatic TNF-α production. Thus, this study demonstrated that endotoxin triggers hepatic TNF-α production during alcohol intoxication.
Oxidative stress, independent of endotoxin, has also been implicated in TNF-α production. For example, hypoxia per se stimulates NF-κB activation and TNF-α gene transcription in macrophages. 39 To address the role of oxidative stress in alcohol-induced TNF-α expression in the liver, several approaches have been used, including exogenous oxidant inhibitors, 40-42 a NADPH oxidase-deficient mouse model, 24 and adenovirus gene transfer. 25,27 Investigations with antioxidants demonstrated that treatment with allopurinol, ebselen, or diphenyleneiodonium sulfate results in the attenuation of NF-κB activation and TNF-α expression. 40-42 All studies suggest that oxidative stress plays an important role in alcohol-induced TNF-α production in the liver. However, a critical time-course study of the dynamic changes between oxidative stress and TNF-α production has not been documented in acute alcohol hepatotoxicity.
In the present study, time-dependent changes of acute alcohol-induced oxidative stress and TNF-α production were compared. Hepatic TNF-α increased at 1.5 hours after a single dose of a high level of alcohol, reaching a peak value at 6 hours. Although oxidative stress also occurred at 1.5 hours after alcohol treatment, this change continued to increase as a function of time and reached a peak value at 12 hours after the treatment. Therefore, there are distinct patterns of changes in oxidative stress and TNF-α production in response to acute alcohol administration. Previous studies have clearly demonstrated that inhibition of oxidative stress leads to attenuation of hepatic TNF-α production. 24-27,40-42 A critical question is how oxidative stress affects TNF-α production. Comparing the dynamic changes of plasma endotoxin, hepatic oxidative stress, and TNF-α production, a significant increase in oxidative stress occurred between the peaks of endotoxin and TNF-α production, indicating that plasma endotoxin may stimulate ROS generation in the liver. To test this hypothesis, the effect of endotoxin neutralization on oxidative stress in the liver was examined. ENP treatment significantly inhibited oxidative stress in addition to attenuating TNF-α production. These results suggest that oxidative stress may mediate endotoxin-triggered TNF-α production in the alcohol-intoxicated liver.
Primary cell cultures have been widely used to test the hypothesis that oxidative stress mediates LPS-induced TNF-α production. Although antioxidant treatment has been known to attenuate LPS-induced TNF-α production in Kupffer cells/macrophages, 43,44 recent studies have shown that ROS are simultaneously generated with TNF-α gene transcription after LPS stimulation. 45,46 Scavenging ROS by NAC led to attenuation of NF-κB activation and TNF-α gene transcription. 47,48 Thus, ROS likely function as second messengers in the signaling transduction of LPS-induced TNF-α production. However, the role of oxidative stress in endotoxin-induced TNF-α production during alcohol intoxication has been difficult to address because the relative contributions of oxidative stress form endotoxin and alcohol metabolism cannot be dissected. To further define the role of oxidative stress in alcohol-induced TNF-α production, inhibition of alcohol-induced oxidative stress was obtained by using NAC and DMSO. Interestingly, both NAC and DMSO attenuated alcohol-induced TNF-α production in the liver without affecting plasma endotoxin elevation. Taken together, these results strongly suggest that oxidative stress at least partially mediates endotoxin-induced hepatic TNF-α production during acute alcohol intoxication.
A number of hypotheses regarding the mechanisms by which alcohol causes cell injury have been suggested with oxidative stress, endotoxin, and proinflammatory cytokine production being leading putative etiological factors. However, the relationship among these factors in the pathogenesis of alcoholic cell injury has not been fully delineated. In the present study, time-course changes in plasma ALT and hepatic histopathology after acute alcohol intoxication were determined. Minor steatotic and necrotic changes in the liver were found at 3 hours after alcohol administration, but both steatosis and necrotic cell death were prominent at 6 and 12 hours. Liver damage was further demonstrated by significant increases in plasma ALT at 6 and 12 hours. In comparison with the time-course changes in endotoxin, oxidative stress, and TNF-α production, the plasma ALT and hepatic necrotic cell death are likely associated with intrahepatic TNF-α production. To further distinguish the role of endotoxin, oxidative stress, and TNF-α production in acute alcohol-induced liver injury, endotoxin neutralization and oxidant inhibition were performed. ENP, NAC, and DMSO treatments abrogated acute alcohol-induced TNF-α production as well as necrotic cell death, suggesting both endotoxin and oxidative stress cause necrotic cell death through TNF-α. However, acute alcohol-induced steatotic liver injury was attenuated by NAC and DMSO, but less affected by ENP. Taken together, these results suggest that acute alcohol-induced necrotic cell death is triggered by TNF-α, while oxidative stress may be more involved in steatosis.
In conclusion, this study demonstrates that acute alcohol administration causes liver injury and biochemical alterations in plasma endotoxin levels, hepatic TNF-α production and oxidative stress. Endotoxin and oxidative stress are involved in hepatic TNF-α production and liver injury under acute alcohol intoxication. Although it may not directly induce intrahepatic TNF-α production, oxidative stress likely mediates endotoxin-induced TNF-α production in acute alcohol hepatotoxicity. This study also suggests that inhibition of oxidative stress may be an important strategy in the prevention/treatment of alcohol-induced liver injury.
Footnotes
Address reprint requests to Dr. Zhanxiang Zhou, University of Louisville School of Medicine, Department of Medicine, 511 South Floyd Street, MDR 525, Louisville, KY. E-mail: z0zhou01@louisville.edu.
Supported in part by National Institutes of Health grants AA13601 (to Z. Z.), AA01762 and AA10496 (to C. J. M.), and HL63760 and HL59225 (to Y. J. K.); by the University of Louisville School of Medicine, the Veterans Administration, and the Jewish Hospital Foundation, Louisville, Kentucky.
Y. J. K. is a Distinguished University Scholar of the University of Louisville.
References
- 1.Dufour MC, Stinson FS, Fe Case M: Trends in cirrhosis morbidity and mortality: United States, 1979–1988. Semin Liver Dis 1993, 13:109-125 [DOI] [PubMed] [Google Scholar]
- 2.Mullen KD, Dasarathy S: Potential new therapies for alcoholic liver disease. Clin Liver Dis 1998, 2:851-881 [Google Scholar]
- 3.Nordmann R, Ribiere C, Rouach H: Implication of free radical mechanisms in ethanol-induced cellular injury. Free Radic Biol Med 1992, 12:219-240 [DOI] [PubMed] [Google Scholar]
- 4.Arteel GE: Oxidants and antioxidants in alcohol-induced liver disease. Gastroenterology 2003, 124:778-790 [DOI] [PubMed] [Google Scholar]
- 5.McClain CJ, Barve S, Deaciuc I, Kugelmas M, Hill D: Tumor necrosis factor and alcoholic liver disease. Semin Liver Dis 1999, 19:205-219 [DOI] [PubMed] [Google Scholar]
- 6.Lieber CS: Ethanol metabolism, cirrhosis and alcoholism. Clin Chim Acta 1997, 257:59-84 [DOI] [PubMed] [Google Scholar]
- 7.Mira L, Maia L, Barreira L, Manso CF: Evidence for radical generation due to NADH oxidation by aldehyde oxidase during ethanol metabolism. Arch Biochem Biophys 1995, 318:53-58 [DOI] [PubMed] [Google Scholar]
- 8.Reinke LE, Lai EK, DuBose CM, McCay PB: Reactive free radical generation in vivo in heart and liver of ethanol-fed rats: correlation with radical formation in vitro. Proc Natl Acad Sci USA 1987, 84:9223-9227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bautista AP, Spitzer JJ: Acute ethanol intoxication stimulates superoxide anion production by in situ perfused rat liver. Hepatology 1992, 15:892-598 [DOI] [PubMed] [Google Scholar]
- 10.Tsukamoto H, Horne W, Kamimura S, Niemela O, Parkkila S, Yla-Herttuala S, Brittenham GM: Experimental liver cirrhosis induced by alcohol and iron. J Clin Invest 1995, 96:620-630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meagher EA, Barry OP, Burke A, Lucey MR, Lawson JA, Rokach J, FitzGerald GA: Alcohol-induced generation of lipid peroxidation products in humans. J Clin Invest 1999, 104:805-813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou Z, Sun X, Lambert JC, Saari JT, Kang YJ: Metallothionein-independent zinc protection from alcoholic liver injury. Am J Pathol 2002, 160:2267-2274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kurose I, Higuchi H, Kato S, Miura S, Watanabe N, Kamegaya Y, Tomita K, Takashi M, Horie Y, Fukuda M, Mizukami K, Ishii H: Oxidative stress on mitochondria and cell membrane of cultured rat hepatocytes and perfused liver exposed to ethanol. Gastroenterology 1997, 112:1331-1343 [DOI] [PubMed] [Google Scholar]
- 14.Rouach H, Fataccioli V, Gentil M, French SW, Mirimoto M, Nordmann R: Effect of chronic ethanol feeding on lipid peroxidation and protein oxidation in relation to liver pathology. Hepatology 1997, 25:351-355 [DOI] [PubMed] [Google Scholar]
- 15.Navasumrit P, Ward TH, Dodd NJ, O’Connor PJ: Ethanol-induced free radicals and hepatic DNA strand breaks are prevented in vivo by antioxidants: effects of acute and chronic ethanol exposure. Carcinogenesis 2000, 21:93-99 [DOI] [PubMed] [Google Scholar]
- 16.Wheeler MD, Kono H, Yin M, Nakagami M, Uesugi T, Arteel GE, Gabele E, Rusyn I, Yamashina S, Froh M, Adachi Y, Iimuro Y, Bradford BU, Smutney OM, Connor HD, Mason RP, Goyert SM, Peters JM, Gonzalez FJ, Samulski RJ, Thurman RG: The role of Kupffer cell oxidant production in early ethanol-induced liver disease. Free Radic Biol Med 2001, 31:1544-1549 [DOI] [PubMed] [Google Scholar]
- 17.McClain CJ, Hill DB, Song Z, Deaciuc I, Barve S: Monocyte activation in alcoholic liver disease. Alcohol 2002, 27:53-61 [DOI] [PubMed] [Google Scholar]
- 18.Nanji AA, Miao L, Thomas P, Rahemtulla A, Khwaja S, Zhao S, Peters D, Tahan SR, Dannenberg AJ: Enhanced cyclooxygenase-2 gene expression in alcoholic liver disease in the rat. Gastroenterology 1997, 112:943-951 [DOI] [PubMed] [Google Scholar]
- 19.Kono H, Wheeler MD, Rusyn I, Lin M, Seabra V, Rivera CA, Bradford BU, Forman DT, Thurman RG: Gender differences in early alcohol-induced liver injury: role of CD14, NF-κB, and TNF-α. Am J Physiol 2000, 278:G652-G661 [DOI] [PubMed] [Google Scholar]
- 20.Lambert JC, Zhou Z, Wang L, Song Z, McClain CJ, Kang YJ: Prevention of alterations in intestinal permeability is involved in zinc inhibition of acute ethanol-induced liver damage in mice. J Exp Phamarcol Ther 2003, 305:880-886 [DOI] [PubMed] [Google Scholar]
- 21.Su GL: Lipopolysaccharides in liver injury: molecular mechanisms of Kupffer cell activation. Am J Physiol 2001, 283:G256-G265 [DOI] [PubMed] [Google Scholar]
- 22.Bautista AP, Spitzer J: Role of Kupffer cells in the ethanol-induced oxidative stress in the liver. Front Biosci 1999, 4:d589-d595 [DOI] [PubMed] [Google Scholar]
- 23.Hasegawa T, Kikuyama M, Sakurai K, Kambayashi Y, Adachi M, Saniabadi AR, Kuwano H, Nakano M: Mechanism of superoxide anion production by hepatic sinusoidal endothelial cells and Kupffer cells during short-term ethanol perfusion in the rat. Liver 2002, 22:321-329 [DOI] [PubMed] [Google Scholar]
- 24.Kono KT, Rusyn I, Yin M, Gable E, Yamashina S, Dikalova A, Kadiiska MB: NADPH oxidase-derived free radicals are key oxidants in alcohol-induced liver disease. J Clin Invest 2000, 106:867-872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wheeler MD, Yamashina S, Froh M, Rusyn I, Thurman RG: Adenoviral gene delivery can inactivate Kupffer cells: role of oxidants in NF-κB activation and cytokine production. J Leukoc Biol 2001, 69:622-630 [PubMed] [Google Scholar]
- 26.Yin M, Gabele E, Wheeler MD, Connor H, Bradford BU, Dikalova A, Rusyn I, Mason R, Thurman RG: Alcohol-induced free radicals in mice: direct toxicants or signaling molecules? Hepatology 2001, 34:935-942 [DOI] [PubMed] [Google Scholar]
- 27.Wheeler MD, Kono H, Yin M, Rusyn I, Froh M, Connor HD, Mason RP, Samulski RJ, Thurman RG: Delivery of the Cu/Zn-superoxide dismutase gene with adenovirus reduces early alcohol-induced liver injury in rats. Gastroenterology 2001, 120:1241-1250 [DOI] [PubMed] [Google Scholar]
- 28.Lee FY, Li Y, Zhu H, Yang S, Lin HZ, Trush M, Diehl AM: Tumor necrosis factor increases mitochondrial oxidant production and induces expression of uncoupling protein-2 in the regenerating mice liver. Hepatology 1999, 29:677-687 [DOI] [PubMed] [Google Scholar]
- 29.Schulze-Osthoff K, Beyaert R, Vandevoorde V, Haegeman G, Fiers W: Depletion of the mitochondrial electron transport abrogates the cytotoxic and gene-inductive effects of TNF-α. EMBO J 1993, 12:3095-3104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yamashina S, Wheeler MD, Rusyn I, Ikejima K, Sato N, Thurman RG: Tolerance and sensitization to endotoxin in Kupffer cells caused by acute ethanol involve interleukin-1 receptor-associated kinase. Biochem Biophys Res Commun 2000, 277:686-690 [DOI] [PubMed] [Google Scholar]
- 31.Wolf D, Schumann J, Koerber K, Kiemer AK, Vollmar AM, Sass G, Papadopoulos T, Bang R, Klein SD, Brune B, Tiegs G: Low-molecular-weight hyaluronic acid induces nuclear factor-κB-dependent resistance against tumor necrosis factor α-mediated liver injury in mice. Hepatology 2001, 34:535-547 [DOI] [PubMed] [Google Scholar]
- 32.Zhou Z, Sun X, Kang YJ: Metallothionein protection against alcoholic liver injury through inhibition of oxidative stress. Exp Biol Med 2002, 227:214-222 [DOI] [PubMed] [Google Scholar]
- 33.Iimuro Y, Gallucci RM, Luster MI, Kono H, Thurman RG: Antibodies to tumor necrosis factor-α attenuate hepatic necrosis and inflammation caused by chronic exposure to ethanol in the rat. Hepatology 1997, 26:1530-1537 [DOI] [PubMed] [Google Scholar]
- 34.Yin M, Wheeler MD, Kono H, Bradford BU, Gallucci RM, Luster MI, Thurman RG: Essential role of tumor necrosis factor-α in alcohol-induced liver injury in mice. Gastroenterology 1999, 117:942-952 [DOI] [PubMed] [Google Scholar]
- 35.Bode C, Kugler V, Bode JC: Endotoxemia in patients with alcoholic and non-alcoholic cirrhosis and in subjects with no evidence of chronic liver disease following acute alcohol excess. J Hepatology 1987, 4:8-14 [DOI] [PubMed] [Google Scholar]
- 36.Enomoto N, Ikejima K, Bradford B, Rivera C, Kono H, Brenner DA, Thurman RG: Alcohol causes both tolerance and sensitization of rat Kupffer cells via mechanisms dependent on endotoxin. Gastroenterology 1998, 115:443-451 [DOI] [PubMed] [Google Scholar]
- 37.Keshavarzian A, Holmes EW, Patel M, Iber F, Fields JZ, Pethkar S: Leaky gut in alcoholic cirrhosis: a possible mechanism for alcohol-induced liver damage. Am J Gastroenterol 1999, 94:200-207 [DOI] [PubMed] [Google Scholar]
- 38.Parlesak A, Schafer C, Schutz T, Bode JC, Bode C: Increased intestinal permeability to macromolecules and endotoxemia in patients with chronic alcohol abuse in different stages of alcohol-induced liver disease. J Hepatol 2000, 32:742-747 [DOI] [PubMed] [Google Scholar]
- 39.Leeper-Woodford SK, Detmer K: Acute hypoxia increases alveolar macrophage tumor necrosis factor activity and alters NF-κB expression. Am J Physiol 1999, 276:L909-L916 [DOI] [PubMed] [Google Scholar]
- 40.Kono H, Rusyn I, Bradford BU, Connor HD, Mason RP, Thurman RG: Allopurinol prevents early alcohol-induced liver injury in rats. J Pharmacol Exp Ther 2000, 293:296-303 [PubMed] [Google Scholar]
- 41.Kono H, Rusyn I, Uesugi T, Yamashina S, Connor HD, Dikalova A, Mason RP, Thurman RG: Diphenyleneiodonium sulfate, an NADPH oxidase inhibitor, prevents early alcohol-induced liver injury in the rat. Am J Physiol 2001, 280:G1005-G1012 [DOI] [PubMed] [Google Scholar]
- 42.Kono H, Arteel GE, Rusyn I, Sies H, Thurman RG: Ebselen prevents early alcohol-induced liver injury in rats. Free Radic Biol Med 2001, 30:403-411 [DOI] [PubMed] [Google Scholar]
- 43.Fox ES, Brower JS, Bellezzo JM, Leingang KA: N-acetylcysteine and α-tocopherol reverse the inflammatory response in activated rat Kupffer cells. J Immunol 1997, 158:5418-5423 [PubMed] [Google Scholar]
- 44.Bellezzo JM, Leingang KA, Bulla GA, Britton RS, Bacon BR, Fox ES: Modulation of lipopolysaccharide-mediated activation in rat Kupffer cells by antioxidants. J Lab Clin Med 1998, 131:36-44 [DOI] [PubMed] [Google Scholar]
- 45.Haddad JJ, Land SC: Redox signaling-mediated regulation of lipopolysaccharide-induced proinflammatory cytokine biosynthesis in alveolar epithelial cells. Antioxid Redox Signal 2002, 4:179-193 [DOI] [PubMed] [Google Scholar]
- 46.Bellezzo JM, Leingang KA, Bulla GA, Britton RS, Bacon BR, Fox ES: Modulation of lipopolysaccharide-mediated activation in rat Kupffer cells by antioxidants. J Lab Clin Med 1998, 131:36-44 [DOI] [PubMed] [Google Scholar]
- 47.Haddad JJ, Land SC: Redox/ROS regulation of lipopolysaccharide-induced mitogen-activated protein kinase (MAPK) activation and MAPK-mediated TNF-α biosynthesis. Br J Pharmacol 2002, 135:520-536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Victor VM, Rocha M, De la Fuente M: Regulation of macrophage function by the antioxidant N-acetylcysteine in mouse-oxidative stress by endotoxin. Int Immunopharmacol 2003, 3:97-106 [DOI] [PubMed] [Google Scholar]