

Abstract
Engagement of immunoglobulin-binding receptors (FcγR) on leukocytes and other cell types is one means by which immunoglobulins and immune complexes activate effector cells. One of these FcγRs, FcγRIIb, is thought to contribute to protection from autoimmune disease by down-regulation of B-cell responsiveness and myeloid cell activation. We assessed the role of FcγRIIb in a mouse model of cryoglobulin-associated membranoproliferative glomerulonephritis induced by overexpression of thymic stromal lymphopoietin (TSLP). TSLP transgenic mice were crossbred with animals deficient for FcγRIIb on the same genetic background (C57BL/6). Renal pathology was assessed in female and male animals (wild-type, FcγRIIb−/−, TSLP transgenic, and combined TSLP transgenic/FcγRIIb−/− mice) after 50 and 120 days, respectively. FcγRIIb−/− mice had no significant renal pathology, whereas overexpression of TSLP induced a membranoproliferative glomerulonephritis, as previously established. TSLP transgenic FcγRIIb−/− mice appeared sick with increased mortality. Kidney function was significantly impaired in male mice corresponding to aggravated glomerular pathology with increases in glomerular matrix and cellularity. This resulted from both a large influx of infiltrating macrophages and increased cellular proliferation. These results emphasize the important role of FcγRIIb in regulating immune responses and suggest that modulation of Fcγ receptor activation or expression may be a useful therapeutic approach for treating glomerular diseases.
Immune complexes represent an important pathogenic mechanism in a variety of autoimmune diseases and trigger inflammatory responses as well as secondary tissue destruction by two main pathways: they bind to complement factor C1q and as a result activate the classical complement cascade leading to the production of the chemoattractants C5a and C3a and the membrane attack complex C5b-9, with its cell lytic and/or activatory properties. 1 The second pathway by which immune complexes can induce tissue injury is via the engagement of cellular receptors for IgG, the Fcγ receptors (FcγR). These receptors represent a diverse family with individual members being able to activate or inhibit cellular responses to immunoglobulins. 2 In the mouse, ligand binding to the multimeric FcγRI or FcγRIII induces cellular activation via the tyrosine-based activation motif (ITAM) of the γ chain and triggers a variety of effector functions including phagocytosis, antibody-dependent cell-mediated cytotoxicity, and the release of cytokines and other inflammatory mediators. 3,4 In contrast, murine FcγRIIb is a single subunit receptor that contains a tyrosine-based inhibitory motif (ITIM). 5 Co-ligation of the inhibitory FcγRIIb receptor with an ITAM-containing receptor or FcγRIIb homoaggregation leads to the abrogation of the activatory signal for inflammatory pathways. 6 Both classes of Fc receptors are co-expressed on cell surfaces and exhibit comparable affinity and specificity for the binding of IgG. The balance between both signaling pathways in an individual cell determines the magnitude of the effector cell response. 7
Cryoglobulins are immunoglobulins or immune complexes in the serum that precipitate in the cold and redissolve after rewarming. 8 One clinically relevant manifestation of the disease takes place in the kidney. Approximately 30% of patients affected by mixed cryoglobulins develop a membranoproliferative glomerulonephritis. 9-11 We have recently described a mouse model of cryoglobulin-associated membranoproliferative glomerulonephritis in which mice overexpressing thymic stromal lymphopoietin (TSLP), an interleukin (IL)-7-like cytokine with B cell-promoting properties, form large amounts of circulating cryoglobulins of mixed IgG-IgM composition. 12 TSLP transgenic mice develop a systemic inflammatory disease that involves the kidneys, lungs, liver, spleen, and skin. The renal injury is an immune complex disease closely resembling human cryoglobulin-associated membranoproliferative glomerulonephritis. 9,10,13 Glomeruli of affected animals have thickened glomerular capillary walls with subendothelial accumulation of immune complexes and a host response that includes reduplication of capillary basement membranes and expansion of the mesangial areas caused by an increase in extracellular matrix and accumulation of immune complexes. Typically, glomeruli show a significant influx of monocytes/macrophages. 12 This predictable animal model enabled us to study the role of activation of the immune system by immune complexes and the subsequent induction of renal injury in cryoglobulin-associated membranoproliferative glomerulonephritis, focusing on the role of the inhibitory arm of the Fc receptor system.
Materials and Methods
Animal Study and Experimental Design
The experimental protocol was reviewed and approved by the Animal Care Committee of the University of Washington in Seattle. Mice for this study were housed in the animal care facility of the University of Washington under standardized specific pathogen-free conditions (25°C, 50% humidity, 12 hour dark/light cycle) with free access to food and water.
C57BL/6 wild-type and TSLP transgenic mice (previously described in detail by Taneda et al 12 ) were crossbred with animals lacking the inhibitory IgG receptor FcγIIb (on the same genetic background) to create TSLP transgenic FcγIIb receptor knockout animals (FcγIIbR−/−). 6 The genotype of the mice used in this study was verified by polymerase chain reaction as previously described for the two mouse strains. 6,12 Eight mice per experimental group (wild-type, FcγIIbR−/−, TSLP transgenic, and TSLP transgenic FcγIIbR−/− animals) were sacrificed at 50 days of age for female mice and 120 days of age for male mice. These time points were chosen because female mice demonstrate faster progression of the disease then male animals and renal pathology reaches a plateau at the times chosen with increasing mortality as mice age further. At the end of the study mice were anesthetized, blood was drawn by cardiac puncture, and organs were collected. Renal tissue was snap-frozen in liquid nitrogen or fixed in either half-strength Karnovsky’s solution for electron microscopy (1% paraformaldehyde and 1.25% glutaraldehyde in 0.1 mol/L Na cacodylate buffer, pH 7.0) or in 10% neutral buffered formalin as well as methyl Carnoy’s solution (60% methanol, 30% chloroform, 10% acetic acid) for standard histology.
Tissue Preparation and Histological Staining
Fixed tissues were processed and embedded in paraffin using routine protocols. Tissues were sectioned at 4-μm thickness for routine staining with hematoxylin and eosin (H&E), periodic acid-Schiff, and immunohistochemistry. Thin sections (2-μm thickness) were used for periodic acid methenamine silver stain (PAM). Immunofluorescence staining was performed on snap-frozen kidneys, sectioned at 6 μm, and fixed in ice-cold acetone for 10 minutes.
Immunohistochemistry
For immunohistochemistry the sections from paraffin-embedded tissues were deparaffinized in xylene and rehydrated in graded ethanol. Antigen retrieval was performed by heating tissue sections in Antigen Unmasking Solution (Vector Laboratories, Burlingame, CA) and endogenous peroxidases were blocked in 3% hydrogen peroxide. Endogenous biotin was blocked using the Avidin/Biotin Blocking kit from Vector Laboratories. Slides were then incubated with the primary antibody diluted in phosphate-buffered saline (PBS) containing 1% bovine serum albumin (Sigma, St. Louis, MO) for 1 hour at room temperature. The sections were washed repeatedly and then incubated with the appropriate secondary antibody. The ABC-Elite Reagent (Vector Laboratories) was used for signal amplification and 3,3′-diaminobenzidine with nickel enhancement was used as chromogen, resulting in black color product. Slides were counterstained in methyl green, dehydrated, and coverslipped.
A Mac-2 antibody from Cederlane (Ontario, Canada) was used to detect macrophages, as previously described. 14 For the detection of mesangial cell activation and smooth muscle-like transformation, an α-smooth muscle actin antibody (clone 1A4; DAKO, Carpinteria, CA) was used as previously described. 15 Cellular proliferation was assessed with a monoclonal mouse Ki67 antibody 16 from Pharmingen (La Jolla, CA) using the DAKO Animal Research kit. Collagen IV was chosen for assessment of extracellular matrix and was stained with a goat polyclonal anti-human collagen IV antibody (Southern Biotechnologies, Birmingham, AL) with a rabbit anti-goat secondary antibody (Vector) as described in the study by Ophascharoensuk and colleagues. 17
Frozen sections were rehydrated in PBS, blocked with normal rabbit serum, and then incubated with fluorescein-conjugated antibodies against IgM, IgG, IgA, and complement factor C3 (all from Cappel Pharmaceuticals, Aurora, OH), coverslipped with Vectashield mounting medium (Vector Laboratories), and viewed with a Zeiss fluorescence microscope, as previously described. 12
Laboratory Data
Urinary protein excretion was assessed with urine test strips (Uristix; Bayer, Elkhart, IN). Blood urea nitrogen was measured using a standard clinical chemistry analyzer (LX-20; Beckman Laboratories, Brea, CA). The formation of cryoprecipitates was checked visually after storage of serum samples at 4°C for 3 to 5 days.
Quantitative Analysis and Statistics
Tissue sections stained with H&E and PAM were used for morphometric analysis. Fifteen random glomerular cross sections were photographed by an examiner blinded for the origin of the sample using a digital camera (Olympus DP11; Olympus America, Melville, NY) and resulting images were imported into the Image Pro Plus Software (Media Cybernetics, Silver Spring, MD). The software was used to quantify the number of nuclei, the amount of extracellular matrix in PAM sections and in sections stained for collagen IV as well as the area occupied by macrophages and the size of the glomerular tuft area. Glomerular proliferation was assessed by counting Ki67-positive cells in at least 20 random glomerular cross sections per animal in a blinded manner. Glomerular α-smooth muscle actin expression was graded semiquantitatively on a scale from 0 (negative) to 4 (strong global mesangial expression) as described previously. 18 A similar method was used to quantify glomerular staining with immunoglobulins and C3. Fluorescence intensity was described on a scale of 0 (negative) to 3 (strong staining) as previously described. 12 Further quantification of the extent of glomerular deposition of immune reactants was achieved by serially diluting the fluorescently labeled detecting antisera throughout a 10-fold range to determine the endpoint-positive titer for each immunoglobulin and for the complement factor C3, as described in the study of Huang and colleagues. 19 Dilutions ranging from 1:200 to 1:2200 were identified for each immune reactant. The mean endpoint titer (eg, 1:1600) was determined for eight animals studied in each experimental group, and results ± SEM compared.
Statistical analysis was performed using the SPSS program (SPSS Inc., Chicago, IL). Means between groups were compared using t-test without assuming equal variances. A P value of <0.05 was considered statistically significant. All data are expressed as mean ± SEM.
Results
FcγRIIb Deficiency Leads to Increased Morbidity and Mortality as Well as Decrease of Renal Function in TSLP Transgenic Mice
C57BL/6 wild-type and FcγRIIb−/− mice had no detectable clinical abnormalities. TSLP transgenic mice occasionally showed ulcerations of their ears, face, and chest that increased with age and were because of cryoglobulin deposition, leukocytoclastic vasculitis, and leukocyte infiltration. These changes also occurred in TSLP transgenic mice with a functional deficiency in the FcγRIIb. Approximately 50% of the animals with the combined mutation developed more severe soft tissue injuries, involving the feet and tail. TSLP transgenic FcγRIIb−/− mice also had an increase in mortality with three male mice dying before the completion of the study at 4 months of age, whereas no spontaneous deaths occurred in the control groups.
TSLP transgenic mice showed systemic inflammatory disease, as previously described, 12 with leukocytosis (see Table 2 ▶ ) and diffuse infiltration of inflammatory cells into a variety of organs including liver, lung, and heart. Spleen and mesenteric lymph nodes were significantly increased in size. Affected organs of TSLP transgenic mice were significantly increased in weight because of marked infiltration with leukocytes as compared to wild-type controls (Table 1) ▶ . Deletion of the FcγRIIb led to a further increase in leukocyte influx and organ weight in livers, spleens, and lungs of TSLP transgenic animals. Peripheral white blood cell count increased further in the doubly mutated female mice, whereas changes in values in male animals did not reach the level of statistical significance. Except for two TSLP transgenic animals, all mice overexpressing TSLP had visible cryoglobulins, independent of their Fcγ receptor status. The lack of visible cryoprecipitates in these two mice is most likely because of the rather low sensitivity of this method and the small amount of serum that is available from each mouse; previous studies have indicated that all TSLP transgenic mice display the cryoglobulinemic phenotype.
Table 2.
Laboratory Data
| Gender | Wild type | FcγRIIb−/− | TSLP transgene | FcγRIIb−/− and TSLP transgene | |
|---|---|---|---|---|---|
| White blood cell count | ♀ | 2.0 ± 0.6 | 2.9 ± 0.6 | 6.5 ± 0.4** | 9.7 ± 1.1# |
| ♂ | 3.1 ± 0.9 | 2.3 ± 0.4 | 9.0 ± 0.8** | 11.6 ± 2.2 | |
| BUN (mg/dl) | ♀ | 22.2 ± 1.2 | 23.0 ± 2.3 | 25.0 ± 2.8 | 31.8 ± 4.7 |
| ♂ | 21.9 ± 0.9 | 20.0 ± 1.3 | 21.1 ± 1.2 | 33.5 ± 3.2# | |
| Proteinuria (see legend) | ♀ | 0.8 ± 0.1 | 0.7 ± 0.1 | 0.8 ± 0.1 | 1.0 ± 0.0 |
| ♂ | 1.1 ± 0.1 | 1.1 ± 0.1 | 0.9 ± 0.1 | 1.6 ± 0.2# |
Renal function at time of sacrifice as assessed by plasma BUN levels and dipstick test for proteinuria. Data expressed as mean ± standard error of the mean. Statistical significances compared to wild-type controls
(**, P < 0.01) or TSLP transgenic animals
(#, P < 0.05).
Proteinuria assessed by dip stick: 1 = 30mg/dl, 2 = 100mg/dl.
Table 1.
Body and Organ Weights
| Gender | Wild type | FcγRIIb−/− | TSLP transgene | FcγRIIb−/− and TSLP transgene | |
|---|---|---|---|---|---|
| Body weight (g) | ♀ | 19.1 ± 0.4 | 20.1 ± 0.6 | 17.6 ± 1.2 | 16.5 ± 1.3 |
| ♂ | 30.9 ± 1.0 | 29.6 ± 0.7 | 27.1 ± 1.0* | 25.5 ± 1.5 | |
| Kidney weight (g) | ♀ | 0.25 ± 0.01 | 0.28 ± 0.01 | 0.27 ± 0.02 | 0.28 ± 0.03 |
| ♂ | 0.38 ± 0.02 | 0.39 ± 0.02 | 0.37 ± 0.02 | 0.47 ± 0.04 | |
| Spleen weight (g) | ♀ | 0.08 ± 0.00 | 0.10 ± 0.01 | 0.32 ± 0.05** | 0.49 ± 0.07 |
| ♂ | 0.09 ± 0.01 | 0.10 ± 0.01 | 0.40 ± 0.03*** | 0.97 ± 0.09### | |
| Liver weight (g) | ♀ | 0.86 ± 0.01 | 1.04 ± 0.05 | 1.17 ± 0.06 | 1.33 ± 0.09 |
| ♂ | 1.39 ± 0.07 | 1.42 ± 0.03 | 1.48 ± 0.04 | 1.96 ± 0.03## | |
| Lung weight (g) | ♀ | 0.15 ± 0.01 | 0.18 ± 0.00 | 0.27 ± 0.02** | 0.36 ± 0.04 |
| ♂ | 0.18 ± 0.01 | 0.18 ± 0.01 | 0.33 ± 0.01*** | 0.54 ± 0.07# | |
| Heart weight (g) | ♀ | 0.12 ± 0.01 | 0.14 ± 0.01 | 0.10 ± 0.01 | 0.14 ± 0.01# |
| ♂ | 0.15 ± 0.01 | 0.17 ± 0.01 | 0.17 ± 0.01 | 0.18 ± 0.01 |
Body and organ weight in different experimental groups at time of sacrifice (day 50 for females and day 120 for males). Data expressed as mean ± standard error of the mean. Statistical significances compared to wild-type controls
(*, P < 0.05;
**, P < 0.01;
***, P < 0.001) or compared to TSLP transgenic animals
(#, P < 0.05;
##, P < 0.01;
###, P < 0.001).
FcγRIIb−/− mice showed no impairment in renal function compared to wild-type controls at the time points assessed in the study (Table 2) ▶ . Female TSLP transgenic mice, with or without deficiency in FcγRIIb, showed no significant changes in kidney function after 50 days. In contrast, male TSLP transgenic FcγRIIb−/− mice had an increase in plasma BUN (34 ± 3.2 mg/dl in FcγRIIb−/− TSLP transgenic mice versus 25 ± 1.2 mg/dl in TSLP transgenic animals, Table 2 ▶ ) and in proteinuria (1.6 ± 0.2 in FcγRIIb−/− TSLP transgenic mice versus 0.9 ± 0.1 in TSLP transgene animals) after 4 months.
TSLP Transgenic Mice without Functional FcγIIb Receptor Demonstrate a Significant Aggravation of Immune Complex-Mediated Renal Disease
TSLP transgenic animals demonstrated typical features of the previously described cryoglobulin-associated membranoproliferative glomerulonephritis 12 (Figure 1) ▶ . Mice showed a significant increase in silver staining glomerular matrix in general (198 ± 25 μm2/glomerular cross-section (gcs) for female wild-type mice versus 471 ± 55 μm2/gcs in TSLP transgenic animals, P < 0.01; male mice: 427 ± 50 μm2/gcs wild-type controls versus 677 ± 47 μm2/gcs in TSLP-overexpressing mice, P < 0.01; Figure 1 ▶ and Table 3 ▶ ) and of collagen IV in particular as an important component of glomerular extracellular matrix. Mesangial cells showed evidence of cellular activation as assessed by semiquantitative grading of glomerular α-smooth muscle actin expression (female mice: 0.3 ± 0.1/gcs in wild-type mice versus 0.9 ± 0.0/gcs in TSLP transgenic animals, P < 0.001; male mice: wild-type mice grading 0.4 ± 0.1/gcs versus 0.7 ± 0.1/gcs in TSLP transgenics, P = 0.56; Figure 2 ▶ ). Glomerular cellularity and proliferation remained unaffected in animals with cryoglobulin-induced immune complex disease. The glomerular area that was occupied by Mac-2 staining in TSLP transgenic animals was significantly increased when quantified morphometrically (0.5 ± 0.1% and 0.3 ± 0.2% for female and male wild-type mice, respectively, versus 2.5 ± 0.7% (female mice) and 3.6 ± 1.2% (male mice), P < 0.05; Figure 3 ▶ ).
Figure 1.
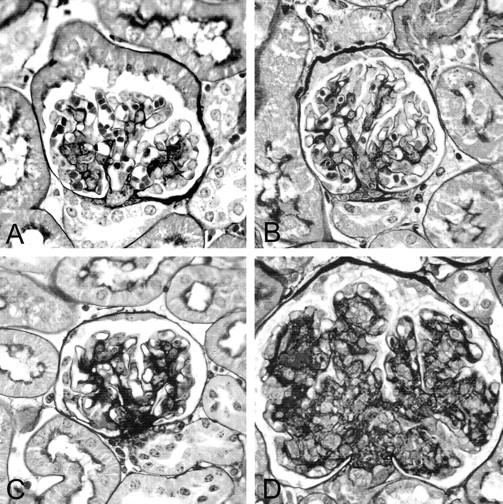
Glomerular architecture. The figure depicts representative glomeruli from animals of each experimental group in a PAM stain. A and B: Normal glomerular architecture of wild-type and FcγIIbR−/− mice, respectively. C: A glomerulus from a TSLP transgenic mouse with increase in glomerular matrix. The glomerulus in D is from a TSLP transgenic animal with a deletion in the FcγIIb receptor and shows a significant increase in glomerular extracellular matrix and cellularity. Original magnifications, ×400.
Table 3.
Morphometric Data and Immunohistochemical Assessment
| Gender | Wild type | FcγRIIb−/− | TSLP transgene | FcγRIIb−/− and TSLP transgene | |
|---|---|---|---|---|---|
| Glomerular size (μm2) | ♀ | 2810 ± 114 | 2846 ± 120 | 3029 ± 176 | 4021 ± 194## |
| ♂ | 3163 ± 148 | 3167 ± 97 | 3399 ± 232 | 5485 ± 387## | |
| Extracellular matrix/gcs (μm2) | ♀ | 198 ± 25 | 241 ± 30 | 473 ± 54*** | 1277 ± 176## |
| ♂ | 427 ± 50 | 433 ± 54 | 677 ± 47*** | 1749 ± 214## | |
| Extracellular matrix/area (% glomerular area) | ♀ | 7 ± 1 | 9 ± 1 | 15 ± 1** | 28 ± 3## |
| ♂ | 14 ± 1 | 13 ± 1 | 21 ± 3** | 32 ± 3## | |
| Collagen IV/gcs (μm2) | ♀ | 257 ± 18 | 261 ± 29 | 442 ± 42** | 1100 ± 145## |
| ♂ | 280 ± 29 | 335 ± 30 | 699 ± 47*** | 1807 ± 179## | |
| Collagen IV/area (% glomerular area) | ♀ | 10 ± 1 | 12 ± 1 | 19 ± 2*** | 32 ± 3## |
| ♂ | 11 ± 1 | 12 ± 1 | 23 ± 1*** | 38 ± 2### | |
| Glomerular cellularity/gcs (cells/gcs) | ♀ | 43 ± 2 | 42 ± 1 | 47 ± 2 | 57 ± 2## |
| ♂ | 42 ± 2 | 40 ± 2 | 48 ± 4 | 84 ± 7## | |
| Glomerular cellularity/area (% glomerular area) | ♀ | 1.5 ± 0.1 | 1.4 ± 0.0 | 1.6 ± 0.1 | 1.5 ± 0.1 |
| ♂ | 1.3 ± 0.1 | 1.3 ± 0.1 | 1.4 ± 0.1 | 1.6 ± 0.1 | |
| Cellular proliferation (Ki67 positive cells/gcs) | ♀ | 0.6 ± 0.1 | 0.6 ± 0.1 | 0.9 ± 0.2 | 1.9 ± 0.4# |
| ♂ | 0.2 ± 0.0 | 0.3 ± 0.2 | 0.3 ± 0.1 | 0.9 ± 0.3# | |
| Macrophage infiltration (μm2/gcs) | ♀ | 15 ± 3 | 14 ± 2 | 76 ± 22* | 278 ± 49## |
| ♂ | 10 ± 3 | 43 ± 13* | 145 ± 62 | 832 ± 169## | |
| Macrophage infiltration (% glomerular area) | ♀ | 0.5 ± 0.1 | 0.6 ± 0.1 | 2.5 ± 0.7* | 6.8 ± 1.1## |
| ♂ | 0.3 ± 0.2 | 1.5 ± 0.5* | 3.6 ± 1.2* | 12.8 ± 2.1## | |
| Glomerular α-smooth muscle actin expression | ♀ | 0.3 ± 0.1 | 0.4 ± 0.0 | 0.9 ± 0.0*** | 1.9 ± 0.3## |
| ♂ | 0.4 ± 0.0 | 0.4 ± 0.1 | 0.7 ± 0.1 | 2.1 ± 0.3## |
Morphometric analysis of kidney sections from wild-type, FcγRIIb−/−, TSLP transgenic, and combined TSLP transgenic FcγRIIb−/− mice. Data expressed as mean ± standard error of the mean. Statistical significances compared to wild type controls
(*P < 0.05;
**, P < 0.01;
***, P < 0.001) or compared to TSLP transgenic animals
(#, P < 0.05;
##, P < 0.01;
###, P < 0.001).
Figure 2.
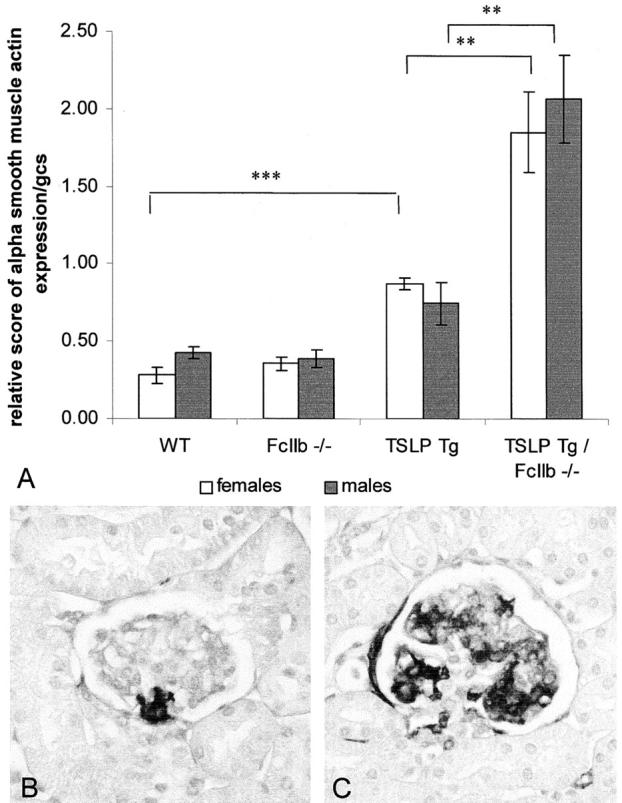
Glomerular α-smooth muscle actin expression. A: Semiquantitative assessment of glomerular α-smooth muscle actin expression. Graphs show mean ± SEM. Statistically significant differences between experimental groups are expressed as **, P < 0.01 and ***, P < 0.001. B: Immunohistochemical stain for α-smooth muscle actin; representative picture of glomerular α-smooth muscle actin expression in a wild-type mouse. C: Representative picture of glomerular α-smooth muscle actin expression in a TSLP transgenic FcγIIbR−/− animal. Original magnification, ×400 (B).
Figure 3.
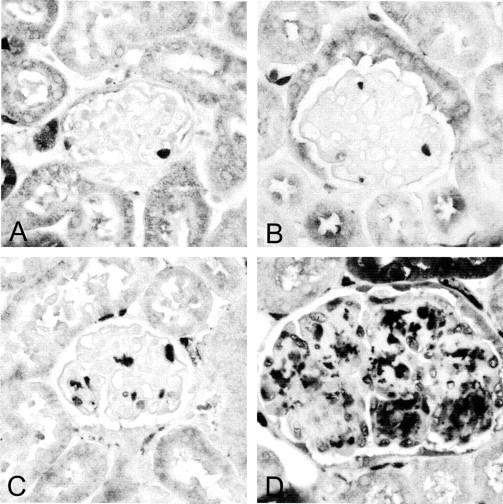
Glomerular macrophages. Immunohistochemical stain for macrophages using a Mac-2 antibody. Glomeruli for wild-type (A) or FcγIIbR−/− (B) mice show occasional infiltration with macrophages (dark stain). In contrast, there is marked glomerular macrophage influx in TSLP transgenic animals as shown in C. FcγIIbR-deficient TSLP transgenic mice show a significant increase in macrophage influx compared to TSLP transgenic mice with functional FcγIIb receptor (D). Original magnification, ×400 (A).
The genetic background of FcγRIIb−/− mice has been shown to be a susceptibility factor for autoimmune disease with C57BL/6 animals, but not BALB/c mice, developing autoantibodies and autoimmune glomerulonephritis. 20 FcγRIIb−/− mice in our study had no significant changes in glomerular matrix or cellularity compared to wild-type C57BL/6 mice (Table 3) ▶ . Glomerular cell proliferation and mesangial α-smooth muscle actin expression were not different from wild-type controls. However, male FcγRIIb−/− mice showed a small increase in glomerular macrophage count (1.5 ± 0.5% versus 0.3 ± 0.2% in wild-type controls, P < 0.05).
TSLP transgenic mice deficient in FcγRIIb showed a significant aggravation of the immune complex-mediated renal disease that corresponded to decreased renal function and an increase in proteinuria as seen in male TSLP transgenic FcγRIIb−/− animals. Glomerular size was significantly increased in female and male FcγRIIb−/− TSLP transgenic animals (3029 ± 176 μm2/gcs and 3399 ± 232 μm2/gcs in female and male TSLP transgenic mice versus 4021 ± 194 μm2/gcs and 5485 ± 387 μm2/gcs in female and male FcγRIIb-deficient TSLP transgenic animals, P < 0.01; Figure 1 ▶ and Table 3 ▶ ). This increased glomerular size was caused by an increase in both glomerular extracellular matrix and glomerular cellularity. Female and male mice showed a comparable increase in glomerular area occupied by black silver stain from 471 ± 55 μm2/gcs to 1277 ± 176 μm2/gcs (females, P < 0.01) and from 677 ± 47 μm2/gcs to 1749 ± 214 μm2/gcs (males, P < 0.01). These findings were confirmed by immunostaining of collagen IV, one of the main constituents of glomerular extracellular matrix (Table 3) ▶ . In addition, glomerular α-smooth muscle actin expression, a marker for mesangial cell activation, was strongly increased in TSLP transgenic mice deficient in FcγRIIb compared to TSLP transgenic animals (P < 0.01). Mesangiolysis was a frequent finding in TSLP transgenic FcγRIIb−/− mice. Glomerular cellularity was also significantly increased in TSLP transgenic mice deficient in the FcγRIIb. Female TSLP-overexpressing mice had an average glomerular cell number of 47 ± 2 cells/gcs whereas combined transgenic FcγRIIb−/− animals had a mean cell number of 57 ± 2 cells/gcs (P < 0.01). Male animals with a much longer life span compared to female mice had an even greater increase in glomerular cell number from 48 ± 4 cells/gcs for TSLP transgenic animals to 84 ± 7 cells/gcs in TSLP transgenic FcγRIIb−/− (P < 0.01). This increase was because of both an increase in proliferating glomerular cells and infiltration of monocytes/macrophages. Both genders demonstrated a significant increase in cells expressing Ki67 as marker of proliferating cells (Table 3 ▶ , P < 0.05). Aside from proliferation of intrinsic glomerular cells, there was a significant increase in the amount of macrophages that infiltrated the glomeruli of TSLP transgenic animals with a deficiency in the FcγRIIb compared to TSLP transgenic mice with functional FcγIIb receptor. The mean glomerular area that was occupied by macrophages increased from 2.5 ± 0.7% in female TSLP transgenic mice to 6.8 ± 1.1% in TSLP transgenic FcγRIIb−/− mice (P < 0.01) and from 3.6 ± 1.2% (TSLP transgenics) to 12.8 ± 2.1% in male TSLP transgenic mice lacking functional FcγIIb receptor (P < 0.01).
Glomerular Immunoglobulin and Complement Deposition in TSLP Transgenic Mice Is Unaffected by Deficiency of the FcγRIIb
C57BL/6 wild-type mice and female FcγRIIb−/− animals had only small amounts of immunoglobulins and complement component C3 deposited in the glomeruli (Figure 4) ▶ , as routinely encountered in many murine strains under normal laboratory living conditions. In contrast, male FcγIIb receptor-deficient mice had significantly more glomerular staining for IgG (mean score of 1.5 ± 0.2/gcs in FcγRIIb−/− mice versus 0.7 ± 0.1/gcs in C57BL/6 wild-type animals) and complement C3 (mean score of 1.5 ± 0.2/gcs in FcγRIIb−/− mice versus 0.6 ± 0.2/gcs in C57BL/6 wild-type animals), which in conjunction with increased glomerular macrophage influx may indicate an early phase of immune-mediated glomerulonephritis.
Figure 4.

Glomerular deposition of immunoglobulins and complement. Graphs show semiquantitative assessment of glomerular immunoglobulin deposition and deposition of complement factor C3. Columns show mean ± SEM. Statistically significant differences between experimental groups are expressed as *, P < 0.05; **, P < 0.01; and ***, P < 0.001.
TSLP transgenic mice showed large amounts of IgG, IgM, and IgA in their glomeruli. In addition, they also demonstrated high levels of complement component C3 in glomeruli (Figure 4) ▶ . Female and male FcγRIIb-deficient TSLP transgenic animals demonstrated strong glomerular immunoglobulin and complement deposition, which were comparable in amount to TSLP transgenic mice with functional FcγIIb receptor. To avoid missing differences in glomerular immunoglobulin deposition that might be obscured by assaying the extent of such deposition at only a single, possibly saturating, titer of detecting antibody, we then repeated our immunofluorescence studies using serial dilutions of the antisera used to detect IgG, IgM, IgA, and complement factor C3. These dilution studies confirmed the patterns seen in the semiquantitative assessment by showing no significant differences between experimental groups in the endpoint concentrations at which each of the different immunoglobulins could be detected (Table 4) ▶ . However, the endpoint dilution at which positive glomerular C3 staining could be demonstrated was slightly, but significantly, higher in TSLP transgenic animals with functional deletion of the FcγIIb receptor (1263 ± 105 in TSLP mice versus 1675 ± 94 in FcγIIbR−/− TSLP transgenic animals, P < 0.05).
Table 4.
Glomerular Immunoglobulin and Complement Factor C3 Deposition
| TSLP transgene | FcγRIIb−/− and TSLP transgene | |
|---|---|---|
| IgG | 1:1643 ± 73 | 1:1675 ± 75 |
| IgM | 1:1643 ± 40 | 1:1600 ± 0 |
| IgA | 1:1300 ± 184 | 1:1075 ± 75 |
| C3 | 1:1263 ± 105 | 1:1675 ± 94* |
Endpoint titers for serial dilutions of fluorescently labeled detecting antisera.
Data expressed as mean of the last dilutions at which glomerular staining could be detected for the experimental groups (n = 8) ± SEM.
*Statistically significant compared to TSLP transgenic animals, P < 0.05.
Discussion
We recently described a mouse model of a membranoproliferative glomerulonephritis associated with mixed cryoglobulinemia in TSLP transgenic mice. TSLP is a cytokine of the IL-2 family, which supports the growth of pre-B cells and promotes the development of pre-B cells to immature B cells. 21,22 Its function is closely related to IL-7, and both cytokines use the IL-7Rα chain for receptor binding. 23 Mice systemically overexpressing TSLP show leukocytosis and systemic inflammatory disease that involves major vital organs such as lung, liver, and kidney. 12 Transgenic mice develop cryoglobulinemia with high systemic levels of immunoglobulins of the IgM and IgG class that precipitate in the cold and lead to a systemic disease similar to human cryoglobulinemia, 11,24 including immune complex-mediated membranoproliferative glomerulonephritis. 13 Renal lesions in this model are characterized by deposition of large amounts of immunoglobulins in the mesangium and in the subendothelial region of glomerular capillary walls, and influx of macrophages into glomeruli. The deposited immune complexes fix complement, a process that may cause some of the ensuing glomerular injury. However, in view of the recent demonstration that Fcγ receptors can also be major mediators of immune complex-mediated injury in target organs, 7,25 we hypothesized that the deposition of immune complexes in glomeruli initiates downstream effects in the kidney involving receptors for the Fc portion of IgG. To address this hypothesis we produced combined TSLP transgenic and FcγR IIb-deficient animals to assess how renal disease in the TSLP model is modified in the absence of this Fc receptor.
In this study we were able to demonstrate that FcγRIIb is important for the inhibition of deleterious levels of activation of the cellular response to immune complexes. Deficiency in this receptor led to a significant aggravation of immune complex-mediated renal disease in TSLP transgenic mice. Renal lesions included an increase in glomerular size, resulting from an increase in glomerular extracellular matrix and cellularity. The increase in glomerular cellularity was caused by increased numbers of infiltrating macrophages and high levels of proliferating glomerular cells. This augmentation of renal pathology was associated with a decline in renal function and increase in proteinuria. These results indicate that FcγIIb receptors are involved in suppressing or limiting immune complex-induced macrophage influx, and are involved directly or indirectly in subsequent processes that result in generation of extracellular matrix and cellular proliferation. FcγRIIb therefore play an important role in balancing the extent of an immune response to a specific stimulus.
Previous studies have emphasized the role of the inhibitory FcγRIIb in the afferent and efferent arm of the host defense system. FcγIIb-deficient animals display significantly elevated immunoglobulin levels in response to thymus-dependent and thymus-independent antigens and are highly susceptible to IgG-triggered mast cell degranulation. 6 Inhibitory FcγRs have also been shown to be involved in the maintenance of peripheral tolerance as demonstrated in a mouse strain with a genetic background resistant to the induction of collagen-induced autoimmune diseases. Deletion of FcγIIb receptor function renders these animals susceptible to collagen IV-mediated Goodpasture’s syndrome or collagen II-induced arthritis. 26,27 Deficiency in the inhibitory FcγRIIb has also been associated with the strain-specific development of autoantibodies and autoimmune glomerulonephritis in C57BL/6 but not in BALB/c mice. 20 At 4 months of age the mutant C57BL/6 mice started to develop proteinuria and by the age of 8 months animals suffered from multiorgan inflammatory disease consistent with systemic vasculitis. Glomeruli displayed glomerulosclerosis, hypercellularity, and IgG deposition. Consistent with this previous report, the young C57BL/6 FcγRIIb−/− mice in our study of ages 50 and 120 days had no increase in urinary protein excretion and no significant renal pathology. However, male mice had a slight increase in glomerular macrophage content and small increase in glomerular deposition of IgG and C3 at 4 months indicating the possibility of a beginning autoimmune injury process.
A key question arising from this study is whether the effects of FcγRIIb on cryoglobulin-associated membranoproliferative glomerulonephritis are mediated by receptors on circulating leukocytes or by FcγIIb receptors on intrinsic renal cells such as mesangial cells. Both Fcγ receptors on myeloid and lymphoid cells as well as on intrinsic renal cells have been shown to interact with circulating immune complexes. 28-30 Stimulation of activating Fcγ receptors on hematopoietic cells triggers effector responses such as macrophage phagocytosis, antibody-dependent cell-mediated cytotoxicity, neutrophil activation, and inhibition of B-cell activation. 31-35 Binding of immune complexes to Fcγ receptors on mesangial cells has been described to be involved in the release of proinflammatory mediators, monocyte recruitment, and the expression of matrix proteins. 28,36-38 A recent study identified FcγRII as an important receptor expressed by murine mesangial cells and demonstrated that selective blockade of this receptor resulted in enhanced neutrophil infiltration and local chemokine production in mice exposed to anti-GBM antibodies. 39 Although our study was not designed to differentiate the contribution of FcγIIb receptors on circulating leukocytes versus those on renal cells, the fact that immunoglobulin deposition was similar in TSLP transgenic animals with and without functional FcγIIb receptor suggests that the response of renal cells to immune complexes was altered leading to increased cell proliferation, activation of mesangial cells, recruitment of monocytes/macrophages, and deposition of extracellular matrix. Glomerular deposition of complement factor C3 was also not substantially affected by deficiency in the FcγIIb receptor. The basis for the slight but statistically significant increase in the dilution endpoint of C3 positivity is not readily apparent. However, potential effects of FcγRIIb deficiency in leukocytes causing either increased production of cryoglobulins or increased secretion of proinflammatory mediators in cryoglobulin-associated renal disease cannot be excluded. Further evidence pointing to the crucial role of FcγRIIb on intrinsic renal cells as the key mediator of immune complex-mediated glomerulonephritis is the fact that the number of circulating white blood cells was not significantly elevated in TSLP transgenic FcγRIIb knockout mice compared to TSLP transgenic animals with functional FcγRIIb, whereas leukocyte infiltration into different organs such as spleen, liver, and lung was significantly augmented in FcγRIIb-deficient TSLP transgenic mice. We were not able to determine exact cryocrit levels because of the small blood volume in mice but the fact that glomerular deposition of immunoglobulins was not increased, suggests that circulating cryoglobulin levels were not elevated in TSLP transgenic mice without FcγRIIb. Studies of mice with nephrotoxic serum nephritis, another form of antibody-mediated glomerular injury, provide conflicting evidence that supports and refutes a primary role for FcγRIIb expression by intrinsic renal cells as the key determinant of disease expression. Studies of Tarzi and colleagues 40 indicate that the renal pathology in this disease model is solely dependent on FcγR on circulating leukocytes. Knockout mice deficient in a different set of FcγR−/− (FcγR I and III), which have opposite biological effects than those of FcγRIIb, receiving bone marrow transplant with wild-type leukocytes were not protected from anti-GBM glomerulonephritis while wild-type mice transplanted with FcγR−/− bone marrow were completely protected from the development of renal disease. Radeke and colleagues 39 demonstrated that blocking of renal FcγIIb receptors in nephrotoxic serum nephritis led to an increase in renal infiltration with neutrophils and an elevation of local chemokine production suggesting a role for renal Fcγ receptors in the development of the disease. Mice chimeric for FcγRIIb are not yet available to directly test the importance of leukocyte versus renal expression of this receptor for mediation of glomerulonephritis.
In conclusion, our results emphasize the role of FcγR receptors in the pathogenesis of cryoglobulin-associated membranoproliferative glomerulonephritis. They highlight the importance of FcγRIIb in limiting the extent of the host response to immune complexes, show exacerbation of glomerulonephritis in the absence of this receptor, and suggest that modulation of expression or activation state of Fc receptors might be a useful therapeutic approach for immune complex-mediated glomerular diseases.
Acknowledgments
We thank Xiangling Yang and Zandro Paredes for excellent technical assistance.
Footnotes
Address reprint requests to Charles E. Alpers, M.D., University of Washington Medical Center, Department of Pathology, Box 356100, 1959 NE Pacific St., Seattle, Washington 98195. E-mail: calp@u.washington.edu.
Supported in part by grants from the Northwest Kidney Centers Foundation, Genzyme, and the National Institute of Health (grants DK 47659 and HL 63652).
References
- 1.Liszewski MK, Farries TC, Lublin DM, Rooney IA, Atkinson JP: Control of the complement system. Adv Immunol 1996, 61:201-283 [DOI] [PubMed] [Google Scholar]
- 2.Ravetch JV, Luster AD, Weinshank R, Kochan J, Pavlovec A, Portnoy DA, Hulmes J, Pan YC, Unkeless JC: Structural heterogeneity and functional domains of murine immunoglobulin G Fc receptors. Science 1986, 234:718-725 [DOI] [PubMed] [Google Scholar]
- 3.Greenberg S, Chang P, Silverstein SC: Tyrosine phosphorylation is required for Fc receptor-mediated phagocytosis in mouse macrophages. J Exp Med 1993, 177:529-534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Clynes R, Ravetch JV: Cytotoxic antibodies trigger inflammation through Fc receptors. Immunity 1995, 3:21-26 [DOI] [PubMed] [Google Scholar]
- 5.Amigorena S, Bonnerot C, Drake JR, Choquet D, Hunziker W, Guillet JG, Webster P, Sautes C, Mellman I, Fridman WH: Cytoplasmic domain heterogeneity and functions of IgG Fc receptors in B lymphocytes. Science 1992, 256:1808-1812 [DOI] [PubMed] [Google Scholar]
- 6.Takai T, Ono M, Hikida M, Ohmori H, Ravetch JV: Augmented humoral and anaphylactic responses in Fc gamma RII-deficient mice. Nature 1996, 379:346-349 [DOI] [PubMed] [Google Scholar]
- 7.Ravetch JV, Bolland S: IgG Fc receptors. Annu Rev Immunol 2001, 19:275-290 [DOI] [PubMed] [Google Scholar]
- 8.Ferri C, Zignego AL, Pileri SA: Cryoglobulins. J Clin Pathol 2002, 55:4-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.D’Amico G, Fornasieri A: Cryoglobulinemic glomerulonephritis: a membranoproliferative glomerulonephritis induced by hepatitis C virus. Am J Kidney Dis 1995, 25:361-369 [DOI] [PubMed] [Google Scholar]
- 10.D’Amico G: Renal involvement in hepatitis C infection: cryoglobulinemic glomerulonephritis. Kidney Int 1998, 54:650-671 [DOI] [PubMed] [Google Scholar]
- 11.Trejo O, Ramos-Casals M, Garcia-Carrasco M, Yague J, Jimenez S, de la Red G, Cervera R, Font J, Ingelmo M: Cryoglobulinemia: study of etiologic factors and clinical and immunologic features in 443 patients from a single center. Medicine (Baltimore) 2001, 80:252-262 [DOI] [PubMed] [Google Scholar]
- 12.Taneda S, Segerer S, Hudkins KL, Cui Y, Wen M, Segerer M, Wener MH, Khairallah CG, Farr AG, Alpers CE: Cryoglobulinemic glomerulonephritis in thymic stromal lymphopoietin transgenic mice. Am J Pathol 2001, 159:2355-2369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tarantino A, De Vecchi A, Montagnino G, Imbasciati E, Mihatsch MJ, Zollinger HU, Di Belgiojoso GB, Busnach G, Ponticelli C: Renal disease in essential mixed cryoglobulinaemia. Long-term follow-up of 44 patients. Q J Med 1981, 50:1-30 [PubMed] [Google Scholar]
- 14.Segerer S, Hudkins KL, Taneda S, Wen M, Cui Y, Segerer M, Farr AG, Alpers CE: Oral interferon-alpha treatment of mice with cryoglobulinemic glomerulonephritis. Am J Kidney Dis 2002, 39:876-888 [DOI] [PubMed] [Google Scholar]
- 15.Hugo C, Shankland SJ, Bowen-Pope DF, Couser WG, Johnson RJ: Extraglomerular origin of the mesangial cell after injury. A new role of the juxtaglomerular apparatus. J Clin Invest 1997, 100:786-794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Key G, Becker MH, Baron B, Duchrow M, Schluter C, Flad HD, Gerdes J: New Ki-67-equivalent murine monoclonal antibodies (MIB 1–3) generated against bacterially expressed parts of the Ki-67 cDNA containing three 62 base pair repetitive elements encoding for the Ki-67 epitope. Lab Invest 1993, 68:629-636 [PubMed] [Google Scholar]
- 17.Ophascharoensuk V, Giachelli CM, Gordon K, Hughes J, Pichler R, Brown P, Liaw L, Schmidt R, Shankland SJ, Alpers CE, Couser WG, Johnson RJ: Obstructive uropathy in the mouse: role of osteopontin in interstitial fibrosis and apoptosis. Kidney Int 1999, 56:571-580 [DOI] [PubMed] [Google Scholar]
- 18.Alpers CE, Hudkins KL, Gown AM, Johnson RJ: Enhanced expression of “muscle-specific” actin in glomerulonephritis. Kidney Int 1992, 41:1134-1142 [DOI] [PubMed] [Google Scholar]
- 19.Huang XR, Tipping PG, Shuo L, Holdsworth SR: Th1 responsiveness to nephritogenic antigens determines susceptibility to crescentic glomerulonephritis in mice. Kidney Int 1997, 51:94-103 [DOI] [PubMed] [Google Scholar]
- 20.Bolland S, Ravetch JV: Spontaneous autoimmune disease in Fc(gamma)RIIB-deficient mice results from strain-specific epistasis. Immunity 2000, 13:277-285 [DOI] [PubMed] [Google Scholar]
- 21.Friend SL, Hosier S, Nelson A, Foxworthe D, Williams DE, Farr A: A thymic stromal cell line supports in vitro development of surface IgM+ B cells and produces a novel growth factor affecting B and T lineage cells. Exp Hematol 1994, 22:321-328 [PubMed] [Google Scholar]
- 22.Ray RJ, Furlonger C, Williams DE, Paige CJ: Characterization of thymic stromal-derived lymphopoietin (TSLP) in murine B cell development in vitro. Eur J Immunol 1996, 26:10-16 [DOI] [PubMed] [Google Scholar]
- 23.Park LS, Martin U, Garka K, Gliniak B, Di Santo JP, Muller W, Largaespada DA, Copeland NG, Jenkins NA, Farr AG, Ziegler SF, Morrissey PJ, Paxton R, Sims JE: Cloning of the murine thymic stromal lymphopoietin (TSLP) receptor: formation of a functional heteromeric complex requires interleukin 7 receptor. J Exp Med 2000, 192:659-670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brouet JC, Clauvel JP, Danon F, Klein M, Seligmann M: Biologic and clinical significance of cryoglobulins. A report of 86 cases. Am J Med 1974, 57:775-788 [DOI] [PubMed] [Google Scholar]
- 25.Ravetch JV: A full complement of receptors in immune complex diseases. J Clin Invest 2002, 110:1759-1761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakamura A, Yuasa T, Ujike A, Ono M, Nukiwa T, Ravetch JV, Takai T: Fcgamma receptor IIB-deficient mice develop Goodpasture’s syndrome upon immunization with type IV collagen: a novel murine model for autoimmune glomerular basement membrane disease. J Exp Med 2000, 191:899-906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yuasa T, Kubo S, Yoshino T, Ujike A, Matsumura K, Ono M, Ravetch JV, Takai T: Deletion of fcgamma receptor IIB renders H-2(b) mice susceptible to collagen-induced arthritis. J Exp Med 1999, 189:187-194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sedor JR, Carey SW, Emancipator SN: Immune complexes bind to cultured rat glomerular mesangial cells to stimulate superoxide release. Evidence for an Fc receptor. J Immunol 1987, 138:3751-3757 [PubMed] [Google Scholar]
- 29.Santiago A, Satriano J, DeCandido S, Holthofer H, Schreiber R, Unkeless J, Schlondorff D: A specific Fc gamma receptor on cultured rat mesangial cells. J Immunol 1989, 143:2575-2582 [PubMed] [Google Scholar]
- 30.Ravetch JV, Kinet JP: Fc receptors. Annu Rev Immunol 1991, 9:457-492 [DOI] [PubMed] [Google Scholar]
- 31.Young JD, Ko SS, Cohn ZA: The increase in intracellular free calcium associated with IgG gamma 2b/gamma 1 Fc receptor-ligand interactions: role in phagocytosis. Proc Natl Acad Sci USA 1984, 81:5430-5434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Anderson CL, Shen L, Eicher DM, Wewers MD, Gill JK: Phagocytosis mediated by three distinct Fc gamma receptor classes on human leukocytes. J Exp Med 1990, 171:1333-1345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Titus JA, Perez P, Kaubisch A, Garrido MA, Segal DM: Human K/natural killer cells targeted with hetero-cross-linked antibodies specifically lyse tumor cells in vitro and prevent tumor growth in vivo. J Immunol 1987, 139:3153-3158 [PubMed] [Google Scholar]
- 34.Chan PL, Sinclair NR: Regulation of the immune response. V. An analysis of the function of the Fc portion of antibody in suppression of an immune response with respect to interaction with components of the lymphoid system. Immunology 1971, 21:967-981 [PMC free article] [PubMed] [Google Scholar]
- 35.Phillips NE, Parker DC: Fc-dependent inhibition of mouse B cell activation by whole anti-mu antibodies. J Immunol 1983, 130:602-606 [PubMed] [Google Scholar]
- 36.Lopez-Armada MJ, Gomez-Guerrero C, Egido J: Receptors for immune complexes activate gene expression and synthesis of matrix proteins in cultured rat and human mesangial cells: role of TGF-beta. J Immunol 1996, 157:2136-2142 [PubMed] [Google Scholar]
- 37.Hora K, Satriano JA, Santiago A, Mori T, Stanley ER, Shan Z, Schlondorff D: Receptors for IgG complexes activate synthesis of monocyte chemoattractant peptide 1 and colony-stimulating factor 1. Proc Natl Acad Sci USA 1992, 89:1745-1749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Neuwirth R, Singhal P, Diamond B, Hays RM, Lobmeyer L, Clay K, Schlondorff D: Evidence for immunoglobulin Fc receptor-mediated prostaglandin2 and platelet-activating factor formation by cultured rat mesangial cells. J Clin Invest 1988, 82:936-944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Radeke HH, Janssen-Graalfs I, Sowa EN, Chouchakova N, Skokowa J, Loscher F, Schmidt RE, Heeringa P, Gessner JE: Opposite regulation of type II and III receptors for immunoglobulin G in mouse glomerular mesangial cells and in the induction of anti-glomerular basement membrane (GBM) nephritis. J Biol Chem 2002, 277:27535-27544 [DOI] [PubMed] [Google Scholar]
- 40.Tarzi RM, Davies KA, Robson MG, Fossati-Jimack L, Saito T, Walport MJ, Cook HT: Nephrotoxic nephritis is mediated by Fcgamma receptors on circulating leukocytes and not intrinsic renal cells. Kidney Int 2002, 62:2087-2096 [DOI] [PubMed] [Google Scholar]