

Abstract
A reduced activity of protein phosphatase 2A (PP2A) has been shown in brains of patients with Alzheimer’s disease (AD), a neurodegenerative disorder characterized histopathologically by amyloid plaques and neurofibrillary tangles. Tau, as the principal component of neurofibrillary tangles, can be hyperphosphorylated by a reduced activity of PP2A in vitro and by pharmacological approaches, suggesting a crucial role of PP2A in tangle formation. To dissect the role of PP2A in vivo, we previously generated transgenic mice with chronically reduced PP2A activity by expressing a dominant-negative mutant form of the PP2A catalytic subunit Cα, L199P, under the control of a neuron-specific promoter. In these mice, endogenous tau is phosphorylated at the epitopes Ser202/Thr205 and Ser422. In vitro, these tau phospho-epitopes can be phosphorylated by the kinases ERK and JNK, and the kinases themselves are negatively regulated by PP2A. In this study, we show that chronic inhibition of PP2A activity in L199P transgenic mice causes the activation of ERK and JNK as demonstrated by the phosphorylation and nuclear accumulation of the ERK and JNK substrates, Elk-1 and c-Jun. TUNEL staining revealed that activated JNK signaling was not associated with cell death. Our findings imply that PP2A is a negative regulator of the ERK and JNK signaling pathways in vivo, suggesting that in AD, tau hyperphosphorylation may be caused in part by PP2A dysfunction.
Protein phosphatase 2A (PP2A) is a serine/threonine-specific protein phosphatase with important roles in development, cell growth, and transformation. It accounts for a significant portion of the total phosphatase activity in many tissues and cell types. 1 All PP2A holoenzymes have a 36-kd catalytic subunit (C) and a 65-kd structural scaffolding subunit (A) in common. These core subunits assemble with regulatory and targeting subunits (B) of various sizes to form functionally distinct heterotrimers which are likely to have different functions in the cell. 2,3
More than 30 protein kinase activities are modulated by PP2A in vitro, and several kinases form stable complexes with PP2A. 4 Even if only a subset of these turn out to be physiological substrates, PP2A must play a major role in kinase regulation: extensive biochemical, pharmacological, and genetic evidence suggests that PP2A controls the activities of several major protein kinase families in the cell, including the mitogen-activated protein kinase (MAPK) cascades. 4-7 MAPKs include the extracellular signal-related kinases (ERKs), that are activated by mitogenic stimuli including growth factors, the c-Jun N-terminal kinases (JNKs), and p38 MAPKs, that are activated by multiple stimuli, including cellular stress and apoptotic signals.
Numerous observations in vitro suggest that PP2A plays a major role in the down-regulation of the ERK pathway, and that PP2A may be active at several levels of the signaling cascade. 4 MAPK dephosphorylation has been attributed to PP2A, in addition to a unique family of dual specificity phosphatases called MAP kinase phosphatases (MKPs). 8-12 In addition, genetic evidence also implicates PP2A in positive and negative regulation of the ERK/MAPK pathway during Drosophila photoreceptor development. 4 Finally, treatment of cells with PP2A-inhibiting toxins, such as okadaic acid (OA), lead to the activation of ERK and JNK. 13,14 In agreement with this, activation of PP2A inhibited JNK activity. JNK co-precipitated with the PP2A structural subunit Aα, supporting the conclusion that PP2A is a key regulator of JNK. 15 However, as several PP2A-like phosphatases are also targets of OA, distinguishing these enzymes from PP2A by the use of OA alone is difficult in studies of intact cells.
PP2A was implicated in pathological processes including human neurodegenerative diseases: 16-18 the activity of PP2A is reduced in Alzheimer’s disease (AD) brains, and PP2A mRNA expression is decreased. 2,19,20 Neurofibrillary tangles in AD are mainly composed of the microtubule-associated protein tau, which is a substrate of PP2A in vitro. In tangles, tau is phosphorylated at a higher degree at physiological sites and at additional pathological sites. 21,22 Tau hyperphosphorylation may precede tangle formation in the disease process. As phosphorylated tau binds less well to microtubules, its soluble levels increase, possibly an initial event in the assembly of abnormal tau filaments. 23,24 In experimental systems, dephosphorylation of tau can be blocked acutely by OA. Experimental attempts to analyze the effects of chronically reduced PP2A activity in vivo by gene targeting were complicated by embryonic lethality of the null mutant. 25,26 Therefore, we generated transgenic mice that express a dominant-negative mutant form of the catalytic subunit Cα of PP2A, L199P, in neurons resulting in a chronic reduction of PP2A activity in total brain homogenates. 27 In these mice, endogenous murine tau protein was hyperphosphorylated at two distinct epitopes, the physiological site Ser-202/Thr-205 and the pathological site Ser-422. Here, we show an activation of the ERK and JNK signaling pathway in L199P transgenic mice, suggesting an additional, indirect role of PP2A in tau hyperphosphorylation, besides the direct dephosphorylation of tau by PP2A.
Materials and Methods
Transgenic Mice
To determine the role of PP2A in tau phosphorylation, we previously generated transgenic mice expressing a dominant-negative mutant form of PP2ACα, L199P, under control of a neuron-specific promoter. 27 The cDNA of the human PP2A Cα mutant L199P was fused to a single hemagglutinin (HA) epitope located immediately downstream of the start codon, and subcloned into the neuron-specific murine Thy1.2 expression vector. Transgenic mice were produced by pronuclear microinjection of B6D2F1 × B6D2F1 embryos, and founder animals were intercrossed with C57BL/6 mice to establish lines. The L199P transgenic mice showed a chronic reduction of PP2A activity to 66%. Endogenous tau protein was hyperphosphorylated at the AT8 epitope Ser-202/Thr-205, as determined by immunohistochemistry, and at the pS422 epitope, as shown by Western blotting, whereas other phospho-epitopes of tau were not phosphorylated. Tau accumulated in the cell bodies and dendrites of cortical neurons and cerebellar Purkinje cells. 27
Antibodies
For detection of the hemagglutinin-epitope by immunohistochemistry (IH), two monoclonal anti-HA antibodies (Roche, Rotkreuz, Switzerland; IH 1:200, WB (Western blotting) 1:1000, and Santa Cruz Inc., Santa Cruz, CA; IH 1:100) and a polyclonal anti-HA antibody (Santa Cruz; IH 1:500) were used. 28
Rabbit antiserum MEK1 (Transduction Laboratories, BD Biosciences, San Diego, CA; phosphorylation-state independent, IH 1:100, WB 1:200) detects MEK1 and MEK2, 29 whereas the phospho-MEK1/2 antiserum detects MEK only when phosphorylated at Ser-217/Ser-221 (Cell Signaling Technology, Beverly, MA; IH 1:1000, WB 1:1000); 30 antiserum ERK1 (K-23) (Santa Cruz; IH 1:100, WB 1:200) and monoclonal antibody A5 (Gibco BRL, Invitrogen, Carlsbad, CA; IH 1:100) detect ERK1 and ERK2; 31 monoclonal antibody E-4 detects ERK1 and ERK2 phosphorylated at Tyr-204 (Santa Cruz; WB 1:100); 32 Elk-1 antiserum (Cell Signaling Technology; IH 1:50, WB 1:1000) detects total, phosphorylation-state independent levels of Elk-1; 33 and mouse monoclonal antibody sc-8406 (Santa Cruz; IH 1:50, WB 1:100) reacts with Elk-1 phosphorylated at Ser-383. 34
Rabbit antiserum JNK1 (C-17;sc-474) (Santa Cruz; IH 1:100, WB 1:200) detects JNK1 p46 and JNK3, 35 mouse monoclonal P-JNK (G-7;sc-6254) (Santa Cruz; IH 1:40, WB 1:100) reacts with phosphorylated JNK1, JNK2, and JNK3, and does not cross-react with ERK1, ERK2, or p38; 36,37 rabbit antiserum sc-45 (Santa Cruz; IH 1:50, WB 1:200) detects c-Jun and does not cross-react with Jun B or Jun D; 38,39 and mouse monoclonal P-c-Jun (KM-1; sc-822) (Santa Cruz; IH 1:50, WB 1:100) reacts with c-Jun phosphorylated at Ser-63. 40
Monoclonal anti-calbindin-D-28K (Sigma, Buchs, Switzerland; IH 1:1000, WB 1:3000) detects calbindin; monoclonal antibody AC-15 detects β-actin (Abcam Limited, Cambridge, UK; WB 1:1000); rabbit antiserum A8717 was raised against the carboxy-terminal portion of the amyloid precursor protein, APP (Sigma; IH 1:1000); phospho-APP (Thr-668) polyclonal antiserum (Cell Signaling Technology; IH 1:50) detects different isoforms of the amyloid precursor protein (APP) only when phosphorylated at Thr668; and the cleaved caspase-3 (Asp-175) antiserum detects the large fragment of activated caspase-3 thatresults from cleavage after Asp-175 (Cell Signaling Technology; IH 1:50).
Secondary Antibodies
For peroxidase/3-3′-diaminobenzidine (DAB) stainings, secondary antibodies (Vectastain ABC kits PK-6101 and PK-6102) were obtained from Vector Laboratories (Burlingame, CA) and Santa Cruz Inc. (biotin-labeled anti-goat IgG, 1:500). For immunofluorescence, secondary Cy3-conjugated rabbit antibodies (1:500) and Cy2-conjugated mouse antibodies (1:500) were obtained from Jackson ImmunoResearch Laboratories (Baltimore, PA).
Immunohistochemistry
Brains from three 12-month-old transgenic and three control mice were transcardially perfused with 4% (w/v) paraformaldehyde. Immunohistochemical and immunofluorescence stainings were done on 4-μm parasaggital paraffin sections, by using standard published procedures. 41 In addition, 40-μm parasaggital vibratome sections were permeabilized with 0.1% (v/v) NP-40 (Calbiochem, San Diego, CA). Some of the sections were pretreated with 5 μg/ml proteinase K in phosphate-buffered saline (PBS) at 37°C for 2.5 minutes for signal enhancement. Sections were dehydrated in an ascending series of ethanol and flat-embedded between glass slides and cover-slips in Eukitt (Kindler, Freiburg, Germany) or Mowiol 4–88 (Roche) containing 2.5% (w/v) diazabicyclo[2.2.2]octane (Sigma). For all stainings, controls were included in which the primary antibody was omitted. Specificity of the commercial antibodies and antisera was confirmed by immunoblot analysis (see below).
Densitometric quantification of the immunohistochemical analysis was performed with the Image Gauge 3.45 Science Lab 99 program (Fuji Foto Film Ltd. and Koshin Graphic Systems). The area of single immunopositive cells was selected and the immunoreactivity per area determined. Each measurement was performed with more than 20 cells and the mean and SD were calculated.
TUNEL Stainings
To detect cells undergoing apoptosis, the POD in situ cell death detection kit (Roche) was used as described. 24 In brief, paraffin-embedded sections were rehydrated, treated with 5 μg/ml proteinase K in PBS for 10 minutes at 37°C, washed with ice-cold PBS four times, incubated in 3% H2O2 in methanol for 5 minutes at room temperature, and washed again. For a positive control, sections were incubated in 100 μg/ml DNase I (Roche) in 20 mmol/L TrisHCl pH 8.0/10 mmol/L MgCl2 for 10 minutes at room temperature. Then, the labeling solution containing the enzyme terminal deoxynucleotidyl transferase (POD Kit) was diluted 1:10 and added to the sections for 30 minutes at 37°C. For a negative control, the enzyme was omitted from the labeling solution. Sample sections were washed, blocked with 2% (w/v) bovine serum albumin (BSA), and incubated with convert solution (POD kit), washed again, and incubated in 1:10 diluted DAB solution (Pierce, Perbio Science, Helsingborg, Sweden) for 10 minutes at room temperature to visualize the fragmented DNA. Sections were dehydrated and mounted in Eukitt.
Immunoprecipitations and Immunoblots
To analyze protein levels, total cerebellar extracts were prepared and normalized for protein contents by using the DC Protein Assay (Bio-Rad, Hercules, CA), as described. 27 To obtain nuclear extracts, cerebelli were dounce homogenized in a hypotonic buffer (10 mmol/L Hepes, 10 mmol/L KCl, 5 mmol/L MgCl2, 1 mmol/L DL-dithiothreitol (DTT), 1 mmol/L phenylmethanesulfonyl fluoride (PMSF), 50 mmol/L NaF, 1 mmol/L sodium vanadate, 10 μmol/L ammonium molybdate, supplemented with Complete protease inhibitors (Roche)) and incubated on ice for 15 minutes. All subsequent steps were done at 4°C. After centrifugation for 10 minutes at 850 ×g, the pellet was resuspended in 1 ml hypotonic buffer and sheared with a 22-gauge syringe. Next, the homogenate was adjusted to 0.25 mol/L sucrose, sheared again, and centrifuged for 5 minutes at 500 ×g. The supernatant (“fraction A”) was saved. The pellet was washed three times with a buffer containing 0.25 mol/L sucrose, 6 mmol/L MgCl2, 10 mmol/L Tris-HCl (pH 7.4), 0.1% (v/v) Triton X-100, 1 mmol/L PMSF, 50 mmol/L NaF, 1 mmol/L sodium vanadate, and 10 μmol/L ammonium molybdate in the presence of Complete protease inhibitors for 5 minutes at 500 ×g to obtain the “nuclear fraction.” The supernatant from the first wash was pooled with “fraction A” to obtain the “cytosolic fraction.” 42 Extracts were separated by polyacrylamide gel electrophoresis, followed by electrophoretic transfer onto a nitrocellulose membrane (Hybond-ECL, Amersham, Buckinghamshire, England). Ponceau stainings were included to confirm loading of comparable amounts of protein. The membranes were blocked and reacted with primary and horseradish peroxidase (HRP)-conjugated secondary antibodies as described. 27
For immunoprecipitations, cerebelli were homogenized in lysis buffer (50 mmol/L Tris (pH 8.0), 150 mmol/L NaCl, 1 mmol/L EDTA, 10 μmol/L ammonium molybdate, 50 mmol/L NaF, 1 mmol/L sodium vanadate, and 1% (v/v) Triton-X-100) in the presence of protease inhibitors (Complete, Roche), and centrifuged at 5000 ×g, followed by a centrifugation step at 20000 ×g. The supernatant fractions were used for the subsequent incubations. Antibodies (specific for calbindin, c-Jun, and phospho-c-Jun) were bound to Protein A/Protein G-Sepharose (Amersham) overnight at 4°C, followed by five washes with lysis buffer, and incubated with lysates normalized for equal protein content overnight at 4°C. A no-lysate control was included. After five washes, the Protein A/Protein G-Sepharose beads were eluted with sample buffer at 70°C. Samples were separated on a 10% NuPage gel (Novex, Invitrogen, Basel, Switzerland), and transferred onto a nitrocellulose membrane. Residual protein-binding sites were blocked by incubation with 5% semi-fat dried milk in Tris-buffered saline (TBS) with 0.1% (v/v) Tween-20 for 1 hour at room temperature, followed by a 3-hour incubation at room temperature with the primary antibody in TBS with 0.1% Tween-20, 1% (w/v) semi-fat fried milk, and 0.02% sodium azide. Antibodies specific for c-Jun, phospho-c-Jun, and calbindin were used at 1:500, 1:500, and 1:3000 dilutions, respectively. Following four washes for a total of 30 minutes in TBS, the membranes were incubated with an HRP-linked sheep anti-rabbit antibody (Amersham, NA931) at 1:4000 dilutions for 1 hour at room temperature, followed by a 30-minute wash in TBS. Then, the membrane was incubated for 1 minute in ECL reagent (Vector Laboratories), excess liquid was removed, and the membrane was exposed to X-ray films. For re-use, membranes were stripped for 30 minutes at 50°C in 100 mmol/L 2-mercaptoethanol, and 2% (w/v) SDS in 62.5 mmol/L Tris (pH 6.8).
Results
Colocalization of Hyperphosphorylated Tau with JNK and MEK in PP2A Cα L199P Mutant Mice
PP2ACα L199P dominant-negative mutant mice express the transgene in hippocampal neurons at low levels and in cortical and cerebellar neurons at high levels (Figure 1) ▶ . Transgenic Purkinje cells tend to be smaller as compared with Purkinje cells in controls. A functional consequence of the chronic reduction of PP2A activity in these mice is the hyperphosphorylation of tau at the AT8 epitope. 27 To determine whether kinases known to be regulated by PP2A in vitro are also affected in vivo in these cells, we co-stained transgenic and control brain sections with the mouse monoclonal antibody AT8 and rabbit antisera specific for JNK and MEK, respectively (Figure 2) ▶ . Immunohistochemical stainings revealed that in AT8-positive transgenic Purkinje cells, JNK levels were increased (Figure 2, A–C) ▶ in comparison to non-transgenic littermate controls (Figure 2, D–F) ▶ . Additionally, cells with high levels of AT8 staining such as brain stem (Figure 2G–I) ▶ and cortical neurons (Figure 2, K–M) ▶ revealed a similar co-localization pattern. Likewise, MEK, the upstream kinase of ERK, was co-localized with AT8 in cortical neurons (Figure 2, N–P) ▶ as well as in Purkinje cells and neurons of the brain stem (data not shown). Together, these findings suggest that L199P expression causes increased levels of kinases in a subset of cells that either express the transgene above a critical level or may be more susceptible to inhibition of PP2A.
Figure 1.
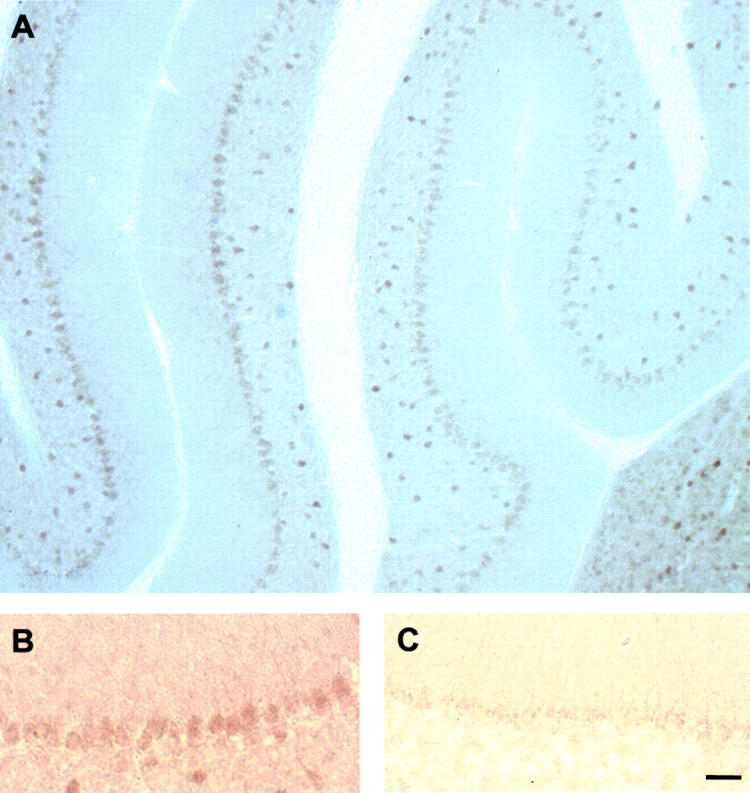
Mutant PP2A Cα L199P expression in cerebellar Purkinje cells. Parasaggital vibratome sections of PP2A Cα L199P transgenic mice (A and B) and age-matched wild-type littermate controls (C) were stained with an antibody directed against the HA tag fused to the Cα L199P protein. Highest expression of Cα L199P was found in Purkinje cells of transgenic mice, and was absent in the control. In addition to Purkinje cells, L199P is expressed in additional cell types including cortical neurons, the brain stem, and, to a much lesser extent, the hippocampus. 27 Bar, 150 μm (A); and 50 μm (B and C).
Figure 2.
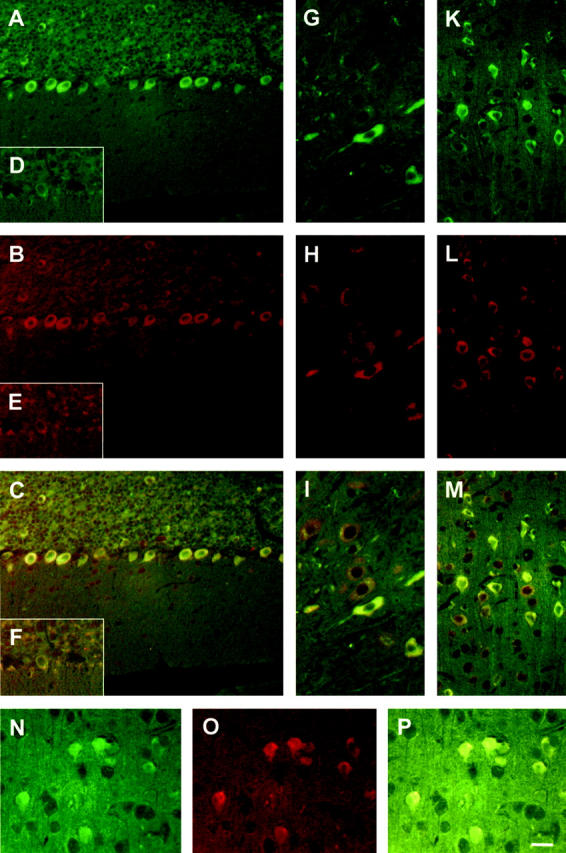
Increased JNK levels in cells with an increased phosphorylation of tau. In cells expressing L199P at high levels, the PP2A substrate tau is phosphorylated at the AT8-epitope Ser202/Thr205. 27 Immunohistochemical stainings using phosphorylation-independent anti-JNK and anti-MEK antisera reveal that in AT8-positive transgenic Purkinje cells (A, Cy2, shown in green), JNK levels are increased (B, Cy3, shown in red; C, merge), whereas in non-transgenic littermate controls, both AT8 and JNK stainings are weak (D–F). Additional cell-types with high levels of AT8 staining such as brain stem (G–I) and cortical neurons (K–M) reveal a similar co-localization pattern. Likewise, MEK (O, in red) is co-localized with AT8 (N, in green) in many brain areas including the cortex (P, merge). Bar, 30 μm (A–M); and 20 μm (N–P).
Inhibition of PP2A Activates the ERK Signaling Pathway
To determine in more detail whether hyperphosphorylation of tau in L199P brains is caused by up-regulated kinases, we analyzed the PP2A-regulated kinases ERK and JNK and their substrates by immunohistochemistry. All immunostainings were done in duplicates with sections obtained from three transgenic mice and three control mice littermates. The transcription factor Elk-1 is a preferred target of activated ERK. 43 Phosphorylation of several sites, in particular Ser-383, of Elk-1, is critical for transcriptional activation. The upstream kinase of ERK, MEK1, is known to accumulate transiently in the nucleus on activation. Compared to control cells, both the phosphorylated form of MEK (Figure 3, A and B) ▶ as well as MEK1 (Figure 3, C–F) ▶ accumulated both in the cytoplasm and the nucleus of transgenic Purkinje cells, possibly due to the chronic inhibition of PP2A. In Purkinje cells, where the transgene was predominantly expressed and where tau was hyperphosphorylated, ERK accumulated significantly in the cytoplasm and nucleus as compared to controls (Figure 3, G–K) ▶ .
Figure 3.
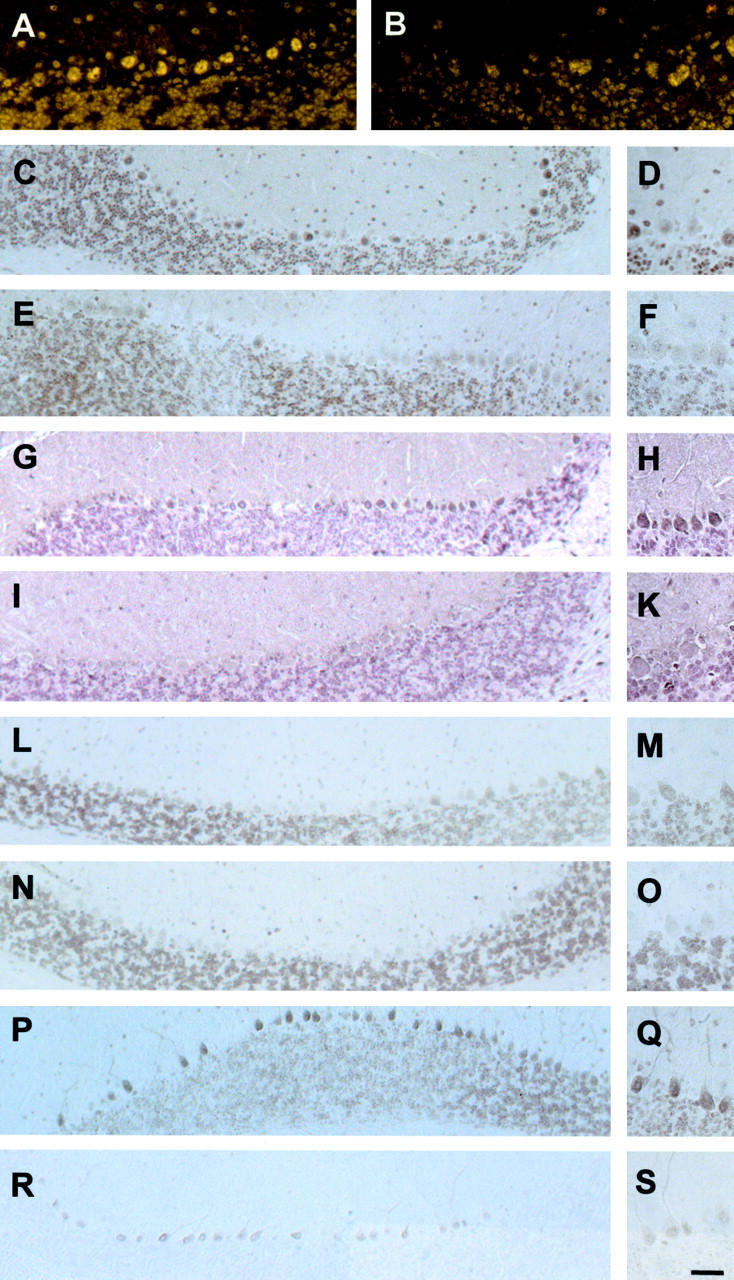
Activated ERK signaling in L199P expressing Purkinje cells. High and low magnification images are shown for images C–S. Transgenic and control sections were taken at identical settings. In transgenic Purkinje cells, the phosphorylated form of the upstream kinase of ERK, MEK1, accumulates both in the cytoplasm and the nucleus of transgenic (A) compared to control cells (B). Using a phosphorylation-independent antibody, stainings reveal increased levels of MEK both in the cytoplasm and nucleus of transgenic (C and D) compared to control cells (E and F). ERK accumulates significantly in the cytoplasm and nucleus of transgenic neurons (G and H) as compared to controls (I and K). Although in transgenic Purkinje cells (L and M), stainings do not reveal significantly higher levels of the transcription factor Elk-1 compared to controls (N and O), an antibody specific for Elk-1 phosphorylated at Ser-383 reveals, in transgenic (P and Q) compared to wild-type control sections (R and S), enriched staining of Purkinje cells that is mainly confined to the nucleus (O). Bar, 30 μm (A and B); 100 μm (C, E, G, I, L, N, P, and R); and 50 μm (D, F, H, K, M, O, Q, and S).
ERK signaling is known to result in the accumulation of phosphorylated Elk-1 in the nucleus. Although in transgenic Purkinje cells stainings revealed only slightly higher levels of the transcription factor Elk-1 compared to controls (Figure 3, L–O) ▶ , an antibody specific for Elk-1 phosphorylated at Ser-383 revealed, in transgenic compared to wild-type control sections, enriched staining of Purkinje cells that was mainly confined to the nucleus (Figure 3, P–S) ▶ .
To exclude that our immunohistochemical findings reflect a generalized increase of phospho-proteins in Purkinje cells of transgenic brains, we used two antisera, one phosphorylation-independent antiserum specific for the carboxy-terminus of APP, the other specific for APP phosphorylated at Thr-668. 44-46 No differences were found when Purkinje cells of transgenic mice were compared with controls (Figure 4, A–D) ▶ . However, there is evidence for an altered dendritic morphology (Figure 4, C–F) ▶ , and the subcellular localization of APP is known to be determined by phosphorylation of Thr-668. 47 To further exclude that transgenic Purkinje cells display increased protein expression, we stained the sections with an antibody directed against calbindin, a protein predominantly expressed by Purkinje cells. Although the Purkinje cells tended to be smaller in transgenic brain, by immunohistochemistry, levels of calbindin were not significantly different compared to controls (Figure 4, E and F) ▶ .
Figure 4.
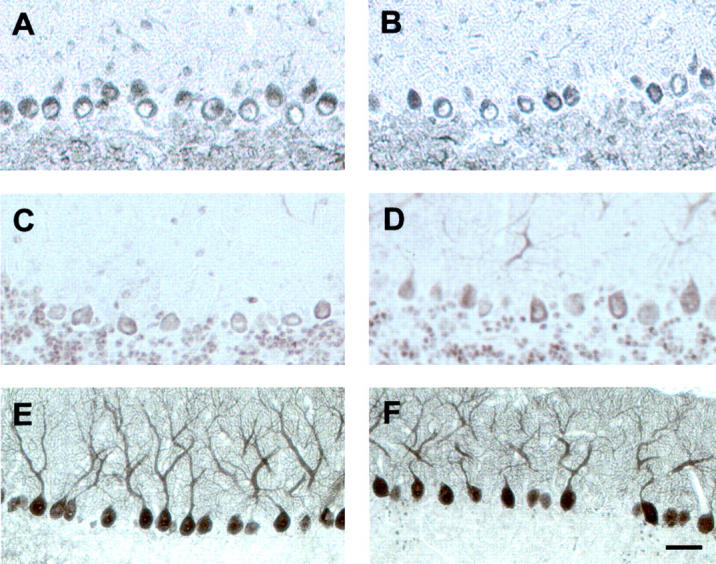
Expression of APP and calbindin are not affected in transgenic Purkinje cells. No generalized increase of phospho-proteins was observed in Purkinje cells of transgenic (A and C) compared with wild-type brains (B and D), as shown with a phosphorylation-independent antiserum specific for the carboxy-terminus of APP (A and B), and an antiserum specific for APP phosphorylated at Thr-668 (C and D). To exclude that transgenic Purkinje cells displayed increased protein expression, we analyzed calbindin, a protein predominantly expressed by Purkinje cells. We could not detect any significant differences in the intensity of immunoreactivity (E and F, see also Figure 5 ▶ ). Bar, 50 μm (A–D); and 75 μm (E and F).
Notably, staining intensities for the different kinases investigated and their substrates varied between individual cells both in transgenic and wild-type mice, possibly due to differences in physiological states. To quantify the immunohistochemical findings, we analyzed between 20 and 50 cells per group by densitometry. Only those cells which were cut in a medial plane and which showed immunoreactivity above background, were counted. Generally, the number of immunopositive cells was much higher in transgenic compared with control animals for most proteins analyzed, including MEK and ERK (Figures 5 and 6) ▶ . For the densitometric analysis, this aspect was not included, as we only determined the signal intensity of immunopositive but not of all Purkinje cells. Therefore, the calculated differences, although significant, are smaller than is actually the case if all Purkinje cells would be computed. For all stainings, signal intensities in the control group were set at 100. Whereas levels of APP, phospho-APP, and calbindin were not affected by the presence of the L199P transgene (APP: control, 100 ± 6.1 relative units (U), transgenic, 98.6 ± 5.1 U, P = 0.517; phospho-APP: control, 100 ± 11.7 U, transgenic, 99.8 ± 17.5 U, P = 0.754; calbindin: control, 100 ± 4.4 U, transgenic, 100.7 ± 3.3 U, P = 0.696; Mann-Whitney U-test), the densitometric analysis revealed up to twofold increases of the phosphorylated forms of MEK and Elk-1 in transgenic Purkinje cells (Figure 5) ▶ . This difference was statistically significant (phospho-MEK: control, 100 ± 23.8 relative units (U), transgenic, 192 ± 14.5 U, P < 0.001; phospho-Elk-1: control, 100 ± 8 U, transgenic, 201.7 ± 9.4 U, P < 0.001; Mann-Whitney U-test). To extend our immunohistochemical findings, we performed a Western blot analysis of cerebellar extracts of L199P mice and non-transgenic littermate controls with a panel of antibodies used in the immunohistochemical analysis (Figure 7A) ▶ . Levels of phosphorylated Elk-1 were higher in transgenic brain extracts. MEK levels were reduced in transgenic extracts. However, the amount of phosphorylated MEK was equal, suggesting an increased ratio of phosphorylated MEK to total MEK. However, with most antibodies no significant differences were observed, possibly reflecting an under-representation of Purkinje cells in the total cerebellar homogenates.
Figure 5.

Quantitative densitometric analysis of the immunohistochemical stainings. For a quantitative analysis of the immunohistochemical data, obtained with antibodies against APP, P-APP, calbindin, MEK, P-MEK, ERK, Elk-1, and P-Elk1, between 20 and 50 cells per group were densitometrically analyzed. Signal intensities in the control group were set at 100. Whereas APP, phospho-APP, and calbindin levels are not affected by the presence of the L199P transgene [Mann-Whitney U-test: not significant (two-tailed exact significance) comparing transgenic with control mice], significant increases are found for levels of the phosphorylated forms of MEK and Elk-1 [Mann-Whitney U-test: P < 0.001 (two-tailed exact significance) comparing transgenic with control mice].
Figure 7.

Western blot analysis and immunoprecipitation of cerebellar extracts. By Western blot analysis of cerebellar extracts with most antibodies, no significant differences are revealed between L199P mice (tg) and non-transgenic littermate controls (ct), possibly reflecting an under-representation of Purkinje cells in the total cerebellar homogenates. However, levels of phosphorylated Elk-1 are higher in transgenic brain extracts. Also, the ratio of phospho-MEK to MEK is increased (A). Immunoprecipitation of c-Jun and phosphorylated c-Jun from total cerebellar lysates, followed by immunoblots with antibodies specific for c-Jun and phosphorylated c-Jun does not reveal differences between genotypes because Purkinje cells proteins are not enriched by this method (B).
Together, these results show that the chronic inhibition of PP2A in transgenic mice causes a significant up-regulation of MEK1 and ERK, as well as a nuclear accumulation of the activated forms of the transcription factor Elk-1 in a phosphorylated form in the nucleus, suggesting that inhibition of PP2A causes an activation of the ERK signaling pathway.
Inhibition of PP2A Activates JNK and c-Jun
In vitro, PP2A has been shown to dephosphorylate the stress-activated kinase JNK. c-Jun is a substrate of JNK. It is activated by phosphorylation at both Ser-63 and Ser-73, and accumulates in the nucleus. 48 To determine whether expression of L199P also affected the JNK signaling pathway in vivo, we analyzed JNK by immunohistochemistry and found increased levels in Purkinje cells of transgenic mice with pronounced staining of the somatodendritic compartment (Figure 6, A and B) ▶ . JNK1 is present in both the cytoplasm and in nuclei as has been shown previously for cerebellar granule cell cultures where JNK1 is mainly extranuclear, whereas JNK2/3 is more abundant in the nucleus. 49 To determine whether JNK was activated, we used an antibody specific for phosphorylated JNK and found that it accumulated in the nucleus of most transgenic Purkinje cells. This only occurred rarely and with lower intensities than that seen in control sections (Figure 6, C and D) ▶ . To determine whether increased levels of JNK resulted in the accumulation of phosphorylated c-Jun in the nucleus, we used an antibody specific for c-Jun phosphorylated at Ser-63. We found that phospho-c-Jun was mainly localized to the nucleus of transgenic Purkinje cells (Figure 6, E and F) ▶ .
Figure 6.

Activated JNK signaling in L199P expressing Purkinje cells. Parasaggital sections were stained with an anti-JNK antibody demonstrating increased levels in Purkinje cells of transgenic mice compared to controls, with pronounced staining of the somatodendritic compartment (A and B). To determine whether JNK was activated, we used an antibody specific for phosphorylated JNK and found that it accumulated in the nucleus of most transgenic Purkinje cells (C and D). This occurred rarely and with lower intensities than that seen in control sections. To facilitate the visualization of the cells and to illustrate the pronounced nuclear staining more clearly, the cell borders of Purkinje cells are marked with a black line. Phospho-c-Jun is mainly localized to the nucleus of transgenic Purkinje cells (E and F). A quantitative densitometric analysis reveals significant increases of JNK, P-JNK, and P-c-jun levels in transgenic compared with control Purkinje cells [Mann-Whitney U-test: JNK: control, 100 ± 38.2 relative units (U), transgenic, 262.5 ± 38.2 U, P < 0.001; phospho-JNK: control, 100 ± 32.7 U, transgenic, 552.7 ± 92.3 U, P = 0.001; phospho-c-jun: control, 100 ± 32.7 U, transgenic, 174.5 ± 32.4 U, P < 0.001 (two-tailed exact significance)] (G). Bar, 50 μm (A and B); 30 μm (C and D); and 25 μm (E and F).
Again, we performed a Western blot analysis of cerebellar extracts of L199P mice and non-transgenic littermate controls (Figure 7, A and B) ▶ . Whereas levels of phosphorylated Elk-1 were elevated in transgenic brain extracts, no significant differences were observed for JNK, phospho-JNK, and c-jun, possibly reflecting an under-representation of Purkinje cells in the total cerebellar homogenates. Analysis of nuclear extracts (data not shown) did not reveal further differences between transgenic and control extracts, possibly for the same reason as outlined above. The slightly lower levels in transgenic total extracts of calbindin, a protein mainly expressed by Purkinje cells, may be due to the finding that a subset of transgenic Purkinje cells tends to be smaller as compared with control cells (Figure 4) ▶ . As phosphorylated c-jun was not detectable in total extracts, we immunoprecipitated c-Jun and phosphorylated c-Jun from total cerebellar lysates, followed by immunoblots with antibodies specific for c-Jun and phosphorylated c-Jun (Figure 7B) ▶ . As this method does not allow for an enrichment of Purkinje cells proteins, the immunoprecipitation did not reveal differences between genotypes. Therefore, we performed a quantitative densitometric analysis which revealed up to fivefold increases of JNK, phosphorylated JNK, and phosphorylated c-jun levels in transgenic compared with control Purkinje cells (Figure 6G) ▶ . This difference was statistically significant (JNK: control, 100 ± 38.2 relative units (U), transgenic, 262.5 ± 38.2 U, P < 0.001; phospho-JNK: control, 100 ± 32.7 U, transgenic, 552.7 ± 92.3 U, P = 0.001; phospho-c-Jun: control, 100 ± 32.7 U, transgenic, 174.5 ± 32.4 U, P < 0.001; Mann-Whitney U-test). Together, our results demonstrate that chronic inhibition of PP2A by expression of a dominant-negative mutant form of PP2A Cα causes the activation of JNK and the phosphorylation and nuclear accumulation of c-Jun, suggesting an activation of the JNK signaling pathway.
Activation of JNK Is Not Correlated with Increased Apoptosis
As JNK has been implicated in apoptosis, we determined whether activation of JNK in Purkinje cells of L199P mice is related to increased apoptosis. We found no increase in TUNEL-positive Purkinje cells in transgenic mice compared to controls (Figure 8, A and B) ▶ . As a negative control, we omitted the POD convert solution (Figure 8C) ▶ and, as a positive control, we pretreated sections with DNase I (Figure 8D) ▶ . As an additional control, we included a spinal cord section of a tau transgenic mouse line with a motor phenotype (Figure 8E) ▶ . Stainings of L199P transgenic brain sections with an antiserum that detects the large fragment of activated caspase-3 resulting from cleavage after Asp-175, revealed no differences compared to controls (data not shown). Together, these data demonstrate that JNK activation can be dissociated from the induction of apoptosis.
Figure 8.

Activation of JNK is not associated with apoptosis. Activation of JNK in L199P brains was not correlated with increased apoptosis as shown by TUNEL stainings. No TUNEL-positive Purkinje cells were found in transgenic mice compared with controls (A and B). As a negative control, the POD convert solution was omitted (C) and, as a positive control, sections were pretreated with DNase I (D). In addition, a spinal cord section of a tau transgenic mouse line with a motor phenotype was included (E). Bar, 50 μm.
Discussion
Chronic Inhibition of PP2A Induces Activation of the ERK and JNK Signaling Pathways
In the present study, we analyzed transgenic mice that express a dominant-negative mutant form of PP2A Cα, L199P. We have previously shown that the PP2A activity in these mice was reduced, and that tau was hyperphosphorylated and accumulated in cortical and cerebellar Purkinje cells. 27 Here, we demonstrated that impairment of PP2A activity in vivo resulted in increased levels of JNK and MEK in cortical neurons, Purkinje cells, and neurons of the brainstem. Those cells that displayed elevated levels of JNK and MEK showed increased phosphorylation of the PP2A substrate tau. Activation of ERK and JNK signaling, as shown by the accumulation of phosphorylated forms of the transcription factors Elk-1 and c-Jun in the nucleus was shown in detail for Purkinje cells of transgenic mice. By that, we extend previous in vitro studies that have shown a role of PP2A in the negative regulation of mammalian ERK and JNK cascades. 11,12,50 Our findings are also in agreement with in vitro studies showing that inhibition of PP2A by expression of SV40 small t antigen stimulates the mitogenic ERK cascade resulting in MEK activation. 30
Our findings suggest that expression of the dominant-negative mutant form of PP2A Cα, L199P, in Purkinje cells prevents the inactivation of the ERK and JNK signaling pathway by maintaining Elk-1 and c-Jun in an activated state as shown by increased levels of phosphorylated Elk-1 and c-Jun in the nucleus. Although we cannot exclude that additional kinases are also activated, the finding that tau is only hyperphosphorylated at two of the multiple potential phosphorylation sites (see below) makes it unlikely that multiple kinase pathways are up-regulated. This notion is further supported by the finding that phospho-epitopes of other proteins, such as Thr-668 of APP, were not phosphorylated to a higher degree in L199P mice.
We showed an activation of the ERK and JNK signaling pathways by PP2A inactivation in cortical neurons, brain stem neurons, and Purkinje cells of the cerebellum. In our analysis, we concentrated on the latter as in Purkinje cells the activation of these pathways was most pronounced. A likely reason is that in these cells the transgene was expressed at highest levels. However, a distinct role of PP2A in cerebellar functions is suggested by the identification of a familiar form of a cerebellar syndrome, spinocerebellar ataxia 12, that is associated with a CAG expansion in the 5′ region of the PP2A regulatory subunit PR55/Bβ, a gene ubiquitously expressed in brain. 51,52
Tau Is Phosphorylated in Vivo at Sites That Are Phosphorylated by ERK and JNK in Vitro
We have previously shown in the L199P transgenic mice with decreased PP2A activity that endogenous murine tau is hyperphosphorylated at the AT8 epitope Ser-202/Thr-205, a physiological tau epitope, and at the pS422 epitope Ser-422, a pathological tau epitope. Theoretically, this hyperphosphorylation could be directly caused by PP2A, which can bind to tau and may dephosphorylate tau at reduced levels, 53 or indirectly by kinases that are known to be either substrates of PP2A or that exist in complexes with PP2A, 4,54 or both. Here we showed that reduction of PP2A activity caused ERK and JNK signaling as shown by the accumulation of phosphorylated transcription factors in the nucleus. In vitro, the AT8 and pS422 epitopes are excellent substrates of ERK and JNK. 21,55 More specifically, in the course of the characterization of a novel phosphorylation-dependent monoclonal antibody against tau, it was shown that Ser-422 is a good in vitro substrate for ERK, but not for glycogen synthase kinase-3 (GSK-3) or neuronal cdc2-like kinase (Cdk5). 21 These studies implied that ERK or an ERK-like enzyme may be involved in the phosphorylation of AD tau. 21 In a related study, it was shown that, in comparison with GSK-3, JNK was more effective at phosphorylating Ser-202/Thr-205 (the AT8 epitope) and especially effective at phosphorylating Ser-422. Consistent with this, JNK-phosphorylated tau was weakly stained by the AT180 antibody directed against phosphorylated Thr-231/Ser-235 (an epitope not phosphorylated in brains of L199P transgenic mice), whereas GSK-3-phosphorylated tau was labeled more powerfully by AT180. 55 It has been shown by the association of c-Jun and phospho-JNK with neurofibrillary tangles that JNK could play a role in neurons undergoing pathological changes. 56 Our data suggest that tau hyperphosphorylation at epitopes Ser-202/Thr-205 and Ser-422 in PP2A mutant mice is due to increased activities of ERK and JNK signaling. These findings are in agreement with those obtained by pharmacological treatment of rat brain slices with okadaic acid, an inhibitor of PP2A and PP1. An analysis by immunohistochemistry and Western blotting revealed that levels of activated ERK and MEK1/2 were dramatically increased. 57 Together with our findings, these results strongly suggest that PP2A can down-regulate the serine and threonine sites on ERK and MEK1/2 and that a reduced PP2A activity in AD brain might cause the abnormal phosphorylation of distinct sites of tau.
The Ser-422 epitope might be of high importance in tau pathogenesis because injection of β-amyloid fibrils (Aβ42) into brains of P301L mutant tau transgenic mice caused phosphorylation of tau at Ser-422 together with a significant increase in the number of neurofibrillary tangles. Together, these data suggest a role of the ERK and JNK signaling pathways in the phosphorylation of the Ser-422 epitope and in neurofibrillary tangle formation. 41
Activation of the JNK Pathways Is Related to Tau Phosphorylation but Not to Apoptosis
JNK has been implicated in apoptosis. Activation of JNK in the Purkinje cells of our L199P mutant mice, however, failed to induce apoptosis in these cells as shown by the absence of TUNEL-positive neurons. Similarly, staining with an antibody directed against activated caspase-3 failed to detect apoptotic cells (data not shown). Our findings model those obtained with human brain material, as strong immunoreactivity for JNK was seen in tau-positive neuronal and glial cells in diseases including AD, Pick’s disease, progressive supranuclear palsy, corticobasal degeneration, and frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17). By double immunohistochemistry, JNK co-localized with tau in the inclusions. 56,58 However, analysis of apoptosis-related changes such as DNA fragmentation (TUNEL stainings) and activation of caspase-3 showed that expression of JNK was unrelated to activation of an apoptotic cascade. 58 Similarly, activated ERK was shown to be specifically increased in neurons vulnerable in AD that are the site of oxidative damage and abnormal phosphorylation. 59
In mice transgenic for human APP with the Swedish familial AD mutation, JNK activation was localized to amyloid deposits, within neurites containing tau phosphorylated at Ser-202/Thr-205. However, c-Jun was not activated. 37 Long-lasting JNK activation has also been reported after axotomy in the central or peripheral nervous system. JNK activation persisted for several days after axotomy. In both studies, despite persistently increased induction of both JNK and c-Jun, there was no neuronal death. 60,61 Both these and our findings are consistent with the notion that there is no widespread apoptosis in AD. 62 There is even evidence that apoptotic features such as DNA fragmentation are rather an effective defense mechanism to prevent apoptotic death. Also, the fact that neuronal cell death in AD occurs over a lengthy period suggests mechanisms distinct from the classical apoptotic process. 62
Ablation of individual subunits of PP2A (and other phosphatases) in Drosophila Schneider 2 cells by RNA interference has shown that regulation of ERK signaling and apoptosis are mediated by distinct regulatory subunits of PP2A. In particular, the B56 regulatory subunit seemed to play a crucial role as either a complete ablation of PP2A or of both B56 isoforms resulted in the induction of apoptotic markers. 7 In L199P Purkinje cells, sufficient complexes of functional B56-containing heterotrimers may form, and thus prevent the induction of apoptosis in these cells. Collectively, these and our data indicate that ERK and JNK are associated with hyperphosphorylation of tau in human diseases and in our mouse model, suggesting a role of these kinases in the development of degenerative diseases with tau pathology. A careful staging of AD brains revealed a chronological and spatial relationship between activated ERK, JNK, and p38 during progression of AD. While all three kinases were shown to be activated in the same susceptible neurons in mild and severe cases (Braak stages III-VI), in non-demented cases with limited pathology (Braak stages I and II) both ERK and JNK/SAPK were activated but p38 was not. However, in non-demented cases lacking any sign of pathology (Braak stage 0), either ERK alone or JNK alone was shown to be activated indicating that MAPK pathways are differentially activated during the course of AD. 63
Our data are consistent with the view that PP2A exerts a regulatory function at multiple sites of the ERK and JNK signaling pathways. In addition, our data suggest that hyperphosphorylation of tau is mediated not only directly by reduced PP2A activity toward tau, but also indirectly by the activation of the ERK and JNK pathway (Figure 9) ▶ . PP2A function may be further assessed by using either mice with deleted regulatory B subunits or transgenic mice that express mutant forms of PP2A Cα subunits which can recruit only a subset of regulatory B subunits into the functional holoenzyme. 64
Figure 9.

Model of hyperphosphorylation of tau caused by the expression of a dominant-negative mutant form of PP2A Cα in transgenic mice. Our data suggest that hyperphosphorylation of tau at distinct phospho-epitopes is mediated not only directly by reduced PP2A activity toward tau, but also indirectly by the activation of the ERK and JNK pathway. As PP2A is a heterotrimeric protein which is formed by at least 20 different subunits, the different actions of PP2A may be mediated by distinct holoenzyme complexes of PP2A.
Acknowledgments
We thank Eva Moritz for excellent technical assistance and Dr. Gerald Radziwill (Institut für Medizinische Virologie, Universität Zürich) for critical reading of the manuscript.
Footnotes
Address reprint requests to Jürgen Götz, Division of Psychiatry Research, University of Zürich, August Forel Str. 1, 8008 Zürich, Switzerland. E-mail: goetz@bli.unizh.ch.
Supported by grants from the Swiss National Science Foundation, the Hartmann Müller Stiftung, the Zentrum für Neurobiologie Zürich (ZNZ), the Bayer Alzheimer Research Network (BARN), and the National Center of Competence in Research on Neural Plasticity and Repair (to J.G).
S.K. and P.K. contributed equally to this study.
Present address of S.K. is Center of Molecular Biology, University of Heidelberg, Im Neuenheimer Feld 282, 69120 Heidelberg, Germany.
References
- 1.Shenolikar S: Protein serine/threonine phosphatases: new avenues for cell regulation. Annu Rev Cell Biol 1994, 10:55-86 [DOI] [PubMed] [Google Scholar]
- 2.Sontag E: Protein phosphatase 2A: the Trojan horse of cellular signaling. Cell Signal 2001, 13:7-16 [DOI] [PubMed] [Google Scholar]
- 3.Schmidt K, Kins S, Schild A, Nitsch RM, Hemmings BA, Gotz J: Diversity, developmental regulation, and distribution of murine PR55/B subunits of protein phosphatase 2A. Eur J Neurosci 2002, 16:2039-2048 [DOI] [PubMed] [Google Scholar]
- 4.Millward TA, Zolnierowicz S, Hemmings BA: Regulation of protein kinase cascades by protein phosphatase 2A. Trends Biochem Sci 1999, 24:186-191 [DOI] [PubMed] [Google Scholar]
- 5.Gomez N, Cohen P: Dissection of the protein kinase cascade by which nerve growth factor activates MAP kinases. Nature 1991, 353:170-173 [DOI] [PubMed] [Google Scholar]
- 6.Anderson NG, Maller JL, Tonks NK, Sturgill TW: Requirement for integration of signals from two distinct phosphorylation pathways for activation of MAP kinase. Nature 1990, 343:651-653 [DOI] [PubMed] [Google Scholar]
- 7.Silverstein AM, Barrow CA, Davis AJ, Mumby MC: Actions of PP2A on the MAP kinase pathway and apoptosis are mediated by distinct regulatory subunits. Proc Natl Acad Sci USA 2002, 99:4221-4226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alessi DR, Gomez N, Moorhead G, Lewis T, Keyse SM, Cohen P: Inactivation of p42 MAP kinase by protein phosphatase 2A and a protein tyrosine phosphatase, but not CL100, in various cell lines. Curr Biol 1995, 5:283-295 [DOI] [PubMed] [Google Scholar]
- 9.Guan KL, Butch E: Isolation and characterization of a novel dual specific phosphatase, HVH2, which selectively dephosphorylates the mitogen-activated protein kinase. J Biol Chem 1995, 270:7197-7203 [DOI] [PubMed] [Google Scholar]
- 10.Hirsch DD, Stork PJ: Mitogen-activated protein kinase phosphatases inactivate stress-activated protein kinase pathways in vivo. J Biol Chem 1997, 272:4568-4575 [DOI] [PubMed] [Google Scholar]
- 11.Tanaka T, Zhong J, Iqbal K, Trenkner E, Grundke-Iqbal I: The regulation of phosphorylation of tau in SY5Y neuroblastoma cells: the role of protein phosphatases. FEBS Lett 1998, 426:248-254 [DOI] [PubMed] [Google Scholar]
- 12.Chung H, Brautigan DL: Protein phosphatase 2A suppresses MAP kinase signaling and ectopic protein expression. Cell Signal 1999, 11:575-580 [DOI] [PubMed] [Google Scholar]
- 13.Gause KC, Homma MK, Licciardi KA, Seger R, Ahn NG, Peterson MJ, Krebs EG, Meier KE: Effects of phorbol ester on mitogen-activated protein kinase kinase activity in wild-type and phorbol ester-resistant EL4 thymoma cells. J Biol Chem 1993, 268:16124-16129 [PubMed] [Google Scholar]
- 14.Sonoda Y, Kasahara T, Yamaguchi Y, Kuno K, Matsushima K, Mukaida N: Stimulation of interleukin-8 production by okadaic acid and vanadate in a human promyelocyte cell line, an HL-60 subline: possible role of mitogen-activated protein kinase on the okadaic acid-induced NF-κB activation. J Biol Chem 1997, 272:15366-15372 [DOI] [PubMed] [Google Scholar]
- 15.Shanley TP, Vasi N, Denenberg A, Wong HR: The serine/threonine phosphatase, PP2A: endogenous regulator of inflammatory cell signaling. J Immunol 2001, 166:966-972 [DOI] [PubMed] [Google Scholar]
- 16.Goedert M, Jakes R, Qi Z, Wang JH, Cohen P: Protein phosphatase 2A is the major enzyme in brain that dephosphorylates tau protein phosphorylated by proline-directed protein kinases or cyclic AMP-dependent protein kinase. J Neurochem 1995, 65:2804-2807 [DOI] [PubMed] [Google Scholar]
- 17.Gong CX, Lidsky T, Wegiel J, Zuck L, Grundke-Iqbal I, Iqbal K: Phosphorylation of microtubule-associated protein tau is regulated by protein phosphatase 2A in mammalian brain: implications for neurofibrillary degeneration in Alzheimer’s disease. J Biol Chem 2000, 275:5535-5544 [DOI] [PubMed] [Google Scholar]
- 18.Gotz J, Tolnay M, Barmettler R, Ferrari A, Burki K, Goedert M, Probst A, Nitsch RM: Human tau transgenic mice: towards an animal model for neuro- and glialfibrillary lesion formation. Adv Exp Med Biol 2001, 487:71-83 [PubMed] [Google Scholar]
- 19.Gong CX, Shaikh S, Wang JZ, Zaidi T, Grundke-Iqbal I, Iqbal K: Phosphatase activity toward abnormally phosphorylated tau: decrease in Alzheimer disease brain. J Neurochem 1995, 65:732-738 [DOI] [PubMed] [Google Scholar]
- 20.Vogelsberg-Ragaglia V, Schuck T, Trojanowski JQ, Lee VM: PP2A mRNA expression is quantitatively decreased in Alzheimer’s disease hippocampus. Exp Neurol 2001, 168:402-412 [DOI] [PubMed] [Google Scholar]
- 21.Hasegawa M, Jakes R, Crowther RA, Lee VM, Ihara Y, Goedert M: Characterization of mAb AP422, a novel phosphorylation-dependent monoclonal antibody against tau protein. FEBS Lett 1996, 384:25-30 [DOI] [PubMed] [Google Scholar]
- 22.Bussiere T, Hof PR, Mailliot C, Brown CD, Caillet-Boudin ML, Perl DP, Buee L, Delacourte A: Phosphorylated serine422 on tau proteins is a pathological epitope found in several diseases with neurofibrillary degeneration. Acta Neuropathol (Berl) 1999, 97:221-230 [DOI] [PubMed] [Google Scholar]
- 23.Gotz J: Tau and transgenic animal models. Brain Res Brain Res Rev 2001, 35:266-286 [DOI] [PubMed] [Google Scholar]
- 24.Gotz J, Chen F, Barmettler R, Nitsch RM: Tau filament formation in transgenic mice expressing P301L tau. J Biol Chem 2001, 276:529-534 [DOI] [PubMed] [Google Scholar]
- 25.Gotz J, Probst A, Ehler E, Hemmings B, Kues W: Delayed embryonic lethality in mice lacking protein phosphatase 2A catalytic subunit Cα. Proc Natl Acad Sci USA 1998, 95:12370-12375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gotz J, Probst A, Mistl C, Nitsch RM, Ehler E: Distinct role of protein phosphatase 2A subunit Cα in the regulation of E-cadherin and β-catenin during development. Mech Dev 2000, 93:83-93 [DOI] [PubMed] [Google Scholar]
- 27.Kins S, Crameri A, Evans DR, Hemmings BA, Nitsch RM, Gotz J: Reduced PP2A activity induces hyperphosphorylation and altered compartmentalization of tau in transgenic mice. J Biol Chem 2001, 276:38193-38200 [DOI] [PubMed] [Google Scholar]
- 28.Manolson MF, Wu B, Proteau D, Taillon BE, Roberts BT, Hoyt MA, Jones EW: STV1 gene encodes functional homologue of 95-kDa yeast vacuolar H(+)-ATPase subunit Vph1p. J Biol Chem 1994, 269:14064-14074 [PubMed] [Google Scholar]
- 29.Jaaro H, Rubinfeld H, Hanoch T, Seger R: Nuclear translocation of mitogen-activated protein kinase kinase (MEK1) in response to mitogenic stimulation. Proc Natl Acad Sci USA 1997, 94:3742-3747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sontag E, Sontag JM, Garcia A: Protein phosphatase 2A is a critical regulator of protein kinase Cζ signaling targeted by SV40 small t to promote cell growth and NF-κB activation. EMBO J 1997, 16:5662-5671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hecquet C, Lefevre G, Valtink M, Engelmann K, Mascarelli F: cAMP inhibits the proliferation of retinal pigmented epithelial cells through the inhibition of ERK1/2 in a PKA-independent manner. Oncogene 2002, 21:6101-6112 [DOI] [PubMed] [Google Scholar]
- 32.Hamilton M, Wolfman A: Oncogenic Ha-Ras-dependent mitogen-activated protein kinase activity requires signaling through the epidermal growth factor receptor. J Biol Chem 1998, 273:28155-28162 [DOI] [PubMed] [Google Scholar]
- 33.Sugimoto T, Stewart S, Han M, Guan KL: The kinase suppressor of Ras (KSR) modulates growth factor and Ras signaling by uncoupling Elk-1 phosphorylation from MAP kinase activation. EMBO J 1998, 17:1717-1727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hans A, Syan S, Crosio C, Sassone-Corsi P, Brahic M, Gonzalez-Dunia D: Borna disease virus persistent infection activates mitogen-activated protein kinase and blocks neuronal differentiation of PC12 cells. J Biol Chem 2001, 276:7258-7265 [DOI] [PubMed] [Google Scholar]
- 35.Benkoussa M, Brand C, Delmotte MH, Formstecher P, Lefebvre P: Retinoic acid receptors inhibit AP1 activation by regulating extracellular signal-regulated kinase and CBP recruitment to an AP1-responsive promoter. Mol Cell Biol 2002, 22:4522-4534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Metzler B, Hu Y, Dietrich H, Xu Q: Increased expression and activation of stress-activated protein kinases/c-Jun NH(2)-terminal protein kinases in atherosclerotic lesions coincide with p53. Am J Pathol 2000, 156:1875-1886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Savage MJ, Lin YG, Ciallella JR, Flood DG, Scott RW: Activation of c-Jun N-terminal kinase and p38 in an Alzheimer’s disease model is associated with amyloid deposition. J Neurosci 2002, 22:3376-3385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nead MA, Baglia LA, Antinore MJ, Ludlow JW, McCance DJ: Rb binds c-Jun and activates transcription. EMBO J 1998, 17:2342-2352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kurokawa T, Katai N, Shibuki H, Kuroiwa S, Kurimoto Y, Nakayama C, Yoshimura N: BDNF diminishes caspase-2 but not c-Jun immunoreactivity of neurons in retinal ganglion cell layer after transient ischemia. Invest Ophthalmol Vis Sci 1999, 40:3006-3011 [PubMed] [Google Scholar]
- 40.Harding TC, Xue L, Bienemann A, Haywood D, Dickens M, Tolkovsky AM, Uney JB: Inhibition of JNK by overexpression of the JNL binding domain of JIP-1 prevents apoptosis in sympathetic neurons. J Biol Chem 2001, 276:4531-4534 [DOI] [PubMed] [Google Scholar]
- 41.Gotz J, Chen F, van Dorpe J, Nitsch RM: Formation of neurofibrillary tangles in P301L tau transgenic mice induced by Aβ 42 fibrils. Science 2001, 293:1491-1495 [DOI] [PubMed] [Google Scholar]
- 42.Graham JM: Graham JM Rickwood D eds. Homogenization of Tissues and Cells in Subcellular Fractionation: A Practical Approach. 1996:pp 1-29 Oxford University Press New York
- 43.Hazzalin CA, Mahadevan LC: MAPK-regulated transcription: a continuously variable gene switch? Nat Rev Mol Cell Biol 2002, 3:30-40 [DOI] [PubMed] [Google Scholar]
- 44.Ouimet CC, Baerwald KD, Gandy SE, Greengard P: Immunocytochemical localization of amyloid precursor protein in rat brain. J Comp Neurol 1994, 348:244-260 [DOI] [PubMed] [Google Scholar]
- 45.Suzuki T, Oishi M, Marshak DR, Czernik AJ, Nairn AC, Greengard P: Cell cycle-dependent regulation of the phosphorylation and metabolism of the Alzheimer amyloid precursor protein. EMBO J 1994, 13:1114-1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Iijima K, Ando K, Takeda S, Satoh Y, Seki T, Itohara S, Greengard P, Kirino Y, Nairn AC, Suzuki T: Neuron-specific phosphorylation of Alzheimer’s β-amyloid precursor protein by cyclin-dependent kinase 5. J Neurochem 2000, 75:1085-1091 [DOI] [PubMed] [Google Scholar]
- 47.Ando K, Oishi M, Takeda S, Iijima K, Isohara T, Nairn AC, Kirino Y, Greengard P, Suzuki T: Role of phosphorylation of Alzheimer’s amyloid precursor protein during neuronal differentiation. J Neurosci 1999, 19:4421-4427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vogt PK: Jun, the oncoprotein. Oncogene 2001, 20:2365-2377 [DOI] [PubMed] [Google Scholar]
- 49.Coffey ET, Smiciene G, Hongisto V, Cao J, Brecht S, Herdegen T, Courtney MJ: c-Jun N-terminal protein kinase (JNK) 2/3 is specifically activated by stress, mediating c-Jun activation, in the presence of constitutive JNK1 activity in cerebellar neurons. J Neurosci 2002, 22:4335-4345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Takekawa M, Maeda T, Saito H: Protein phosphatase 2Cα inhibits the human stress-responsive p38 and JNK MAPK pathways. EMBO J 1998, 17:4744-4752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Strack S, Zaucha JA, Ebner FF, Colbran RJ, Wadzinski BE: Brain protein phosphatase 2A: developmental regulation and distinct cellular and subcellular localization by B subunits. J Comp Neurol 1998, 392:515-527 [PubMed] [Google Scholar]
- 52.Holmes SE, O’Hearn EE, McInnis MG, Gorelick-Feldman DA, Kleiderlein JJ, Callahan C, Kwak NG, Ingersoll-Ashworth RG, Sherr M, Sumner AJ, Sharp AH, Ananth U, Seltzer WK, Boss MA, Vieria-Saecker AM, Epplen JT, Riess O, Ross CA, Margolis RL: Expansion of a novel CAG trinucleotide repeat in the 5′ region of PPP2R2B is associated with SCA12. Nat Genet 1999, 23:391-392 [DOI] [PubMed] [Google Scholar]
- 53.Sontag E, Nunbhakdi-Craig V, Lee G, Brandt R, Kamibayashi C, Kuret J, White CL, III, Mumby MC, Bloom GS: Molecular interactions among protein phosphatase 2A, tau, and microtubules: implications for the regulation of tau phosphorylation and the development of tauopathies. J Biol Chem 1999, 274:25490-25498 [DOI] [PubMed] [Google Scholar]
- 54.Drewes G, Lichtenberg-Kraag B, Doring F, Mandelkow EM, Biernat J, Goris J, Doree M, Mandelkow E: Mitogen activated protein (MAP) kinase transforms tau protein into an Alzheimer-like state. EMBO J 1992, 11:2131-2138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Reynolds CH, Utton MA, Gibb GM, Yates A, Anderton BH: Stress-activated protein kinase/c-jun N-terminal kinase phosphorylates tau protein. J Neurochem 1997, 68:1736-1744 [DOI] [PubMed] [Google Scholar]
- 56.Zhu X, Raina AK, Rottkamp CA, Aliev G, Perry G, Boux H, Smith MA: Activation and redistribution of c-Jun N-terminal kinase/stress activated protein kinase in degenerating neurons in Alzheimer’s disease. J Neurochem 2001, 76:435-441 [DOI] [PubMed] [Google Scholar]
- 57.Pei JJ, Gong CX, An WL, Winblad B, Cowburn RF, Grundke-Iqbal I, Iqbal K: Okadaic-acid-induced inhibition of protein phosphatase 2A produces activation of mitogen-activated protein kinases ERK 1/2, MEK 1/2, and p70 S6, similar to that in Alzheimer’s disease. Am J Pathol 2003, 163:845-858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Atzori C, Ghetti B, Piva R, Srinivasan AN, Zolo P, Delisle MB, Mirra SS, Migheli A: Activation of the JNK/p38 pathway occurs in diseases characterized by tau protein pathology and is related to tau phosphorylation but not to apoptosis. J Neuropathol Exp Neurol 2001, 60:1190-1197 [DOI] [PubMed] [Google Scholar]
- 59.Perry G, Roder H, Nunomura A, Takeda A, Friedlich AL, Zhu X, Raina AK, Holbrook N, Siedlak SL, Harris PL, Smith MA: Activation of neuronal extracellular receptor kinase (ERK) in Alzheimer disease links oxidative stress to abnormal phosphorylation. Neuroreport 1999, 10:2411-2415 [DOI] [PubMed] [Google Scholar]
- 60.Herdegen T, Claret FX, Kallunki T, Martin-Villalba A, Winter C, Hunter T, Karin M: Lasting N-terminal phosphorylation of c-Jun and activation of c-Jun N-terminal kinases after neuronal injury. J Neurosci 1998, 18:5124-5135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kenney AM, Kocsis JD: Peripheral axotomy induces long-term c-Jun amino-terminal kinase-1 activation and activator protein-1 binding activity by c-Jun and junD in adult rat dorsal root ganglia in vivo. J Neurosci 1998, 18:1318-1328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Perry G, Nunomura A, Lucassen P, Lassmann H, Smith MA: Apoptosis and Alzheimer’s disease. Science 1998, 282:1268-1269 [DOI] [PubMed] [Google Scholar]
- 63.Zhu X, Castellani RJ, Takeda A, Nunomura A, Atwood CS, Perry G, Smith MA: Differential activation of neuronal ERK, JNK/SAPK, and p38 in Alzheimer disease: the “two hit” hypothesis. Mech Ageing Dev 2001, 123:39-46 [DOI] [PubMed] [Google Scholar]
- 64.Evans DR, Myles T, Hofsteenge J, Hemmings BA: Functional expression of human PP2Ac in yeast permits the identification of novel C-terminal and dominant-negative mutant forms. J Biol Chem 1999, 274:24038-24046 [DOI] [PubMed] [Google Scholar]