

Abstract
Chronic myocardial ischemia is the leading cause of impaired myocardial contractility and heart failure. To identify differentially expressed genes in human ischemic cardiomyopathy (ICM), we constructed a subtracted cDNA library using specimens of ICM compared to normal human heart. Among 100 randomly sequenced clones, seven sequences represented recently identified candidate genes for differential expression in cardiac hypertrophy. A further clone without a known hypertrophy-association coded for the adhesion molecule NCAM(CD56). RNase protection assay, immunohistochemistry, and Western blotting revealed strong overexpression of NCAM(CD56) in all hearts with ICM (n = 14) compared to normal hearts (n = 8), whereas in congestive cardiomyopathy (CCM) (n = 8), hypertrophic obstructive cardiomyopathy (n = 2), myocarditis (n = 4), and sarcoidosis (n = 2), at most slight overexpression of NCAM(CD56) was observed. NCAM(CD56) overexpression abnormally involved the whole cell membrane and the cytoplasma of cardiomyocytes only inside and adjacent to ischemia-induced cardiac scars. Normal or hypertrophic fibers at a distance from ischemic scars were devoid of NCAM overexpression. Identical alterations were observed in an experimental rat ICM model, but not in normal nor in spontaneously hypertensive rat hearts. In search of NCAM(CD56)-related transcription factors we found RUNX1(AML1) up-regulation in ICM and detected RUNX1(AML1) binding within the NCAM(CD56) promoter by electromobility shift assay. We concluded that strong overexpression of NCAM(CD56) and RUNX1(AML1) is a constant and characteristic feature of cardiomyocytes within or adjacent to scars in ICM.
The most common cause of chronic heart failure is coronary artery disease (CAD), which results in left ventricular dysfunction. 1,2 The morphological changes of the heart in chronic heart failure due to CAD have been termed ischemic cardiomyopathy (ICM). 1,3-6
Among the earliest events during ischemia-induced ventricular dysfunction, the renin-angiotensin system and secretion of atrial natriuretic peptide (ANP) are activated. 7,8 In addition, in the endothelin system, 9,10 cytokines such as IL-1, IL-6, and tumor necrosis factor-α, 11-14 stress-proteins, 15 and anti-oxidants 16 change their expression pattern. However, these changes generally are not characteristic for ICM.
To identify differentially overexpressed genes in ICM compared to normal hearts we used a PCR-based technique to construct a subtracted cDNA library. We find that strong overexpression of NCAM (CD56) and the transcription factor RUNX1(AML1) is a highly sensitive and characteristic marker of cardiomyocytes within or adjacent to scars in ICM compared to normal hearts, while at most slight overexpression is observed in CCM, hypertrophic obstructive cardiomyopathy (HOCM), and myocarditis, including sarcoidosis. This molecular response to ischemic heart damage appears to be phylogenetically conserved, because analogous alterations occurred in an experimental rat model of ischemic heart disease compared to normal and spontaneously hypertensive rats.
Materials and Methods
Human Tissue
Heart tissue obtained from autopsies within 6 hours after death was shock-frozen in liquid nitrogen and stored at −80°C. Ischemic heart disease of the 14 patients in this study had been diagnosed either by coronary angiography (n = 8), medical history of myocardial infarction with typical electrocardiogram signs (n = 3), or by clinical features of acute myocardial infarction (n = 3). In the eight patients with congestive cardiomyopathy (CCM) and the two patients with HOCM, absence of coronary artery disease was angiographically confirmed. Normal human hearts (n = 8) as well as heart specimens from patients with myocarditis (n = 4) and sarcoidosis (n = 2) were without a history or autopsy findings suggestive of cardiovascular disease and hypertension. The study was approved by an institutional review committee, with all procedures following institutional guidelines.
Animal Tissue
Myocardial infarction was induced by ligation of the left coronary artery in adult (250 to 290 g) female Wistar rats (Charles River, Sulzfeld, FRG) as described. 17 Sham-operated rats served as controls. Eight weeks after surgery, infarcted (n = 6) and sham-operated hearts (n = 6) were removed. Spontaneously hypertensive rats (SHR; Charles River) (310 to 330 g) were used as non-ischemic cardiomegaly controls. Samples were fixed in 4% paraformaldehyde, sliced perpendicular to the long axis of the heart, and embedded in paraffin.
Subtracted cDNA Library
2 μg of mRNA from an ICM (case number S280/96) and a normal heart (case S62/98) (Table 1) ▶ were used for polymerase chain reaction (PCR)-based cDNA subtraction (PCR-Select Subtraction kit; Clontech, Heidelberg, Germany) following the manufacturer’s instructions. Following a single subtraction reaction, cDNA fragments from the forward and reverse reaction were cloned into pGEMT-vector (Promega, Heidelberg, Germany) and transformed in competent JM105 Escherichia coli. Plasmid DNA of 100 randomly selected clones from the forward reaction was sequenced by the cycle-sequencing method using dye terminators and the ABI 373 sequencer (Applied Biosystems, Weiterstadt, Germany).
Table 1.
Clinical and Pathological Data of Patients Investigated (n = 28)
| Case no. | Sex/age | Clinical and pathological diagnoses | Heart weight |
|---|---|---|---|
| 188/95 | M/63 | AH, severe CA with two VD, recurrent MI | 620 g |
| 313/95 | F/62 | AH, recurrent MI, severe CA with three VD | 580 g |
| S233/96 | M/68 | DM, AH, severe CA with three VD, liver cirrhosis, septicopyemia | 520 g |
| S234/96 | M/88 | AH, severe CA with three VD, bronchopneumonia | 550 g |
| S280/96 | M/68 | AH, recurrent MI, severe CA with three VD | 720 g |
| S130/97 | M/78 | AH, recurrent MI, severe CA with two VD (RCX, RCA) | 680 g |
| S190/98 | M/45 | AH, recurrent MI, severe CA with three VD | 510 g |
| S196/98 | M/70 | AH, severe CA with two VD, recurrent MI | 440 g |
| S236/98 | M/63 | AH, DM, recurrent MI, severe CA | 700 g |
| S252/98 | M/69 | DM, recurrent MI, severe CA with two VD (LCA, RCA) | 450 g |
| S258/99 | M/63 | AH, DM, acute MI, severe CA, recurrent MI | 530 g |
| S7/99 | M/74 | Recurrent MI, severe CA with three VD | 310 g |
| S52/99 | M/51 | AH, recurrent MI, moderate CA | 440 g |
| S04/00 | F/60 | AH, recurrent MI, moderate CA | 680 g |
| S111/97 | M/62 | CCM, no CA | 710 g |
| E22500/90 | M/54 | CCM, no CA | 630 g |
| S141/97 | M/32 | CCM, no CA | 570 g |
| S100/99 | F/59 | CCM, no CA | 680 g |
| S18/00 | F/57 | CCM, no CA | 570 g |
| S254/99 | M/71 | CCM, no CA | 720 g |
| S132/00 | M/44 | CCM, no CA | 650 g |
| S208/01 | M/67 | CCM, no CA | 860 g |
| S183/99 | M/33 | HOCM, no CA | 720 g |
| S196/93 | M/60 | HOCM, no CA | 490 g |
| A15076/01 | F/36 | Sarcoidosis, no CA | 440 g |
| A15051/01 | M/51 | Sarcoidosis, no CA | 360 g |
| A17995/02 | M/7 | Parvovirus B19 myocarditis, no CA | NA |
| A12978/01 | M/71 | Eosinophilic myocarditis, no CA | 680 g |
| A12021/01 | F/39 | Giant cell myocarditis, no CA | 450 g |
| S 256/99 | M/31 | Giant cell myocarditis, no CA | 320 g |
| S244/96 | F/24 | Moschcowitz syndrome, no CA | 250 g |
| S55/97 | F/61 | Rupture of arteriovenous angioma, AH, no CA | 360 g |
| S31/98 | F/52 | Craniocerebral trauma, no CA | 280 g |
| S62/98 | M/43 | Anaphylactic shock, no CA | 300 g |
| S200/98 | M/61 | Malignant melanoma, no AH, no CA | 290 g |
| S114/99 | M/26 | Anaphylactic shock, no CA | 270 g |
| S160/99 | M/29 | Aplastic anemia, liver dystrophy, no CA | 280 g |
| S133/00 | F/63 | Adenocarcinoma of the ovary, no CA | 270 g |
AH, Arterial hypertension; CA, Coronary arteriosclerosis DM, Diabetes mellitus; MI, Myocardial infarction; VD, Vessel disease;
Western Blot
Total protein extracts were isolated after homogenization of shock-frozen heart tissue in 2% sodium dodecyl sulfate (SDS), 50 mmol/L Tris (pH 6.8), 100 mmol/L DTT, 0.01% bromophenol blue, and separated on SDS-polyacrylamide gel electrophoresis (PAGE). 18 After blotting on nitrocellulose membrane, proteins were stained by Ponceau red. Blocking of membranes and incubations with the first and second antibody (2 hours each; Table 2 ▶ ) were carried out using 1X TBS/1X Tween 20/10% milk powder. Immunocomplexes were detected by the enhanced chemiluminescence system (Gibco, Heidelberg, Germany).
Table 2.
First and Second Antibodies Applied in this Study
| First (F) or second (S) antibody, source | Concentration |
|---|---|
| ALCAM (CD166), Santa Cruz (N-21), F | 1:400 |
| BLCAM (CD22), Serotec (MCA1809), F | 1:200 |
| BLCAM (CD22), Santa Cruz (H-221), F | 1:400 |
| C-CAM1 (CD66), Santa Cruz (N-19), F | 1:400 |
| HCAM (CD44), Santa Cruz (DF 1485), F | 1:400 |
| HCAM (CD44), Santa Cruz (F-4), F | 1:200 |
| ICAM-1 (CD54), Santa Cruz (G-5), F | 1:400 |
| ICAM-2 (CD102), Santa Cruz (F-5), F | 1:400 |
| ICAM-3 (CD50), Oncogene (Ab-2), F | 1:600 |
| ICAM-3 (CD50), Santa Cruz (3.1), F | 1:400 |
| KALIG-1, Santa Cruz (C-20), F | 1:400 |
| NCAM(CD56), Novocastra (1B6), F | 1:100 |
| NCAM(CD56), Santa Cruz (123C3), F | 1:400 |
| PECAM-1 (CD31), Novocastra, F | 1:100 |
| VCAM-1 (CD106), Serotec (IE5), F | 1:100 |
| VCAM-1(CD106), Santa Cruz (C-19), F | 1:400 |
| Muscle-specific actin, Novocastra (HHF35), F | 1:400 |
| RUNX1 (AML1), Santa Cruz (N-20) (N terminus), F | 1:400 |
| RUNX1 (AML1), Santa Cruz (C-19) (C terminus), F | 1:400 |
| CREB-1, Santa Cruz (C-21), F | 1:400 |
| Hox D9, Santa Cruz (H-342), F | 1:400 |
| PAX 2/5/8, Santa Cruz (F-19), F | 1:400 |
| PAX 3/7, Santa Cruz, (C-20), F | 1:400 |
| rabbit-anti-mouse Ig, DAKO, S | 1:2000 |
| goat-anti-rabbit Ig, DAKO, S | 1:2000 |
| rabbit-anti-goat Ig, DAKO, S | 1:2000 |
RNase Protection Assay
Total RNA of human heart was processed according to PharMingen’s (San Diego, CA) RiboQuant protocol using anti-sense RNA as probe. Probes were generated using the following PCR primers: NCAM(CD56) UP: 5′-GAC GGC GGC TCC CCC ATC AGA-3′; NCAM(CD56) LP: 5′-ATG ACG ATG AGG ATG CCC ACG-3′; RUNX1(AML1) UP: 5′-TGC GCA CCG ACA GCC CCA ACT-3′; RUNX1(AML1) LP: 5′-GCT GTG TCT TCC TCC TGC ATC-3′; GAPDH UP: 5′-CAA CAG CGA CAC CCA CTC CTC-3′; GAPDH LP: 5′-CAT GTG GGC CAT GAG GTC CAC CAC-3′). PCR products were cloned into pGEM T-vector (Promega) and sequenced. Plasmid DNA was linearized with NotI RUNX1(AML1) and NCAM(CD56) or SalI (GAPDH), labeled by in vitro transcription (PharMingen) with [32Pα]UTP and separated by PAGE on a 6% acrylamide gel. Labeled RNA was extracted for 1 hour in 1 mol/L ammonium acetate at 60°C.
The predicted length of the transcripts were: NCAM(CD56), unprotected 360 bp/protected 283 bp; RUNX1(AML1), unprotected 592 bp/protected 527 bp; GAPDH, unprotected 233 bp/protected 156 bp.
Immunohistochemistry
The anti-human-NCAM(CD56) monoclonal mouse antibodies 1B6 (Novocastra, Newcastle, UK) and 123C3 (Santa Cruz Biotechnology, Santa Cruz, CA) and the anti-rat-NCAM(CD56) rabbit polyclonal antibody H-300 (Santa Cruz) were used at 1:100 dilution in a four-step immunoperoxidase labeling for single antigens in formalin-fixed, paraffin-embedded sections (2-μm-thick). 19
Electromobility Shift Assay
Jurkat cell nuclear extracts were isolated as described. 18 As probes, synthetic oligonucleotides containing a WT or mutant RUNX1(AML1) consensus sequence from the NCAM(CD56) and GMCSF promoter 20 (Table 3) ▶ were radioendlabeled with [γ32P]ATP (Amersham Biosciences, Inc., Heidelberg, Germany). As controls, non-radioendlabeled mutant or WT consensus oligonucleotides were mixed with nuclear extracts either with or without anti-RUNX1(AML1) antibody, incubated with 1X binding buffer and 1 μg/μl poly(dI-dC) for 30 minutes on ice and analyzed on a 6% polyacrylamide gel in 0.25 TBE for 3 hours at room temperature. Autoradiography of vacuum-dried gels was carried out for 24 to 96 hours at −70°C.
Table 3.
WT and Mutant (MUT) Oligonucleotides Used in EMSA
| Oligonucleotide | Sequence |
|---|---|
| NCAM(CD56)- WT-sense | 5′-TAA TAA AGA GAC CAC AGA TTT CAG AA-3′ |
| NCAM(CD56)- WT-antisense | 5′-TTC TGA AAT CTG TGG TCT CTT TAT TA-3′ |
| NCAM(CD56)-MUT-sense | 5′-TAA TAA AGA GAC GCC AGA TTT CAG AA-3′ |
| NCAM(CD56)-MUT-antisense | 5′-TTC TGA AAT CTG GCG TCT CTT TAT TA-3′ |
| GMCSF-WT-sense | 5′-GGC ATT TTG TGG TCA CCA TTA ATC-3′ |
| GMCSF-WT-antisense | 5′-GAT TAA TGG TGA CCA CAA AAT GCC-3′ |
| GMCSF-WT-sense | 5′-GGC ATT TTG GCG TCA CCA TTA ATC-3′ |
| GMCSF-WT-antisense | 5′-GAT TAA TGG TGA CGC CAA AAT GCC-3′ |
Results
NCAM is Overexpressed in ICM
Among 100 sequenced clones from the PCR-subtract forward reaction, 7 clones (coding for Myoglobulin, Myosin regulatory light chain, Myosin light chain, CD59, SLIM-1, actin, ubiquitin) were identical to recently reported strong candidates for differentially expressed genes in cardiac hypertrophy. 21 In addition we isolated a 286-bp fragment homologous to the nucleotide sequence 1950–2236 of the 3′ end of NCAM(CD56). Since NCAM(CD56) had not been identified among the candidate “cardiac hypertrophy genes” in the previous extensive search by Hwang et al, 21 we concluded that NCAM(CD56) might be a candidate “ICM gene”.
Strong overexpression of NCAM(CD56) mRNA occurred in all human ICM samples compared to normal hearts irrespective of the presence of previous myocardial infarction, 5,6 whereas CCM showed only a slight up-regulation (Figure 1a) ▶ . By Western blot, all ICM samples strongly overexpressed the 125-kd NCAM(CD56) isoform, while normal heart samples expressed only traces of NCAM(CD56), and again CCM samples revealed slight but significant overexpression. (Figure 1b) ▶ . By immunohistochemistry, NCAM(CD56) protein in normal hearts was detectable only at the disci intercalcares (Figure 2, a and b) ▶ , while NCAM(CD56) in ICM was overexpressed on the whole cell membrane and in the cytoplasm (Figure 2, c and d) ▶ . NCAM(CD56) overexpression was restricted to cardiomyocytes inside and adjacent to scars in ICM, while apparently normal and hypertrophic cardiomyocytes without contact to scars expressed NCAM(CD56) at control levels (Figure 2e) ▶ . In CCM a focal and slight increase of NCAM(CD56) in the cytoplasma but not on the cell membrane was seen (Figure 2f) ▶ , and either slight but significant, or no up-regulation was seen on cardiomyocytes inside or adjacent to formations of larger regions of interstitial fibrosis (Figure 2, g and h) ▶ . The pattern of NCAM(CD56) overexpression in ICM is strikingly similar to the pattern observed in tumor-infiltrated skeletal muscle (Figure 3) ▶ . Like in cardiomyocytes inside and directly adjacent to ischemic scars, NCAM(CD56) overexpression is strictly confined to skeletal muscle fibers surrounded by and in direct contact with carcinoma infiltrates.
Figure 1.
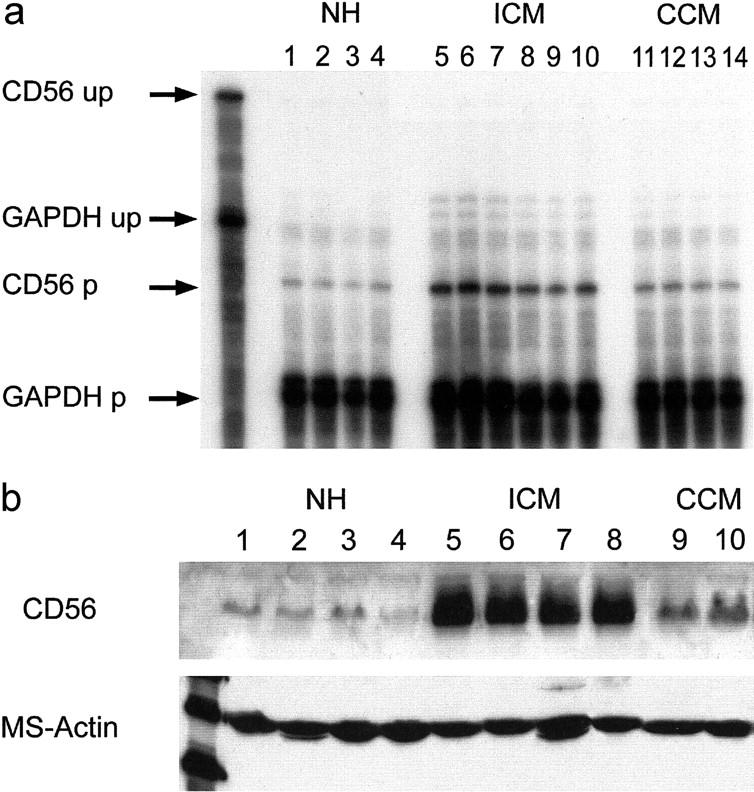
RNase Protection Assay (a) and Western blot (b) of normal human control hearts (lanes 1 to 4 in a and b) and ICM hearts (a, lanes 5 to 10; b, lanes 5 to 8). Strong overexpression of NCAM(CD56) message and protein in ICM. Slight but significant NCAM(CD56) mRNA and protein overexpression in CCM (a, lanes 11 to 14; b, lanes 9 to 10). Equal loading of mRNA or protein, respectively, per lane checked by GAPDH (a) and muscle-specific actin Western blot (b). up, unprotected radioactive probe; p, protected radioactive probe.
Figure 2.
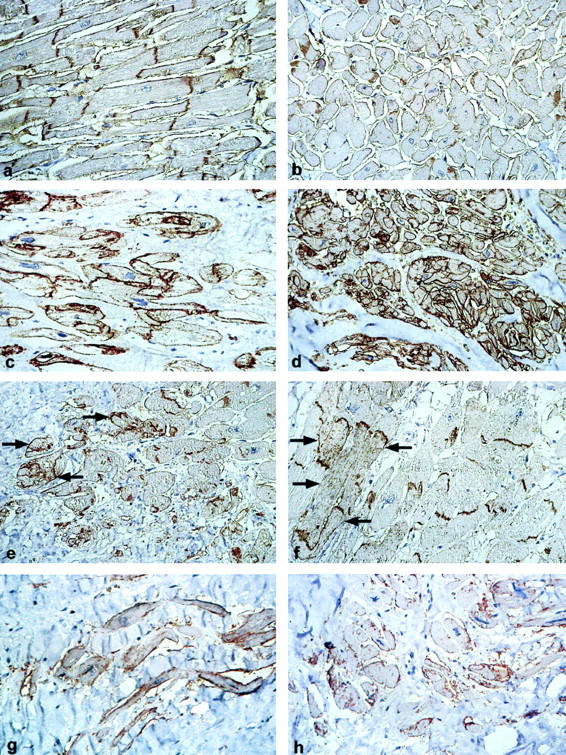
NCAM(CD56) expression in normal human heart at the disci intercalcares in longitudinal (a) and cross-sections (b). Overexpression and abnormal membrane and cytoplasmatic staining in ICM in longitudinal (c) and cross-sections (d). Restriction of NCAM(CD56) overexpression to cardiomyocytes inside and in contact with a scar (arrows) in ICM (e). Focal and slightly increased expression of NCAM(CD56) in the cytoplasma of cardiomyocytes (f) as well as in cardiomyocytes inside and adjacent to scars in longitudinal (g) and cross-sections (h) in CCM. Immunoperoxidase, ×200.
Figure 3.

Immunohistochemical staining for NCAM(CD56) (a) and cytokeratin 7 (b) of a breast carcinoma infiltrating the musculus pectoralis major. Muscle fibers surrounded by or directly adjacent to carcinoma cells overexpress NCAM(CD56) protein. No NCAM(CD56) overexpression in muscle fibers at a distance from the carcinoma cells.
NCAM(CD56) Overexpression in a Rat Model of ICM
To further investigate the role of NCAM(CD56) as a candidate “ICM gene”, we analyzed its expression in well-established rat models of ICM and cardiac hypertrophy. 17 An identical pattern of NCAM(CD56) overexpression occurred in experimental ICM of rat hearts (Figure 4b) ▶ compared to sham-operated and spontaneously hypertensive rat hearts (Figure 4, a and c) ▶ .
Figure 4.
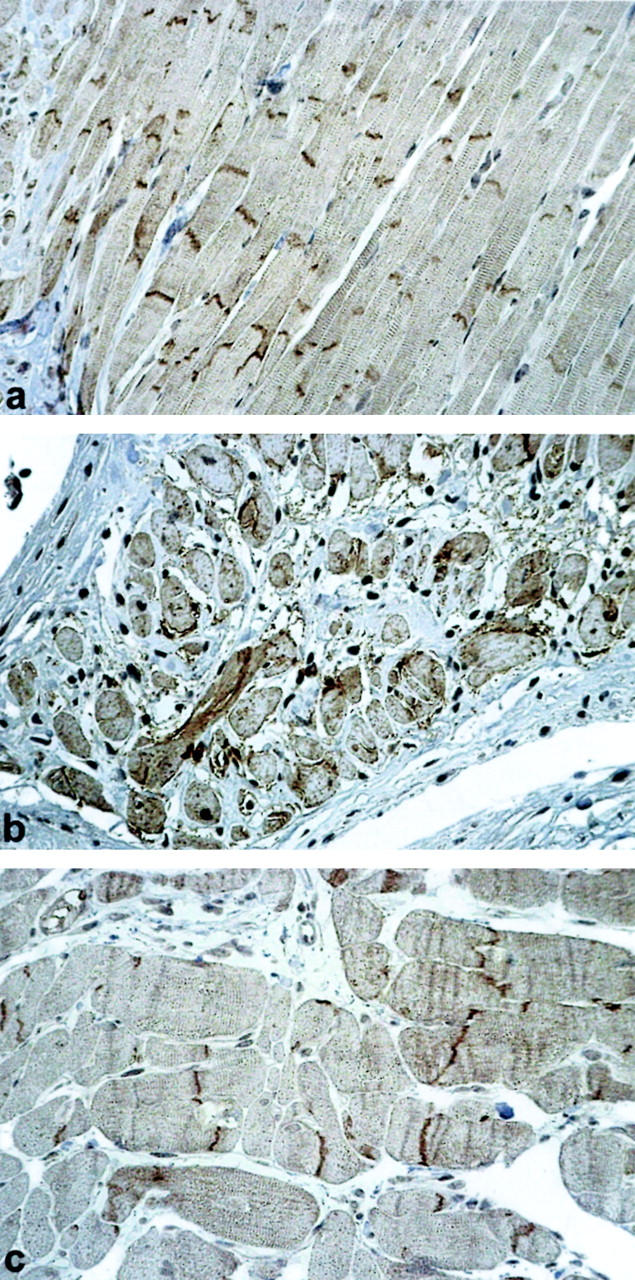
Immunohistochemical detection of NCAM(CD56) at the disci intercalcares in a normal rat heart (a). NCAM(CD56) overexpression on the whole cell membrane and in the cytoplasm of cardiomyocytes adjacent to a cardiac scar in rat heart with experimental ICM (b). Normal cardiac NCAM(CD56) expression in a spontaneously hypertensive rat (c). Immunoperoxidase, ×200.
NCAM(CD56) Is Not or Only Slightly Overexpressed in Heart Disorders Other than ICM
Tissue samples from patients suffering from heart diseases other than ICM (Figure 5a, c, e, g) ▶ , including sarcoidosis (n = 2), myocarditis (n = 4) and HOCM (n = 2), were immunohistochemically stained for NCAM(CD56) (Figure 5b, d, f, h) ▶ . Either slight (5b, sarcoidosis) or no recognizable overexpression (5 days, eosinophilic myocarditis; 5 hours, HOCM) of NCAM(CD56) was observed in these disease states. Furthermore, NCAM(CD56) was not overexpressed but even down-regulated on acutely infarcted cardiomyocytes (Figure 5f) ▶ as compared to normal cardiomyocytes. By contrast, viable cardiomyocytes within an ischemic scar adjacent to the acute infarct clearly showed up-regulated NCAM(CD56) expression (Figure 5f) ▶ .
Figure 5.
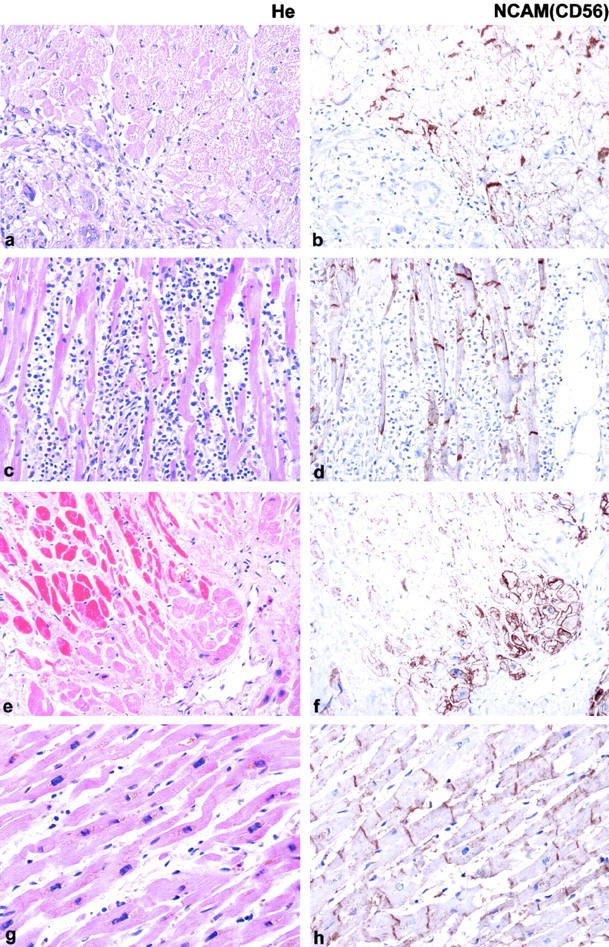
Various cardiac pathologies and associated NCAM(CD56) expression. Autopsy samples of heart tissue from patients suffering from sarcoidosis (a and b), eosinophilic myocarditis (c and d), acute heart infarction adjacent to an old ischemic scar (e and f) and HOCM (g and h). Either very slight or no up-regulation of NCAM(CD56) is seen on the cardiomyocytes adjacent to or within the inflammatory infiltrate in sarcoidosis, eosinophilic myocarditis, and HOCM. In a case of ICM (case 313/95, e and f) clear overexpression of NCAM(CD56) is detected in the cytoplasma and on the whole cell membrane of viable cardiomyocates within or adjacent to scars (lower right half) whereas no NCAM(CD56) expression is detectable on non-viable, hypereosinophilic cardiomyocytes in a region of acute cardiac infarct (upper left half). HE, Immunoperoxidase ×200.
Other Cell Adhesion Molecules in ICM
By Western blot, PECAM (CD31) revealed slight overexpression in ICM compared to controls (Figure 6) ▶ . ICAM-1 (CD54) was expressed at high levels, and BL-CAM (CD22) (Figure 6) ▶ , VCAM (CD106), and ALCAM (CD166) (not shown) were expressed at low levels in most cardiomyocytes of all samples. HCAM (CD44), ICAM-2 (CD102), ICAM-3 (CD50), KALIG, and C-CAM (CD66) proteins were undetectable (not shown).
Figure 6.
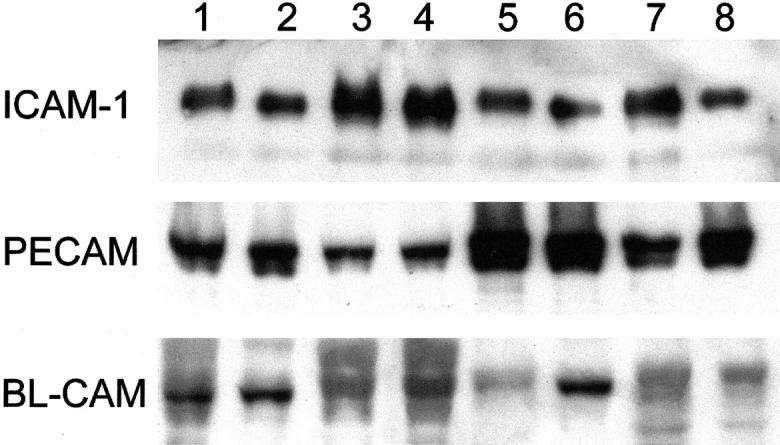
Western blots of normal human hearts (lanes 1 to 4) and ICM (lanes 5 to 8) with antibodies against ICAM-1 (CD54), PECAM (CD31), and BL-CAM (CD22). Slight increase only of PECAM in ICM compared to control.
RUNX1(AML1) is Overexpressed in ICM and Binds to the NCAM(CD56) Promoter
By Western blots of total protein extracts, the NCAM(CD56)-targeting transcription factors NF-kappa B p65 and p50, HOXD9, and PAX 2/5/8 were detected in ICM and normal hearts in similar amounts (Figure 7a) ▶ . PAX3/7 protein was undetectable. By contrast, the 52-kd RUNX1(AML1) isoform, which is up-regulated in parallel to NCAM(CD56) in denervated mouse skeletal muscle, 22 and RUNX1(AML1) mRNA were overexpressed in all ICM specimens with only slight overexpression in CCM samples (Figure 7, b and c) ▶ . In addition, a 38-kd band was detected in all heart extracts although down-regulated in ICM (Figure 7c) ▶ .
Figure 7.
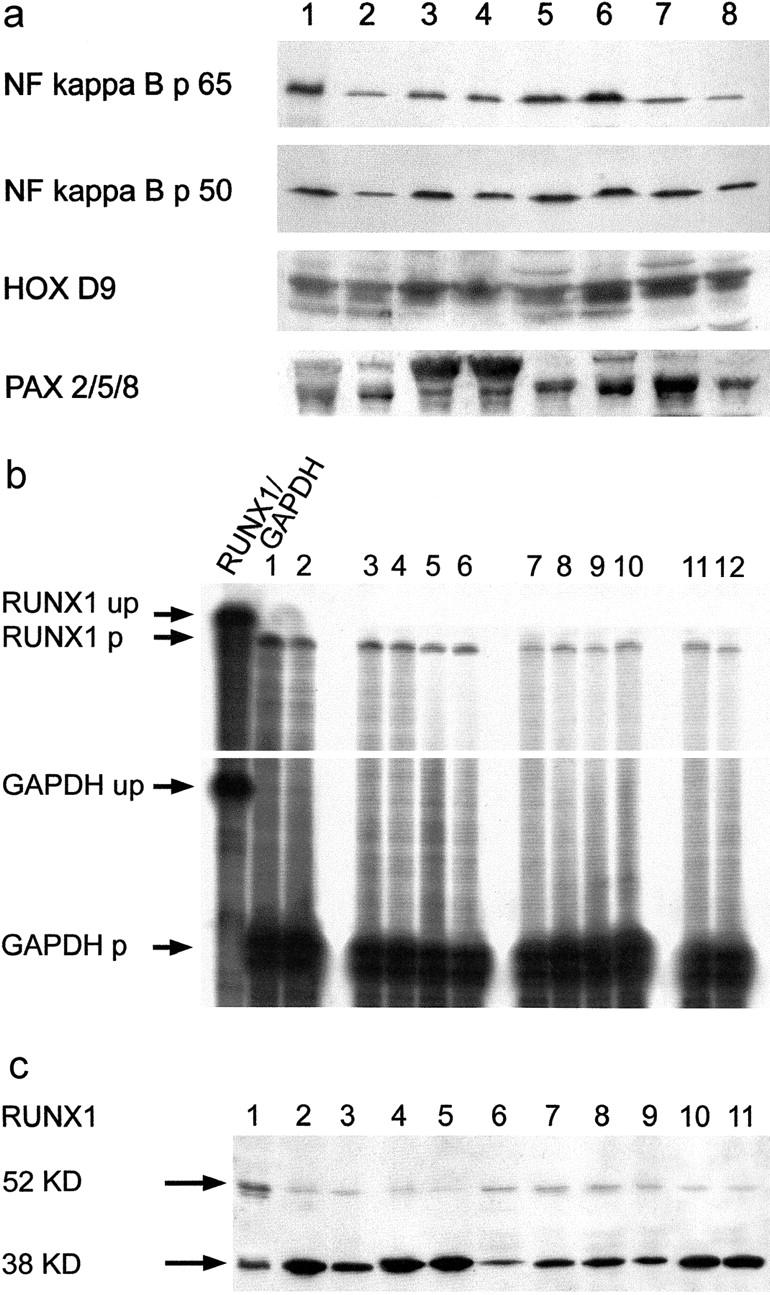
Detection of NCAM(CD56)-related transcription factors NF-kappa B, HOXD9, PAX (a), and RUNX1(AML1) (b and c) in normal and ICM hearts. a: Similar amounts of NF-kappa B p65 and p50, HOXD9, and PAX 2/5/8 in normal (lanes 1 to 4) and ICM hearts (lanes 5 to 8) (Western blots). b: Overexpression of RUNX1(AML1) mRNA in ICM (lanes 3 to 6) compared to normal human hearts (lanes 7 to 10) and slight overexpression in CCM (lanes 11 to 12). Jurkat-derived (lane 1) and TE671 rhabdomyosarcoma-derived (lane 2) control mRNAs (RNase protection assays). c: Overexpression of the 52-kd RUNX1(AML1) isoform 30 in ICM (lanes 6 to 9) compared to normal human hearts (lanes 2 to 5) and again either very slight or no overexpression in CCM (lanes 10 to 11) (Western blots). Jurkat cell extract (lane 1, positive control). Strong expression of an additional 38-kd band in all samples, with down-regulation in ICM (lanes 6 to 9) and CCM (lanes 10 to 11). up, unprotected radioactive probe; p, protected radioactive probe
To examine RUNX1(AML1) binding to the human NCAM(CD56) promoter, we performed electromobility shift assays (EMSAs) using Jurkat nuclear extracts as source of RUNX1(AML1) protein. RUNX1(AML1) formed a radioactive complex when mixed with radiolabeled WT NCAM(CD56) consensus probe (Figure 8) ▶ . The specificity of the complex was checked severalfold: 1) complex formation did not occur with a radioactive mutant NCAM(CD56) probe; 2) the complex disappeared when non-radioendlabeled competitor NCAM(CD56) or GM-CSF consensus probes were added in excess; 3) a supershift of the DNA-protein complex occurred when anti-RUNX1(AML1) antibody was added to Jurkat nuclear extract mixed with radioendlabeled WT NCAM(CD56) consensus probe.
Figure 8.
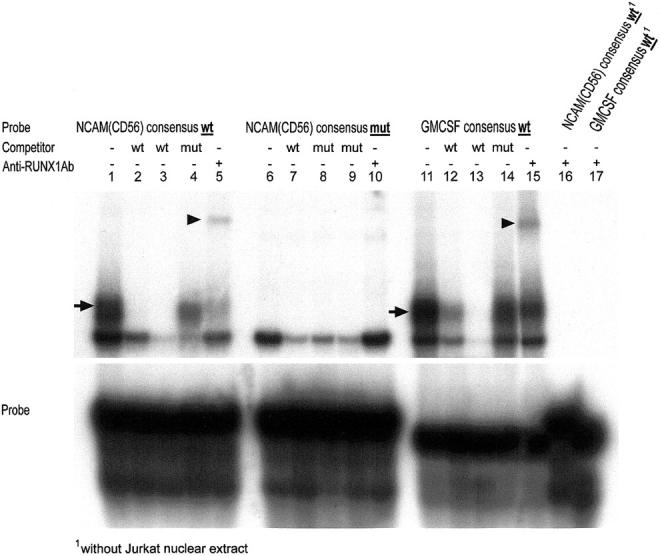
Detection of specific RUNX1(AML1) protein binding within the human NCAM(CD56) promoter by electromobility shift assay (EMSA). Radioactive DNA-protein complex formation by mixing RUNX1(AML1) protein-expressing Jurkat cell extract with [γ-32P]ATP radioendlabeled WT NCAM(CD56) consensus probe (lanes 1, 4, and 5, arrow) and as control with [γ-32P]ATP radioendlabeled WT GMCSF consensus probe (lanes 11, 12, 14 and 15, arrow) but not with [γ32P]ATP radioendlabeled mutant (mt) NCAM(CD56) probe (lanes 6 to 10). Supershifts of the specific complexes by adding anti-RUNX1(AML1) mAb (lanes 5 and 15, arrowheads). No complex formation between the anti-RUNX1(AML1) antibody and radioendlabeled WT NCAM(CD56) (lane 16) or WT GMCSF (lane 17) consensus probes in the absence of RUNX1(AML1) (ie, Jurkat nuclear extract).
Discussion
NCAM(CD56) is a member of the immunoglobulin superfamily, mediating intercellular adhesion in the nervous system and skeletal muscle. 23-25 NCAM(CD56) expression was also found at the disci intercalcares in normal human hearts and cardiac allografts. 26,27 A correlation between NCAM(CD56) expression levels and heart pathology has not been reported. Therefore, it is our main finding that strong overexpression of NCAM(CD56) is consistently associated with ICM (Figures 1, 2, 4 and 5) ▶ . Strong NCAM(CD56) protein overexpression was restricted to cardiomyocytes associated with ischemia-induced scars and was qualitatively different from expression in normal hearts since it involved the whole cardiomyocyte cell membrane and cytoplasm (Figure 2, c and d) ▶ . This NCAM(CD56) expression pattern appears characteristic for cardiomyocytes within or adjacent to scars in ICM, while at most slight overexpression was found in hearts from patients with CCM, HOCM, sarcoidosis, and myocarditis (Figures 2 and 5) ▶ . The relationship of the findings to ischemia is supported by identical observations in a rat model of ICM 17 (Figure 4b) ▶ but not in hearts of spontaneously hypertensive rats (Figures 4, a and c) ▶ . The findings imply that NCAM(CD56) overexpression is neither a marker for cardiac hypertrophy nor for contractile insufficiency.
The second new finding is the identification of RUNX1(AML1) as a transcription factor that is up-regulated in parallel to NCAM(CD56) overexpression in ICM (Figure 7) ▶ . Overexpression of the 52-kd RUNX1(AML1) isoform in parallel to NCAM(CD56) in ICM and the demonstration that RUNX1(AML1) has a binding site within the NCAM(CD56) promoter suggest that RUNX1(AML1) plays a role in NCAM(CD56) regulation. This hypothesis is supported by two additional findings. First, other transcription factors with potential binding sites within the NCAM(CD56) promoter (like NF-kappa B 28 ) did not exhibit differential expression in ICM as compared to controls (Figure 6a) ▶ . The findings do not exclude, however, that constitutively expressed NF-kappa B, HOX, PAX, or other transcription factors contribute to ICM-related NCAM(CD56) expression. Second, hyperexpression of the 52-kd RUNX1(AML1) isoform in ICM was accompanied by down-regulation of a 38-kd band (Figure 6c) ▶ that reacted with an antibody against the N terminus of RUNX1(AML1). This band might correspond to the 38-kd RUNX1(AML1) delta N isoform 29 that has a known dominant negative function. Thus, the findings suggest that the active RUNX1(AML1) isoform is up-regulated while a dominant negative isoform is down-regulated in ICM. Since there are other RUNX1(AML1) isoforms with dominant negative effects, 30-32 it appears warranted to analyze ICM hearts for the whole spectrum of RUNX1(AML1) isoforms.
The functional significance of NCAM(CD56) overexpression in ICM is unknown, as are the trigger(s) underlying RUNX1(AML1) up-regulation. One possible indication of the trigger(s) is the restriction of NCAM(CD56) overexpression to the vicinity of ischemia-induced scars. This expression pattern is strikingly similar to that encountered in tumor-infiltrated human skeletal muscle (Figure 3) ▶ . Skeletal muscle fibers separated from each other by tumor cells up-regulate NCAM(CD56), while adjacent tumor-free muscle fibers do not. Therefore, we suggest that loss of cell-cell interaction might trigger NCAM(CD56) gene expression in cardiac and skeletal muscle, respectively. This hypothesis is analogous to the findings of Saffitz et al, 33,34 who showed that down-regulation of the gap junction protein connexin43 is due to loss of cell-cell interaction. Since this effect is mediated by stress-induced activation of c-JUN N-terminal kinase, 35 it will be interesting to analyze NCAM expression in relation to c-JUN kinase activity.
According to this hypothesis, concomitant up-regulation of NCAM(CD56) and RUNX1(AML1) is not only a feature of ICM (Figures 2, 3, and 7) ▶ and tumor-infiltrated skeletal muscle (Figure 3) ▶ but also of muscle after denervation. 22 Interestingly, identical triggers target AChR-γ-subunit gene expression in skeletal muscle. 19,26,36,37 Thus, as a common theme, “communication failures” by either neuromuscular blockade or disruption of (cardio)myocyte-(cardio)myocyte contacts stereotypically are associated with increased transcription of the NCAM(CD56), RUNX1(AML1), and AChR-γ-subunit genes. Therefore, it will be interesting to determine whether denervation-related genes in addition to RUNX1(AML1) and NCAM(CD56) play a role in ICM.
In summary, we identified NCAM(CD56) hyperexpression as a local response of cardiomyocytes to scar formation in ICM. This response is probably regulated by various RUNX1(AML1) isoforms that we suspect to act as positive or dominant-negative effectors targeting the NCAM(CD56) promoter.
Acknowledgments
We thank Mrs. Margrit Bonengel, Mrs. Sabine Roth, and Mr. Erwin Schmitt for expert technical assistance.
Footnotes
Address reprint requests to Dr. Stefan Gattenlöhner, Institute of Pathology, University of Würzburg, Josef-Schneiderstr.2, 97080 Würzburg, Germany. E-mail: stefan.gattenloehner@mail.uni-wuerzburg.de.
Supported by the Sander-Stiftung, grant 99.112.1.
References
- 1.Gheorghiade M, Bonow RO: Chronic heart failure in the United States: a manifestation of coronary artery disease. Circulation 1998, 97:282-289 [DOI] [PubMed] [Google Scholar]
- 2.Bourassa MG, Gurne O, Bangdiwala SI, Ghali JK, Young JB, Rousseau M, Johnstone DE, Yusuf S: Natural history and patterns of current practice in heart failure: the Studies of Left Ventricular Dysfunction (SOLVD) Investigators. J Am Coll Cardiol 1993, 22:14A-19A [DOI] [PubMed] [Google Scholar]
- 3.Williams JF BM, Fowler MB, Francis GS, Garson A, Jr, Gersh BJ, Hammer DF, Hlatky MA, Leier CV, Packer M, Pitt B, Ullyot DJ, Wexler LF, Winters W, Jr, Ritchie JL, Cheitlin MD, Eagle KA, Gardner TJ, Gibbons RJ, Lewis RP, O’Rourke RA, Ryan TJ: Guidelines for the evaluation and management of heart failure: report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee on Evaluation and Management of Heart Failure). Circulation 1995, 92:27647586389 [Google Scholar]
- 4.Richardson P, McKenna W, Bristow M, Maisch B, Mautner B, O’Connell J, Olsen E, Thiene G, Goodwin J, Gyarfas I, Martin I, Nordet P: Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the definition and classification of cardiomyopathies. Circulation 1996, 93:841-842 [DOI] [PubMed] [Google Scholar]
- 5.Felker GM, Shaw LK, O’Connor CM: A standardized definition of ischemic cardiomyopathy for use in clinical research. J Am Coll Cardiol 2002, 39:210-218 [DOI] [PubMed] [Google Scholar]
- 6.Tenenbaum A, Fisman EZ, Motro M: Toward a redefinition of ischemic cardiomyopathy: is it an indivisible entity? J Am Coll Cardiol 2002, 40:205-206 [DOI] [PubMed] [Google Scholar]
- 7.Benedict CR, Johnstone DE, Weiner DH, Bourassa MG, Bittner V, Kay R, Kirlin P, Greenberg B, Kohn RM, Nicklas JM, McIntyre K, Quinones MA, Yusut S, : for the SOLVD Investigators: Relation of neurohumoral activation to clinical variables and degree of ventricular dysfunction: a report from the registry of Studies of Left Ventricular Dysfunction (SOLVD) Investigators. J Am Coll Cardiol 1994, 23:1410-1420 [DOI] [PubMed] [Google Scholar]
- 8.Benedict CR: Neurohumoral aspects of heart failure. Cardiol Clin 1994, 12:9-23 [PubMed] [Google Scholar]
- 9.Lerman A, Hildebrand FL, Jr, Aarhus LL, Burnett JC, Jr: Endothelin has biological actions at pathophysiological concentrations. Circulation 1991, 83:1808-1814 [DOI] [PubMed] [Google Scholar]
- 10.McMurray JJ, Ray SG, Abdullah I, Dargie HJ, Morton JJ: Plasma endothelin in chronic heart failure. Circulation 1992, 85:1374-1379 [DOI] [PubMed] [Google Scholar]
- 11.Mann DL, Young JB: Basic mechanisms in congestive heart failure: recognizing the role of proinflammatory cytokines. Chest 1994, 105:897-904 [DOI] [PubMed] [Google Scholar]
- 12.Torre-Amione G, Kapadia S, Lee J, Durand JB, Bies RD, Young JB, Mann DL: Tumor necrosis factor-α and tumor necrosis factor receptors in the failing human heart. Circulation 1996, 93:704-711 [DOI] [PubMed] [Google Scholar]
- 13.Schreiner GF: Immune modulation of cardiac cell function. Trans Am Clin Climatol Assoc 1998, 109:39-49 [PMC free article] [PubMed] [Google Scholar]
- 14.Vadlamani L, Abraham WT: Insights into pathogenesis and treatment of cytokines in cardiomyopathy. Curr Cardiol Rep 2000, 2:120-128 [DOI] [PubMed] [Google Scholar]
- 15.Kloner RA, Jennings RB: Consequences of brief ischemia: stunning, preconditioning, and their clinical implications: part 2. Circulation 2001, 104:3158-3167 [DOI] [PubMed] [Google Scholar]
- 16.Heads RJ, Latchman DS, Yellon DM: The molecular basis of adaptation to ischemia in the heart: the role of stress proteins and anti-oxidants in the ischemic and reperfused heart. EXS 1996, 76:383-407 [DOI] [PubMed] [Google Scholar]
- 17.Pfeffer MA, Pfeffer JM, Fishbein MC, Fletcher PJ, Spadaro J, Kloner RA, Braunwald E: Myocardial infarct size and ventricular function in rats. Circ Res 1979, 44:503-512 [DOI] [PubMed] [Google Scholar]
- 18.Sambrook JFE, Maniatis T: Molecular Cloning 1989, vol 3.:pp 18.60-18.78 Cold Spring Harbor Press New York [Google Scholar]
- 19.Gattenloehner S, Vincent A, Leuschner I, Tzartos S, Muller-Hermelink HK, Kirchner T, Marx A: The fetal form of the acetylcholine receptor distinguishes rhabdomyosarcomas from other childhood tumors. Am J Pathol 1998, 152:437-444 [PMC free article] [PubMed] [Google Scholar]
- 20.Takahashi A, Satake M, Yamaguchi-Iwai Y, Bae SC, Lu J, Maruyama M, Zhang YW, Oka H, Arai N, Arai K, Ito Y: Positive and negative regulation of granulocyte-macrophage colony- stimulating factor promoter activity by AML1-related transcription factor, PEBP2. Blood 1995, 86:607-616 [PubMed] [Google Scholar]
- 21.Hwang DM, Dempsey AA, Lee CY, Liew CC: Identification of differentially expressed genes in cardiac hypertrophy by analysis of expressed sequence tags. Genomics 2000, 66:1-14 [DOI] [PubMed] [Google Scholar]
- 22.Zhu X, Yeadon JE, Burden SJ: AML1 is expressed in skeletal muscle and is regulated by innervation. Mol Cell Biol 1994, 14:8051-8057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grumet M, Rutishauser U, Edelman GM: Neural cell adhesion molecule is on embryonic muscle cells and mediates adhesion to nerve cells in vitro. Nature 1982, 295:693-695 [DOI] [PubMed] [Google Scholar]
- 24.Edelman GM: Cell adhesion molecules in neural histogenesis. Annu Rev Physiol 1986, 48:417-430 [DOI] [PubMed] [Google Scholar]
- 25.Rutishauser U, Grumet M, Edelman GM: Neural cell adhesion molecule mediates initial interactions between spinal cord neurons and muscle cells in culture. J Cell Biol 1983, 97:145-152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gordon L, Wharton J, Moore SE, Walsh FS, Moscoso JG, Penketh R, Wallwork J, Taylor KM, Yacoub MH, Polak JM: Myocardial localization and isoforms of neural cell adhesion molecule (N-CAM) in the developing and transplanted human heart. J Clin Invest 1990, 86:1293-1300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.al-Mahdawi S, Shallal A, Wyse RK: Neural cell adhesion molecule (N-CAM) in fetal and mature human heart. FEBS Lett 1990, 267:183-185 [DOI] [PubMed] [Google Scholar]
- 28.Krushel LA, Cunningham BA, Edelman GM, Crossin KL: NF-κ B activity is induced by neural cell adhesion molecule binding to neurons and astrocytes. J Biol Chem 1999, 274:2432-2439 [DOI] [PubMed] [Google Scholar]
- 29.Zhang YW, Bae SC, Huang G, Fu YX, Lu J, Ahn MY, Kanno Y, Kanno T, Ito Y: A novel transcript encoding an N-terminally truncated AML1/PEBP2 αB protein interferes with transactivation and blocks granulocytic differentiation of 32Dcl3 myeloid cells. Mol Cell Biol 1997, 17:4133-4145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Levanon D, Glusman G, Bangsow T, Ben-Asher E, Male DA, Avidan N, Bangsow C, Hattori M, Taylor TD, Taudien S, Blechschmidt K, Shimizu N, Rosenthal A, Sakaki Y, Lancet D, Groner Y: Architecture and anatomy of the genomic locus encoding the human leukemia-associated transcription factor RUNX1/AML1. Gene 2001, 262:23-33 [DOI] [PubMed] [Google Scholar]
- 31.Aziz-Aloya RB, Levanon D, Karn H, Kidron D, Goldenberg D, Lotem J, Polak-Chaklon S, Groner Y: Expression of AML1-d, a short human AML1 isoform, in embryonic stem cells suppresses in vivo tumor growth and differentiation. Cell Death Differ 1998, 5:765-773 [DOI] [PubMed] [Google Scholar]
- 32.Tanaka T, Tanaka K, Ogawa S, Kurokawa M, Mitani K, Nishida J, Shibata Y, Yazaki Y, Hirai H: An acute myeloid leukemia gene, AML1, regulates hemopoietic myeloid cell differentiation and transcriptional activation antagonistically by two alternative spliced forms. EMBO J 1995, 14:341-350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kanno S, Saffitz JE: The role of myocardial gap junctions in electrical conduction and arrhythmogenesis. Cardiovasc Pathol 2001, 10:169. [DOI] [PubMed] [Google Scholar]
- 34.Kanno S, Kovacs A, Yamada KA, Saffitz JE: Connexin43 as a determinant of myocardial infarct size following coronary occlusion in mice. J Am Coll Cardiol 2003, 41:681-686 [DOI] [PubMed] [Google Scholar]
- 35.Petrich BG, Gong X, Lerner DL, Wang X, Brown JH, Saffitz JE, Wang Y: c-Jun N-terminal kinase activation mediates down-regulation of connexin43 in cardiomyocytes. Circ Res 2002, 91:640-647 [DOI] [PubMed] [Google Scholar]
- 36.Witzemann V, Barg B, Nishikawa Y, Sakmann B, Numa S: Differential regulation of muscle acetylcholine receptor γ- and ε-subunit mRNAs. FEBS Lett 1987, 223:104-112 [DOI] [PubMed] [Google Scholar]
- 37.Gattenlohner S, Schneider C, Thamer C, Klein R, Roggendorf W, Gohlke F, Niethammer C, Czub S, Vincent A, Muller-Hermelink HK, Marx A: Expression of foetal type acetylcholine receptor is restricted to type 1 muscle fibres in human neuromuscular disorders. Brain 2002, 125:1309-1319 [DOI] [PubMed] [Google Scholar]