

Abstract
Anti-microtubule agents such as vinorelbine and paclitaxel, which are extensively used in the treatment of lung cancers, activate mitotic spindle checkpoint. Although defects of the mitotic spindle checkpoint are thought to play a role in the genesis of chromosome instability, we previously reported its frequent impairment in human lung cancer cell lines. In this study, we examined a panel of 13 human cancer cell lines comprising 11 lung and 2 other cancers and found a significant difference in the resistance to apoptosis induced by anti-microtubule agents between mitotic spindle checkpoint-impaired and -proficient cancer cell lines. This finding was in marked contrast to a lack of such correlation with a DNA damaging agent, cis-platin. Interestingly, anti-microtubule agent-induced apoptosis in mitotic spindle checkpoint-proficient cell lines, NCI-H460 and A549, was shown to be markedly reduced by staurosporine treatment in association with the shortened mitotic arrest, whereas various inhibitors of caspases seemed to have very modest effects. Taken together, these findings suggest the potential involvement of mitotic spindle checkpoint in the induction of apoptosis by anti-microtubule agents in human lung cancers, warranting further studies on the underlying mechanisms.
Cell-cycle checkpoints are a monitoring system that arrests cells at a particular phase of the cell cycle in response to a lack of appropriate signals for progression, thereby maintaining genetic stability and ensuring a smooth progression through the cell cycle. 1 Recent molecular and cellular biological studies have revealed that cell-cycle checkpoints are often impaired in cancer cells, leading to the failure of normal growth control and the generation of genetic instability in cancer cells. 2 We previously reported on the frequent impairment of the mitotic spindle checkpoint in human lung cancer cell lines. 3 The mitotic spindle checkpoint is activated by the presence of unattached kinetochores and impedes the cell cycle at the prometaphase. 4 This checkpoint thus prevents abnormal chromosome segregation because of the lack of bipolar attachment of kinetochores to mitotic spindles, and its dysfunction in cancer cells is thought to play a role in the development of chromosome instability. 5,6
Anti-microtubule agents including nocodazole, vinorelbine, and paclitaxel activate the mitotic spindle checkpoint, while many of them are extensively used as chemotherapeutic agents for the treatment of various cancers including human lung cancers. Although a possible link between integrity of the mitotic spindle checkpoint and sensitivity to the apoptosis induced by anti-microtubule agents is presumed to be present, very few studies have dealt with this question directly, 7,8 and extensive studies have not yet been conducted using a panel of human cancer cell lines. Thus, it has remained uncertain whether such a relationship is indeed present in human cancer cells with frequent impairment of the mitotic spindle checkpoint.
In this study, we examined a panel of 13 human cancer cell lines comprising 11 lung and 2 other cancers and found a significant difference in the resistance to apoptosis induced by anti-microtubule agents between mitotic spindle checkpoint-impaired and -proficient cancer cell lines in contrast to a lack of such a difference in the treatment with the DNA damaging agent, cis-platin. In addition, initial attempts were made to elucidate how signals for the induction of apoptosis are transduced in mitotic checkpoint-proficient cells.
Materials and Methods
Cell Lines and Culture
ACC-LC-176 was established in our laboratory 9 and NCI-H460 and A-427 were purchased from American TypeCulture Collection (Manassas, VA). Other cell lines were generously provided by Dr. L. J. Old (Memorial Sloan-Kettering Cancer Center, New York, NY) and Dr. Y. Hayata (Tokyo Medical College, Tokyo, Japan). Cells were cultured in RPMI 1640 medium supplemented with 5% fetal calf serum (Life Technologies, Inc., Grand Island, NY), 100 U/ml penicillin, and 100 μg/ml streptomycin.
Chemicals
PD98059, LY294002, and SB203580 were purchased from Calbiochem (Darmstadt, Germany). Staurosporine was obtained from Biomol Res. Inc. (Plymouth Meeting, PA), wortmannin and vinorelbine ditartrate was from Kyowa Medics Inc. (Tokyo, Japan), docetaxel from Chugai Pharmaceutical Co. (Tokyo, Japan), and cis-platin from Bristol-Myers Squibb Co. (Tokyo, Japan). Other chemicals were obtained from Sigma (St. Louis, MO).
Measurement of Mitotic Indices and Apoptosis
For cell biological analyses, 1 × 105 cells were inoculated in 60-mm dishes the day before the experiments. Cells were washed with Dulbecco’s modified phosphate-buffered saline (PBS) once, then placed into the RPMI 1640 medium containing anti-microtubule agents at the concentrations indicated. After incubation with anti-microtubule agents for the periods of time indicated, the cells were harvested by trypsinization, followed by fixation with 4% formalin in PBS for 30 minutes. Cells were then attached to glass slides by centrifugation using an AutoSmear CF-12D (Sakura, Tokyo, Japan) and stained with 0.1 μg/ml of 4′,6-diamidino-2-phenylindole (DAPI). To measure the mitotic cells and apoptotic cells, at least 300 cells were counted for each slide with fluorescence microscopy. Cells having condensed chromosomes without a nuclear membrane were regarded as mitotic cells, which corresponded very well to positive staining with an anti-phosphohistone H3 antibody 10 (data not shown). Mitotic indices were calculated as the percentage of mitotic cells among the total viable cells. Cells having fragmented and uniformly condensed nuclei were regarded as apoptotic cells, and this criterion showed a good agreement with cells positively stained with an anti-cleaved caspase-3 antibody (data not shown). The terminal dUTP nick-end labeling (TUNEL) assay using an in situ cell death detection kit (Roche Diagnostics, Mannheim, Germany) was used to confirm the results of the morphology-based assay for the detection of apoptotic cells.
Immunocytochemistry
Cells, which were attached to glass slides as described above, were incubated with an anti-cleaved caspase-3-specific polyclonal antibody (1:1000 dilution; Cell Signaling Technology, Beverly, MA) or an anti-phosphohistone H3-specific antibody (1:200 dilution; Upstate Biotechnology, Lake Placid, NY) for 1 hour at room temperature, followed by a 1-hour incubation with an Alexa 488-conjugated secondary antibody (Molecular Probes, Eugene, OR). Cell nuclei were then stained with DAPI as described above. For staining of BrdU that had been incorporated into nuclear DNA harvested cells were serially fixed with 70% and 100% ethanol before making cytospin preparations as described above. The resultant slides were treated with 2 N HCl for denaturation of DNA and then neutralized with 0.1 mol/L of Tris-HCl (pH 8.0), followed by staining with a fluorescein isothiocyanate-conjugated anti-BrdU antibody (1:20 dilution; Becton & Dickinson, Bedford, MA) and DAPI.
Flow Cytometry
The harvested cells were fixed with 70% ethanol for more than 2 hours, then washed with PBS once, and stained with an anti-cyclin B monoclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA) or an anti-CENP-F polyclonal antibody (Novus Biologicals, Inc., Littleton, CO), followed by incubation with an Alexa 488-conjugated secondary antibody (Molecular Probes). After further staining with propidium iodide, flow cytometric analysis was performed using FACSCalibur and Cellquest software (Becton & Dickinson).
Western Blot Analysis
Ten μg of total cell lysate solubilized in Laemmli’s sample buffer was electrophoresed on 12.5 or 15% sodium dodecyl sulfate-polyacrylamide gels and transferred to Immobilon-P filters (Millipore Corp., Bedford, MA). The filters were first incubated with either a primary antibody against Bcl-2 or those specific to caspase-3, -8, or -9 (Cell Signaling Technology), and then with a horseradish peroxidase-conjugated secondary antibody (Cell Signaling Technology). For visualization, an enhanced chemiluminescence system (Amersham, Buckinghamshire, UK) was used.
Results
Characterization and Affirmation of Mitotic Spindle Checkpoint-Impaired Human Lung Cancer Cell Lines
In our previous study, we used a conventional protocol to examine lung cancer cell lines for the presence of mitotic checkpoint impairment, ie, measurement of mitotic indices in the presence of nocodazole, a microtubule-disrupting agent. To further ascertain the presence of mitotic checkpoint impairment in lung cancer cell lines, we characterized three lung cancer cell lines in greater detail, all of which had wild-type p53 (Table 1) ▶ . As shown in Figure 1A ▶ , the majority of cells in all these cell lines became rapidly arrested at 4n DNA content by treatment with 200 nmol/L of nocodazole for 18 hours. NCI-H460 showed a rapid increase in cells arrested at prometaphase, which reached ∼90% after 18 hours of incubation and gradually decreased because of adaptation, whereas the increases in the mitotic indices in A-427 and PC-1 were very modest (Figure 1B) ▶ . The rise in the mitotic indices of the latter two cell lines plateaued, even if we added more than 100 nmol/L of nocodazole, and no further rises in mitotic indices were detected at least up to 600 nmol/L of nocodazole (Figure 1C) ▶ . We further ascertained that the observed lack of clear increases in mitotic indices in A-427 and PC-1 was indeed because of their failure in mitotic arrest, and this was not caused by cell-cycle arrest before entering prometaphase in response to the nocodazole treatment. For this purpose, we measured mitotic indices in synchronized cells (Figure 2A) ▶ as well as by counting BrdU-labeled mitoses in asynchronous cultures by simultaneous treatment with BrdU and nocodazole (Figure 2B) ▶ . Further, the expression of cell-cycle phase markers was examined in these cell lines treated with nocodazole for 18 hours by means of two-color flow cytometry. As shown in Figure 2C ▶ , the majority of NCI-H460 cells with 4n-DNA content expressed cyclin B, a cell-cycle marker from late S to metaphase. In contrast, accumulation of cyclinB1-negative cells with 4n DNA content was observed in A-427 and PC-1, suggesting that a large proportion of the cells was in the so-called psuedo-G1 state. Similar results were obtained by using an antibody against CENP-F, another cell-cycle marker for G2 and metaphase (data not shown). Collectively, these results clearly indicated that A-427 and PC-1 were not arrested before the mitotic phase and were arrested at prometaphase for significantly shorter periods of time than NCI-H460 in the presence of nocodazole.
Table 1.
Summary of the Relationship between Nocodazole-Induced Apoptosis and Checkpoint Integrity
| Cell line | Sensitivity to nocodazole | p53 status | Mitotic spindle checkpoint* | Reference for p53 status |
|---|---|---|---|---|
| A-427 | Resistant | Wild type | Impaired | 29 |
| PC-1 | Resistant | Wild type | Impaired | 30 |
| SK-LU-1 | Resistant | His to Arg (codon 193) | Impaired | 31 |
| Calu-1 | Resistant | Homozygous deletion | Partially impaired | 30 |
| ACC-LC-176 | Resistant | Wild type | Normal | 30 |
| QG56 | Resistant | Pro to Ser (codon 249) | Normal | 30 |
| NCI-H460 | Sensitive | Wild type | Normal | 30 |
| SBC-3 | Sensitive | Wild type | Normal | 32 |
| A549 | Sensitive | Wild type | Normal | 31 |
| PC-10 | Sensitive | Gly to Cys (codon 245) | Normal | 30 |
| SK-MES-1 | Sensitive | Glu to stop (codon 298) | Normal | 30 |
| HCT116 | Sensitive | Wild type | Normal | 30 |
| HeLa | Sensitive | Wild type | Normal | 33 |
*Mitotic spindle checkpoint response was examined as described previously and graded for mitotic indices at 18 hours after the initiation of nocodazole treatment: impaired, <20%; Partially impaired, 20 to 40%; normal, >40%.
Figure 1.
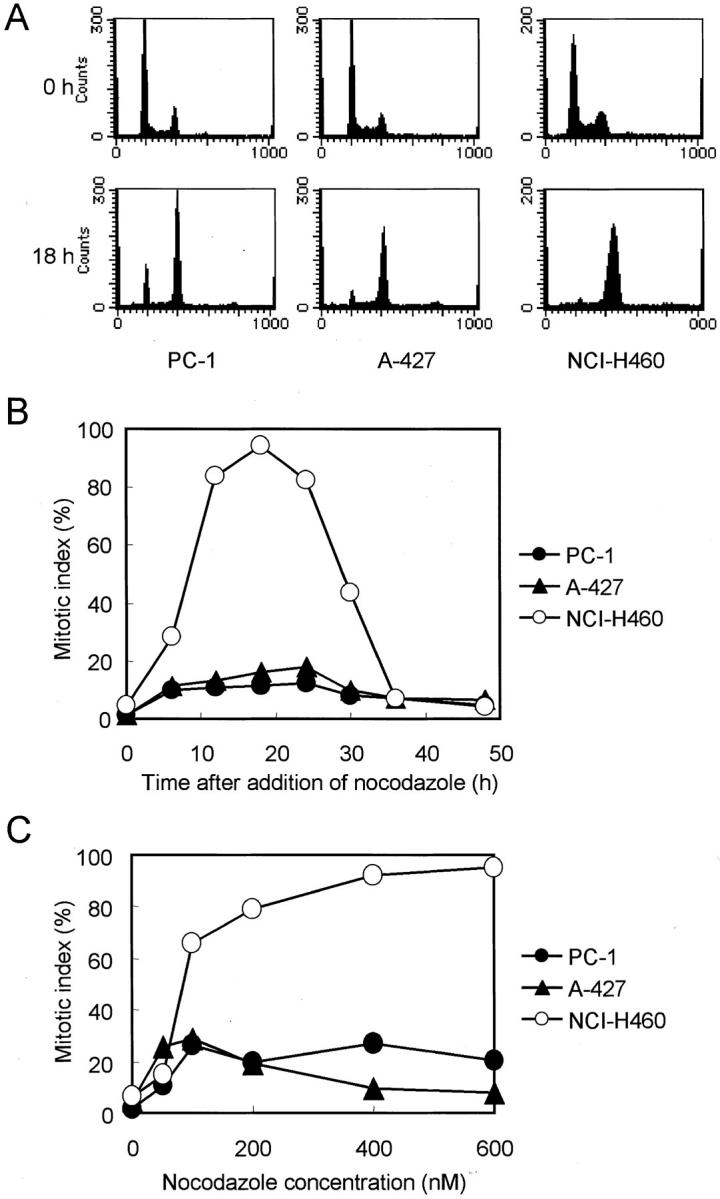
Response of mitotic spindle checkpoint-proficient (NCI-H460) and -impaired (PC-1 and A-427) human lung cancer cell lines to nocodazole. A: Flow cytometric analysis of the cell cycle. Majority of cells are arrested at 4n DNA content in all cell lines when treated with 200 nmol/L of nocodazole for 18 hours. B: Time-dependent changes in mitotic indices. Mitotic index of mitotic spindle checkpoint-proficient NCI-H460 rises rapidly and peaks at 18 hours after the initiation of 200 nmol/L of nocodazole treatment and then declines by adaptation. In contrast, those of the mitotic spindle checkpoint-impaired cell lines show only slight increases. C: Changes in mitotic indices with respect to nocodazole concentrations. Mitotic indices of the mitotic checkpoint-impaired lines are plateaued by 100 nmol/L of nocodazole.
Figure 2.

Affirmation of mitotic arrest in response to nocodazole. A: Inhibition of entry into mitoses in synchronized cells. Cells that had been arrested at the S phase or G1/S boundary by 16 hours of treatment with 5 μmol/L of aphidicolin were released by replacing the culture media. The initial increases in the mitotic indices are similar regardless of the presence or absence of 200 nmol/L of nocodazole, indicating the absence of entry into mitosis in certain cell lines. B: Inhibition of entry into mitoses in BrdU-labeled asynchronous cells. Simultaneous treatment with 10 μmol/L of BrdU and 200 nmol/L of nocodazole results in the similar initial increases in mitotic cells labeled with BrdU, indicating that cell lines are not differentially arrested before entering mitosis. Note that scales of the y axes are different in each cell line. C: Flow cytometric analyses of expression of cyclin B. Upper figures represent the dot plot, and lower figures represent the percentages of cells residing in each quadrant. The majority of cells having 4n DNA content express cyclin B in a mitotic spindle checkpoint-proficient cell line, but not in the impaired cell lines.
Correlation between Mitotic Spindle Checkpoint Status and Induction of Apoptosis by Anti-Microtubule Agents
Anti-microtubule agents are known to induce apoptosis in human cancer cells. 11,12 We therefore studied the relationship between the integrity of the mitotic spindle checkpoint and susceptibility to anti-microtubule-induced apoptosis. As shown in Figure 3A ▶ , apoptotic cells became detectable 18 hours after addition of nocodazole in NCI-H460, and the proportion of apoptotic cells steeply increased with time, reaching ∼60% 48 hours later. In marked contrast, only a small percentage of cells fell into apoptosis in the mitotic spindle checkpoint-impaired A-427 and PC-1 cell lines. To exclude the possibility that the correlation between the two biological characteristics might have arisen by coincidence, we further examined a panel of 13 human cancer cell lines comprising 11 lung and 2 other cancers. Induction of nocodazole-induced apoptosis in cancer cell lines with impaired mitotic spindle checkpoint was found to be significantly lower than that in mitotic spindle checkpoint-proficient cell lines [7.1 ± 1.4 versus 38.0 ± 6.9 (mean ± SE), P = 0.01 by Student’s t-test; Figure 3B ▶ ]. It was noteworthy that all of the mitotic spindle checkpoint-impaired cell lines were highly resistant to the induction of apoptosis. Induction of apoptosis was also measured with the TUNEL assay and showed good concordance of percentages of apoptotic cells between the two types of assays. Specifically, the respective percentages of apoptotic cells determined with the morphology-based and the TUNEL assays were: 55% and 54% in NCI-H460, 70% and 64% in HeLa, 51% and 41% in SBC3, 40% and 42% in A549, 4% and 6% in PC1, 9% and 12% in A427, 5% and 3% in SK-Lu-1, and 10% and 3% in Calu1. On the contrary, susceptibility to apoptosis induced by the DNA damaging anti-cancer agent, cis-platin, did not show any significant differences with the integrity of the mitotic spindle checkpoint (Figure 3C) ▶ .
Figure 3.

Induction of apoptosis by the treatment with nocodazole or cis-platin. A: Time-dependent accumulation of apoptotic cells by the treatment with 200 nmol/L of nocodazole. Apoptotic cells become detectable at 18 hours and progressively accumulate with time in mitotic spindle checkpoint-proficient cell line, but not in the impaired cell lines. B: Significantly lower susceptibility to nocodazole-induced apoptosis in mitotic spindle checkpoint-impaired cell lines in a panel of 13 human cancer cell lines [7.1 ± 1.4 versus 38.0 ± 6.9 (mean ± SE), P = 0.01 by Student’s t-test; B]. The susceptibilities of cell lines are shown as the percentage of apoptotic cells 48 hours after the addition of 200 nmol/L of nocodazole. C: Lack of a significant relationship between the mitotic spindle checkpoint integrity and susceptibility to cis-platin-induced apoptosis in the same panel of cell lines [44.9 ± 19.7 versus 69.7 ± 7.2 (mean ± SE), NS]. Susceptibility to cis-platin is shown as the percentage of apoptotic cells at 48 hours after addition of 10 μg/ml of cis-platin.
We next asked whether a similar relationship was present when lung cancer cells were treated with other agents affecting microtubule spindle formation. Two representative mitotic spindle checkpoint-proficient lines, NCI-H460 and SBC3, were exposed to either a microtubule-depolymerizing agent, vinorelbine, or a microtubule-stabilizing agent, docetaxel, for 48 hours. Vinorelbine and docetaxel induced, respectively, 55% and 53% of apoptotic cells in NCI-H460 and 50% and 72% in SBC3. In contrast, SK-Lu-1 and A-427 with an impaired mitotic spindle checkpoint were highly resistant to both of the two distinct types of anti-microtubule agents, showing induction of apoptosis in only 1% and 3% of the SK-Lu-1 cells and 13% and 25% of the A-427. We note that susceptibility to anti-microtubule agents-induced apoptosis was unrelated to the p53 status of each cell line (Table 1) ▶ . Taken together, these results strongly suggested that the status of mitotic spindle checkpoint integrity might affect the cellular susceptibility of each lung cancer cell line to the induction of apoptosis by anti-microtubule agents.
Signal Transduction Pathways for Nocodazole-Induced Apoptosis
Initial attempts were undertaken to explore the underlying mechanisms of anti-microtubule agent-induced apoptosis. NCI-H460, a representative cell line with intact mitotic spindle checkpoint was treated with a variety of inhibitors, which included PD98059 (a selective MEK inhibitor), LY294002 (a specific phosphatidylinositol 3-kinase inhibitor), wortmannin (phosphatidylinositol 3-kinase and ATM kinase inhibitor), SB203580 (a specific p38 kinase inhibitor), thymidine (DNA synthesis blocker), staurosporine (a broad specific Ser/Thr-kinase inhibitor), Z-VAD-fmk (a broad specific caspase inhibitor), Z-LEHD-fmk (a specific caspase-9 inhibitor), and Z-IETD-fmk (a specific caspase-8 inhibitor) (Figure 4) ▶ . Tne most notable inhibition of apoptosis was observed with the addition of 30 nmol/L of staurosporine, yielding nearly complete suppression of both nocodazole-induced apoptosis and an increase in mitotic index (data not shown). In addition, 12-hour pretreatment of cells with 2.0 mmol/L of thymidine, which blocks cells at the G1/S boundary and within the S phase, before the addition of nocodazole, modestly reduced apoptosis, although this reduction may have been considerably underestimated because of the toxic effect of prolonged thymidine treatment itself. Unexpectedly, the effect of Z-IETD-fmk, a caspase-8-specific inhibitor, and of Z-LEHD-fmk, a caspase-9-specific caspase inhibitor, was negligible, while Z-VAD-fmk, a caspase inhibitor with broad specificity, had only a limited inhibitory effect on apoptosis in NCI-H460. We did not observe any significant reduction in nocodazole-induced apoptosis as a result of adding other inhibitors except for the PD98059 MEK inhibitor, which yielded a modest effect. In contrast to the limited apoptotic effects of caspase inhibitors on NCI-H460, their effect on A-427, which has an impaired mitotic spindle checkpoint, was significant. Z-VAD-fmk suppressed residual activity of nocodazole-induced apoptosis in this cell line by 94%, while Z-IETD-fmk and Z-LEHD-fmk yielded 39% and 42% reduction of apoptosis, respectively.
Figure 4.

Effects of various metabolic inhibitors on the nocodazole-induced apoptosis in mitotic spindle checkpoint-proficient NCI-H460 cells. PD98059, LY294002, wortmannin, SB203580, and caspase inhibitors were added simultaneously with 200 nmol/L of nocodazole treatment, while thymidine and staurosporine were added 12 hours before and after the initiation of nocodazole treatment, respectively. Note that staurosporine treatment markedly reduces apoptotic cells 48 hours after the initiation of nocodazole treatment.
Association between Mitotic Arrest and Anti-Microtubule Agent-Induced Apoptosis
Staurosporine, which is a broad specificity inhibitor of serine/threonine kinases, is known to force cells to exit from mitotic arrest imposed by anti-microtubule agents. 13 We asked whether nocodazole-induced apoptosis could be reduced by delayed exposure to staurosporine after cells started to accumulate at the mitotic phase. Staurosporine was added to the mitotic spindle checkpoint-proficient NCI-H460 and A549 cell lines 12 hours after the initiation of nocodazole treatment, at which time the increase in the mitotic index had become evident. This delayed exposure to staurosporine effectively caused rapid exit from mitosis in association with a striking reduction in apoptosis (Figure 5; A to D) ▶ . Further examination revealed that such reduction of apoptosis could be attained, if staurosporine was added within 18 hours after the initiation of nocodazole treatment, but was found to be no longer effective after 24 hours (Figure 5E) ▶ . Taken together, these results suggest that mitotic arrest for a prolonged period of time, ie, longer than 6 to 12 hours, might destine cells to fall into apoptosis by a mechanism involving a staurosporine-sensitive molecule.
Figure 5.

Effects of staurosporine on the reduction of mitotic indices (A and C) and inhibition of the apoptosis induced by nocodazole (B and D) in mitotic checkpoint-proficient cell lines. Staurosporine at concentrations sufficient to block progression toward the mitotic phase (30 nmol/L) was subsequently added to NCI-H460 (A and B) and A549 (C and D) 12 hours after the initiation of 200 nmol/L nocodazole treatment. Note that staurosporine markedly reduced apoptosis in both cell lines, while staurosporine induces rapid reduction of the mitotic index. E: Effect of timing of the addition of staurosporine on the inhibition of the nocodazole-induced apoptosis in NCI-H460. Note that staurosporine effectively inhibits apoptosis when added early on, but that later addition of staurosporine was not effective for the suppression of apoptosis in cells that had been already exposed to nocodazole for longer than 24 hours.
It is generally accepted that one of the important targets for staurosporine is the cyclin B/cdc2 kinase. We therefore examined whether olomoucine, a specific cyclin B/cdc2 kinase inhibitor, could also reduce nocodazole-induced apoptosis as was observed with staurosporine. Olomoucine dose dependently prevented cells from entering mitosis, but a reduction in apoptosis was not induced even at a dose of 200 μmol/L (Figure 6, A and B) ▶ . These results suggest that cyclin B/cdc2 kinase may not be the sole target for staurosporine in terms of apoptosis-reducing activity and that there may well be another staurosporine-sensitive kinase(s) involved in the process.
Figure 6.

No reduction of nocodazole-induced apoptosis by simultaneous treatment with olomoucine in NCI-H460. A: Dose-dependent inhibition of the mitotic entry by olomoucine. Mitotic indices were counted 18 hours after the initiation of simultaneous treatment with nocodazole and olomoucine. B: No reduction in nocodazole-induced apoptosis by olomoucine. Apoptotic cells were examined after 48 hours of the treatment with nocodazole and olomoucine.
Limited Involvement of the Bcl-2/Caspase Pathway in the Anti-Microtubule Agent-Induced Apoptosis
It has been suggested that the induction of apoptosis in response to anti-microtubule agents may result from inactivation of bcl-2 because of its phosphorylation and consequential activation of caspases, although significance of this pathway remains a matter of controversy. We therefore studied on the relationship in our system of susceptibility to nocodazole with phosphorylation of bcl-2 as well as with caspase activation. As shown in Figure 7 ▶ , degrees of bcl-2 phosphorylation tightly corresponded to the integrity of the mitotic spindle checkpoint. We observed that bcl-2 was extensively phosphorylated at 18 hours in all three mitotic spindle checkpoint-proficient cell lines examined, including nocodazole-resistant QG56. On the other hand, bcl-2 was hardly phosphorylated in any of the mitotic spindle checkpoint-impaired cell lines. Further analysis using mitotic cell fractions collected from NCI-H460 and A-427, however, revealed that phosphorylated bcl-2 was detectable in parallel to the proportion of mitotic cells (data not shown). These findings suggest that low degrees of bcl-2 phosphorylation in the checkpoint-impaired cell lines may reflect the presence of small fractions of mitotic cells.
Figure 7.

Induction of phosphorylation of bcl-2 by nocodazole treatment. Apparent phosphorylation of bcl-2 is present in mitotic spindle checkpoint-proficient cell lines including nocodazole-resistant QG56.
It is generally accepted that inactivation of bcl-2 induces activation of caspases through the release of cytochrome c from mitochondria. The activation of caspases was analyzed by monitoring the appearance of their respective cleaved active forms. As shown in Figure 8 ▶ , caspases-8, -9, and -3 were clearly activated at 48 hours in NCI-H460, and similarly, but albeit at considerably lower levels, in A-427. It was noted that cleavage of caspase-3, an effector caspase, was completely inhibited by the addition of Z-VAD-fmk, while partially blocked cleavage of caspases-8 and -9 was also detected. Together with our finding described above that Z-VAD-fmk inhibited nocodazole-induced apoptosis only to a very limited extent, the bcl-2/caspase pathway seemed to play a limited role in the anti-microtubule agent-induced apoptosis in mitotic checkpoint-proficient NCI-H460, suggesting the involvement of an additional staurosporine-sensitive pathway(s).
Figure 8.

Induction of activating cleavages of caspases by nocodazole treatment. Note that complete inhibition of caspase 3 cleavage is seen despite a very modest reduction in apoptosis (see Figure 4 ▶ ) in nocodazole-sensitive, mitotic checkpoint-proficient NCI-H460.
Discussion
Anti-microtubule agents have been shown to be active in the treatment of lung cancers. However, it is rather poorly understood how cancer cells respond to these agents and what determines cellular susceptibility. It has been suggested that response to paclitaxel is related to the expression levels of her2/neu and bcl-2, 14 whereas others have claimed that increased microtubule dynamics are associated with resistance to paclitaxel. 15 In this study, we have shown that the status of the mitotic spindle checkpoint exhibits a significant relationship with susceptibility to anti-microtubule agent-induced apoptosis in human lung cancer cell lines and that mitotic spindle checkpoint-impaired cell lines are highly resistant to anti-microtubule agents. In this connection, it is of note that impairment of the mitotic spindle checkpoint is present in ∼40% of human lung cancer cell lines. 3
Our results are in line with a previous experimental study by Taylor and McKeon. 7 They used a dominant-negative mutant of Bub1, a key component of the mitotic spindle checkpoint and showed that dominant-negative Bub1 impaired the mitotic spindle checkpoint and allowed cells to escape from cell death induced by nocodazole. It is interesting that a similar relationship was recently repeated in adult T-cell leukemia, 16 in which HTLV-1 Tax protein was shown to affect subcellular localization of the MAD1 and MAD2 proteins, leading to the induction of the failure of mitotic spindle checkpoint response and chemoresistance to microtubule inhibitors. As for lung cancer, Weitzel and Vandré 8 observed that a lung cancer cell line with an impaired mitotic spindle checkpoint was more resistant to CI-980, an anti-microtubule agent, than another lung cancer cell line with an intact checkpoint, but comparison merely between two cell lines make it difficult to draw any reliable conclusions. In our study with a panel of 13 human cancer cell lines and representative anti-microtubule agents including both stabilizer and depolymerizers of microtubules, the presence of statistically significant association between the two biological characteristics was clearly shown. In contrast to the status of the mitotic spindle checkpoint, the status of p53 showed no correlation with the susceptibility to nocodazole-induced apoptosis. In this regard, it was once suggested that loss of normal p53 function sensitizes cancer cells to paclitaxel by increasing G2/M arrest and apoptosis, 17 but further studies failed to find such a correlation in human lung cancer. 18
Anti-microtubule agents bind microtubules and cause either destabilization or stabilization in microtubule dynamics. Their anti-tumor activities are thought to be manifested through consequential cell-cycle arrest at the mitotic phase and subsequent apoptosis. 19 However, the biochemical pathways downstream of their binding to microtubules are not well understood, 11,12 and previous reports varied in the way they emphasized the importance of different pathways, although such discrepancies may have resulted from differences in the cell types used. One of the major focuses in this area of research has been phosphorylation of bcl-2 in response to the treatment of anti-microtubule agents in association with the loss of its anti-apoptotic function. 20,21 However, it should be noted that the significance of bcl-2 phosphorylation still remains controversial in terms of anti-microtubule agent-induced apoptosis. Some studies have suggested that phosphorylation of bcl-2 may be a molecular determinant of cell survival or death, 21,22 whereas others have claimed that its phosphorylation is a mere marker of mitotic phase events and is not directly related to the induction of apoptosis. 23 In the present study, the degrees of phosphorylation of bcl-2 were shown to be related to the integrity to the mitotic spindle checkpoint, pointing to a possible dependence on the duration of mitotic arrest. Inactivation of bcl-2 is generally thought to induce apoptosis through release of cytochrome c followed by the activation of caspases. Notably, the addition of a broad specificity caspase inhibitor, however, showed a very weak inhibitory effect on the nocodazole-induced apoptosis in mitotic spindle checkpoint-proficient NCI-H460, at a concentration sufficient to completely inhibit activation of caspase-3. Collectively, our results suggest the involvement of an additional apoptotic pathway(s) in the induction of apoptosis by anti-microtubule agents. We note that Huisman and colleagues 24 similarly suggested the importance of an as yet unidentified, non-bcl-2/cytochrome c/caspase pathway in the manifestation of the cytotoxic effects of paclitaxel. Consistent results have been obtained in a study analyzing the cytotoxic effect of paclitaxel in NCI-H460.
One of the most striking findings observed in this study is the marked suppression by staurosporine in nocodazole-induced apoptosis. Staurosporine is known to inhibit a wide range of serine/threonine kinases 25,26 and that a brief staurosporine treatment of mitotic cells triggers premature exit from mitosis and polyploid cell formation. 13 This study clearly demonstrates that staurosporine not only enhances mitotic exit but also inhibits the induction of apoptosis in nocodazole-treated cells. Although a molecular target(s) of staurosporine in this phenomenon remains to be identified, cyclin B/cdc2 kinase did not appear to be playing a major role, because olomoucine, a specific inhibitor of cyclin B/cdc2, failed to show a comparable effect on the suppression of apoptosis. In this connection, it is interesting that a phosphatase inhibitor, calyculin A, has been shown to inhibit staurosporine-induced effects by directly inhibiting phosphatase responsible for nuclear reassembly and histone H1 dephosphorylation 27 and that farnesyl transferase inhibitors enhance the induction of metaphase block and apoptosis in response to paclitaxel. 28
In conclusion, this study is the first clear demonstration of a possible association between the induction of anti-microtubule agent-induced apoptosis and the presence of intact mitotic spindle checkpoint and the consequential mitotic arrest in human lung cancer cell lines. Considering the frequent impairment of the mitotic spindle checkpoint in lung cancers, further studies are warranted to better understand the underlying mechanisms of the relationships, aiming at the development of novel means to overcome resistance to anti-microtubule chemotherapeutic agents.
Footnotes
Address reprint requests to Takashi Takahashi, M.D., Division of Molecular Oncology, Aichi Cancer Center Research Institute, 1-1 Kanokoden, Chikusa-ku, Nagoya 464-8681, Japan. E-mail: tak@aichi-cc.jp.
Supported in part by a Grant-in-Aid for Scientific Research on Priority Areas from the Ministry of Education, Science, Sports, and Culture, Japan, and a Grant-in-Aid for the Second Term Comprehensive Ten-Year Strategy for Cancer Control from the Ministry of Health and Welfare, Japan.
A. M.’s current address is Cellseed Inc., Fukuchi Bldg.1F, Shinjuku, Shinjuku-ku, Tokyo 160-0022, Japan.
References
- 1.Hartwell LH, Weinert TA: Checkpoints: controls that ensure the order of cell cycle events. Science 1989, 246:629-634 [DOI] [PubMed] [Google Scholar]
- 2.Elledge SJ: Cell cycle checkpoints: preventing an identity crisis. Science 1996, 274:1664-1672 [DOI] [PubMed] [Google Scholar]
- 3.Takahashi T, Haruki N, Nomoto S, Masuda A, Saji S, Osada H, Takahashi T: Identification of frequent impairment of the mitotic checkpoint and molecular analysis of the mitotic checkpoint genes, hsMAD2 and p55CDC, in human lung cancers. Oncogene 1999, 18:4295-4300 [DOI] [PubMed] [Google Scholar]
- 4.Rudner AD, Murray AW: The spindle assembly checkpoint. Curr Opin Cell Biol 1996, 8:773-780 [DOI] [PubMed] [Google Scholar]
- 5.Lengauer C, Kinzler KW, Vogelstein B: Genetic instabilities in human cancers. Nature 1998, 396:643-649 [DOI] [PubMed] [Google Scholar]
- 6.Masuda A, Takahashi T: Chromosome instability in human lung cancers: its possible underlying mechanisms and potential consequences in the pathogenesis. Oncogene 2002, 21:6884-6897 [DOI] [PubMed] [Google Scholar]
- 7.Taylor SS, McKeon F: Kinetochore localization of murine Bub1 is required for normal mitotic timing and checkpoint response to spindle damage. Cell 1997, 89:727-735 [DOI] [PubMed] [Google Scholar]
- 8.Weitzel DH, Vandre DD: Differential spindle assembly checkpoint response in human lung adenocarcinoma cells. Cell Tissue Res 2000, 300:57-65 [DOI] [PubMed] [Google Scholar]
- 9.Hida T, Ariyoshi Y, Kuwabara M, Sugiura T, Takahashi T, Hosoda K, Niitsu Y, Ueda R: Glutathione S-transferase pi levels in a panel of lung cancer cell lines and its relation to chemo-radiosensitivity. Jpn J Clin Oncol 1993, 23:14-19 [PubMed] [Google Scholar]
- 10.Juan G, Traganos F, James WM, Ray JM, Roberge M, Sauve DM, Anderson H, Darzynkiewicz Z: Histone H3 phosphorylation and expression of cyclins A and B1 measured in individual cells during their progression through G2 and mitosis. Cytometry 1998, 32:71-77 [DOI] [PubMed] [Google Scholar]
- 11.Wang LG, Liu XM, Kreis W, Budman DR: The effect of antimicrotubule agents on signal transduction pathways of apoptosis: a review. Cancer Chemother Pharmacol 1999, 44:355-361 [DOI] [PubMed] [Google Scholar]
- 12.Wang TH, Wang HS, Soong YK: Paclitaxel-induced cell death: where the cell cycle and apoptosis come together. Cancer 2000, 88:2619-2628 [DOI] [PubMed] [Google Scholar]
- 13.Hall LL, Th’ng JP, Guo XW, Teplitz RL, Bradbury EM: A brief staurosporine treatment of mitotic cells triggers premature exit from mitosis and polyploid cell formation. Cancer Res 1996, 56:3551-3559 [PubMed] [Google Scholar]
- 14.Perez-Soler R, Kemp B, Wu QP, Mao L, Gomez J, Zeleniuch-Jacquotte A, Yee H, Lee JS, Jagirdar J, Ling YH: Response and determinants of sensitivity to paclitaxel in human non-small cell lung cancer tumors heterotransplanted in nude mice. Clin Cancer Res 2000, 6:4932-4938 [PubMed] [Google Scholar]
- 15.Goncalves A, Braguer D, Kamath K, Martello L, Briand C, Horwitz S, Wilson L, Jordan MA: Resistance to taxol in lung cancer cells associated with increased microtubule dynamics. Proc Natl Acad Sci USA 2001, 98:11737-11742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kasai T, Iwanaga Y, Iha H, Jeang KT: Prevalent loss of mitotic spindle checkpoint in adult T-cell leukemia confers resistance to microtubule inhibitors. J Biol Chem 2002, 277:5187-5193 [DOI] [PubMed] [Google Scholar]
- 17.Wahl AF, Donaldson KL, Fairchild C, Lee FY, Foster SA, Demers GW, Galloway DA: Loss of normal p53 function confers sensitization to taxol by increasing G2/M arrest and apoptosis. Nat Med 1996, 2:72-79 [DOI] [PubMed] [Google Scholar]
- 18.King TC, Akerley W, Fan AC, Moore T, Mangray S, Hsiu Chen M, Safran H: p53 mutations do not predict response to paclitaxel in metastatic nonsmall cell lung carcinoma. Cancer 2000, 89:769–773 [DOI] [PubMed]
- 19.Jordan MA, Toso RJ, Thrower D, Wilson L: Mechanism of mitotic block and inhibition of cell proliferation by taxol at low concentrations. Proc Natl Acad Sci USA 1993, 90:9552-9556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Basu A, Haldar S: Microtubule-damaging drugs triggered bcl2 phosphorylation-requirement of phosphorylation on both serine-70 and serine-87 residues of bcl2 protein. Int J Oncol 1998, 13:659-664 [DOI] [PubMed] [Google Scholar]
- 21.Haldar S, Jena N, Croce CM: Inactivation of Bcl-2 by phosphorylation. Proc Natl Acad Sci USA 1995, 92:4507-4511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haldar S, Basu A, Croce CM: Bcl2 is the guardian of microtubule integrity. Cancer Res 1997, 57:229-233 [PubMed] [Google Scholar]
- 23.Ling YH, Tornos C, Perez-Soler R: Phosphorylation of Bcl-2 is a marker of M phase events and not a determinant of apoptosis. J Biol Chem 1998, 273:18984-18991 [DOI] [PubMed] [Google Scholar]
- 24.Huisman C, Ferreira CG, Broker LE, Rodriguez JA, Smit EF, Postmus PE, Kruyt FA, Giaccone G: Paclitaxel triggers cell death primarily via caspase-independent routes in the non-small cell lung cancer cell line NCI-H460. Clin Cancer Res 2002, 8:596-606 [PubMed] [Google Scholar]
- 25.Davis PD, Hill CH, Keech E, Lawton G, Nixon JS, Sedgwick AD, Wadsworth J, Westmacott D, Wilkinson SE: Potent selective inhibitors of protein kinase C. FEBS Lett 1989, 259:61-63 [DOI] [PubMed] [Google Scholar]
- 26.Herbert JM, Seban E, Maffrand JP: Characterization of specific binding sites for [3H]-staurosporine on various protein kinases. Biochem Biophys Res Commun 1990, 171:189-195 [DOI] [PubMed] [Google Scholar]
- 27.Paulson JR, Patzlaff JS, Vallis AJ: Evidence that the endogenous histone H1 phosphatase in HeLa mitotic chromosomes is protein phosphatase 1, not protein phosphatase 2A. J Cell Sci 1996, 109:1437-1447 [DOI] [PubMed] [Google Scholar]
- 28.Moasser MM, Sepp-Lorenzino L, Kohl NE, Oliff A, Balog A, Su DS, Danishefsky SJ, Rosen N: Farnesyl transferase inhibitors cause enhanced mitotic sensitivity to taxol and epothilones. Proc Natl Acad Sci USA 1998, 95:1369-1374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gualberto A, Aldape K, Kozakiewicz K, Tlsty TD: An oncogenic form of p53 confers a dominant, gain-of-function phenotype that disrupts spindle checkpoint control. Proc Natl Acad Sci USA 1998, 95:5166-5171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Haruki N, Harano T, Masuda A, Kiyono T, Takahashi T, Tatematsu Y, Shimizu S, Mitsudomi T, Konishi H, Osada H, Fujii Y, Takahashi T: Persistent increase in chromosome instability in lung cancer: possible indirect involvement of p53 inactivation. Am J Pathol 2001, 159:1345-1352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lehman TA, Bennett WP, Metcalf RA, Welsh JA, Ecker J, Modali RV, Ullrich S, Romano JW, Appella E, Testa JR, Gerwin BI, Harris CC: p53 mutations, ras mutations, and p53-heat shock 70 protein complexes in human lung carcinoma cell lines. Cancer Res 1991, 51:4090-4096 [PubMed] [Google Scholar]
- 32.Jia LQ, Osada M, Ishioka C, Gamo M, Ikawa S, Suzuki T, Shimodaira H, Niitani T, Kudo T, Akiyama M, Kimura N, Matsuo M, Mizusawa H, Tanaka N, Koyama H, Namba M, Kanamaru R, Kuroki T: Screening the p53 status of human cell lines using a yeast functional assay. Mol Carcinog 1997, 19:243-253 [DOI] [PubMed] [Google Scholar]
- 33.Scheffner M, Munger K, Byrne JC, Howley PM: The state of the p53 and retinoblastoma genes in human cervical carcinoma cell lines. Proc Natl Acad Sci USA 1991, 88:5523-5527 [DOI] [PMC free article] [PubMed] [Google Scholar]