

Abstract
Previous studies using a cardiac-specific metallothionein (MT)-overexpressing transgenic mouse model have demonstrated that MT inhibits ischemia/reperfusion-induced myocardial injury. The present study was undertaken to test the hypothesis that the MT inhibition is associated with suppression of apoptosis mediated by mitochondrial cytochrome c release and caspase-3 activation. An open-chest coronary artery occlusion and reperfusion procedure to produce ischemia/reperfusion-induced left ventricle infarction was used in MT-overexpressing transgenic mice and non-transgenic controls. After 30 minutes of ischemia, the left ventricle was reperfused to allow blood flow through the previously occluded coronary artery bed. Myocardial infarction produced after reperfusion for 4 hours was significantly reduced in the MT transgenic mice. This inhibition correlated with the antiapoptotic effect of MT, as determined by a terminal deoxynucleotidyl transferase-mediated deoxyuridine 5-triphosphate nick-end labeling assay, mitochondrial cytochrome c release and caspase-3 activation. Ischemia/reperfusion-induced lipid peroxidation was also significantly inhibited in the MT-transgenic heart. Dimethylsulfoxide, a chemical scavenger for reactive oxygen species, was used to confirm the antioxidant effect of MT and found to suppress myocardial infarction and lipid peroxidation just as MT did. This study thus demonstrates that MT suppresses ischemia/reperfusion-induced myocardial apoptosis through, at least in part, the inhibition of cytochrome c-mediated caspase-3 activation pathway. The antiapoptotic effect of MT likely results from the suppression of oxidative stress and correlates with the inhibition of myocardial infarction.
Previous studies using a cardiac-specific metallothionein (MT)-overexpressing transgenic (MT-TG) mouse model 1 and a Langendorff heart perfusion preparation have demonstrated that MT functions in myocardial protection against ischemia/reperfusion injury. 2 MT concentrations were about 10-fold higher in the MT-TG hearts than in the wild-type (WT) controls. In MT-TG hearts, a significant improvement of the suppressed contractile force post ischemia was observed. The efflux of creatine kinase from the MT-TG hearts was reduced by more than 50%. In addition, the zone of infarction induced by ischemia/reperfusion at the end of reperfusion was suppressed by about 40% in the MT-TG hearts. 2
Mechanisms for myocardial protection from ischemia/reperfusion injury by MT are not fully understood. MT is a potent antioxidant in the heart 3-7 and oxidative stress is a critical mediator for myocardial damage induced by ischemia/reperfusion. 8-10 Most importantly, oxidative stress causes myocardial apoptosis, which is closely associated with ischemia/reperfusion injury. 11,12 Recent studies have shown that mitochondria play an important role in apoptosis. 13 Mitochondrial cytochrome c release occurs in a variety of pro-apoptotic conditions, particularly under oxidative stress. 14-16 Cytochrome c, through a series of cascade reactions, activates caspase-3 and leads to apoptosis. 17,18 Mitochondrial dysfunction is one of the most critical events associated with myocardial ischemia/reperfusion injury. 19 It is possible that MT protects the heart from ischemia/reperfusion injury through inhibition of oxidative stress-mediated mitochondrial cytochrome c release and caspase-3 activation, which has been demonstrated in the MT protection from Adriamycin-induced myocardial injury. 5,6
This study was thus undertaken to investigate possible mechanisms by which MT functions in cardioprotection against ischemia/reperfusion injury focusing on the effect of MT on myocardial apoptosis induced by ischemia/reperfusion. We used an open-chest coronary artery occlusion and reperfusion model to produce regional ischemia/reperfusion to the left ventricle. We present evidence to show that ischemia/reperfusion-induced apoptosis was associated with myocardial infarction. The apoptotic effect was significantly suppressed and the infarct area was markedly reduced in the MT-TG myocardium. Moreover, mitochondrial cytochrome c release and caspase-3 activation induced by ischemia/reperfusion were inhibited in the MT-TG myocardium. These results thus demonstrate that MT suppresses ischemia/reperfusion-induced myocardial apoptosis through, at least in part, the inhibition of the cytochrome c-mediated caspase-3 activation pathway.
Materials and Methods
Materials
ApopTag in situ apoptosis detection kit was purchased from Intergen (Purchase, NY). Monoclonal mouse anti-cytochrome c and polyclonal rabbit anti-active caspase-3 antibodies were purchased from BD PharMingen (San Diego, CA). Biotinylated goat anti-rabbit IgG antibody and horseradish peroxidase (HRP)-streptavidin were obtained from Zymed Laboratories, Inc. (San Francisco, CA). Caspase-3 substrate I (Ac-DEVD-pNA) and p-nitroaniline were the products of Calbiochem Corp. (La Jolla, CA). A bicinchoninic acid protein assay kit was the product of Pierce (Rockford, IL). All other chemicals and reagents were obtained from Sigma Chemical Co. (St. Louis, MO) unless otherwise stated.
Animals
MT-TG mice and the WT C57BL/6 controls were obtained from The Jackson Laboratory (Bar Harbor, ME). Detailed descriptions for the MT-TG mouse lines were reported previously. 20 The animals were chosen without regard to sex because our preliminary studies showed no significant difference in cardiac response to ischemia/reperfusion between sexes. They were housed in quarters maintained at 22°C to 23°C in a 12-hour light/dark cycle. Mice had free access to rodent chow and tap water. All animal procedures were approved by the Institutional Animal Care and Use Committee, which is certified by the American Association for Accreditation of Laboratory Animal Care.
Myocardial MT Concentrations
MT concentrations in myocardial tissues were determined by a cadmium-hemoglobin affinity assay. 21 Briefly, the left ventricle was homogenized in 4 volumes of 10 mmol/L Tris-HCl buffer (pH 7.4) at 4°C. After centrifugation of the homogenate at 10,000 × g for 15 minutes, a 200-μl aliquot of the supernatant was transferred to microtubes for MT analysis. MT concentrations in the myocardial tissue are expressed as micrograms per gram of heart tissue.
Open-Chest Coronary Artery Occlusion and Reperfusion
Both male and female MT-TG and WT mice aged 8 to 12 weeks old (20 to 25 g body weight) were used. Anesthesia was given by an intraperitoneal injection of pentobarbital sodium (4 mg/ml in 10 μl/g body weight). The mouse open-chest coronary artery occlusion and reperfusion procedure described previously 22 was followed with some modifications. In brief, mice were placed in a supine position and an endotracheal polyethylene (PE) 90 tubing was used to provide ventilation via a rodent ventilator (Harvard, South Natick, MA) at a rate of 100 cycles per min. Oxygen (100%) was provided to the in-flow of the ventilator. In preliminary studies, we have compared infarct sizes between mice under the normoxia conditions and those exposed to 100% oxygen and found no significant differences between the two groups. However, the survival rates for the mice under normoxia and exposed to 100% oxygen were 50% and 95%, respectively; thus, 100% oxygen was used. The chest was opened by a lateral cut along the up-margin of the fourth rib. The left auricle was slightly retraced to expose the entire left coronary artery system. Ligation was done using a 7−0 silk suture and a tapered needle passed underneath the left anterior descending (LAD) branch, a 1-mm section of PE-10 tubing was placed on top of the vessel, and a knot was tied on top of the tubing to occlude the coronary artery, no veins were occluded with this maneuver, which was ensured by observation under microscope and by the success of ischemia following coronary artery ligation. If veins were ligated, the ligation-affected myocardium would not change color (index of ischemia) due to blood retention. Coronary artery occlusion lasted 30 minutes and reperfusion was established by cutting the knot on top of the PE-10 tubing. The chest wall was closed and the animal was removed from the respirator and kept warm by a heat lamp and allowed to breath 100% oxygen via a nasal cone. Reperfusion of the previously occluded coronary bed was allowed for 1, 2, 3, or 4 hours.
Assessment of Area at Risk and Infarct Size
After varying time periods of reperfusion following a 30-minute ischemia of the heart, the chest was reopened, and LAD coronary artery was reoccluded through the previous ligation site. The aorta was cannulated using a PE-10 tubing, and 1% Evans blue was perfused into the aorta and coronary arteries to allow distribution throughout the ventricular wall proximal to the coronary artery ligature. The LV was then removed and sectioned transversely into five sections with one section being made at the site of the ligature, and the sections were weighed. Sections of the ventricle were then incubated in 1.5% triphenyltetrazolium chloride (TTC) for 15 minutes at 37°C. After the procedures, the myocardium was stained blue, brick red, and pale white. Each slice was imaged, the digital images were acquired using a microscope and a Spot camera via the accompanying image analysis software (Diagnostic Instruments Inc., Sterling Heights, MI) and stored as JPG data files for analysis. Pseudo-color was used to accentuate the differences between areas that were stained blue or red, or that remained pale. The size of infarction was determined by the following equations: weight of infarction = (A1 × WT1) + (A2 × WT2) + (A3 × WT3) + (A4 × WT4), where A is percent area of infarction by planimetry from subscripted numbers 1 to 4 representing sections, and WT is weight of the same numbered sections; the percentage of infarct in the LV is (WT of infarction/WT of LV) × 100; the area at risk was expressed as percentage of the LV and calculated by (WT of LV − WT of LV stained blue)/WT of LV; and the weight of the LV stained blue was calculated in a similar fashion by the sum of the products of the percent area of each slice multiplying the weight of the respective slice.
Determination of Myocardial Apoptosis
A separate set of parallel experimental animals were used for immunohistochemical studies. The LV removed from mice was fixed in 10% neutral buffered formalin in 0.01 mol/L phosphate-buffered saline (pH 7.3), for 24 hours and embedded in paraplast. Tissue blocks were cut into 5-μm-thick sections and mounted on salinized slides and processed for a terminal deoxynucleotidyl transferase-mediated deoxyuridine 5-triphosphate nick-end labeling (TUNEL) assay to detect fragmented nuclei in the myocardium. An ApopTag in situ detection kit was used according to the manufacturer’s instructions. Briefly, the slides were pretreated with H2O2 and incubated with the reaction mixture containing TdT and digoxigenin-conjugated dUTP for 1 hour at 37°C. Labeled DNA was visualized with peroxidase-conjugated, anti-digoxigenin antibody using 3,3′-diaminobenzidene (DAB) as the chromogen. Rat mammary gland tissue provided in the kit was used as positive control. For negative control, TdT was omitted from the reaction mixture. The TUNEL-positive cells were counted against negative cells under a light microscope at a magnification of ×40, the risk areas were searched, and six visual fields were chosen through an orderly shifting (epicardial, middle, and endocardial) on each slide, and three sections from each animal and six animals from each group were examined. An apoptotic index (the number of myocardial nuclei labeled by the TUNEL method/the number of total myocardial nuclei) was calculated.
Determination of Caspase-3 Activation by Immunohistochemical Method
After treatment with 3% H2O2, the LV sections were incubated overnight at 4°C with polyclonal rabbit anti-active caspase-3 antibody. Sections were then incubated for 30 minutes in biotinylated goat anti-rabbit IgG antibody, followed by incubation with HRP-streptavidin for 20 minutes. The antibody binding sites were visualized by incubation with DAB-H2O2 solution using a DAB kit. Finally, sections were counterstained with 0.5% methyl green.
Enzymatic Assay of Caspase-3
Fresh LV was homogenized with Teflon homogenizer in the extraction buffer [25 mmol/L HEPES buffer, pH 7.4, containing 5 mmol/L ethylenediaminetetraacetate (EDTA), 2 mmol/L dithiothreitol (DTT) and 0.1% CHAPS]. The homogenate was centrifuged at 20,000 × g for 30 minutes. The supernatant was diluted with the assay buffer (50 mmol/L HEPES, 10 mmol/L DTT, 1.0 mmol/L EDTA, 100 mmol/L NaCl, 0.1% CHAPS, and 10% glyceral, pH 7.4) and incubated at 37°C with 200 μmol/L caspase-3 substrate I (Ac-DEVD-pNA). p-Nitroaniline was used as the standard. Cleavage of the substrate was monitored at 405 nm and the specific activity was expressed as picomoles of the product, nitroaniline, per minute per mg protein.
Measurement of Mitochondrial Cytochrome c Release
Fresh LV was homogenized gently using a glass tissue grinder in a suspension buffer (20 mmol/L HEPES, pH 7.4, 1.5 mmol/L MgCl2, 10 mmol/L KCl, 1.0 mmol/L EDTA, 1.0 mmol/L EGTA, 1.0 mmol/L DTT, and 1% protease inhibitor cocktail) with 0.25 mol/L sucrose. The crude homogenate was centrifuged at 750 × g for 10 minutes at 4°C, then at 8000 × g for 20 minutes at 4°C. The pellet centrifuged at 8000 × g was homogenized using a Teflon homogenizer in the suspension buffer without sucrose and was used as the mitochondrial fraction. The supernatant was further centrifuged at 100,000 × g for 60 minutes at 4°C and the pellet was used as the cytosolic fraction. Measurements of lactate dehydrogenase, a cytosolic marker enzyme, and citrate synthase, a mitochondrial enzyme, were done in both fractions. Cross-contamination between the fractions was not detectable. Aliquots of 25 μg of protein from each fraction were loaded on a 15% sodium dodecyl sulfate (SDS)-polyacrylamide gel. After electrophoresis, the protein was transferred onto a nitrocellulose membrane. To further ensure equal loading of the gel and efficient transfer, the filter membranes were stained with Ponceau’s solution (proteins were stained pink) before process for antibody reaction. The membrane was blocked using 5% nonfat milk in Tris-buffered saline (pH 7.5) and probed using monoclonal mouse anti-cytochrome c antibody. The membrane was then processed using an HRP anti-mouse IgG. The protein bands were visualized by an enhanced chemiluminescence detection system (Amersham Biosciences, Piscataway, NJ) and quantified by a Bio-Rad Multi-Analyst (Bio-Rad, Hercules, CA).
Detection of Ischemia/Reperfusion-Induced Lipid Peroxidation
Lipid peroxidation induced by ischemia/reperfusion in the LV was quantified by measuring the malondialdehyde (MDA) concentrations as described previously. 23 Briefly, fresh LV was homogenized in 9 volumes of 50 mmol/L Tris-HCl buffer (pH 7.4) containing 180 mmol/L KCl, 10 mmol/L EDTA, and 0.02% butylated hydroxytoluene. The tissue homogenate was processed for thiobarbituric acid reaction following the procedure described previously. 23 The reaction mixture was centrifuged and the resulting lower phase was extracted and measured at 532 nm. The MDA concentration was calculated using 1,1,5,5-tetraethoxypropane as standard.
Dimethylsulfoxide (DMSO) Treatment
Only C57BL/6 WT mice were used for the DMSO treatment procedure. The mice were subjected to the same ischemia/reperfusion procedure as described above for the open-chest LAD coronary artery ligation model, except that they received DMSO (2 g/kg) injection via the tail vein 10 minutes before the LAD coronary artery reopening. The same amount of vehicle (saline) was administered to the control mice. The effect of DMSO on ischemia/reperfusion-induced infarction and lipid peroxidation were determined at 4 hours after reperfusion.
Statistical Analysis
The data are presented as mean ± SD values from the indicated number of mice and initially analyzed by two-way analysis of variance. Scheffé’s F-test was used to determine the significance of differences. A value of P < 0.05 was considered statistically significant.
Results
Reduction in Myocardial Infarct Size in the MT-TG Mouse Heart
Ischemia/reperfusion-induced myocardial infarction was assessed by TTC staining as described in Materials and Methods. The slices from both experimental groups showed that infarct and non-infarct areas were clearly discernible. The staining patterns of slices from the MT-TG and WT LV subjected to 30 minutes of ischemia and 4 hours of reperfusion were compared (Figure 1A) ▶ . Large continuous infarct zones were observed in the WT myocardium. However, only small, scattered infarct zones were observed in the MT-TG heart. Analysis of the slices indicated that the total volume of myocardial infarction was significantly reduced in the MT-TG myocardium (Figure 1B) ▶ . The total MT concentrations were 60.6 μg/g tissue in the MT-TG myocardium and 5.4 μg/g tissue in the WT myocardium.
Figure 1.
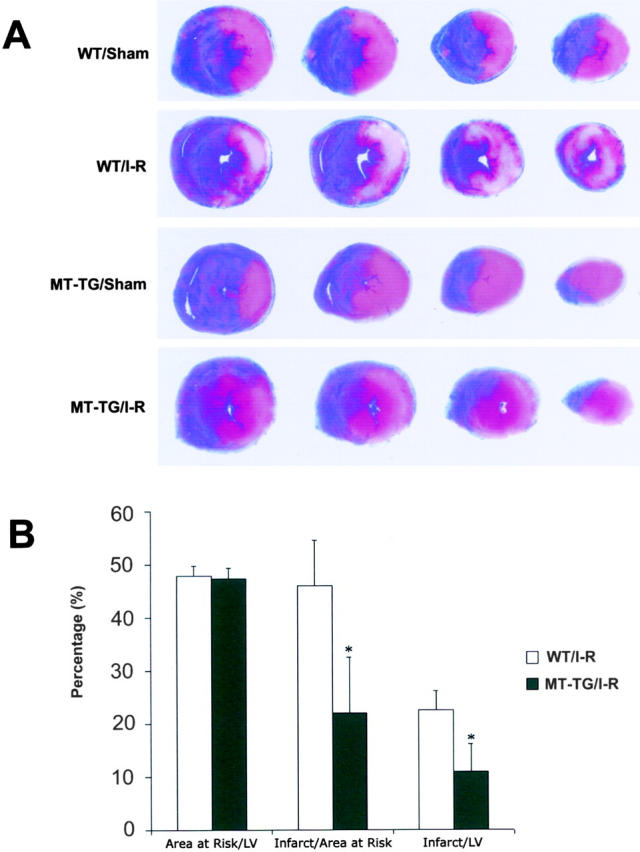
Effect of MT on ischemia/reperfusion-induced myocardial infarction. A: The area at risk and the area of infarction were stained by Evans blue and TTC in both MT-TG and WT group. Transverse sections of hearts were from the level where ligation started as shown on the left to apex on the right. All areas that were stained blue were not at risk. Areas that appeared pale white after TTC staining were infarcted. Areas that remained red were not infarcted but were in the area at risk. B: The results of infarct size and area at risk by using Sigmascanpro software. Data are mean ± SD. *, P < 0.05.
Effect of MT on Myocardial Apoptosis and Caspase-3 Activation Induced by Ischemia/Reperfusion
To define the effect of MT on ischemia/reperfusion-induced myocardial apoptosis, MT-TG and WT LV were subjected to ischemia for 30 minutes, followed by 1, 2, 3, or 4 hours of reperfusion. A TUNEL assay was used to examine the effect of MT on ischemia/reperfusion-induced apoptosis. As shown in Figure 2A ▶ , numerous TUNEL-positive cells were identified in the WT myocardium. In contrast, the number of TUNEL-positive cells was significantly reduced in the MT-TG myocardium. Quantitative analysis showed that the number of the TUNEL-positive cells reached a peak value at 3 hours after reperfusion (26.3 ± 4.9% of the total populations in the risk area of the WT myocardium), the same trend was observed in the transgenic myocardium, however, only 14.4 ± 2.6% of the total populations underwent apoptosis at the same time-point (Figure 2B) ▶ . The basal levels of apoptosis in either MT-TG or WT myocardium under sham-operation control conditions were less than 0.1%.
Figure 2.

Effect of MT on ischemia/reperfusion-induced apoptosis and caspase-3 activation in the heart. A: Myocardial tissues obtained from both WT and MT-TG under sham operation were TUNEL-negative. Numerous TUNEL-positive cells in the WT myocardium after 30 minutes ischemia followed by reperfusion for 4 hours are shown in the infarct region. The numbers of TUNEL-positive cells in the MT-TG myocardium under the same operation procedure were significantly decreased. B: Apoptotic index indicates that MT-TG myocardium had significantly less TUNEL-positive cells than that in WT myocardium. C: Detection of active form of caspase-3 in the TUNEL-positive region, the control hearts of MT-TG and WT showed negative staining, but massive positive staining in the same region that showed TUNEL-positive was observed in the WT heart. A moderate positive staining was also observed in the TUNEL-positive region in the MT-TG heart. D: Enzymatic assay of caspase-3 activity. Caspase-3 activity was significantly inhibited in MT-TG myocardium compared to that in WT myocardium. The time-course changes in the caspase-3 activities were closely correlated with that observed in the TUNEL assay. Data presented are mean ± SD values from five mouse hearts of each group. *, P < 0.05; **, P < 0.01.
The TUNEL-positive cells may not all be apoptotic because the staining procedure also recognizes DNA breaks due to other causes such as necrosis. To verify the apoptotic cells, an immunohistochemical procedure to recognize active caspase-3 was used. The co-localization of TUNEL-positive and caspase-3 staining would provide confirmation of the apoptotic myocytes. As shown in Figure 2C ▶ , the caspase-3-positive staining area is closely overlapped with the TUNEL-positive area. Furthermore, the activation of caspase-3 was examined by an enzymatic assay (Figure 2D) ▶ . Ischemia/reperfusion increased caspase-3 activity in the WT myocardium. This elevation was significantly suppressed in the MT-TG myocardium. As demonstrated in Figure 2 ▶ , the time-dependent changes in caspase-3 activation and TUNEL-positive staining induced by ischemia/reperfusion were closely correlated and the same inhibitory effect of MT on both caspase-3 activation and TUNEL-positive staining was observed.
Mitochondrial Cytochrome c Release by Ischemia/Reperfusion
Mitochondrial cytochrome c release plays an important role in caspase-3 activation. We tested whether cytochrome c release was involved in the ischemia/reperfusion-induced apoptosis in the myocardium. As shown in Figure 3 ▶ , Western blot analysis revealed that most of the cellular cytochrome c in the myocardium was found in the mitochondria under sham-operation control conditions. Ischemia/reperfusion significantly increased cytosolic concentrations of cytochrome c with a concomitant decrease of the content in mitochondria in the WT myocardium. Quantitative analysis showed that ischemia/reperfusion reduced total mitochondrial cytochrome c by 11.3% in the WT myocardium. This effect was significantly suppressed in the MT-TG myocardium: 1.4% of total cytochrome c was released from mitochondria.
Figure 3.

The effect of MT on mitochondrial cytochrome c release induced by ischemia/reperfusion. The animals were subjected to 30 minutes ischemia followed by 4 hours reperfusion. Cytochrome c was isolated from cytosolic (Cyto) and mitochondrial (Mito) fractions. Protein (25 μg) was subjected to SDS-PAGE immunoblot analysis by using an anti-cytochrome c antibody.
Effect of MT on Ischemia/Reperfusion-Induced Lipid Peroxidation
MDA is a product of lipid peroxidation induced by a diversity of oxidative injury. It has been used as a biomarker of myocardial oxidative damage. The result presented in Figure 4 ▶ shows the effect of MT on MDA concentrations in the myocardium. Ischemia/reperfusion dramatically elevated the cardiac MDA concentrations in both MT-TG and WT mice 4 hours after reperfusion. However, the concentration of MDA in the MT-TG myocardium was significantly lower than in the WT under the same ischemia/reperfusion treatment.
Figure 4.

Effect of MT on ischemia/reperfusion-induced increase in MDA concentrations in the heart. The animals were subjected to 30 minutes ischemia followed by reperfusion for 4 hours. Each value was obtained from six animals and the data are expressed as mean ± SD. **, P < 0.01.
Effects of DMSO on Ischemia/Reperfusion-Induced Myocardial Infarction
To demonstrate the role of oxidative stress in ischemia/reperfusion-induced infarction, DMSO, a free-radical scavenger, was intravenously administrated 10 minutes before the animals (WT mice only) were subjected to reperfusion. As shown in Figure 5A ▶ , DMSO significantly reduced ischemia/reperfusion-induced infarct area. Analysis of all of the slices indicated that the total volume of myocardial infarction was reduced by 49.3% in the DMSO-treated group. DMSO also efficiently decreased MDA concentrations (by 70.2%) in the heart (Figure 5B) ▶ .
Figure 5.

Effects of DMSO pretreatment on myocardial infarction and MDA elevation induced by ischemia/reperfusion. The animals were treated with DMSO 10 minutes before they were subjected to 30 minutes ischemia followed by reperfusion for 4 hours. A: The infarct size and area at risk were analyzed in ischemia/reperfused heart pretreated with saline or DMSO. B: MDA concentrations in ischemia/reperfused hearts pretreated with saline or DMSO. Data are mean ± SD values. *, P < 0.05; **, P < 0.01.
Discussion
Our results demonstrate that MT suppresses ischemia/reperfusion-induced myocardial infarction and apoptosis. MT also inhibited mitochondrial cytochrome c release and caspase-3 activation. Ischemia reperfusion caused myocardial lipid peroxidation. The lipid peroxidation level in the MT-TG myocardium was significantly reduced, which was in an agreement with the known antioxidant function of MT in the heart. 3 Pretreatment with DMSO, a chemical free-radical scavenger, also suppressed ischemia/reperfusion-induced myocardial infarction as well as lipid peroxidation, thus demonstrating the role of oxidative stress in the myocardial infarction and providing evidence that MT protection may result from its antioxidant action.
Oxyradicals have been shown to induce apoptosis in many cell types, including cardiomyocytes. 11,12,24,25 Apoptosis, a gene-regulated death pathway, allows the tissue to rid itself of damaged cells. The involvement of apoptosis in the ischemia/reperfusion process has been pursued actively over the last decade and has been thought to be a critical cellular event involved in the pathogenesis of myocardial ischemia/reperfusion injury. 24 In the present study, a TUNEL assay was used to identify cells containing fragmented nuclei, an indication of apoptosis. Numerous TUNEL-positive cells were found in the WT myocardium subjected to ischemia/reperfusion. To confirm the TUNEL result, an immunohistochemical assay of active caspase-3 was done to identify co-localization of the TUNEL-positive and caspase-3 activation cardiac cells, which thus defined myocardial apoptosis. In the time-course study, there was no indication of apoptosis at 0 hours of reperfusion, which was in agreement with most studies in which apoptosis was not detected after short periods of myocardial ischemia. 24,25 Myocardial apoptosis was detected at 1 hour after reperfusion and reached a peak value 3 hours after reperfusion. This result suggests that apoptotic process is initiated during early reperfusion and the removal of apoptotic cells begins after 3 hours of reperfusion. It has been demonstrated that the formation of reactive oxygen species (ROS) increases significantly after a few minutes of reperfusion. 26 The increased lipid peroxide levels, in association with the appearance of apoptosis, suggested that ROS were possibly involved in the initiation of apoptosis. An important observation is that the number of TUNEL-positive cells was significantly reduced in the MT-TG myocardium. The results that ischemia/reperfusion-induced lipid peroxidation and apoptosis were significantly suppressed in the MT-TG myocardium suggest a link between the MT antioxidant action and myocardial protection from ischemia/reperfusion-induced apoptosis.
In searching possible action sites of MT inhibition of ischemia/reperfusion-induced myocardial apoptosis, we focused on the mitochondrial cytochrome c release and caspase-3 activation pathway. The release of cytochrome c from mitochondria into cytosol is a critical initiation step in ROS-triggered apoptosis. 13 The detection of changes in cytochrome c concentrations between mitochondria and cytosol by Western blot showed that cytosolic cytochrome c increased in the WT myocardium post ischemia/reperfusion. This change was suppressed in the MT-TG myocardium. Corresponding to this alteration, caspase-3 was also activated in the WT myocardium; this activation was again suppressed in the MT-TG myocardium. These results thus indicate that MT attenuates ischemia/reperfusion-induced apoptosis in the myocardium by, at least in part, inhibiting mitochondrial cytochrome c release and subsequent caspase-3 activation. A direct interaction between MT and ROS or RNS has been demonstrated in cell-free experiments. 27-29 We demonstrated in this study that lipid peroxide levels increased by ischemia/reperfusion were dramatically decreased by MT.
To elucidate the significance of ROS in the ischemia/reperfusion-induced myocardial infarction, we chose a chemical scavenger for free radicals to abolish the accumulation of ROS during early reperfusion. Several studies have demonstrated that DMSO is a very effective free radical scavenger. 30-32 Treatment with DMSO 10 minutes before reperfusion efficiently suppressed ischemia/reperfusion-induced lipid peroxidation. This inhibitory effect was associated with the reduction of myocardial infarct size. These results thus provide further evidence that ROS plays a critical role in myocardial infarction and the myocardial protection of MT results, at least in part, from inhibition of oxidative stress.
Based on the results of this study and others, 1,2,4-7 we speculate that MT may be useful in preventing heart injury under oxidative stress conditions. It is important to note that MT is highly inducible under a wide variety of stress conditions, including oxidative stress. The regulation of MT expression has been well studied, and several agents have been identified to selectively elevate MT levels in the heart, such as bismuth subnitrate, 33 isoproterenol, 34 and tumor necrosis factor-α. 35 Therefore, the basis for developing pharmaceutical agents to increase MT concentrations in the heart already exists. Exploring the potential of MT in protection against ischemia/reperfusion injury would likely result in novel approaches to this clinical problem and could positively influence clinical outcomes.
Footnotes
Address reprint requests to Dr. Y. James Kang, Department of Medicine, University of Louisville School of Medicine, 511 South Floyd Street, MDR 530, Louisville, KY 40202. E-mail: yjkang01@athena.louisville.edu.
Supported in part by National Institutes of Health grants HL63760 and HL59225 (to Y. J. K.).
Y. J. K. is a distinguished University Scholar of the University of Louisville.
References
- 1.Kang YJ, Chen Y, Yu A, Voss-McCowan M, Epstein PN: Overexpression of metallothionein in the heart of transgenic mice suppresses doxorubicin cardiotoxicity. J Clin Invest 1997, 100:1501-1506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kang YJ, Li G, Saari JT: Metallothionein inhibits ischemia-reperfusion injury in mouse heart. Am J Physiol 1999, 276:H993-H997 [DOI] [PubMed] [Google Scholar]
- 3.Kang YJ: The antioxidant function of metallothionein in the heart. Proc Soc Exp Biol Med 1999, 222:263-273 [DOI] [PubMed] [Google Scholar]
- 4.Sun X, Zhou Z, Kang YJ: Attenuation of doxorubicin chronic toxicity in metallothionein-overexpressing transgenic mouse heart. Cancer Res 2001, 61:3382-3387 [PubMed] [Google Scholar]
- 5.Wang GW, Klein JB, Kang YJ: Metallothionein inhibits doxorubicin-induced mitochondrial cytochrome c release and caspase-3 activation in cardiomyocytes. J Pharmacol Exp Ther 2001, 298:461-468 [PubMed] [Google Scholar]
- 6.Kang YJ, Zhou ZX, Wang GW, Buridi A, Klein JB: Suppression by metallothionein of doxorubicin-induced cardiomyocyte apoptosis through inhibition of p38 mitogen-activated protein kinases. J Biol Chem 2000, 275:13690-13698 [DOI] [PubMed] [Google Scholar]
- 7.Wang GW, Zhou Z, Klein JB, Kang YJ: Inhibition of hypoxia/reoxygenation-induced apoptosis in metallothionein-overexpressing cardiomyocytes. Am J Physiol Heart Circ Physiol 2001, 280:H2292-H2299 [DOI] [PubMed] [Google Scholar]
- 8.Curello S, Ceconi C, de Giuli F, Panzal AF, Milanesi B, Calarco M, Pardini A, Marzollo P, Alfieri O, Messineo F, Ferrari R: Oxidative stress during reperfusion of human hearts; potential sources of oxygen free radicals. Cardiovasc Res 1995, 29:118-125 [PubMed] [Google Scholar]
- 9.Jeroudi MO, Hartley CJ, Bolli R: Myocardial reperfusion injury: role of oxygen radicals and potential therapy with antioxidant. Am J Cardiol 1994, 73:2B-7B [DOI] [PubMed] [Google Scholar]
- 10.Lucchesi BR: Myocardial ischemia, reperfusion and free radical injury. Am J Cardiol 1990, 65:14I-23I [DOI] [PubMed] [Google Scholar]
- 11.Fliss H, Gattinger D: Apoptosis in ischemic and reperfused rat myocardium. Circ Res 1996, 79:949-956 [DOI] [PubMed] [Google Scholar]
- 12.Gottlieb RA, Burleson KO, Kloner RA, Babior BM, Engler RL: Reperfusion injury induces apoptosis in rabbit cardiomyocytes. J Clin Invest 1994, 94:1621-1628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Green DR, Reed JC: Mitochondria and apoptosis. Science 1998, 281:1309-1312 [DOI] [PubMed] [Google Scholar]
- 14.Higuchi M, Proske RJ, Yeh ET: Inhibition of mitochondrial respiratory chain complex I by TNF results in cytochrome c release, membrane permeability transition, and apoptosis. Oncogene 1998, 17:2515-2524 [DOI] [PubMed] [Google Scholar]
- 15.Pan G, Humke EW, Dixit VM: Activation of caspases triggered by cytochrome c in vitro. FEBS Lett 1998, 426:151-154 [DOI] [PubMed] [Google Scholar]
- 16.Ma Y, Ogino T, Kawabata T, Li J, Eguchi K, Okada S: Cupric nitrilotriacetate-induced apoptosis in HL-60 cells association with lipid peroxidation, release of cytochrome c from mitochondria, and activation of caspase-3. Free Radic Biol Med 1999, 27:227-233 [DOI] [PubMed] [Google Scholar]
- 17.Liu X, Kim CN, Yang J, Jemmerson R, Wang X: Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell 1996, 86:147-157 [DOI] [PubMed] [Google Scholar]
- 18.Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X: Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 1997, 91:479-489 [DOI] [PubMed] [Google Scholar]
- 19.Jassem W, Fuggle SV, Rela M, Koo DD, Heaton ND: The role of mitochondria in ischemia/reperfusion injury. Transplantation 2002, 73:493-499 [DOI] [PubMed] [Google Scholar]
- 20.Palmiter RD, Sandgren EP, Koeller DM, Brinster RL: Distal regulatory elements from the mouse metallothionein locus stimulate gene expression in transgenic mice. Mol Cell Biol 1993, 13:5266-5275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eaton DL, Cherian MG: Determination of metallothionein in tissues by cadmium-hemoglobin affinity assay. Methods Enzymol 1991, 205:83-88 [DOI] [PubMed] [Google Scholar]
- 22.Michael LH, Entman ML, Hartley CJ, Youker KA, Zhu J, Hall SR, Hawkins HK, Berens K, Ballantyne CM: Myocardial ischemia and reperfusion: a murine model. Am J Physiol 1995, 269:H2147-H2154 [DOI] [PubMed] [Google Scholar]
- 23.Ohkawa H, Ohishi N, Yagi K: Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal Biochem 1979, 95:351-358 [DOI] [PubMed] [Google Scholar]
- 24.Gottlieb RA, Engler RL: Apoptosis in myocardial ischemia-reperfusion. Ann NY Acad Sci 1999, 874:412-426 [DOI] [PubMed] [Google Scholar]
- 25.Maulik N, Yoshida T, Das DK: Oxidative stress developed during the reperfusion of ischemic myocardium induces apoptosis. Free Radic Biol Med 1998, 24:869-875 [DOI] [PubMed] [Google Scholar]
- 26.Henry TD, Archer SL, Nelson D, Weir EK, From AH: Enhanced chemiluminescence as a measure of oxygen-derived free radical generation during ischemia and reperfusion. Circ Res 1990, 67:1453-1461 [DOI] [PubMed] [Google Scholar]
- 27.Cai L, Klein JB, Kang YJ: Metallothionein inhibits peroxynitrite-induced DNA and lipoprotein damage. J Biol Chem 2000, 275:38957-38960 [DOI] [PubMed] [Google Scholar]
- 28.Abel J, de Ruiter N: Inhibition of hydroxyl-radical-generated DNA degradation by metallothionein. Toxicol Lett 1989, 47:191-196 [DOI] [PubMed] [Google Scholar]
- 29.Thomas JP, Bachowski GJ, Girotti AW: Inhibition of cell membrane lipid peroxidation by cadmium- and zinc-metallothioneins. Biochim Biophys Acta 1986, 884:448-461 [DOI] [PubMed] [Google Scholar]
- 30.Finney JW, Urschel HC, Balla GA, Race GA, Jay BE, Pingree HP, Dorman HL, Mallams JT: Protection of the ischemic heart with DMSO alone or DMSO with hydrogen peroxide. Ann NY Acad Sci 1967, 141:231-241 [DOI] [PubMed] [Google Scholar]
- 31.Hatipoglu AR, Temiz E, Yuksel M, Hoscoskun Z, Coskun I, Huseyinova G: The comparison of electron microscopy and scintigraphy in determining the protective effect of dimethylsulphoxide (DMSO) on ischemia/reperfusion injury through Pringle maneuver. Hepatogastroenterology 2001, 48:799-802 [PubMed] [Google Scholar]
- 32.Yuksel M, Hatipoglu A, Temiz E, Salihoglu YS, Huseyinova G, Berkarda S: The role of hepatobiliary scintigraphy in the evaluation of the protective effects of dimethylsulphoxide in ischaemic/reperfusion injury of liver. Nucl Med Commun 2000, 21:775-780 [DOI] [PubMed] [Google Scholar]
- 33.Naganuma A, Satoh M, Imura N: Specific reduction of toxic side effects of Adriamycin by induction of metallothionein in mice. Jpn J Cancer Res 1988, 79:406-411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Namikawa K, Okazaki Y, Nishida S, Kimoto S, Akai F, Tomura T, hashimoto S: Changes in myocardial metallothionein on isoproterenol-induced myocardial injury. Yakugaku Zasshi 1993, 113:591-595 [PubMed] [Google Scholar]
- 35.Sato M, Sasaki M, Hojo H: Tissue-specific induction of metallothionein synthesis by tumor necrosis factor-α. Res Commun Chem Pathol Pharmacol 1992, 75:159-172 [PubMed] [Google Scholar]