

Abstract
To determine the methylation profile of multiple tumor-related genes during multistep hepatocarcinogenesis, we investigated the methylation status of CpG islands of 9 genes, using methylation-specific polymerase chain reaction for 60 paired hepatocellular carcinoma (HCC) and non-HCC liver tissue samples, 22 dysplastic nodule (DN), 30 liver cirrhosis (LC), 34 chronic hepatitis (CH) and 20 normal liver samples. The methylation status of 9 genes was correlated to the clinicopathological findings of HCC patients. All HCC samples showed methylation of at least one gene, whereas it was shown in 72.7% of DN and 40% of LC, but was not shown in CH and normal liver samples (P < 0.001). The number of genes methylated showed a stepwise increase with the progression of stages (0 for normal liver and CH, 0.5 for LC, 1.5 for DN, and 3.7 for HCC (P < 0.001)). The genes frequently methylated in HCC were APC (81.7%), GSTP1 (76.7%), RASSF1A (66.7%), p16 (48.3%), COX-2 (35%), and E-cadherin (33.3%). COX-2, p16, RASSF1A, and TIMP-3 were not methylated in LC and CH from patients without concurrent HCC. Chronic liver diseases with concurrent HCC showed higher methylation frequencies of the tested genes, and a higher number of methylated genes than those without concurrent HCC. HCC patients with methylation of E-cadherin or GSTP1 showed poorer survival than those without (P = 0.034 and 0.043, respectively). In conclusion, our results indicated that CpG island methylation of tumor-related genes is an early and frequent event, and accumulates step-by-step during a multistep hepatocarcinogenesis. CpG island methylation of E-cadherin or GSTP1 might serve as a potential biomarker for prognostication of HCC patients.
Hepatocellular carcinoma (HCC) ranks as the fifth most common cancer in the world. 1 In Korea, an endemic area of HCC, most HCCs are associated with chronic hepatitis B or C viral infections, and the HCC-associated death rate is high, 21.3/100,000 persons. 2 The development and progression of HCC is a multistep process whereby the normal hepatocytes undergo inflammation, fibrosis by the hepatitis virus or other stimuli, followed by liver cirrhosis (LC), which then progresses to HCC or dysplastic nodule (DN) and subsequent HCC. The understanding of the molecular pathways of hepatocarcinogenesis is limited, although recent molecular biological studies have led to rapid progress in the understanding of the molecular events involved. Most previous studies have concentrated on the documentation of mutational events leading to the activation of oncogenes or the inactivation of tumor suppressor genes in hepatocarcinogenesis. 3 However, recent advances in the field of epigenetics have brought an awareness, where not only genetic, but also epigenetic changes, play roles in carcinogenesis. 4,5 DNA methylation is one of the best-understood epigenetic mechanisms. It has been firmly established that aberrant hypermethylation of CpG islands in gene promoter regions correlate with the lack of gene transcription. 6
In HCCs, a growing number of genes have been recognized as undergoing aberrant CpG island hypermethylation, which is associated with the transcriptional inactivation and loss of gene function, suggesting that CpG island hypermethylation is an important molecular mechanism for the development of HCC. Most studies have focused on single target genes, 7-11 and a few have attempted to analyze the hypermethylation of multiple genes in HCCs and associated chronic liver diseases. 12-14 However, DNA methylation has not yet been investigated in DN. Thus, information relating to CpG island hypermethylation during multistep hepatocarcinogenesis is quite limited.
In the present study, we determined the methylation status of CpG islands, including 9 genes and/or 5 MINT loci in normal liver, CH, LC, DN, and HCC, and correlated the methylation status to the clinicopathological data of HCC patients. We compared the methylation frequency of 9 genes in chronic liver diseases with respect to the association of HCC. The 9 genes were selected for their involvement in carcinogenesis and frequent epigenetic inactivation in other tumor types. The present study aimed to determine the chronological pattern of CpG island hypermethylation of multiple genes along the multistep process of hepatocarcinogenesis, and to identify useful epigenetic biomarkers for the disease progression or outcome of HCCs.
Materials and Methods
Tissue Samples and DNA Extraction
A total of 226 liver samples were obtained from surgically resected (60 HCC, 10 DN, and 30 LC), or needle-biopsied (12 DN and 34 CH), specimens from patients treated at the Seoul National University Hospital, Seoul, Korea. The tissue samples were formalin-fixed, paraffin-embedded tissues, consisting of 60 paired specimens of primary HCC and non-HCC liver tissues (mean age, 53.8 years; 47 males and 13 females; 54 hepatitis B virus (HBV)-positive and 6 hepatitis C virus (HCV)-positive), 22 DN (57.8 years; 14 males and 8 females; 16 HBV-positive and 4 HCV-positive), 30 LC (46.5 years; 20 males and 10 females; 27 HBV-positive, 2 HCV-positive, and 1 autoimmune etiology), 34 CH (31.5 years; 30 males and four females; 19 HBV-positive, 13 HCV-positive, and 2 autoimmune hepatitis) and 20 normal liver tissue samples (58.4 years; seven males and 13 females). 30 LC and 34 CH, which appear in Tables 2 and 4 ▶ , were obtained from patients without HCCs and paired non-tumorous liver tissues from HCC patients were divided into two groups, 29 liver cirrhosis with concurrent HCC, and 31 chronic hepatitis with concurrent HCC, which appear in Table 4 ▶ . DN was classified according to the International Working Party’s criteria into low-grade DN and high-grade DN. 15 Low-grade DN was composed of minimally atypical hepatocytes with slightly increased cellularity, whereas high-grade DN showed cellular atypia, with an irregular trabecular and/or pseudoglandular arrangement, but insufficient for the diagnosis of malignancy. In the present study, low-grade DN was not included in the study material because low-grade DN is not well discriminated from large regenerative cirrhotic nodule.
Table 2.
The Frequency of CpG Island Hypermethylation in Neoplastic Liver Samples and Non-Neoplastic Liver Samples without Concurrent Hepatocellular Carcinoma
| Diagnosis | No. of cases | No. of cases methylated for at least one gene (%)* | Average no. of methylated gene† |
|---|---|---|---|
| Hepatocellular carcinoma | 60 | 60 (100) | 3.7 |
| Dysplastic nodule | 22 | 16 (72.7) | 1.5 |
| Liver cirrhosis | 30 | 12 (40) | 0.5 |
| Chronic hepatitis | 34 | 0 | 0 |
| Normal liver | 20 | 0 | 0 |
*Analyzed by χ2 test, P < 0.001.
†Analyzed by one-way ANOVA test, P < 0.001.
Table 4.
Methylation Frequency for Nine Genes and the Average Number of Methylated Genes in Chronic Hepatitis and Liver Cirrhosis Samples with or without Hepatocellular Carcinoma
| Gene | Frequency of methylation (%) | |||
|---|---|---|---|---|
| LC-HCC (n = 29) | CH-HCC (n = 31) | LC (n = 30) | CH (n = 34) | |
| APC | 13 (44.8) | 6 (19.4) | 4 (13.3) | 0 |
| COX-2 | 6 (20.7) | 8 (25.8) | 0 | 0 |
| DAP-kinase | 3 (10.3) | 0 | 3 (10) | 0 |
| E-cadherin | 2 (6.9) | 2 (6.5) | 2 (6.7) | 0 |
| GSTP1 | 11 (37.9) | 5 (16.1) | 5 (16.7) | 0 |
| hMLH1 | 0 | 0 | 0 | 0 |
| p16 | 5 (17.2) | 6 (19.4) | 0 | 0 |
| RASSF1A | 0 | 0 | 0 | 0 |
| TIMP3 | 2 (6.9) | 0 | 0 | 0 |
| Average no. of methylated genes* | 2.3 | 1.8 | 1.2 | 0.4 |
*Analyzed by two-tailed t-test, P < 0.001.
LC-HCC, liver cirrhosis with concurrent hepatocellular carcinoma; CH-HCC, chronic hepatitis with concurrent HCC.
After identification of the tumorous lesion on the hematoxylin-eosin-stained slides of HCC or DN patients, portions of tumors, where tumor cells comprised more than 80% of the cells, were scraped from 20-μm-thick paraffin sections. Paired non-tumorous liver tissues were scraped in the areas remote from the tumor, and free from the intravascular tumor emboli. The collected materials were dewaxed by washing in xylene, and rinsed in ethanol. The dried tissues were digested using proteinase K and subjected to classical DNA extraction using phenol/chloroform/isoamylalcohol and ethanol precipitation.
Sodium Bisulfite Modification and Methylation-Specific (MSP) PCR
Sodium bisulfite modification of the DNA from 226 samples was performed as previously described. 17 Briefly, 10 μl (5 μg) of genomic DNA was heat-denatured for 6 minutes at 97°C, followed by incubation with 0.2 mol/L NaOH for 10 minutes at room temperature. The denatured DNA was treated with 3.5 mol/L sodium bisulfite and 1 mmol/L hydroquinone (pH 5.0) for 16 hours at 55°C. The reaction mixture was purified with a JETSORB gel extraction kit (Genomed, Bad Oeynhausen, Germany), and desulphonated with 0.3 mol/L NaOH for 10 minutes at room temperature. The DNA was then precipitated with three volumes of cold ethanol, dissolved in H2O, and stored at −20°C.
MSP was performed to examine the methylation status at CpG islands of APC, COX-2, DAP-kinase, E-cadherin, GSTP1, hMLH1, p16, RASSF1A, TIMP-3, MINT1, MINT12, MINT25, MINT31 and MINT32 loci. The primer sequences of each locus, for both the methylated and unmethylated reactions are described in Table 1 ▶ . To amplify the bisulfite-modified promoter sequence of p16, E-cadherin, COX-2 and hMLH1, a polymerase chain reaction (PCR) mixture, containing 1X PCR buffer [10 mmol/L Tris (pH 8.3), 50 mmol/L KCl and 1.5 mmol/L MgCl2], deoxynucleotide triphosphates (each at 0.2 mmol/L), primers (10 pmol each), and bisulfite-modified DNA (30–50 ng), in a final volume of 25 μl, was used. For amplification of the APC, DAP-kinase, GSTP1, RASSF1A, TIMP3, MINT1, MINT12, MINT25, MINT31, and MINT32 clones, a PCR mixture containing 1X PCR buffer [16.6 mmol/L (NH4)2SO4, 67 mmol/L Tris (pH 8.8), 6.7 mmol/L MgCl2 and 10 mmol/L β-mercaptoethanol], deoxynucleotide triphosphates (each at 1 mmol/L), primers (10 pmol each), and bisulfite-modified DNA (30–50 ng), in a final volume of 25 μl, was used. The reactions were hot-started at 98°C for 5 minutes, followed by the addition of 0.75 U of Taq polymerase (Takara Shuzo Co., Kyoto, Japan). The amplifications were carried out in a thermal cycler (PerkinElmer, Foster City, CA) for 33 cycles (40 seconds at 95°C, 50 seconds at variable temperatures according to primer, and 50 seconds at 72°C), with a final 10-minute extension. The PCR products underwent electrophoresis on 2.5% agarose gels, and were visualized under UV illumination after ethidium bromide staining.
Table 1.
Primer Sequences and PCR Conditions for MSP Analysis
| Primer name | Primer sequence (5′–3′) forward | Primer sequence (5′–3′) reverse | Product size (bp) | Annealing temp. (°C) | References | |
|---|---|---|---|---|---|---|
| APC | M | TATTGCGGAGTGCGGGTC | TCGACGAACTCCCGACGA | 98 | 55 | 17 |
| U | GTGTTTTATTGTGGAGTGTGGGTT | CCAATCAACAAACTCCCAACAA | 108 | 60 | ||
| COX-2 | M | TTAGATACGGCGGCGGCGGC | TCTTTACCCGAACGCTTCCG | 161 | 61 | 18 |
| U | ATAGATTAGATATGGTGGTGGTGGT | CACAATCTTTACCCAAACACTTCCA | 171 | 61 | ||
| DAP-kinase | M | GGATAGTCGGATCGAGTTAACGTC | CCCTCCCAAACGCCGA | 98 | 60 | 19 |
| U | GGAGGATAGTTGGATTGAGTTAATGTT | CAAATCCCTCCCAAACACCAA | 98 | 60 | ||
| E-cadherin | M | TTAGGTTAGAGGGTTATCGCGT | TAACTAAAAATTCACCTACCGAC | 115 | 57 | 16 |
| U | TAATTTTAGGTTAGAGGGTTATTGT | CACAACCAATCAACAACACA | 97 | 53 | ||
| GSTP1 | M | TTCGGGGTGTAGCGGTCGTC | GCCCCAATACTAAATCACGACG | 91 | 59 | 20 |
| U | GATGTTTGGGGTGTAGTGGTTGTT | CCACCCCAATACTAAATCACAACA | 97 | 59 | ||
| hMLH1 | M | TATATCGTTCGTAGTATTCGTGT | TCCGACCCGAATAAACCCAA | 153 | 60 | 21 |
| U | TTTTGATGTAGATGTTTTATTAGGGTTGT | ACCACCTCATCATAACTACCCACA | 124 | 60 | ||
| p16 | M | TTATTAGAGGGTGGGGCGGATCGC | GACCCCGAACCGCGACCGTAA | 150 | 65 | 16 |
| U | TTATTAGAGGGTGGGGTGGATTGT | CAACCCCAAACCACAACCATAA | 151 | 60 | ||
| RASSF1A | M | GTGTTAACGCGTTGCGTTGCGTATC | AACCCCGCGAACTAAAAACGA | 93 | 60 | 22 |
| U | TTTGGTTGGAGTGTGTTAATGTG | CAAACCCCACAAACTAAAAACAA | 105 | 60 | ||
| TIMP3 | M | CGTTTCGTTATTTTTTGTTTTCGGTTTTC | CCGAAAACCCCGCCTCG | 116 | 59 | 23 |
| U | TTTTGTTTTGTTATTTTTTGTTTTTGGTTTT | CCCCCCAAAAACCCCACCTCA | 122 | 59 | ||
| MINT1 | M | AAAAAAAAACACCTAAAACTCA | CTACTTCGCCTAACCTAACG | 102 | 64 | 24 |
| U | GGGGTTGAGGTTTTTTGTTAGT | TTCACAACCTCAAATCTACTTCA | 117 | 64 | ||
| MINT12 | M | TTGGGAGTTTATTTAGGTCG | ACAACGATCTTCCGAATTTA | 152 | 55 | 25 |
| U | TGGGAGTTTATTTAGGTTGG | AAACACAACAATCTTCCAAAT | 155 | 55 | ||
| MINT25 | M | GTTCGTTAGAGTAATTTTGCG | TTATAACTAACGAAACACCGC | 128 | 55 | 25 |
| U | AGTAATTTTGTGGTGGAAGG | ACTAACAAAACACCACACCC | 114 | 55 | ||
| MINT31 | M | TTGAGACGATTTTAATTTTTTGC | AAAACCATCACCCCTAAACG | 100 | 62 | 24 |
| U | GAATTGAGATGATTTTAATTTTTTGT | CTAAAACCATCACCCCTAAACA | 105 | 64 | ||
| MINT32 | M | GATGTTAGAGGAATTTAGGC | AAAACGAACGAAACGTCCG | 126 | 64 | 24 |
| U | GAGTGGTTAGAGGAATTTAGGT | CTAAAAAAACAAACAAAACATCCA | 133 | 62 | ||
M, methylated sequence; U, unmethylated sequence.
Results
Results of MSP in HCC and Premalignant Stages
We examined the methylation status of the CpG islands of 9 tumor-related genes and/or 5 MINT loci, known to be frequently methylated in other cancers, in 60 paired HCC and non-HCC liver tissue samples, 22 DN, 30 LC, 34 CH, and 20 normal liver samples (Figure 1) ▶ . Samples giving negative results in the PCR with specific primer sequences for the unmethylated forms of p16 gene were excluded from the study, because the presence of an unmethylated p16 gene was considered to ensure the integrity of the bisulfite-modified DNA in the samples. The detailed results of the methylation for 9 genes in a large series of liver samples are shown in Figure 2 ▶ . All 60 HCC samples had methylation of one or more genes, ranging from 1 to 7, whereas none of the samples from the normal liver tissues and CH were methylated. CpG island methylation was detected for at least one of the tested gene in 40, 72.7, and 68.3% of LC, DN, and non-HCC liver tissue from HCC patients, respectively.
Figure 1.

Representative examples of MSP analysis of APC, COX-2, E-cadherin, DAP-kinase, GSTP1, p16, RASSF1A, TIMP3, and hMLH1 in HCC, corresponding non-cancerous liver tissue, DN, LC, and CH samples. DNA extracted from 226 liver tissues was amplified with primers specific to the unmethylated (U) or the methylated (M) CpG islands of each gene after modification with sodium bisulfite.
Figure 2.
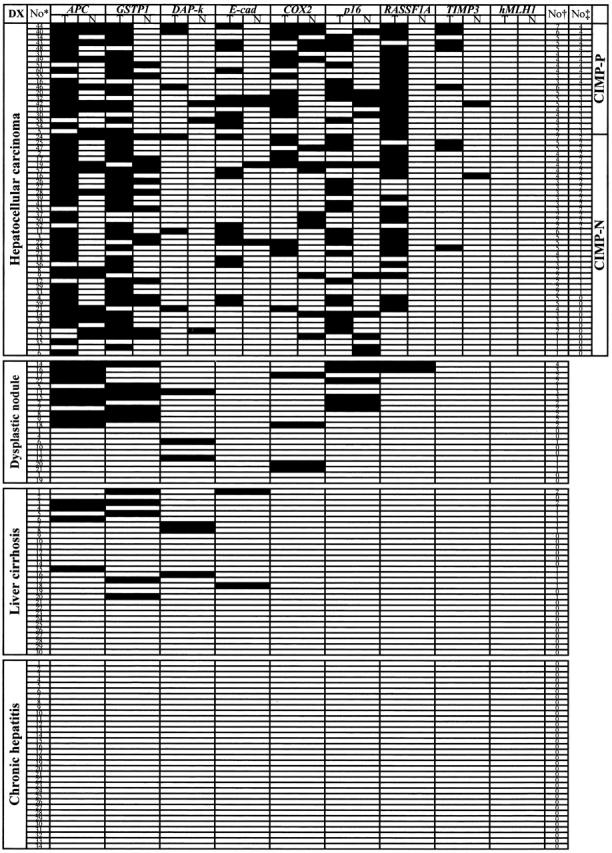
Summary of methylation analysis of APC, COX-2, E-cadherin, DAP-kinase, GSTP1, p16, RASSF1A, TIMP3, and hMLH1 in 226 liver samples. Filled boxes indicates the presence of methylation and open boxes indicates the absence of methylation. DX, diagnosis; T, tumor; N, paired non-tumorous liver tissue; No*, case number; No†, number of genes methylated; No‡, number of MINT loci methylated; CIMP-P, CpG island methylator phenotype-positive cases; CIMP-N, CIMP-negative cases; DAP-k, DAP-kinase; E-cad, E-cadherin.
When the number of genes methylated (n = 9) in each step lesion was compared, with the exclusion of the non-tumorous tissue samples of HCC patients (Table 2) ▶ , the average number of genes methylated showed a stepwise increase during the progression of the lesion (0 for normal liver or CH, 0.5 for LC, 1.5 for DN, and 3.7 for HCC), and the differences between each step lesions were statistically significant (P < 0.001, one-way analysis of variance test). The number of methylated genes was remarkably higher in HCC than in the corresponding non-HCC liver samples (average 3.7 vs. 1.2, P < 0.001, analyzed by 2-tailed t-test).
Frequency of Methylation of Each Gene in Different Stages of Multistep Hepatocarcinogenesis
The frequency of aberrant methylation for each gene is summarized in Table 3 ▶ . Of the 9 genes tested in this study, the genes most frequently methylated in HCCs were APC (81.7%), GSTP1 (76.7%), RASSF1A (66.7%), and p16 (48.3%). COX-2 and E-cadherin were methylated at a frequencies of 35% and 33.3%, respectively. DAP-kinase and TIMP3 were methylated in less than 10% of HCC samples, and hMLH1 was not methylated at all.
Table 3.
Methylation Frequency for Nine Genes in Hepatocellular Carcinoma and Preneoplastic Lesions
| Gene | Frequency of methylation (%) | ||||||
|---|---|---|---|---|---|---|---|
| HCC (n = 60) | DN (n = 22) | LC (n = 30) | CH (n = 34) | Significance* | |||
| HCC vs. DN | DN vs. LC | HCC vs. LC | |||||
| APC | 49 (81.7) | 10 (45.5) | 4 (13.3) | 0 | 0.002 | 0.013 | <0.001 |
| COX-2 | 21 (35) | 4 (18.2) | 0 | 0 | NS | 0.027 | <0.001 |
| DAP-kinase | 6 (10) | 3 (13.6) | 3 (10) | 0 | NS | NS | NS |
| E-cadherin | 20 (33.3) | 0 | 2 (6.7) | 0 | 0.001 | NS | 0.008 |
| GSTP1 | 46 (76.7) | 7 (31.8) | 5 (16.7) | 0 | <0.001 | NS | <0.001 |
| hMLH1 | 0 | 0 | 0 | 0 | |||
| p16 | 29 (48.3) | 6 (27.3) | 0 | 0 | 0.130 | 0.004 | <0.001 |
| RASSF1A | 40 (66.7) | 2 (9.1) | 0 | 0 | <0.001 | NS | <0.001 |
| TIMP3 | 8 (13.3) | 0 | 0 | 0 | NS | 0.048 | |
*Analyzed by Fisher’s exact test.
HCC, hepatocellular carcinoma; DN, dysplastic nodule; LC, liver cirrhosis; CH, chronic hepatitis; NS, not significant.
When the methylation frequency of an individual gene in lesions of various steps was compared, with exclusion of the HCC-associated non-tumorous liver samples, the tested genes, with the exception of the hMLH1 and DAP-kinase, generally showed an increase in the methylation frequency and different methylation behaviors along the multistep carcinogenesis. hMLH1 was not methylated in any of the samples of the four step lesions, and DAP-kinase was methylated at a similar frequency in HCC, DN, and LC, but not in CH. APC, E-cadherin, and GSTP1 were methylated in LC, as well as in the neoplastic lesions. A significant difference in the methylation frequency between HCC and DN, or between HCC and LC, was found in all three genes, but only APC showed a significant difference between DN and LC (P = 0.013; Fisher’s exact test). COX-2, p16 and RASSF1A were methylated in the neoplastic lesions (HCC and DN), but not in chronic liver diseases (LC and CH), and TIMP-3 was methylated in HCC only. The methylation frequency of RASSF1A was significantly different between HCC and DN (P < 0.001; Fisher’s exact test), but that of COX-2 or p16 was not (P > 0.05; Fisher’s exact test). A temporal order was noted in the timing of the methylation of the tested individual genes along the multistep hepatocarcinogenesis; the methylation of APC, DAP-kinase, E-cadherin, or GSTP1 preceded that of COX-2, p16, RASSF1A, or TIMP-3, along the multistep hepatocarcinogenesis (Figure 3) ▶ .
Figure 3.

Frequencies of CpG island methylation of APC, COX-2, DAP-kinase, E-cadherin, GSTP1, hMLH1, p16, RASSF1A, and TIMP3 in normal liver tissue, preneoplastic lesions, and hepatocellular carcinoma samples.
Association of HCC versus Methylation Frequency in Chronic Liver Diseases
Table 4 ▶ shows the difference in the methylation frequency of the tested genes between LC or CH samples, associated with and without HCC. The number of genes methylated was significantly higher in LC (n = 29) or CH samples (n = 31) obtained from the patients with HCC than in LC or CH without HCC (average number of methylated genes, 1.5 vs. 0.5, and 0.9 vs. 0, respectively, P < 0.001, analyzed by two-tailed t-test). While CH without concurrent HCC showed methylation for none of the genes tested, CH with concurrent HCC showed methylation for COX-2, APC, p16, GSTP1, and E-cadherin in a decreasing order of the methylation frequency. LC without an associated HCC harbored no methylation of COX-2, p16, or TIMP-3, which were methylated in LC with a concurrent HCC. RASSF1A was methylated in the neoplastic lesions only (HCC and DN), but not in chronic liver diseases, regardless of the association of HCC.
Clinicopathological Correlations and Survival Analysis
We tried to explore the clinicopathological significance of the methylation status of the genes tested. Small HCC, defined as 2 cm or less (n = 4), showed less frequent methylation than HCC larger than 2 cm (n = 55). The average number of genes methylated in small and large HCCs was 2.5 and 4.7, respectively (P = 0.004, two-tailed t-test). The methylation of GSTP1 was closely associated with tumor size (average 6.8 cm vs. 4.7 cm in methylation-positive and -negative samples, respectively, P = 0.025, two-tailed t-test). No other associations were found between the methylation status of specific genes and the clinicopathological findings, including age, sex, type of hepatitis virus, gross type, histological differentiation, and pTNM stage.
We explored the survival of HCC patients, and tried to find whether the methylation status of a specific gene is significantly associated. Survival data were available for 55 of 60 HCC patients. The follow-up period ranged from 8 to 36 months (mean, 24 months). Of the 55 patients, 14 (23%) died of disease (mean, 10 months), and 41 (68%) were still alive (mean, 23 months). Overall, the patients survived between 1 and 36 months, with a mean of 20 months. The follow-up results revealed a significant difference between the overall survival and the methylation-positive or methylation-negative cases for the E-cadherin or GSTP1 (Figure 4) ▶ . Thirty-six patients, negative for methylation of the E-cadherin in the HCC samples showed favorable outcomes compared to the 19 patients with methylation of this gene (mean survival time, 32 vs. 23 months, respectively; P = 0.034, log rank test). For GSTP1, 44 methylation-positive patients had a shorter survival period than the 11 methylation-negative patients (P = 0.043, log rank test), and the methylation-negative patients were all still living at the time of the follow-up.
Figure 4.
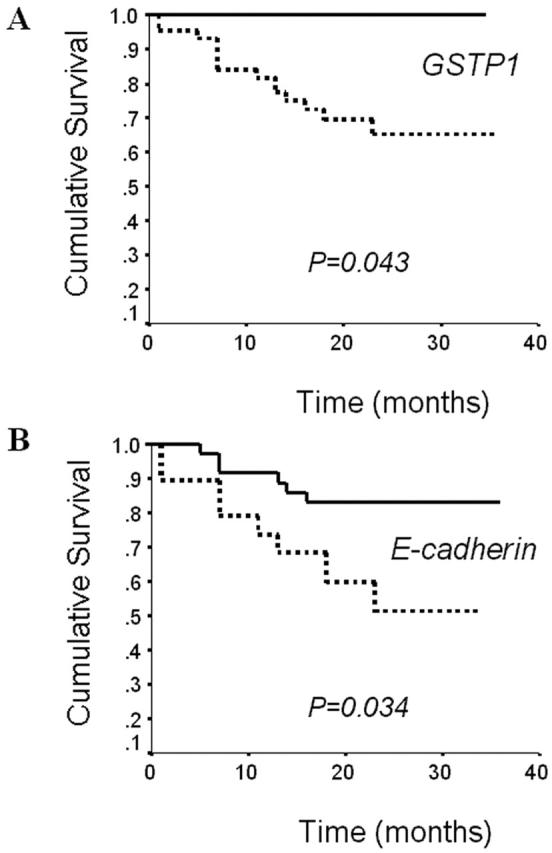
Overall survival of hepatocellular carcinoma patients according to the methylation status of GSTP1 (A) and E-cadherin (B). Solid line, patients without methylation of examined gene; dotted line, patients with methylation of examined gene.
CpG Island Methylator Phenotype (CIMP) in HCC
To determine the CIMP in a subset of HCCs, we investigated the methylation status of 5 cancer-specific MINT loci in HCC samples. The methylation frequencies were 46.7, 40, 66.7, 23.3, and 18.3% for the MINT31, MINT1, MINT12, MINT32, and MINT25, respectively. 20 (33.3%) of HCCs constituted CIMP-positive tumors when these were arbitrarily defined as cases where three or more loci of the 5 MINT loci tested were methylated. With respect to the methylation of the 9 genes tested, CIMP+ HCCs had a greater number of methylated genes than CIMP-negative tumors (4.4 vs. 3.3, respectively; P = 0.004, two-tailed t-test). There was no significant association between the CIMP status and the clinicopathological findings, including age, sex, gross type, histological differentiation, type of hepatitis virus infected and condition of the adjacent non-cancerous liver tissue.
Discussion
CpG island hypermethylation is an important mechanism for loss of function of tumor suppressor genes in human cancer. A growing number of genes have been reported to undergo CpG island hypermethylation in HCCs, which indicates the potential role of CpG island hypermethylation in hepatocarcinogenesis. HCC develops, and progresses, via a multistep process. DN, which arises in LC, is a precancerous lesion, with a high risk for further progression into HCCs. In contrast to the accumulating series of studies on the CpG island hypermethylation of tumor suppressor genes in HCC, no data are available for DN on the methylation status of the genes that have shown methylation in HCCs. If CpG island hypermethylation is an important mechanism for the development of HCC, it would be expected to occur in DN with considerable frequency. The present study has demonstrated that CpG island hypermethylation is a frequent event in DN, and that the number of genes methylated in DN was significantly higher than in LC, but lower than in HCC. These findings suggest that CpG island hypermethylation contributes to the development of DN, and acts in the early stages of the multistep process of HCCs.
There have been previous studies investigating CpG island hypermethylation in chronic liver diseases associated with HCCs, but only few on CpG island hypermethylation in chronic liver diseases from patients without HCCs. 7-14 In the present study, we found that the methylation profiles in chronic liver diseases associated with HCC were quite different from those free of HCC. Compared with chronic liver diseases without an associated HCC, those with a concurrent HCC showed significant increases in the methylation frequency of an individual gene, and the number of genes methylated. The occurrence of CpG island hypermethylation of APC, COX-2, E-cadherin, GSTP1 or p16 in CH with a concurrent HCC suggests that these methylation markers could be used for the risk assessment of a malignant transformation. In LC, the methylation of COX-2, p16 or TIMP-3 may serve as such biomarkers, although the methylation frequency of TIMP-3 was low (less than 10%), and therefore, less informative.
The significant difference of genes methylated between chronic liver diseases with and without an associated HCC suggests that HCC may arise in the liver with a methylation field defect. However, the possibility that the methylation events in chronic liver diseases with a concurrent HCC might be due to contamination of adjacent malignant cells cannot be excluded, although most of the non-tumorous samples obtained were more than 3 cm from HCC, and were confirmed to be free of microscopic tumor emboli with microscopic examination. Against the possibility of contamination are the findings of the discordant methylation patterns of the tested genes, where the specific genes were methylated in non-tumorous liver samples, but not in the corresponding HCC. This discordance can be seen in the results of Zöchbauer-Müller et al, 26 who studied CpG island hypermethylation of multiple genes in non-small cell lung cancers, and described that the genes methylated in the nonmalignant lung tissues were not methylated in the corresponding lung cancers from the same patients. A possible explanation for this discordance is that hepatocytes of CH or LC might have genetic or epigenetic changes, 12 but individual hepatocytes from the same samples might be heterogeneous with respect to these changes, 27,28 and a methylation-negative-clone for a certain gene among heterogeneous hepatocytes may acquire a growth advantage, and develops into HCC.
E-cadherin mediates homotypic cell-cell adhesion of epithelial cells and reduction of E-cadherin may be related to the acquisition of cell motility and tumor cell invasion. 29 The down-regulation of E-cadherin is related to allelic deletion of the gene or CpG island hypermethylation of the promoter CpG islands and both mechanisms have been demonstrated in HCC. 29-31 In the present study, E-cadherin was methylated in premalignant lesions at a frequency less than 7% and the methylation frequency increased markedly to 30% in HCC, the difference being statistically significant. The results suggest that E-cadherin methylation may participate in malignant transformation of HCC. Reduced expression of E-cadherin decreases intercellular adhesiveness, which may result in initiation of invasion and destruction of normal tissue morphology. Recently, two groups of researchers have analyzed the methylation status of E-cadherin for the non-tumorous liver tissues from patients with HCC using restriction analysis, 29,30 but the methylation frequency was quite different from each other (5% vs. 81%). The former lower frequency was consistent with the result of the present study. This discrepancy may be related to the difference of the sensitivity of the methods detecting methylation or the difference of CpG sites analyzed in restriction analysis of these researchers and MSP of the present study because a striking variability between the methylation level of individual CpG site has been demonstrated in the normal epithelial cell or cancer cell line with reduced expression of E-cadherin. 27,32
The findings of the present study indicate that HCC is one of the tumors with a high frequency of CpG island hypermethylation. Although the underlying mechanism of aberrant methylation in cancer cells remains unclear, a series of studies have suggested a relationship between viral oncogenesis and aberrant methylation, which was identified in SV40-positive malignant mesotheliomas and EBV-positive gastric carcinomas. 33,34 Such a relationship is supported by in vitro studies that have demonstrated frequent methylation of both viral DNA and adjacent host DNA segment following viral integration into the host DNA. 35-37 However, viral integration into the host DNA does not seem to be necessary for the activation of aberrant methylation because HCV does not insert into the host DNA, in contrast to HBV, and there was no difference in the methylation frequency between HBV-positive and HCV-positive HCCs in our study (data not shown). The relationship between hepatitis virus and aberrant methylation was suggested in a recent study 13 that demonstrated a higher frequency of CpG island methylation in hepatitis-virus-positive HCCs than in hepatitis-virus-negative ones.
To summarize, we studied the CpG island hypermethylation of nine tumor-related genes, and determined the frequency and chronology of methylation events of specific genes during the multistep hepatocarcinogenesis from CH to HCC. Our results demonstrated that CpG island hypermethylation occurs in the premalignant stages, and tends to accumulate during multistep hepatocarcinogenesis. Our data suggested that the CpG island hypermethylation of COX-2 or p16 might be potential molecular markers for the identification of patients with chronic liver disease at high risk for progression into HCC, and the CpG island hypermethylation of E-cadherin or GSTP1 might serve as potential biomarker for the prognostication of HCC.
Footnotes
Address reprint requests to Gyeong Hoon Kang, M.D., Department of Pathology, Seoul National University College of Medicine, 28 Yongon-dong, Chongno-gu, Seoul, 110–744, Korea. E-mail address: ghkang@snu.ac.kr.
Supported in part by grant number 03–2001-012 from the Seoul National University Hospital Research Fund, and by the 2002 BK21 project for Medicine, Dentistry, and Pharmacy, Seoul, Korea.
References
- 1.Bosch X, Ribes J, Borras J: Epidemiology of primary liver cancer. Semin Liver Dis 1999, 19:271-285 [DOI] [PubMed] [Google Scholar]
- 2.: Ministry of Health and Welfare Republic of Korea: Annual Report of Cancer Registry Program in the Republic of Korea. 2000:pp 1-191 Ministry of Health and Welfare Republic of Korea Seoul
- 3.Feitelson MA, Sun B, Tufan NLS, Liu J, Pan J, Lian Z: Genetic mechanisms of hepatocarcinogenesis. Oncogene 2002, 21:2593-2604 [DOI] [PubMed] [Google Scholar]
- 4.Jones PA, Laird PW: Cancer epigenetics comes of age. Nat Genet 1999, 21:163-167 [DOI] [PubMed] [Google Scholar]
- 5.Baylin SB, Herman JG: DNA hypermethylation in tumorigenesis: epigenetics joins genetics. Trends Genet 2000, 16:168-174 [DOI] [PubMed] [Google Scholar]
- 6.Baylin SB, Herman JG, Graff JR, Vertino PM, Issa JPJ: Alterations in DNA methylation: a fundamental aspect of neoplasia. Adv Cancer Res 1998, 72:141-196 [PubMed] [Google Scholar]
- 7.Iwata N, Yamamoto H, Sasaki S, Itoh F, Suzuki H, Kikuchi T, Kaneto H, Iku S, Ozeki I, Karino Y, Satoh T, Toyota J, Satoh M, Endo T, Imai K: Frequent hypermethylation of CpG islands and loss of expression of the 14–3-3 σ gene in human hepatocellular carcinoma. Oncogene 2000, 19:5298-5302 [DOI] [PubMed] [Google Scholar]
- 8.Kaneto H, Sasaki S, Yamamoto H, Itoh F, Toyota M, Suzuki H, Ozeki I, Iwata N, Ohmura T, Satoh T, Karino Y, Satoh T, Toyota J, Satoh M, Endo T, Omata M, Imai K: Detection of hypermethylation of the p16INK4A gene promoter in chronic hepatitis and cirrhosis associated with hepatitis B or C virus. Gut 2001, 48:327-377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liew CT, Li HM, Lo KW, Leow CK, Chan JY, Hin LY, Lau WY, Lai PBS, Lim BK, Huang J, Leung WT, Wu S, Lee JKC: High frequency of p16INK4A gene alterations in hepatocellular carcinoma. Oncogene 1999, 18:789-795 [DOI] [PubMed] [Google Scholar]
- 10.Kanai Y, Ushijima S, Hui AM, Ochiai A, Tsuda H, Sakamoto M, Hirohashi S: The E-cadherin gene is silenced by CpG methylation in human hepatocellular carcinomas. Int J Cancer 1997, 71:355-359 [DOI] [PubMed] [Google Scholar]
- 11.Tchou JC, Lin X, Freije D, Isaacs WB, Brooks JD, Rashid A, De Marzo AM, Kanai Y, Hirohashi S, Nelson WG: GSTP1 CpG island DNA hypermethylation in hepatocellular carcinomas. Int J Oncol 2000, 16:663-676 [DOI] [PubMed] [Google Scholar]
- 12.Kondo Y, Kanai Y, Sakamoto M, Mizokami M, Ueda R, Hirohashi S: Genetic instability and aberrant DNA methylation in chronic hepatitis and cirrhosis: a comprehensive study of loss of heterozygosity and microsatellite instability at 39 loci and DNA hypermethylation on 8 CpG islands in microdissected specimens from patients with hepatocellular carcinoma. Hepatology 2000, 32:970-979 [DOI] [PubMed] [Google Scholar]
- 13.Saito Y, Kanai Y, Sakamoto M, Saito H, Ishii H, Hirohashi S: Expression of mRNA for DNA methyltransferases and methyl-CpG binding proteins and DNA methylation status on CpG islands and pericentromeric satellite regions during human hepatocarcinogenesis. Hepatology 2001, 33:561-568 [DOI] [PubMed] [Google Scholar]
- 14.Shen L, Ahuja N, Shen Y, Habib NA, Toyota M, Rashid A, Issa JP: DNA methylation and environmental exposures in human hepatocellular carcinoma. J Natl Cancer Inst 2002, 94:755-761 [DOI] [PubMed] [Google Scholar]
- 15.: International Working Party: Terminology of nodular hepatocellular lesions Hepatology 1995, 22:983-993 [DOI] [PubMed] [Google Scholar]
- 16.Hermans JG, Graff JR, Myöhänen S, Nelkin BD, Baylin SB: Methylation-specific PCR. A novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA 1996, 93:9821-9826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsuchiya T, Tamura G, Sato K, Endoh Y, Sakata K, Jin Z, Motoyama T, Usuba O, Kimura W, Nishizuka S, Wilson KT, James SP, Yin J, Fleisher AS, Zou T, Silverberg SG, Kong D, Meltzer SJ: Distinct methylation patterns of two APC gene promoters in normal and cancerous gastric epithelia. Oncogene 2000, 19:3642-3646 [DOI] [PubMed] [Google Scholar]
- 18.Akhtar M, Cheng Y, Magno RM, Ashktorab H, Smoot DT, Meltzer SJ, Wilson KT: Promoter methylation regulates Helicobacter pylori-stimulated cyclooxygenase-2 expression in gastric epithelial cells. Cancer Res 2001, 61:2399-2403 [PubMed] [Google Scholar]
- 19.Katzenellenbogen RA, Baylin SB, Herman JG: Hypermethylation of the DAP-kinase CpG island is a common alteration in B-cell malignancies. Blood 1999, 93:4347-4353 [PubMed] [Google Scholar]
- 20.Esteller M, Corn PG, Urena JM, Gabrielson E, Baylin SB, Herman JG: Inactivation of glutathione s-transferase P1 gene by promoter hypermethylation in human neoplasia. Cancer Res 1998, 58:4515-4518 [PubMed] [Google Scholar]
- 21.Kang GH, Shim YH, Ro JY: Correlation of methylation of the hMLH1 promoter with lack of expression of hMLH1 in sporadic gastric carcinomas with replication error. Lab Invest 1999, 79:903-909 [PubMed] [Google Scholar]
- 22.Lo KW, Kwong J, Hui ABU, Chan SYY, To KF, Chan ASC, Chow LSN, Teo PML, Johnson PJ, Huang DP: High frequency of promoter hypermethylation of RASSF1A in nasopharyngeal carcinoma. Cancer Res 2001, 61:3877-3881 [PubMed] [Google Scholar]
- 23.Bachman KE, Herman JG, Corn PG, Merlo A, Costello JF, Cavenee WK, Baylin SB, Graff JR: Methylation-associated silencing of the tissue inhibitor of metalloproteinase-3 gene suggests a suppressor role in kidney, brain, and other human cancers. Cancer Res 1999, 59:798-802 [PubMed] [Google Scholar]
- 24.Ueki T, Toyota M, Skinner H, Walter KM, Yeo CJ, Issa JP, Hruban RH, Goggins M: Identification and characterization of differentially methylated CpG islands in pancreatic carcinoma. Cancer Res 2001, 61:8540-8546 [PubMed] [Google Scholar]
- 25.Lee S, Kim WH, Jung HY, Yang MH, Kang GH: Aberrant CpG island methylation of multiple genes in intrahepatic cholangiocarcinoma. Am J Pathol 2002, 161:1015-1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zöchbauer-Müller S, Fong KM, Virmani AK, Geradts J, Gazdar AF, Minna JD: Aberrant promoter methylation of multiple genes in non-small cell lung cancers. Cancer Res 2001, 61:249-255 [PubMed] [Google Scholar]
- 27.Graff JR, Gabrielson E, Fujii H, Baylin SB, Herman JG: Methylation patterns of the E-cadherin 5′ CpG island are unstable and reflect the dynamic, heterogeneous loss of E-cadherin expression during metastatic progression. J Biol Chem 2000, 275:2727-2732 [DOI] [PubMed] [Google Scholar]
- 28.Matsumura T, Makino R, Mitamura K: Frequent down-regulation of E-cadherin by genetic and epigenetic changes in the malignant progression of hepatocellular carcinoma. Clin Cancer Res 2001, :594-599 [PubMed] [Google Scholar]
- 29.Mareel M, Boterberg T, Noe V, Van Hoorde L, Vermeulen S, Bruyneel E, Bracke M: E-cadherin/catenin/cytoskeleton complex: a regulator of cancer invasion. J Cell Physiol 1997, 173:271-274 [DOI] [PubMed] [Google Scholar]
- 30.Kanai Ushijima S, Tsuda H, Sakamoto M, Hirohashi S: Aberrant DNA methylation precedes loss of heterozygosity on chromosome 16 in chronic hepatitis and liver cirrhosis. Cancer Lett 2000, 148:73-80 [DOI] [PubMed] [Google Scholar]
- 31.Wei Y, Nhieu JTV, Prigent S, Srivatanakul P, Tiollais P, Buendia MA: Altered expression of E-cadherin in hepatocellular carcinoma: correlations with genetic alterations, β-catenin expression, and clinical features. Hepatology 2002, 36:692-701 [DOI] [PubMed] [Google Scholar]
- 32.Graff JR, Herman JG, Myöhänen S, Baylin SB, Vertino PM: Mapping patterns of CpG island methylation in normal and neoplastic cells implicates both upstream and downstream regions in de novo methylation. J Biol Chem 1997, 272:22322-22329 [DOI] [PubMed] [Google Scholar]
- 33.Toyooka S, Pass HI, Shivapurkar N, Fukuyama Y, Maruyama R, Toyooka KO, Gilcrease M, Farinas A, Minna JD, Gazdar AF: Aberrant methylation and simian virus 40 tag sequences in malignant mesothelioma. Cancer Res 2001, 61:5727-5730 [PubMed] [Google Scholar]
- 34.Kang GH, Lee S, Kim WH, Lee HW, Kim JC, Rhyu MG, Ro JY: Epstein-Barr virus-positive gastric carcinoma demonstrates frequent aberrant methylation of multiple genes and constitutes CpG island methylator phenotype-positive gastric carcinoma. Am J Pathol 2002, 160:787-794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Doefler W: The insertion of foreign DNA into mammalian genomes and its consequences: a concept in oncogenesis. Adv Cancer Res 1995, 66:313-344 [DOI] [PubMed] [Google Scholar]
- 36.Heller H, Kammer C, Wilgenbus P, Doerfler W: Chromosomal insertion of foreign (adenovirus type 12, plasmid, or bacteriophage λ) DNA is associated with enhanced methylation of cellular DNA segments. Proc Natl Acad Sci USA 1995, 92:5515-5519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Remus R, Kammer C, Heller H, Schmitz B, Schell G, Doerfler W: Insertion of foreign DNA into an established mammalian genome can alter the methylation of cellular DNA sequences. J Virol 1999, 73:1010-1022 [DOI] [PMC free article] [PubMed] [Google Scholar]