

Abstract
Transforming growth factor-β1 (TGF-β1) and the renin-angiotensin-aldosterone system are key mediators in kidney fibrosis. Integrin αvβ6, a heterodimeric matrix receptor expressed in epithelia, binds and activates latent TGF-β1. We used β6 integrin-null mice (β6−/−) to determine the role of local TGF-β1 activation in renal fibrosis in the unilateral ureteral obstruction (UUO) model. Obstructed kidneys from β6−/− mice showed less injury than obstructed kidneys from wild-type (WT) mice, associated with lower collagen I, collagen III, plasminogen activator inhibitor (PAI-1), and TGF-β1 mRNA levels and lower collagen content. Infusion with either angiotensin II (Ang II) or aldosterone (Aldo) or combination in β6−/− UUO mice significantly increased collagen contents to levels comparable to those in identically treated WT. Active TGF-β protein expression in β6−/− mice was less in UUO kidneys with or without Ang II infusion compared to matched WT mice. Activated Smad 2 levels in β6−/− obstructed kidneys were lower than in WT UUO mice, and did not increase when fibrosis was induced in β6−/− UUO mice by Ang II infusion. Anti-TGF-β antibody only partially decreased this Ang II-stimulated fibrosis in β6−/− UUO kidneys. In situ hybridization and immunostaining showed low expression of PAI-1 mRNA and protein in tubular epithelium in β6−/− UUO kidneys, with increased PAI-1 expression in response to Ang II, Aldo, or both. Our results indicate that interruption of αvβ6-mediated activation of TGF-β1 can protect against tubulointerstitial fibrosis. Further, the robust induction of tubulointerstitial fibrosis without increase in activated Smad 2 levels in obstructed β6−/− mice by Ang II suggests the existence of a TGF-β1-independent pathway of induction of fibrosis through angiotensin.
Transforming growth factor (TGF)-β has been implicated as a key molecule in fibrosis and immune regulation, and its expression is increased in numerous fibrotic conditions. 1 Mammals express three isoforms (TGF-β1, TGF-β2, and TGF-β3) that are synthesized in the latent form and must be activated before they can bind to their receptors and induce TGF-β-mediated effects. The noncovalent association of mature TGF-β with an N-terminal fragment of the same gene product [termed latency-associated protein (LAP)] is responsible for maintaining TGF-β isoforms in latent form. 1
Integrins are composed of α and β subunits that govern cell-cell and cell-extracellular matrix interactions, thus influencing growth, differentiation, and development. The integrin αvβ6 is expressed principally on epithelial cells and serves as a receptor for the RGD motif of the extracellular matrix proteins fibronectin, tenascin, and vitronectin. 2 The αvβ6 integrin is highly expressed in the lung, skin, and kidney during organogenesis. 3 In the mouse, β6 integrin subunit mRNA expression is very low at day 11 of gestation, increasing dramatically by E14 and E17, and plateauing by 2 weeks after birth. Expression of β6 integrin mRNA in the adult rat is present in proximal tubules, cortical thick ascending limb, inner and outer medullary collecting duct, and macula densa. 4 Expression of αvβ6 integrin is increased in response to inflammation or repair in the kidney, such as in chronic pyelonephritis or transplant rejection. 3 Mice devoid of αvβ6 integrin show significant inflammation in skin and lungs, but do not have renal structural abnormalities. 5
TGF-β1 LAP is a ligand for the integrin αvβ6, and its binding induces activation of TGF-β1. 6 Further, in a bleomycin model of lung epithelial cell injury, knockout mice devoid of β6 integrin were completely protected against the fibrosis that developed in the wild type (WT). 6 Interestingly, this protection against injury occurred despite persistent macrophage infiltration in the knockout that was similar in magnitude to that seen in the WT. This protection was linked to prevention of TGF-β1 activation by absence of αvβ6 integrin.
Activated TGF-β facilitates matrix accumulation in at least four ways, including increased synthesis of matrix proteins, increased synthesis of integrins involved in matrix assembly at the cell surface, decreased synthesis of matrix metalloproteinases, and increased synthesis of proteinase inhibitors, including plasminogen activator inhibitor-1 (PAI-1), thereby decreasing matrix degradation. PAI-1 is the major physiological inhibitor of tissue-type plasminogen activator (t-PA) and urokinase-like plasminogen activator (u-PA), both of which activate plasminogen to plasmin, thus promoting fibrinolysis and proteolysis. 7,8 Plasmin also activates other latent matrix metalloproteinases. Important interactions of the renin-angiotensin-aldosterone system (RAAS) with PAI-1 have been demonstrated in vitro and in vivo, supporting a direct induction of PAI-1 by angiotensin, and enhancement of this induction by aldosterone (Aldo). 9-12 However, in vitro data suggest that angiotensin induction of PAI-1 has an early TGF-β-independent stage with later TGF-β-dependent increase of PAI-1. 13 Thus, the RAAS, TGF-β, and PAI-1 have been intricately linked in injury response and fibrosis. 14 In this study, we used β6−/− mice to examine possible TGF-β-dependent and -independent mechanisms of renal fibrosis.
Materials and Methods
Animals
Adult male WT and β6 integrin-null mice (β6−/−) with C57BL/6 or 129Sv background were used. Mice were housed in microisolator cages in a pathogen-free barrier facility. All protocols were approved by the Vanderbilt University Institutional Animal Care and Use Committee.
Experimental Protocol
Groups of WT and β6−/− mice, age 10 to 12 weeks, underwent unilateral ureteral obstruction (UUO) (WT UUO, n = 12; β6−/− UUO, n = 12) under sterile conditions as described previously. 15 Left ureters of mice were double-ligated with 6-0 silk and cut between the two ligated points. The upper ligation was consistently placed at the level of lower pole of the kidney. In pilot studies, identical degree of fibrosis developed on 129 and C57BL/6 background after UUO, and results are therefore combined. To investigate the effects of RAAS on fibrosis, angiotensin II (Ang II), Aldo, or both were administered to additional mice (WT UUO + Ang II, n = 6; β6−/− UUO + Ang II, n = 6; WT UUO + Aldo, n = 4; β6−/− UUO + Aldo, n = 3; WT UUO + Ang II/Aldo, n = 6; β6−/− UUO + Ang II/Aldo, n = 6). Miniosmotic pumps (Alzet, model 1002; Durect Corp., Cupertino, CA) filled with Ang II (concentration of 40 μg/kg/hour; Bachem Bioscience Inc., Torrance, CA) were inserted into the peritoneal space. The contents of the miniosmotic pump were delivered at a rate of 0.25 μl/hour for 2 weeks. Aldo implantable pellets (Innovative Research of America, Sarasota, FL) were inserted subcutaneously for 2 weeks releasing at 140 μg/kg/day. 16 Doses for Ang II or Aldo were based on previous long-term studies showing only moderate hypertension with mild fibrosis in the rat with these levels, and our unpublished data of mild hypertension, induction of PAI-1 but only minimal fibrosis after up to 8 weeks with these doses in the mouse. 17
To determine the TGF-β-dependent and -independent pathways for inducing fibrosis, monoclonal anti-TGF-β antibody (Ab) (1D11; Genzyme Corp., Framingham, MA) or isotype-matched irrelevant control antibody (13C4, Genzyme Corp.) were given to WT or β6−/− mice with UUO with or without Ang II infusion. Low and high doses of 1D11 (low, 0.5 mg/kg/body weight; high, 5 mg/kg/body weight) and control antibody (13C4, 5 mg/kg BW) were administered every other day by intraperitoneal injections. The doses of anti-TGF-β antibody were chosen based on inhibition of biological activity of TGF-β in the mouse without producing histopathology in any organs of the body. 18 The first TGF-β antibody injection was given 1 day before UUO. WT UUO mice were treated with antibodies as follows: control antibody 13C4 (WT UUO + CONT Ab, n = 5; WT UUO + Ang II + CONT Ab, n = 3), high-dose anti-TGF-β antibody (WT UUO + H-TGF-β Ab, n = 5; WT UUO + Ang II + H-TGF-β Ab, n = 5), or low-dose anti-TGF-β antibody (WT UUO + L-TGF-β Ab, n = 4; WT UUO + Ang II + L-TGF-β Ab, n = 4). β6−/− UUO mice infused with Ang II were treated with antibodies as follows: control antibody 13C4 (β6−/− UUO + Ang II + CONT Ab, n = 4), high-dose anti-TGF-β antibody (β6−/− UUO + Ang II + H-TGF-β Ab, n = 8), or low-dose anti-TGF-β antibody (β6−/− UUO + Ang II + L-TGF-β Ab, n = 5). At 5 or 14 days after UUO surgery, mice for the above groups were sacrificed and both obstructed and nonobstructed contralateral kidneys (used as normal control) were harvested for morphological, immunostaining, biochemical, and molecular assessment.
Morphological Assessment
The degree of tubulointerstitial fibrosis was scored on Masson’s trichrome-stained coronal sections. All cortical and medullary fields were scored from 0 to 4+ for each field, with an average for each kidney calculated. Fibrosis was assessed as 0 for 0%, 1 for <25%, 2 for 25 to 50%, 3 for >50 to 75%, and 4 for >75% of each field occupied by tubulointerstitial fibrosis. All sections were examined without knowledge of the treatment protocol.
Blood Pressure Measurement
Systolic blood pressure was measured using a tail-cuff blood pressure monitor for rats and mice (model 2000; Muromachi Kikai Co., Ltd., Tokyo, Japan) in conscious, trained mice at room temperature.
Immunohistochemistry
Kidney tissues were fixed overnight in 4% paraformaldehyde at 4°C, and embedded in paraffin using standard techniques. Four-μm sections were dewaxed and rehydrated. Endogenous peroxidase was quenched with 3% hydrogen peroxidase for 10 minutes and exposed to Power Block (BioGenex Laboratories, San Ramon, CA) for 45 minutes.
Proliferating cells were identified by proliferating cell nuclear antigen (PCNA) cyclin polypeptide immunohistochemistry using the Mouse on Mouse kit (Innogenex, San Ramon, CA). Monoclonal mouse anti-human PCNA (1:100; DAKO Corp., Carpinteria, CA) was applied, followed by rabbit anti-mouse antibody (DAKO), biotinylated goat anti-rabbit Ig (BioGenex), and peroxidase-conjugated streptavidin. PCNA-positive tubulointerstitial cells in the cortex were semiquantified using a 0 to 4+ scale by scoring all high-power cortical fields per section (0 to 4 represent: 0%, <25%, 25 to 50%, >50 to 75%, and >75% of the tubulointerstitial nuclei in the field showing positive nuclear staining).
For immunostaining of PAI-1, rabbit anti-rat PAI-1 antibody (1:50; American Diagnostica, Inc., Greenwich, CT) was used at 4°C overnight followed by biotinylated goat anti-rabbit Ig (BioGenex) and peroxidase-conjugated streptavidin.
For detection of active TGF-β, sections were treated with hyaluronidase (Sigma Chemical Co., St. Louis, MO) at 1 mg/ml in 0.1 mol/L sodium acetate, pH 5.5, containing 0.85% NaCl at 37°C for 30 minutes. After blocking with 5% normal goat serum, a rabbit polyclonal antibody to active TGF-β (LC 1-30-1; gift from Dr. Kathy Flanders, National Cancer Institute) at 6.5 μg/μl in Tris-buffered saline/0.4%Triton buffer was used overnight at 4°C. 19 Biotinylated goat anti-rabbit Ig (BioGenex) was added and followed by peroxidase-conjugated streptavidin.
TSP-1 was stained with primary goat anti-human TSP-1 (1:100, N-20, sc12312; Santa Cruz Biotechnology Inc., Santa Cruz, CA) overnight at 4°C and secondary horseradish peroxidase-conjugated anti-goat antibody (1:100). Diaminobenzidine was added as a chromogen.
Infiltration of macrophages was detected by rat anti-mouse F4/80 antibody (1:20; Serotec Inc., Raleigh, NC). Biotinylated anti-rat secondary antibody (Vector Laboratories, Burlingame, CA) was used and followed by streptavidin-conjugated alkaline phosphatase (Innogenex). After blocking with 0.25% levamisole for 10 minutes, Sigma Fast Red TR/Naphtol AS-Mx tablets (Sigma) were added as a chromogen. Slides were counterstained with hematoxylin. Infiltrating macrophages in the interstitium were counted and data expressed as macrophage number per high-power field. Control slides treated with nonspecific antisera instead of primary antibodies showed no staining.
Total Kidney Collagen Content
Part of the kidney tissue was frozen, and relative collagen content as a percentage of total protein was calculated from the concentration of hydroxyproline and proline, measured as their phenylisothiocyanate derivatives by reverse phase-high performance liquid chromatography. 20 Briefly, tissue homogenates were hydrolyzed in vacuo in constant boiling HCl (110°C, 16 hours), dried, taken up in 1 ml of redrying solution [ethanol/water/(C2H5)3N, 2:2:1], and 100 μl were redried under nitrogen. Twenty μl of derivatization reagent [methanol/water/(C2H5)3N/-phenylisothiocyanate, 7:1:1:1] were added, and the reaction was allowed to proceed at room temperature for 20 minutes. The sample was then lyophilized during centrifugation to remove the reagents. Samples were either frozen at this stage or diluted to 1 ml with buffer (5% acetonitrile in 5 mmol/L disodium phosphate, pH 7.4). A solution containing amino acid standards was dried and coupled in the same manner. The coupling efficiency was >99% under these conditions. Recovery and efficiency were normalized by including norleucine in each homogenate.
Northern Blot Hybridization
Northern blot was performed as described. 21 Total RNA from kidney was extracted by Trizol reagent (Life Technologies, Grand Island, NY). Twenty μg of total RNA were loaded and fractionated by electrophoresis in 1% agarose gel and transferred to a nylon membrane. The blots were hybridized with the following cDNA probes: mouse PAI-1 (365 bp), 21 mouse TGF-β1 (974 bp), human collagen I (American type Culture Collection, Manassas, VA), and mouse collagen III (355 bp). To evaluate RNA loading, blots were reprobed with rat housekeeping gene GAPDH cDNA. The ratio of specific message to GAPDH was used to quantify expression for each tissue sample.
In Situ Hybridization
[35S]-labeled sense and anti-sense riboprobes for PAI-1 were prepared as previously described by transcription of the pCR II plasmid (Invitrogen, Carlsbad, CA) combining a PAI-1 cDNA insert by SP6 or T7 RNA polymerase (Ambion Inc., Austin, TX). 22 After treatment by proteinase K and triethanolamine/acetic anhydride, sections were dehydrated in ethanol and air-dried. Hybridization was done as described previously. A control in situ hybridization done with sense probes showed no specific signal.
Western Blot Analysis of Smad 2
Frozen kidney tissues were transferred in RIPA plus buffer containing 150 mmol/L NaCl, 50 mmol/L Tris-HCl, pH 7.5, 5 mmol/L ethylenediamine tetraacetic acid, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 100 μg/ml phenylmethyl sulfonyl fluoride, 1:100 phosphatase inhibitor cocktail I, 1:100 phosphatase inhibitor cocktail II (Sigma), 1:100 proteinase inhibitor cocktail tablet (Roche Diagnostics GmbH, Mannheim, Germany). Tissue samples were homogenized in 1.5 ml of homogenizer (Pellet Pestle Dips; Kontes Glass Company, Vineland, NJ) on ice, and centrifuged at 13,000 rpm for 15 minutes at 4°C. The protein concentration was measured using Dc Protein Assay kit (Bio-Rad Laboratories, Hercules, CA). Fifty μg of protein from samples were loaded and separated on 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto a 0.2 μmol/L nitrocellulose membrane. Phosphorylated-Smad 2 (P-Smad 2) was detected using a specific rabbit anti-phosphorylated-Smad2 (P-Smad 2) polyclonal antibody (Upstate Biotechnology, Lake Placid, NY) overnight at 4°C, 1:500 dilutions. After washing in Tris-buffered saline with 0.1% Tween 20 (TBS-T), horseradish peroxidase-labeled donkey anti-rabbit IgG secondary antibody (1:2500 dilution in 5% milk TBS-T) was added and incubated at room temperature for 50 minutes. Protein bands on Western blots were visualized by ECL Plus (Amersham, Arlington Heights, IL) according to the manufacturer’s instructions and were developed on film. The membranes were stripped with 100 mmol/L β-mercaptoethanol, 2% sodium dodecyl sulfate, 62.5 mmol/L Tris-HCL, pH 6.7. Total Smad 2 (T-Smad 2) was detected using rabbit anti-Smad 2 polyclonal antibody (Zymed Laboratories Inc., San Francisco, CA). Cell lysates obtained from human colon carcinoma cell line FET stimulated with TGF-β1 (12 ng/ml for 1 hour, kindly provided by Dr. Pran Datta, Vanderbilt University Medical Center) were used as a positive control.
Statistical Analysis
Results are expressed as mean ± SEM. Statistical difference was assessed by a single factor variance (analysis of variance) followed by unpaired Student’s t-test as appropriate. Nonparametric data were compared by Mann-Whitney U-test. A P value <0.05 was considered to be significant.
Results
Tubulointerstitial Fibrosis in β6−/− UUO Kidney
Tubular dilation, tubular atrophy, and widening of interstitial space were present in obstructed kidneys in WT mice at 5 days. Tubulointerstitial damage progressed in both cortex and medulla by day 14 after obstruction in WT mice. In contrast, obstructed kidneys from β6−/− mice had significantly less injury (Figure 1) ▶ . Nonobstructed kidneys in both WT and β6−/− mice showed few PCNA-positive cells. There was significant tubular epithelial cell proliferation in WT, but not β6−/− mice, after obstruction (Figure 2) ▶ . Macrophage infiltration after UUO was more prominent in β6−/− than in WT, but this difference was not statistically significant (Figure 3) ▶ . Nonobstructed control kidneys in both WT and β6−/− mice expressed similar trace to low levels of mRNA for collagen I, collagen III, PAI-1, and TGF-β1 (Figure 4) ▶ . Renal mRNA levels for collagen I (Figure 4, A and B) ▶ , collagen III (Figure 4, A and C) ▶ , PAI-1 (Figure 4, A and D) ▶ , and TGF-β1 (Figure 4, A and E) ▶ were increased in response to UUO in WT mice. In contrast, β6−/− mice showed mRNA expression levels only one-third to one-half of those in obstructed WT at day 14 (collagen I: WT UUO 1.2 ± 0.1 versus β6−/− UUO 0.3 ± 0.02, P < 0.01; collagen III: WT UUO 1.3 ± 0.1 versus β6−/− 0.4 ± 0.1, P < 0.01; PAI-1: WT UUO 1.1 ± 0.1 versus β6−/− 0.03 ± 0.1, P < 0.01, TGF-β1: WT + UUO 3.6 ± 0.2 versus β6−/− 1.9 ± 0.2, all expressed as density ratios relative to GAPDH mRNA) (Figure 4) ▶ .
Figure 1.
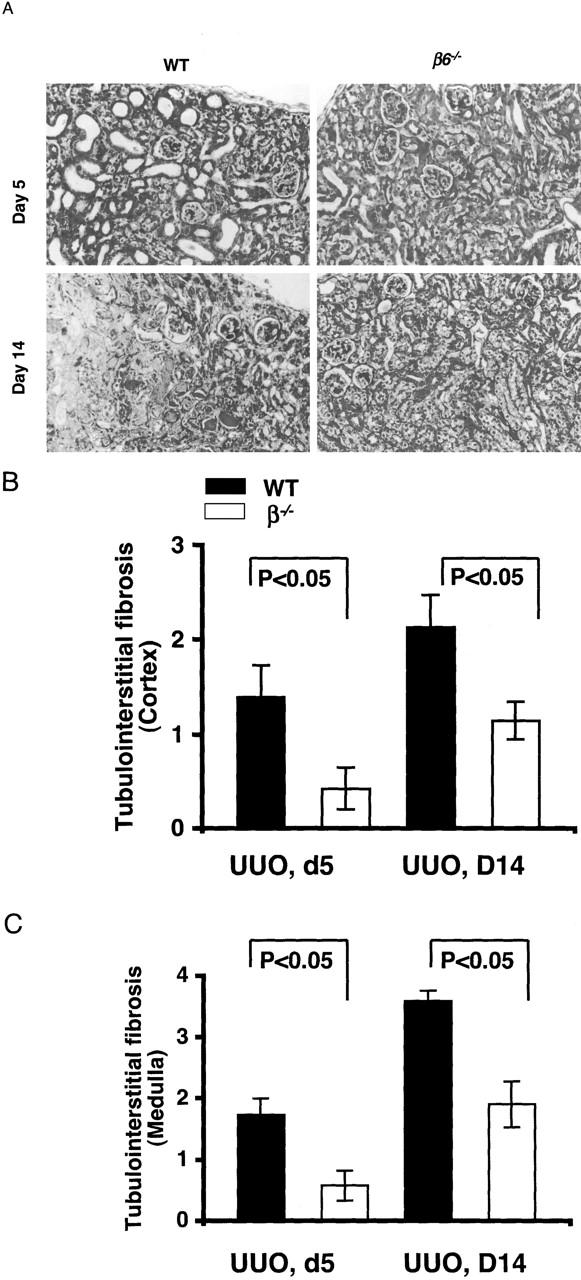
Kidney morphological changes in obstructed kidneys. A: β6−/− mice had less fibrosis at days 5 and 14 after UUO than time-matched WT. B and C: Semiquantitative scoring of fibrosis (0 to 4 + scale) showed less injury in β6−/− UUO in cortex (B) and medulla (C) compared to WT UUO (n = 4 for each group at day 5, n = 8 for each group at day 14). Data are shown as mean ± SEM. Masson’s trichrome stain; original magnifications, ×200.
Figure 2.

Tubular cell proliferation after UUO. A: PCNA immunostaining in tubules and interstitium in WT and β6−/− UUO kidneys at day 5. B: Semiquantitative scoring of PCNA-positive tubulointerstitial cells in cortex at day 5 (n = 4 for each group). Data are shown as mean ± SEM. Original magnifications, ×400.
Figure 3.

F4/80 + interstitial macrophages in obstructed kidneys at day 14. A: Macrophages (in red) in WT and β6−/− UUO kidneys. B: Quantitation of average macrophage number per high-power field (n = 8 for each group). Data are shown as mean ± SEM. Original magnification, ×400.
Figure 4.
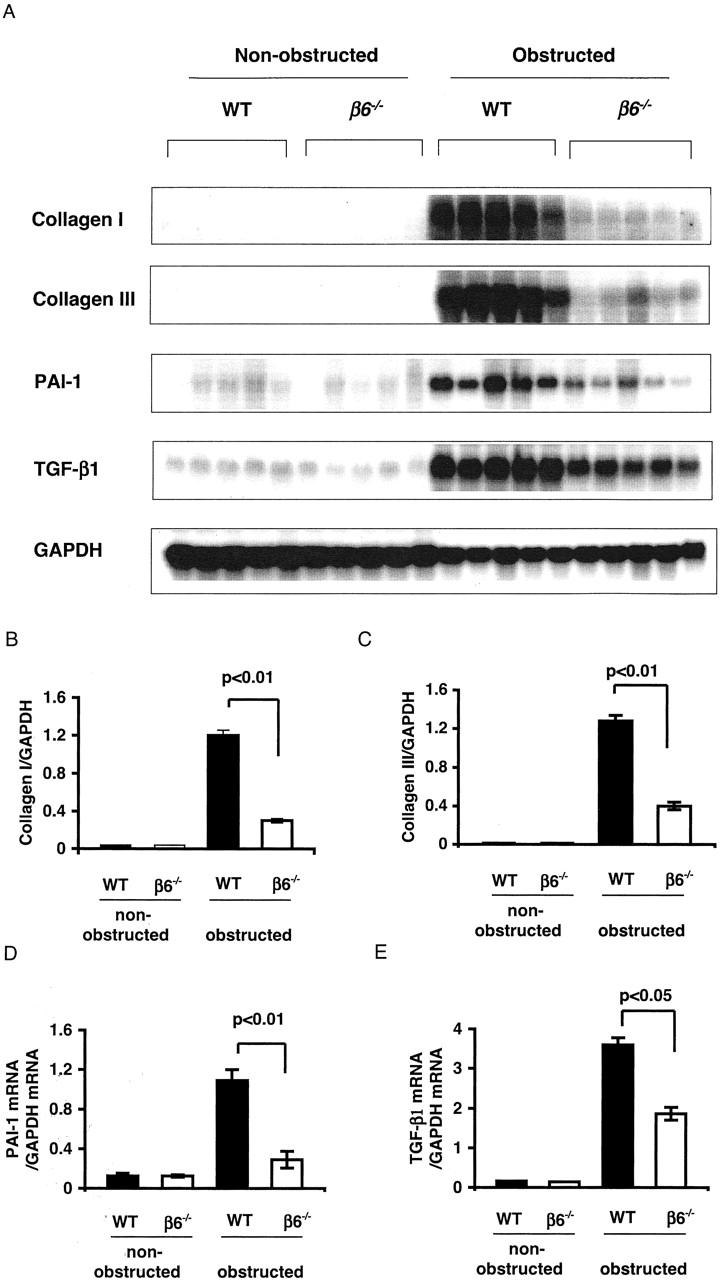
Northern blot analysis of kidney mRNA expression. Collagen I (A, B), collagen III (A, C), PAI-1 (A, D), and TGF-β1 (A, E) mRNA expressions in kidneys were significantly decreased in β6−/− UUO versus WT UUO at day 14 (n = 5 in each group). Data are shown as mean ± SEM.
Systolic Blood Pressure
Blood pressure remained normal in sham-operated control mice (104 ± 6 mmHg) and all other animals with UUO alone (Figure 5) ▶ . Blood pressure increased significantly and comparably in both UUO WT and UUO β6−/− mice infused with Ang II for 14 days (Figure 5) ▶ . Infusion with Aldo alone led to a slight, but not significant, increase in blood pressure compared to mice with UUO alone. Blood pressure in both WT and β6−/− mice with UUO was further augmented in response to a combination infusion of angiotensin and Aldo compared to either alone (Figure 5) ▶ .
Figure 5.

Systolic blood pressure (SBP). Ang II infusion alone or in combination with Aldo for 14 days increased systolic blood pressure significantly in both UUO WT and UUO β6−/− mice. Infusion with Aldo alone slightly increased systolic blood pressure (n = 3 to 8 in each group). Data are shown as mean ± SEM.
Induction of Fibrosis in β6−/− UUO Kidney by the RAAS
Collagen content of obstructed kidneys in WT UUO was significantly increased compared to β6−/− UUO at day 14 (percent collagen/total protein 8.8 ± 0.9 versus 2.7 ± 0.8, P < 0.01; Figure 6 ▶ ). In contrast, obstructed β6−/− showed no significant increase in collagen content compared to baseline (Figure 6) ▶ . In β6−/− UUO mice, infusion of either Ang II or Aldo or combination for 14 days significantly increased collagen contents to levels comparable to those in infused WT UUO (Figure 6) ▶ . Ang II or Aldo infusion alone in β6−/− UUO mice did not affect the macrophage infiltration. Combined Ang II and Aldo infusion in β6−/− UUO mice significantly decreased macrophage infiltration. In contrast, in WT UUO, Ang II infusion led to increased macrophage infiltration to a level similar to obstructed β6−/− kidney (macrophages/hpf: β6−/− UUO 41 ± 7 versus WT UUO 23 ± 9, pNS; β6−/− UUO + Ang II 32 ± 9, β6−/− UUO + Aldo 36 ± 8, β6−/− UUO + Ang II/Aldo 21 ± 6; β6−/− UUO versus β6−/− UUO + Ang II/Aldo, P < 0.05; WT UUO + Ang II 51 ± 14, WT UUO + Aldo 39 ± 6, WT UUO + Ang II/Aldo 34 ± 1, WT UUO versus WT UUO + Ang II, P < 0.05).
Figure 6.

Kidney collagen contents. Total collagen content (expressed as percent total protein) was increased in WT UUO at day 14 compared to the nonobstructed, contralateral kidney, in contrast to no significant change in β6−/− in response to UUO. Infusion of Ang II or Aldo or combination restored full fibrotic response in β6−/− UUO (n = 8 each for WT and β6−/− with UUO only; n = 3 to 8 each for WT and β6−/− with UUO plus infusion). Data are shown as mean ± SEM.
Thrombospondin (TSP)-1 Expression
TSP-1 protein, a potential activator of TGF-β, was similarly abundant in smooth muscle cells of arterioles and arteries in nonobstructed kidneys in both WT and β6−/− mice. TSP-1 was markedly increased in obstructed WT in cortical tubular epithelial cells, with a diffuse and strong granular pattern. In contrast, less TSP-1 was present in injured tubules in obstructed β6−/− kidneys. Infusion with Ang II in obstructed β6−/− mice led to a stronger and more diffuse granular pattern of TSP-1 expression, similar to that seen in Ang II-infused WT UUO (Figure 7) ▶ .
Figure 7.
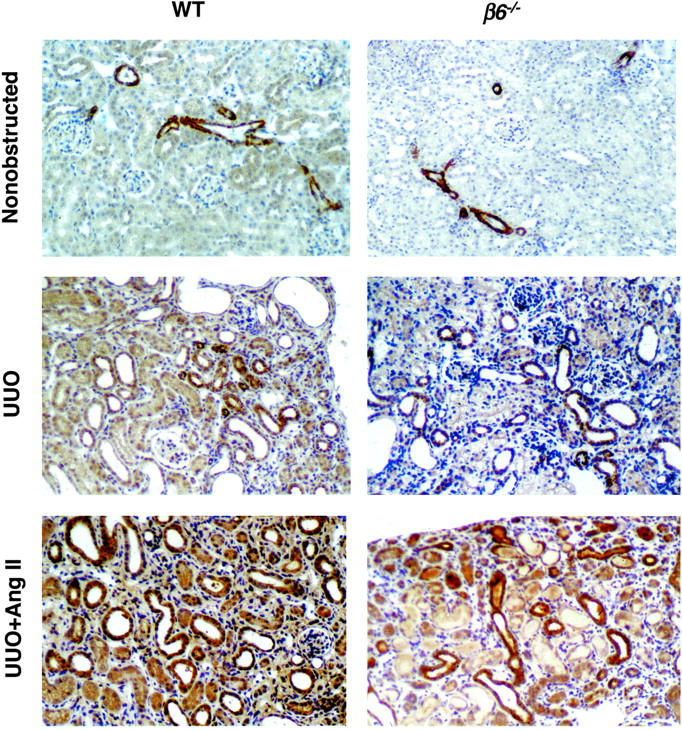
TSP-1 protein expression. TSP-1 protein was identified in nonobstructed kidneys in smooth muscle cells of arterioles and arteries in both WT and β6−/− mice. TSP-1 was markedly increased in obstructed WT kidneys in cortical tubular epithelial cells, with a diffuse and strong granular pattern at day 14. In contrast, less TSP-1 was present in injured tubules in obstructed β6−/−. Infusion with Ang II in obstructed β6−/− lead to a stronger, more diffuse granular pattern of TSP-1 expression, similar to that seen in Ang II-infused WT UUO (n = 3 to 6 for each group). Data are shown as mean ± SEM. Original magnifications, ×200.
Activity of TGF-β and Fibrosis
Active TGF-β immunostaining was present at low level in nonobstructed kidneys in both WT (Figure 8A) ▶ and β6−/− mice (Figure 8B) ▶ . Active TGF-β, mostly expressed in tubular epithelial cells, as well as interstitial cells and glomerular parietal cells, was markedly increased in WT UUO kidneys (Figure 8C) ▶ . In contrast, active TGF-β was only focally present in some tubules in β6−/− UUO kidneys (Figure 8D) ▶ , indicating dampened local activation of TGF-β from these parenchymal tubular epithelial and interstitial cells. Angiotensin infusion substantially increased active TGF-β expression in WT UUO kidney (Figure 8E) ▶ , but only mildly increased active TGF-β in β6−/− UUO mice (Figure 8F) ▶ .
Figure 8.
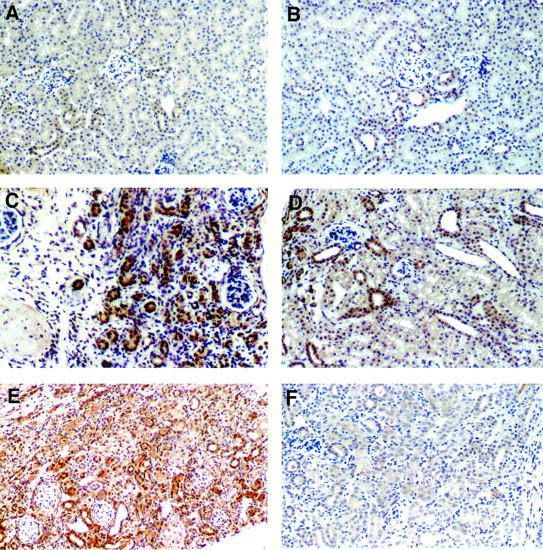
Active TGF-β expression. Active TGF-β was expressed in low levels in nonobstructed kidneys from both WT (A) and β6−/− mice (B). At day 14, active TGF-β, mostly expressed in tubular epithelial cells, as well as interstitial cells and glomerular parietal cells, was markedly increased in WT UUO kidney (C). In contrast, much less active TGF-β was detected in tubules in β6−/− UUO kidney (D). Ang II infusion resulted in fully activated TGF-β expression in WT UUO kidney (E), but only mild increase of active TGF-β in β6−/− UUO mice at day 14 (F). n = 3 to 6 for each group. Original magnifications, ×200.
Total Smad 2 was equally expressed at high levels in nonobstructed contralateral and obstructed kidneys at day 5 and day 14 after UUO in WT mice (Figure 9A) ▶ . Phosphorylated Smad 2, a marker of TGF-β signaling activation, was dramatically increased from day 5 to 14 after obstruction in WT (Figure 9A) ▶ . In contrast, phosphorylated Smad 2 was significantly lower in β6−/− obstructed kidneys and did not increase when fibrosis was induced by infusing Ang II in obstructed β6−/− kidneys (Figure 9, B and C) ▶ . Abundantly expressed phosphorylated-Smad 2 in PET cell lysate stimulated with TGF-β1 served as a positive control (Figure 9B) ▶ .
Figure 9.
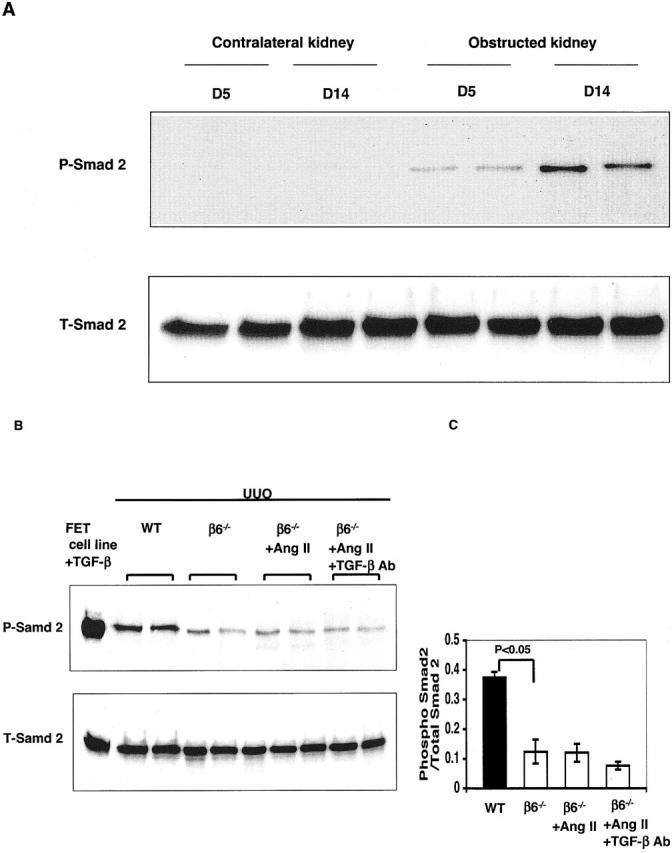
Representative Western blot analysis of Smad 2 in UUO kidneys. A: Phosphorylated Smad 2 (P-Smad 2) and total Smad 2 (T-Smad 2) in nonobstructed contralateral and obstructed kidneys at day 5 and day 14 after UUO in WT mice. P-Smad 2 in obstructed WT kidneys was dramatically increased from day 5 to day 14, contrasting with lower P-Smad 2 in obstructed β6−/− kidneys (B, C). Ang II infusion in β6−/− UUO mice did not increase P-Smad 2 levels (B, C). Anti-TGF-β antibody treatment in β6−/− UUO mice infused with Ang II only numerically decreased P-Smad 2 (B, C). PET cell lysate stimulated with TGF-β1 served as a positive control (B) (n = 5 for each animal group). Data are shown as mean ± SEM.
To further examine mechanisms of kidney fibrosis, we treated mice with anti-TGF-β antibody. The effects of anti-TGF-β antibody on fibrosis were first examined in WT mice with UUO alone. Collagen content in obstructed WT kidneys was significantly decreased by low doses of anti-TGF-β antibody and further decreased with high dose of anti-TGF-β antibody compared to control antibody (Figure 10) ▶ . However, anti-TGF-β antibody alone, even with high dose, only partially inhibited (less than 50%) increased collagen in WT UUO kidneys (Figure 10) ▶ , with or without Ang II infusion (Figure 11) ▶ , suggesting other mechanisms may play a role in fibrosis. Similarly, both low and high doses of anti-TGF-β antibody, but not control antibody, significantly but only partially decreased the Ang II-stimulated collagen increase in β6−/− UUO kidneys (Figure 11) ▶ . Anti-TGF-β antibody treatment in β6−/− UUO mice infused with Ang II only numerically decreased phosphorylated Smad 2 (Figure 9, B and C) ▶ .
Figure 10.

. Effect of TGF-β antibody on kidney collagen content in WT UUO mice. Control antibody had no effect on total collagen content (expressed as percent total protein) increased in obstructed (UUO) WT kidney versus nonobstructed contralateral kidney (C). Total collagen content was partially but significantly inhibited after either low (L) or high (H) doses of TGF-β antibody (Ab) treatment (n = 4 to 5 for each group). Data are shown as mean ± SEM.
Figure 11.

Effect of TGF-β antibody on kidney collagen content in Ang II-infused UUO mice. Total collagen content (expressed as percent total protein) in β6−/− UUO kidney was increased after Ang II infusion, and was only partially inhibited after either high (H) or low (L) doses of TGF-β antibody (Ab) treatment. High dose of TGF-β antibody had similar effects on kidney collagen content in Ang II-infused WT UUO kidney (n = 3 to 8 for each group). *, P < 0.01. Data are shown as mean ± SEM.
PAI-1 Localization and Expression
WT UUO showed moderate PAI-1 mRNA expression in tubules in cortex and medulla and in transitional epithelial cells by in situ hybridization, compared to low-level PAI-1 mRNA expression in obstructed β6−/− kidneys. PAI-1 mRNA expression was increased in obstructed β6−/− kidney after infusion with Ang II, Aldo, or combination, with similar intensities and locations as in identically treated WT mice (Figure 12) ▶ . There was no change in PAI-1 mRNA expression, as detected by Northern blot and in situ hybridization, in response to TGF-β antibody treatments compared to treatment with control antibody (data not shown).
Figure 12.
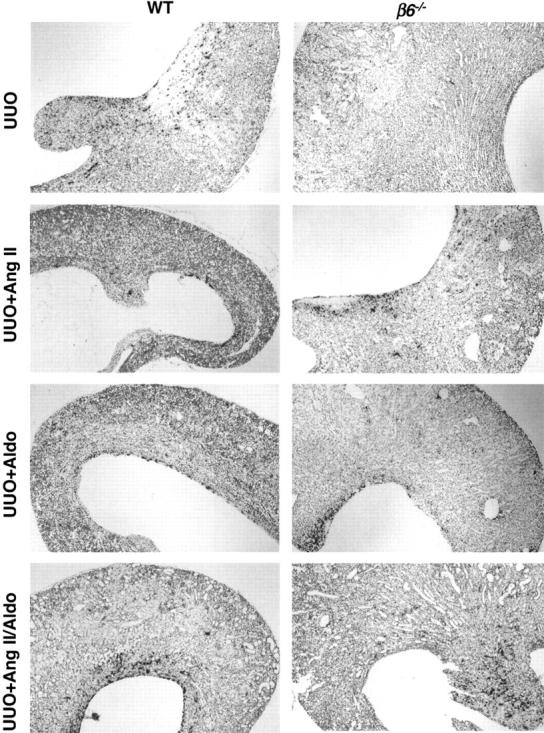
PAI-1 mRNA expression by in situ hybridization. PAI-1 mRNA expression was marked in WT UUO, contrasting very little expression in β6−/− UUO. After Ang II or Aldo or combination infusion, PAI-1 mRNA expression was similar in β6−/− UUO and WT UUO, correlating with similar degree of collagen accumulation (n = 3 to 6 for each group). Original magnifications, ×40.
PAI-1 protein was not present in nonobstructed WT kidneys (Figure 13A) ▶ . However, PAI-1 was highly expressed in WT UUO kidney at day 14, primarily in cortical proximal tubular epithelial cells (Figure 13B) ▶ and glomerular parietal epithelial cells (data not shown). Infusion of Ang II or Aldo (Figure 13, C and D) ▶ in obstructed WT did not alter PAI-1 staining intensity or distribution compared to WT UUO alone (Figure 13B) ▶ . In contrast, β6−/− UUO mice at day 14 showed much less PAI-1 in proximal tubules and none in glomeruli (Figure 13E) ▶ , with focal strong PAI-1 staining in inner medullary tubules (not shown). However, infusion of Ang II in β6−/− UUO mice markedly increased PAI-1 expression to a pattern similar to that seen in WT, with increased proximal tubule PAI-1 expression (Figure 13F) ▶ .
Figure 13.
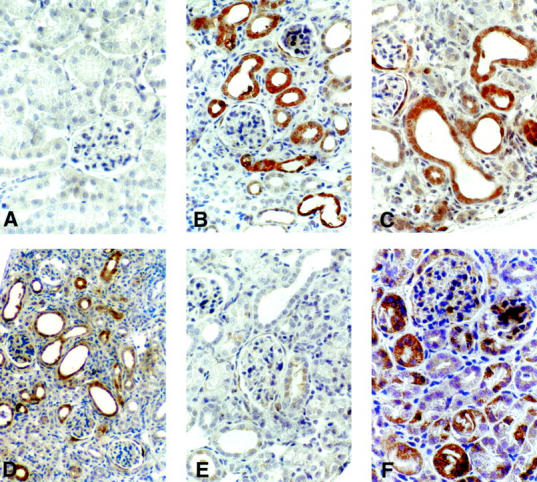
PAI-1 expression in UUO kidney. PAI-1 immunoreactivity in WT nonobstructed kidneys (A); WT UUO kidneys (B); WT UUO kidneys after infusion with Ang II (C) or Aldo (D); and in β6−/− UUO kidney without (E) or with (F) Ang II infusion (n = 3 to 6 for each group). Original magnifications: ×400 (A–C, E, F); ×200 (D).
Discussion
TGF-β and the RAAS have been intricately linked to development of fibrosis, and they are interrelated. Both activate fibrosis in part by inducing matrix synthesis, and in part by inhibiting matrix degradation. A powerful mediator of the latter mechanism is induction of PAI-1. 23 Although angiotensin can directly induce PAI-1 through the type 1 receptor (AT1) in vivo and in vitro, some in vitro evidence points to a later, TGF-β-dependent phase of this induction of PAI-1 by angiotensin. 13 We therefore used β6−/− mice to determine fibrosis mechanisms that are dependent on local TGF-β activation and those that are independent of these TGF-β-mediated events. We chose the β6−/− mouse because it has a normal phenotype, unlike TGF-β1 knockout mice, which succumb to disease related to marked immune cell infiltration. Because αvβ6 integrin is only one mechanism of activation of TGF-β1, other mechanisms of TGF-β1 activation remain potentially active, and thus, the β6−/− mice do not have baseline renal abnormalities. We further chose to use the β6−/− mice particularly for the study of development of tubulointerstitial fibrosis. β6 integrin is expressed in epithelia of skin, lung, and tubular epithelium of kidney. 4 No expression has been detected in the glomerulus. Thus, the local, renal epithelial cell activation of TGF-β1 via β6 and its importance for fibrosis can be determined by inducing tubulointerstitial injury in these mice.
This study indeed demonstrated that αvβ6-mediated TGF-β activation is pivotal for development of full injury after UUO. Tubulointerstitial fibrosis in response to UUO was markedly less in β6−/− mice compared to WT mice, indicating that interruption of β6 mechanisms can protect against tubulointerstitial fibrosis.
Activation of TGF-β can occur through several mechanisms, including plasmin cleavage. 24 However, additional mechanisms must exist in vivo for activation of TGF-β, because knockout of plasminogen, the substrate yielding plasmin, results in mice that do not share the phenotype of TGF-β1 knockout mice. Other mechanisms for activating TGF-β in vitro including reactive oxygen species and TSP-1. 25 Indeed TSP-1 knockout mice have a similar pattern of inflammation as TGF-β1 knockout mice, supporting the concept that thrombospondin plays a major role in activation of TGF-β in vivo. 26 In the present study, there was increased TSP-1 in tubular epithelial cells in WT UUO kidneys. However, in response to angiotensin infusion in obstructed β6−/− mice, there was increased TSP-1 but less activated TGF-β by immunostaining, suggesting that TSP-1-dependent TGF-β activation does not explain the induction of fibrosis by angiotensin in obstructed β6−/− kidneys.
We then examined potential downstream mechanisms for the protective effect of β6 deletion. We examined local, active TGF-β1 expression in WT and β6−/− mice UUO kidney using a specific antibody for detection of active TGF-β 19 and found markedly increased active TGF-β in tubular epithelial cells and some interstitial cells in WT UUO kidneys, contrasting with very little active TGF-β expression in β6−/− UUO. This observation supports that knockout of β6 integrin dampens the local activation of TGF-β in these parenchymal cells in the tubulointerstitium. We also investigated TGF-β activation by assessing its downstream signaling pathways, the Smad proteins. 27,28 Smad-2 in its phosphorylated state has been used as a specific marker of TGF-β activation. Further evidence of the key role of Smad 2 in TGF-β signaling is seen by inhibition of TGF-β-induced fibrosis when it is inactivated by Smad 7. 29 In the obstructed, fibrotic kidneys in the WT mice, we observed, as expected, concomitant enhancement of active TGF-β1 as indirectly assessed by immunostaining and activation of Smad 2. Low expression of phosphorylated Smad 2 in the obstructed, but nonfibrotic kidneys in β6−/− mice indicates that prevention of TGF-β1 activation is a key protective mechanism against fibrosis in β6−/− mice, as previously also observed in the bleomycin pulmonary fibrosis model.
We next determined whether direct RAAS stimuli could induce fibrosis in β6−/− UUO mice. A contribution of angiotensin in interstitial fibrosis in obstructive nephropathy was recently documented in angiotensin type 1 receptor (AT1a) knockout mice. 30 In our model, blood pressure effects may have augmented and contributed to the fibrotic effects of RAAS stimulation. However, fibrosis was not induced by Ang II or Aldo infusion in nonobstructed kidneys, indicating that systemic hemodynamic effects alone were not sufficient to cause injury. In this regard, studies of mice with 0 to 4 copies of the angiotensinogen gene demonstrated that angiotensin regulates at least 50% of the renal interstitial fibrotic response in obstructive nephropathy through nonhemodynamic effects. 31 Indeed, we observed that infusion with either Ang II, Aldo, or their combination for 14 days restored fibrosis in β6−/− UUO mice. The lack of complete prevention of Ang II- or Aldo-induced fibrosis in these mice by treatment with neutralizing antibody to TGF-β supports the existence of a component of fibrosis that is TGF-β-independent. This fibrosis in β6−/− UUO mice with angiotensin was not linked to increased TGF-β activation as assessed by phospho-Smad 2, further supporting that angiotensin can induce TGF-β-independent mechanisms of tubulointerstitial fibrosis in the obstructed β6−/− mice.
Angiotensin is a strong inducer of PAI-1. Increased local PAI-1 expression is directly linked to sites of sclerosis and tubulointerstitial fibrosis. 10,21,23,32 In addition, PAI-1 knockout mice are protected against fibrosis induced by ureteral obstruction. 33 These results suggest that both decreased extracellular matrix synthesis, because of reduced TGF-β activation, and increased matrix degradation, mediated by decreased PAI-1, contribute to the lesser collagen accumulation in β6−/− mice. Indeed, the protected β6−/− UUO mice had very low levels of PAI-1 compared to WT with UUO. Further, the fibrotic response in β6−/− UUO mice treated with RAAS stimuli correlated with increased PAI-1 expression, suggesting that this pathway could be in part TGF-β-independent. Importantly, despite treatment with antibody to TGF-β, both fibrosis and PAI-1 expression were primarily maintained in RAAS-stimulated β6−/− UUO mice. We postulate that modulation of matrix degradation via PAI-1 is an important component of the fibrotic response. PAI-1 increases were similar in intensity and location in both RAAS-stimulated obstructed WT and β6−/− mice. These findings support our hypothesis that the development of fibrosis is tightly linked to local PAI-1 expression that can, in the absence of αvβ6, be TGF-β-independent.
Macrophages have diverse role in tissue injury, mediating repair and remodeling after injury. Persistence of activated macrophages is tightly correlated with progressive fibrosis in the kidney. 34 However, the macrophage may not mediate fibrosis per se. Our data show that despite robust macrophage infiltration after UUO, interstitial fibrosis was blunted in the absence of parenchymal cell responsiveness in β6−/− mice, in parallel to previous observations in the lung bleomycin model. 6
Macrophages bear AT1 receptors, and they respond to angiotensin with chemotaxis and activation. 35 However, we have shown that AT1 receptor-mediated actions of macrophages may have a protective role in some settings of fibrosis. Adoptive transfer of AT1a-null bone marrow resulted in more injury after UUO although the number of macrophages was not altered. 36 In the current study, we also observed lack of correlation of macrophages with fibrosis in β6−/− UUO mice given RAAS stimulation. This lack of correlation of macrophages and fibrosis was also previously observed in UUO mice treated with AT1 receptor antagonist. 37 In the current study, macrophage infiltration was reduced although fibrosis was fully restored by angiotensin infusion, pointing to nonmacrophage-dependent fibrosis. We speculate that parenchymal cell PAI-1 expression could be one such mechanism.
In summary, we have shown that integrin αvβ6 is a critical determinant of kidney fibrosis. Absence of β6 confers remarkable protection against tubulointerstitial fibrosis despite robust macrophage infiltration. Both angiotensin and Aldo induce fibrosis in β6−/− mice with UUO without increased TGF-β activation. This fibrosis was linked to increased PAI-1. These data suggest a TGF-β-independent component of induction of PAI-1. Our findings raise the possibility that targeting integrin αvβ6 and/or PAI-1 may provide novel clinical strategies in treatment of kidney fibrosis.
Acknowledgments
We thank Dr. Steve Ledbetter at Genzyme and Dr. Kathy Flanders at the National Cancer Institute for their help in providing TGF-β antibodies.
Footnotes
Address reprint requests to Agnes B. Fogo, M.D., MCN C3310, Department of Pathology, Vanderbilt University Medical Center, 21st and Garland Ave., Nashville, TN 37232-2561. E-mail: agnes.fogo@vanderbilt.edu.
Supported in part by the National Institutes of Health (grants DK44757 and DK56942 to A. F. and L. J. M., and AG-06528 to J. D.), the Department of Veterans Affairs (to J. D.), and the National Heart, Lung and Blood Institute (grant HL53949 to D. S.).
References
- 1.Sharma K, Ziyadeh FN: The emerging role of transforming growth factor-beta in kidney diseases. Am J Physiol 1994, 266:F829-F842 [DOI] [PubMed] [Google Scholar]
- 2.Munger JS, Harpel JG, Giancotti FG, Rifkin DB: Interactions between growth factors and integrins: latent forms of transforming growth factor-beta are ligands for the integrin alphavbeta1. Mol Biol Cell 1998, 9:2627-2638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Breuss JM, Gallo J, DeLisser HM, Klimanskaya IV, Folkesson HG, Pittet JF, Nishimura SL, Aldape K, Landers DV, Carpenter W, Gillett N, Sheppard D, Matthay MA, Albelda SM, Kramer RH, Pytela R: Expression of the beta 6 integrin subunit in development, neoplasia and tissue repair suggests a role in epithelial remodeling. J Cell Sci 1995, 108:2241-2251 [DOI] [PubMed] [Google Scholar]
- 4.Arend LJ, Smart AM, Briggs JP: Mouse beta(6) integrin sequence, pattern of expression, and role in kidney development. J Am Soc Nephrol 2000, 11:2297-2305 [DOI] [PubMed] [Google Scholar]
- 5.Huang X-Z, Wu JF, Cass D, Erle DJ, Corry D, Young SG, Farese RVJ, Sheppard D: Inactivation of the integrin b6 subunit gene reveals a role of epithelial integrins in regulating inflammation in the lungs and skin. J Cell Biol 1996, 133:921-928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Munger JS, Huang X, Kawakatsu H, Griffiths MJ, Dalton SL, Wu J, Pittet JF, Kaminski N, Garat C, Matthay MA, Rifkin DB, Sheppard D: The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell 1999, 96:319-328 [DOI] [PubMed] [Google Scholar]
- 7.Fogo AB: The role of angiotensin II and plasminogen activator inhibitor-1 in progressive glomerulosclerosis. Am J Kidney Dis 2000, 35:179-188 [DOI] [PubMed] [Google Scholar]
- 8.Ma L-J, Fogo AB: Role of angiotensin II in glomerular injury. Semin Nephrol 2001, 21:544-553 [DOI] [PubMed] [Google Scholar]
- 9.Vaughan DE, Lazos SA, Tong K: Angiotensin II regulates expression of plasminogen activator inhibitor-1 in cultured endothelial cells. J Clin Invest 1995, 95:995-1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oikawa T, Freeman M, Lo W, Vaughan DE, Fogo A: Modulation of plasminogen activator inhibitor-1 in vivo: a new mechanism for the anti-fibrotic effect of renin-angiotensin inhibition. Kidney Int 1997, 51:164-172 [DOI] [PubMed] [Google Scholar]
- 11.Nakamura S, Nakamura I, Ma L-J, Vaughan DE, Fogo AB: Plasminogen activator inhibitor-1 expression is regulated by the angiotensin type 1 receptor in vivo. Kidney Int 2000, 58:251-259 [DOI] [PubMed] [Google Scholar]
- 12.Brown NJ, Kim KS, Chen YQ, Blevins LS, Nadeau JH, Meranze SG, Vaughan DE: Synergistic effect of adrenal steroids and angiotensin II on plasminogen activator inhibitor-1 production. J Clin Endocrinol Metab 2000, 85:336-344 [DOI] [PubMed] [Google Scholar]
- 13.Kagami S, Kuhara T, Okada K, Kuroda Y, Border WA, Noble NA: Dual effects of angiotensin II on the plasminogen/plasmin system in rat mesangial cells. Kidney Int 1997, 51:664-671 [DOI] [PubMed] [Google Scholar]
- 14.Ketteler M, Noble NA, Border WA: Transforming growth factor-beta and angiotensin II: the missing link from glomerular hyperfiltration to glomerulosclerosis? Annu Rev Physiol 1995, 57:279-295 [DOI] [PubMed] [Google Scholar]
- 15.Ma J, Nishimura H, Fogo AB, Kon V, Inagami T, Ichikawa I: Accelerated fibrosis and collagen deposition develop in the renal interstitium of angiotensin type 2 receptor null mutant mice during ureteral obstruction. Kidney Int 1998, 53:937-944 [DOI] [PubMed] [Google Scholar]
- 16.Robert V, Silvestre JS, Charlemagne D, Sabri A, Trouve P, Wassef M, Swynghedauw B, Delcayre C: Biological determinants of aldosterone-induced cardiac fibrosis in rats. Hypertension 1995, 26:971-978 [DOI] [PubMed] [Google Scholar]
- 17.Johnson RJ, Alpers CE, Yoshimura A, Lombardi D, Pritzl P, Floege J, Schwartz SM: Renal injury from angiotensin II mediated hypertension. Hypertension 1992, 19:464-474 [DOI] [PubMed] [Google Scholar]
- 18.Miyajima A, Chen J, Lawrence C, Ledbetter S, Soslow RA, Stern J, Jha S, Pigato J, Lemer ML, Poppas DP, Vaughan ED, Felsen D: Antibody to transforming growth factor-beta ameliorates tubular apoptosis in unilateral ureteral obstruction. Kidney Int 2000, 58:2301-2313 [DOI] [PubMed] [Google Scholar]
- 19.Barcellos-Hoff MH, Ehrhart EJ, Kalia M, Jirtle R, Flanders K, Tsang ML: Immunohistochemical detection of active transforming growth factor-b in situ using engineering tissues. Am J Pathol 1995, 147:1228-1237 [PMC free article] [PubMed] [Google Scholar]
- 20.Davidson JM, Aquino AM, Woodward SC, Wilfinger WW: Sustained microgravity reduces intrinsic wound healing and growth factor responses in the rat. EMBO J 1998, 13:325-329 [DOI] [PubMed] [Google Scholar]
- 21.Ma L-J, Nakamura S, Whitsitt JS, Marcantoni C, Davidson JM, Fogo AB: Regression of sclerosis in aging by an angiotensin inhibition-induced decrease in PAI-1. Kidney Int 2000, 58:2425-2436 [DOI] [PubMed] [Google Scholar]
- 22.Ma LJ, Marcantoni C, Linton MF, Fazio S, Fogo AB: Peroxisome proliferator-activated receptor-gamma agonist troglitazone protects against nondiabetic glomerulosclerosis in rats. Kidney Int 2001, 59:1899-1910 [DOI] [PubMed] [Google Scholar]
- 23.Fogo AB: Progression and potential regression of glomerulosclerosis (Nephrology Forum). Kidney Int 2001, 59:804-819 [DOI] [PubMed] [Google Scholar]
- 24.Lyons RM, Gentry LE, Purchio AF, Moses HL: Mechanism of activation of latent recombinant transforming growth factor-beta 1 by plasmin. J Cell Biol 1990, 110:1361-1367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schultz-Cherry S, Chen H, Mosher DF, Misenheimer TM, Krutzsch HC, Roberts DD, Murphy-Ullrich JE: Regulation of transforming growth factor-beta activation by discrete sequences of thrombospondin 1. J Biol Chem 1995, 270:7304-7310 [DOI] [PubMed] [Google Scholar]
- 26.Crawford SE, Stellmach V, Murphy-Ullrich JE, Ribeiro SM, Lawler J, Hynes RO, Boivin GP, Bouck N: Thrombospondin-1 is a major activator of TGFbeta1 in vivo. Cell 1998, 93:1159-1170 [DOI] [PubMed] [Google Scholar]
- 27.Heldin C, Miyazono K, ten Dijke P: TGF-β signaling from cell membrane to nucleus through SMAD proteins. Nature 1997, 390:465-471 [DOI] [PubMed] [Google Scholar]
- 28.Derynck R, Feng X: Smads: transcriptional activators of TGF-beta responses. Cell 1998, 95:737-740 [DOI] [PubMed] [Google Scholar]
- 29.Li JH, Zhu HJ, Huang XR, Lai KN, Johnson RJ, Lan HY: Smad7 inhibits fibrotic effect of TGF-beta on renal tubular epithelial cells by blocking Smad2 activation. J Am Soc Nephrol 2002, 13:1464-1472 [DOI] [PubMed] [Google Scholar]
- 30.Guo G, Morrissey J, McCracken R, Tolley T, Liapis H, Klahr S: Contributions of angiotensin II and tumor necrosis factor-alpha to the development of renal fibrosis. Am J Physiol 2001, 280:F777-F785 [DOI] [PubMed] [Google Scholar]
- 31.Fern R, Yesko CM, Thornhill BA, Kim HS, Smithies O, RL C: Reduced angiotensinogen expression attenuates renal interstitial fibrosis in obstructive nephropathy in mice. J Clin Invest 1999, 103:39-46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brown NJ, Nakamura S, Ma L-J, Nakamura I, Donnert E, Freeman M, Vaughan DE, Fogo AB: Aldosterone modulates plasminogen activator inhibitor-1 and glomerulosclerosis in vivo. Kidney Int 2000, 58:1219-1227 [DOI] [PubMed] [Google Scholar]
- 33.Oda T, Jung YO, Kim HS, Cai X, Lopez-Guisa JM, Ikeda Y, Eddy AA: PAI-1 deficiency attenuates the fibrogenic response to ureteral obstruction. Kidney Int 2001, 60:587-596 [DOI] [PubMed] [Google Scholar]
- 34.Diamond JR: Macrophages and progressive renal disease in experimental hydronephrosis. Am J Kidney Dis 1995, 26:133-140 [DOI] [PubMed] [Google Scholar]
- 35.Okamura A, Rakugi H, Ohishi M, Yanagitani Y, Takiuchi S, Moriguchi K, Fennessy PA, Higaki J, Ogihawa T: Upregulation of renin-angiotensin system during differentiation of monocyte to macrophages. J Hypertens 1999, 17:537-545 [DOI] [PubMed] [Google Scholar]
- 36.Fujinaka H, Yoshida H, Matsusaka T, Miyazaki T, Nishida M, Linton M, Fogo AB, Fazio S, Ichikawa I: Protective role for the angiotensin II type 1A receptor on bone marrow-derived macrophages in the formation of renal fibrosis. J Am Soc Nephrol 2000, 11:618a [Google Scholar]
- 37.Ishidoya S, Morrissey J, McCracken R, Reyes A, Klahr S: Angiotensin II receptor antagonist ameliorates renal tubulointerstitial fibrosis caused by unilateral ureteral obstruction. Kidney Int 1995, 47:1285-1294 [DOI] [PubMed] [Google Scholar]