

Abstract
The recognition of biologically distinct tumor subsets is fundamental to understanding tumorigenesis. This study investigated the mutational status of the serine/threonine kinase BRAF and the cyclin E regulator FBXW7 (CDC4, FBW7, AGO, SEL10) related to two distinct pancreatic carcinoma subsets: the medullary KRAS2-wild-type and the cyclin E overexpressing tumors, respectively. Among KRAS2-wild-type carcinomas, 33% (3 of 9) contained BRAF V599E mutations; one of which was identified in the pancreatic cancer cell line COLO357. Among 74 KRAS2-mutant carcinomas, no BRAF mutations were identified. Among the KRAS2/BRAF wild-type carcinomas, no mutations within pathway members MEK1, MEK2, ERK1, ERK2, RAP1B, or BAD were found. Using pancreatic cancer microarrays and immunohistochemistry, we determined that 6% (4 of 46 and 5 of 100 in two independent panels) of pancreatic adenocarcinomas overexpress cyclin E. We identified two potential mechanisms for this overexpression including the amplification/gain of CCNE1 gene copies in the Panc-1 and Su86.86 cell lines and a novel somatic homozygous mutation (H460R, in one of 11 pancreatic cancer xenografts having allelic loss) in FBXW7, which was accompanied by cyclin E overexpression by immunohistochemistry. Both BRAF and FBXW7 mutations functionally activate kinase effectors important in pancreatic cancer and extend the potential options for therapeutic targeting of kinases in the treatment of phenotypically distinct pancreatic adenocarcinoma subsets.
Some mutations in carcinomas are highly patterned. As one example, the simultaneous accumulation of mutations within different members of a particular linear signaling pathway is seldom seen within the same neoplasm, perhaps because multiple mutations are unlikely to yield further selective advantages. Studies of the mutational status of RB1, cyclin-dependent kinase 4 (CDK4) and the Cdk4 inhibitor p16 (CDKN2A) support this theory. These three genes lie within a well-described cell cycle control pathway and exhibit mutually exclusive mutations, that is, tumors with mutant forms of one of these genes invariably retain wild-type copies of the others. 1-3 Similar findings have been reported for the genes TP53 and MDM2 4 in the Tp53 suppressive pathway, platelet-derived growth factor receptor α (PDGFRA), and the tyrosine kinase receptor KIT 5 in the platelet-derived growth factor pathway, β-catenin (CTNNB1) and its regulator APC 6 in the Wnt signaling pathway, and most recently for KRAS2 and BRAF 7,8 in what is presumably a major regulatory system for mitogen-activated protein kinases. As another example, subsets of neoplasms with unique phenotypic characteristics often harbor specific mutational patterns. A medullary histology in pancreatic cancer is often associated with DNA mismatch repair abnormalities as well as with wild-type KRAS2 status. 9,10 In ovarian cancer, phenotypic subsets defined by cyclin E overexpression result from cyclin E amplification or from mutations of the F box and tryptophan aspartic acid repeat unit (WD) domain-containing gene, FBXW7. 11 The Fbxw7 protein is the ubiquitin ligase that targets cyclin E for degradation 12 after cyclin E catalyzes the transition from the G1 to S phase of the cell cycle.
This study investigated the relationship of two potential mutational targets, BRAF and FBXW7, which may associate with distinct subsets of pancreas carcinomas. Both of these genes may be particularly attractive for therapeutic targeting using small molecule inhibitors. To date, the role of kinase oncogene pathways in pancreas cancer is relatively underexplored, with studies mainly focusing on AKT2 and epidermal growth factor receptor signaling. 13,14 Examination of other kinase signaling pathways may help elucidate novel mechanisms of tumorigenesis in pancreatic cancer and attractive therapeutic targets. Here, we report the mutational status of BRAF and FBXW7, as well as the genomic amplification of cyclin E (CCNE1), in relation to two distinct subsets of pancreas carcinomas having unique histological and immunohistologic phenotypes.
Materials and Methods
Tissues
Primary adenocarcinomas of the pancreas and non-neoplastic tissues were collected from surgical specimens obtained from patients treated at The Johns Hopkins Medical Institute. Surgically resected cancers were either implanted and propagated in mice as described previously 15 or formalin-fixed and paraffin-embedded for use in the construction of a tissue microarray (array 1). A second cancer tissue array was also created from samples resected at Wayne State University, Detroit, Michigan (V. A., array 2). Sample collection and tissue studies were approved by the Institutional Review Boards.
Cell Lines
The pancreatic cancer cell lines Su86.86, AsPC-1, and Panc-1 were purchased from the American Type Culture Collection (Manassas, VA). The COLO357 pancreatic cell line was obtained from the European Collection of Animal Cell Cultures (Salisbury, United Kingdom). The KRAS2 mutational status was confirmed as wild-type at codons 12, 13, and 61 in COLO357 cell lines by direct sequencing. 10 Genomic DNA was isolated from cell lines, as well as from harvested xenografts and non-neoplastic tissues for the sequencing studies.
Gene Sequencing
PCR primers for BRAF, MEK1, MEK2, ERK1, ERK2, RAP1B, BAD, and FBXW7 were designed using Primer3 (http://www-genome.wi.mit.edu/cgi-bin/primer/primer3_www.cgi) from reference sequences at the National Center for Biotechnology Information website (http://www.ncbi.nlm.nih.gov). The exons of FBXW7 were amplified by PCR from 11 samples known to have loss of heterozygosity (Iacobuzio-Donahue et al, unpublished data) near 4q31.3 while the exons of BRAF, MEK1, MEK2, ERK1, ERK2, RAP1B, and BAD were amplified from nine samples having only wild-type RAS genes. Automated sequencing was performed on amplified fragments and all sequence variants identified were confirmed by the sequencing of independent PCR products. Primer sequences used in this study are available on request.
Fluorescence in Situ Hybridization (FISH)
FISH was performed as described previously. 16 AKT2 (19q13.2) amplification was evaluated using the BAC clone 127D1. CCNE1 (19q12) amplification was evaluated using either the BAC clone 246K7 or phage clones 25 or 26 (described elsewhere 17 ). Signals were evaluated with respect to the control gene, TCF3 (19p13.3, P1 clone 8542). Lymphocytes from a normal donor served as a control.
Southern Blot Analysis
Cyclin E genomic amplification was evaluated by standard Southern blot technologies. The cyclin E hybridization probe was created from a NotI digested fragment of the IMAGE clone, 357807. The blots were then stripped and probed with Clone WI-12306 (19q) as a loading control.
Immunohistochemistry
Immunolabeling was performed as previously described 18 using an anti-cyclin E primary antibody (clone CYE05; Lab Vision, Fremont, CA) and the DAKO (Carpinteria, CA) EnVision+ peroxidase-linked secondary antibody. Optimal cyclin E antibody dilutions were predetermined using known positive control tissues included in each run. Immunopositivity was evaluated by two observers (S. E. K. and R. H. H.).
Results
KRAS2 Signaling Pathway
Exons 11 and 15 of BRAF were sequenced from a unique collection of rare pancreatic adenocarcinomas (n = 9) retaining only wild-type copies of the KRAS2, NRAS, and HRAS genes. Two xenografted pancreatic tumors and the COLO357 cell line were each found to harbor the BRAF codon V599E mutation (Figure 1 ▶ ; Table 1 ▶ ) previously shown to stimulate the kinase activity of Braf. 19 Sequencing of BRAF in the two available constitutional DNA samples from these patients, as well as in an additional 74 typical KRAS2-mutant xenografted pancreatic carcinomas, revealed no genetic alterations within exons 11 and 15 (Figure 1 ▶ ; data not shown). The coding sequences of MEK1, MEK2, ERK1, ERK2, RAP1B, and BAD were sequenced in the KRAS/BRAF wild-type pancreatic cancer cases. These genes are proposed to play a role in the effector arms of Ras and Raf signaling, and might be additional targets of oncogenic disruption in tumors. We, however, failed to identify any mutations in these genes.
Figure 1.
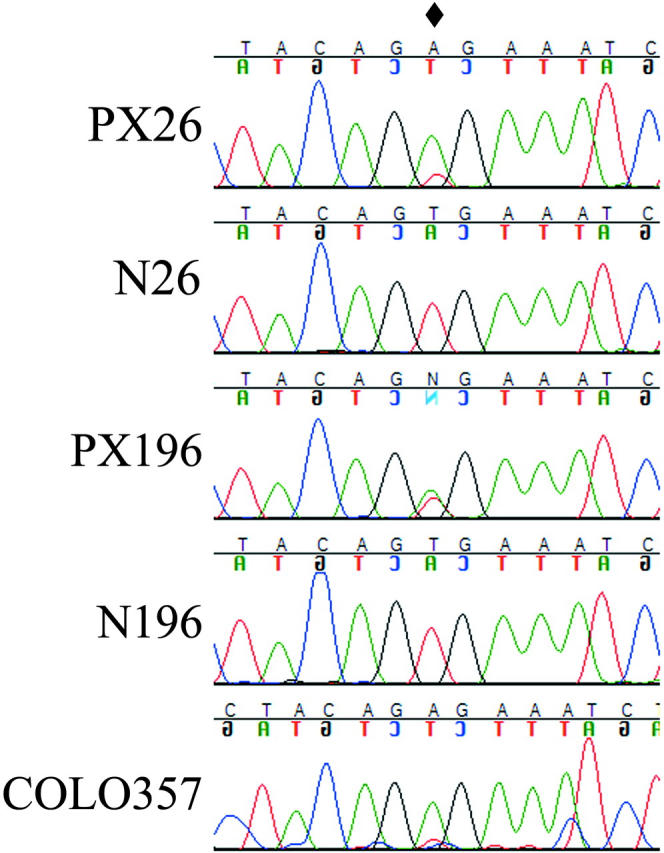
BRAF mutations in pancreatic cancer. Tumors (PX) and the COLO357 cell line display the V599E mutation (diamond). Constitutional DNA samples (N) verify mutation is somatic.
Table 1.
BRAF and FBXW7 Mutations in Subsets of Pancreatic Cancer
| Subsets and samples | Prevalence | Result | Note |
|---|---|---|---|
| Cyclin E overexpression subset | |||
| Tissue array 1 | 4/46 (9%) | Overexpression by IHC | Array created at Johns Hopkins |
| Tissue array 2 | 5/100 (5%) | Overexpression by IHC | Array created at Harper Hospital, Wayne State University |
| PX221 | FBWX7 mutation (H460R, CAT to CGT, homozygous) | Overexpression of cyclin E subsequently determined by IHC | |
| KRAS2 wild-type Subset | |||
| Tumor panel | 7/77 (9%)* | Mutations of K-, N-, and H-ras excluded by sequencing | |
| PX26 | BRAF mutation (V599E, GTG to GAG, homozygous) | Known MSI, medullary histology | |
| PX196 | BRAF mutation (V599E, GTG to GAG, heterozygous) | Known MSI, medullary histology | |
| COLO357 | BRAF mutation (V599E, GTG to GAG, homozygous) | Not MSI, commercial cell line |
*From consecutive xenografted pancreatic adenocarcinomas, previously reported. 10
IHC, immunohistochemistry; MSI, microsatellite instability.
Frequency and Mechanisms of Cyclin E Overexpression
To estimate the frequency of cyclin E overexpression, two pancreatic adenocarcinoma tissue microarrays, created from tissues collected from two separate institutions, were studied by immunohistochemistry. We found that 6% (4 of 46 and 5 of 100) of the pancreatic carcinomas had immunohistochemically detectable levels of nuclear cyclin E when compared to normal cells within the same tissue cores (Figure 2) ▶ . To investigate the mechanism of this overexpression, Southern blot analysis was performed on the pancreatic cancer cell lines Panc-1, AsPC-1, and Su86.86. The results suggest an increase in CCNE1 copy number (Figure 3E) ▶ when compared to levels in the normal N57. FISH analysis confirmed the Southern data showing a low-level amplification of CCNE1 (separate from AKT2 amplification) in Panc-1 cells while ectopic copies of CCNE1 were identified in the Su86.86 cell line (Figure 3) ▶ .
Figure 2.
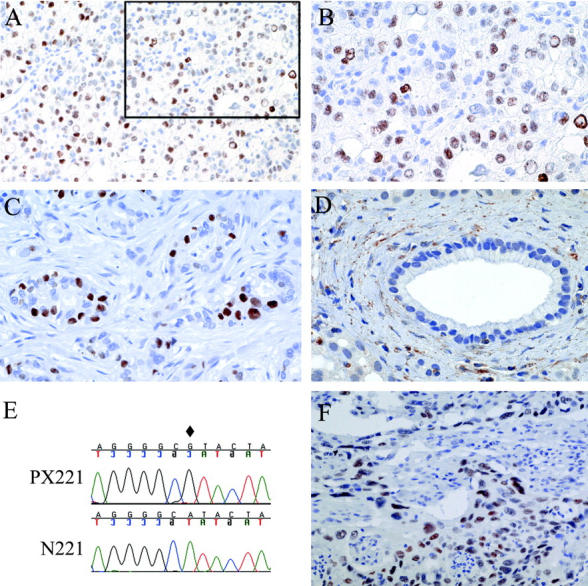
Cyclin E overexpression in pancreatic adenocarcinoma. Pancreatic carcinoma tissue arrays 1 (A and B) and 2 (C) studied for cyclin E expression. B: Higher magnification of A inset. Normal ductal structures (D) failed to express nuclear cyclin E. E: The pancreatic tumor xenograft, PX221, carries a homozygous CAT to CGT somatic missense mutation within exon 9 (diamond). F: Immunohistochemistry confirms the predicted overexpression of cyclin E in PX221. Magnifications: A, ×40; B-D and F, ×100.
Figure 3.

CCNE1 and AKT2 fluorescence in situ hybridization (FISH). A: Interphase nuclei of the Su86.86 cell line showing a marker of 19p (TCF3/E2A, red) and extra copies of CCNE1 (green). B: Metaphase spread of Su86.86 showing TCF3 (red, 19p) and CCNE1 (green). Ectopic CCNE1 is shown with an arrow. C: Metaphase spread of normal cells showing single-copy signals for TCF3 (red, 19p), CCNE1 (green, 19q) and AKT2 (red, 19q). D: Metaphase spread showing amplification of AKT2 (red, q arm) and its centromeric neighbor, CCNE1 (green), in the Panc-1 cell line. TCF3 (red) is also shown as a marker of 19p. E: Southern blots of CCNE1 in the Su86.86, AsPC-1, and Panc-1 cell lines compared to levels in N57 (normal). The WI-12306 clone served as a loading control.
To investigate another potential mechanism for cyclin E overexpression, the exons of FBXW7 were amplified and sequenced from pancreatic cancer xenografts (n = 11) that had known loss of heterozygosity at 4q31.3. We identified a novel, single nucleotide somatic alteration that resulted in a histidine to arginine missense mutation at codon 460 (H460R) in tumor PX221 (Figure 2E ▶ ; Table 1 ▶ ). The formalin-fixed, paraffin-embedded carcinoma of this patient was immunohistochemically studied, and strong immunopositivity was observed for cyclin E specifically in the neoplastic cell nuclei (Figure 2F) ▶ . This confirmed the H460R mutant to be functionally inactive.
Discussion
As many as 90% to 95% of ductal adenocarcinomas of the pancreas have KRAS2 mutations, a finding suggestive of a virtually necessary role in the development of pancreatic cancer. By reverse analogy, however, this suggests that a relatively large percentage (as much as 10%) of pancreatic carcinomas might use alternative methods to stimulate this pathway. Recent reports by Davies et al 19 and Rajagopalan et al 8 have shown that Braf, a serine/threonine kinase located immediately downstream in Ras signaling, is a frequent mutational target in several cell lines and primary cancers including 66% of melanomas and 10% of colorectal carcinomas. In the colorectal carcinomas studied, BRAF mutations were exclusively found in neoplasms having wild-type RAS (half of which also displayed abnormalities in DNA mismatch repair). The current study provides the first evidence of BRAF mutations in pancreatic cancer and reaffirms the mutually exclusive nature of KRAS2/BRAF mutations as well as the apparent requirement for KRAS2-related signal activation during most instances of pancreatic ductal carcinogenesis.
The second signaling pathway investigated in this report focuses on the cell cycle regulator cyclin E. Cyclin E is a known protooncogene that is overexpressed in a variety of cancers. The mechanism of overexpression is reportedly due to amplification at 19q13.1 in some neoplasms. 20-23 Recently, however, several groups 11,24,25 have described mutations in FBXW7, which codes for the cyclin E/ubiquitin ligase conjugating protein, which appear to impair the ability of cells to degrade cyclin E in ovarian, endometrial, and breast cancer. Such observations provide a mechanism for the overexpression of cyclin E in these cancers and for the consequential effects that would disregulate cell cycle control.
For pancreatic cancer, however, the frequency of cyclin E overexpression and therefore its role in tumorigenesis remained uncharacterized. Using two separate tissue microarrays created from geographically distinct tumor banks, we estimated that the overall frequency of cyclin E overexpression in pancreatic cancer is near 6%. Mechanistically, the overexpression of cyclin E appears to be attributable in part to the amplification of 19q13.1 or the mutation of its negative regulator, FBXW7. The FBXW7 mutation identified in this study occurred near the site of other reported mutations suggesting exons 8 and 9 to be critical hotspots for mutations that inactivate this protein. 11,24,25 Indeed, the site of our mutation occurred within the fifth WD domain and is conserved between Homo sapiens, Drosophila melanogaster, Caenorhabditis elegans and Saccharomyces cerevisiae. 25
The current study identified two new mutational targets in pancreatic cancer: the genes encoding the serine/threonine kinase Braf and the cyclin E/ubiquitin ligase conjugating protein Fbxw7. Their identification contributes to a satisfying orderliness of specific mutations in phenotypic subsets of pancreatic cancers as demonstrated by the presence of specific mutations in pancreatic carcinomas with a medullary phenotype and often with microsatellite instability (including ACVR2, TGFBR2, and BRAF 8,26,27 ) and in at least some of those that overexpress cyclin E. In addition, these mutations extend the knowledge of kinase and cyclin abnormalities to genes not previously reported in pancreatic cancer. These findings are of potential therapeutic importance as each of the mutations appears to result in the increased activity of effector kinases important for tumor development. As such, they may be sensitive to small molecule inhibitors currently under development. The clinical recognition of qualitatively distinct tumor subsets may in the future dictate more effective treatment strategies against this deadly disease.
Footnotes
Address reprint requests to Scott E. Kern, M.D., Department of Oncology, Sidney Kimmel Comprehensive Cancer Center, Johns Hopkins Medical Institute, 1650 Orleans St, Baltimore, MD 21231. E-mail: sk@jhmi.edu.
Supported by the National Institutes of Health grants CA92624, CA92117, the Cancer Center Support CORE grant CA21765, and the American Lebanese Syrian Associated Charities (ALSAC).
References
- 1.Otterson GA, Kratzke RA, Coxon A, Kim YW, Kaye FJ: Absence of p16INK4 protein is restricted to the subset of lung cancer lines that retains wild-type Rb. Oncogene 1994, 9:3375-3378 [PubMed] [Google Scholar]
- 2.Shapiro GI, Edwards CD, Kobzik L, Godleski J, Richards W, Sugarbaker DJ, Rollins BJ: Reciprocal Rb inactivation and p16INK4 expression in primary lung cancers and cell lines. Cancer Res 1995, 55:505-509 [PubMed] [Google Scholar]
- 3.Reis RM, Konu-Lebleblicioglu D, Lopes JM, Kleihues P, Ohgaki H: Genetic profile of gliosarcomas. Am J Pathol 2000, 156:425-432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Capoulade C, Bressac-de Paillerets B, Lefrere I, Ronsin M, Feunteun J, Tursz T, Wiels J: Overexpression of MDM2, due to enhanced translation, results in inactivation of wild-type p53 in Burkitt’s lymphoma cells. Oncogene 1998, 16:1603-1610 [DOI] [PubMed] [Google Scholar]
- 5.Heinrich MC, Corless CL, Duensing A, McGreevey L, Chen CJ, Joseph N, Singer S, Griffith DJ, Haley A, Town A, Demetri GD, Fletcher CD, Fletcher JA: PDGFRA activating mutations in gastrointestinal stromal tumors. Science 2003, 299:708-710 [DOI] [PubMed] [Google Scholar]
- 6.Gerstein AV, Almeida TA, Zhao G, Chess E, Shih Ie M, Buhler K, Pienta K, Rubin MA, Vessella R, Papadopoulos N: APC/CTNNB1 (β-catenin) pathway alterations in human prostate cancers. Genes Chromosomes Cancer 2002, 34:9-16 [DOI] [PubMed] [Google Scholar]
- 7.Singer G, Oldt R, 3rd, Cohen Y, Wang BG, Sidransky D, Kurman RJ, Shih Ie M: Mutations in BRAF and KRAS characterize the development of low-grade ovarian serous carcinoma. J Natl Cancer Inst 2003, 95:484-486 [DOI] [PubMed] [Google Scholar]
- 8.Rajagopalan H, Bardelli A, Lengauer C, Kinzler KW, Vogelstein B, Velculescu VE: Tumorigenesis: rAF/RAS oncogenes and mismatch-repair status. Nature 2002, 418:934. [DOI] [PubMed] [Google Scholar]
- 9.Goggins M, Offerhaus GJ, Hilgers W, Griffin CA, Shekher M, Tang D, Sohn TA, Yeo CJ, Kern SE, Hruban RH: Pancreatic adenocarcinomas with DNA replication errors (RER+) are associated with wild-type K-ras and characteristic histopathology: poor differentiation, a syncytial growth pattern, and pushing borders suggest RER+. Am J Pathol 1998, 152:1501-1507 [PMC free article] [PubMed] [Google Scholar]
- 10.Wilentz RE, Goggins M, Redston M, Marcus VA, Adsay NV, Sohn TA, Kadkol SS, Yeo CJ, Choti M, Zahurak M, Johnson K, Tascilar M, Offerhaus GJ, Hruban RH, Kern SE: Genetic, immunohistochemical, and clinical features of medullary carcinoma of the pancreas: a newly described and characterized entity. Am J Pathol 2000, 156:1641-1651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moberg KH, Bell DW, Wahrer DC, Haber DA, Hariharan IK: Archipelago regulates cyclin E levels in Drosophila and is mutated in human cancer cell lines. Nature 2001, 413:311-316 [DOI] [PubMed] [Google Scholar]
- 12.Koepp DM, Schaefer LK, Ye X, Keyomarsi K, Chu C, Harper JW, Elledge SJ: Phosphorylation-dependent ubiquitination of cyclin E by the SCFFbw7 ubiquitin ligase. Science 2001, 294:173-177 [DOI] [PubMed] [Google Scholar]
- 13.Cheng JQ, Ruggeri B, Klein WM, Sonoda G, Altomare DA, Watson DK, Testa JR: Amplification of AKT2 in human pancreatic cells and inhibition of AKT2 expression and tumorigenicity by antisense RNA. Proc Natl Acad Sci USA 1996, 93:3636-3641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kalthoff H, Roeder C, Gieseking J, Humburg I, Schmiegel W: Inverse regulation of human ERBB2 and epidermal growth factor receptors by tumor necrosis factor α. Proc Natl Acad Sci USA 1993, 90:8972-8976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Caldas C, Hahn SA, da Costa LT, Redston MS, Schutte M, Seymour AB, Weinstein CL, Hruban RH, Yeo CJ, Kern SE: Frequent somatic mutations and homozygous deletions of the p16 (MTS1) gene in pancreatic adenocarcinoma. Nat Genet 1994, 8:27-32 [DOI] [PubMed] [Google Scholar]
- 16.Lahti JM, Valentine M, Xiang J, Jones B, Amann J, Grenet J, Richmond G, Look AT, Kidd VJ: Alterations in the PITSLRE protein kinase gene complex on chromosome 1p36 in childhood neuroblastoma. Nat Genet 1994, 7:370-375 [DOI] [PubMed] [Google Scholar]
- 17.Li H, Lahti JM, Valentine M, Saito M, Reed SI, Look AT, Kidd VJ: Molecular cloning and chromosomal localization of the human cyclin C (CCNC) and cyclin E (CCNE) genes: deletion of the CCNC gene in human tumors. Genomics 1996, 32:253-259 [DOI] [PubMed] [Google Scholar]
- 18.Toyooka KO, Toyooka S, Maitra A, Feng Q, Kiviat NC, Smith A, Minna JD, Ashfaq R, Gazdar AF: Establishment and validation of real-time polymerase chain reaction method for CDH1 promoter methylation. Am J Pathol 2002, 161:629-634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, Floyd Y, Gray K, Hall S, Hawes R, Hughes J, Kosmidou V, Menzies A, Mould C, Parker A, Stevens C, Watt S, Hooper S, Wilson R, Jayatilake H, Gusterson BA, Cooper C, Shipley J, Hargrave D, Pritchard-Jones K, Maitland N, Chenevix-Trench G, Riggins GJ, Bigner DD, Palmieri G, Cossu A, Flanagan A, Nicholson A, Ho JW, Leung SY, Yuen ST, Weber BL, Seigler HF, Darrow TL, Paterson H, Marais R, Marshall CJ, Wooster R, Stratton MR, Futreal PA: Mutations of the BRAF gene in human cancer. Nature 2002, 417:949-954 [DOI] [PubMed] [Google Scholar]
- 20.Lin L, Prescott MS, Zhu Z, Singh P, Chun SY, Kuick RD, Hanash SM, Orringer MB, Glover TW, Beer DG: Identification and characterization of a 19q12 amplicon in esophageal adenocarcinomas reveals cyclin E as the best candidate gene for this amplicon. Cancer Res 2000, 60:7021-7027 [PubMed] [Google Scholar]
- 21.Enders GH: Cyclins in breast cancer: too much of a good thing. Breast Cancer Res 2002, 4:145-147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jung YJ, Lee KH, Choi DW, Han CJ, Jeong SH, Kim KC, Oh JW, Park TK, Kim CM: Reciprocal expressions of cyclin E and cyclin D1 in hepatocellular carcinoma. Cancer Lett 2001, 168:57-63 [DOI] [PubMed] [Google Scholar]
- 23.Richter J, Wagner U, Kononen J, Fijan A, Bruderer J, Schmid U, Ackermann D, Maurer R, Alund G, Knonagel H, Rist M, Wilber K, Anabitarte M, Hering F, Hardmeier T, Schonenberger A, Flury R, Jager P, Fehr JL, Schraml P, Moch H, Mihatsch MJ, Gasser T, Kallioniemi OP, Sauter G: High-throughput tissue microarray analysis of cyclin E gene amplification and overexpression in urinary bladder cancer. Am J Pathol 2000, 157:787-794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Spruck CH, Strohmaier H, Sangfelt O, Muller HM, Hubalek M, Muller-Holzner E, Marth C, Widschwendter M, Reed SI: hCDC4 gene mutations in endometrial cancer. Cancer Res 2002, 62:4535-4539 [PubMed] [Google Scholar]
- 25.Strohmaier H, Spruck CH, Kaiser P, Won KA, Sangfelt O, Reed SI: Human F-box protein hCdc4 targets cyclin E for proteolysis and is mutated in a breast cancer cell line. Nature 2001, 413:316-322 [DOI] [PubMed] [Google Scholar]
- 26.Markowitz S, Wang J, Myeroff L, Parsons R, Sun L, Lutterbaugh J, Fan RS, Zborowska E, Kinzler KW, Vogelstein B, Brattain M, Willson JKV: Inactivation of the type II TGF-β receptor in colon cancer cells with microsatellite instability. Science 1995, 268:1336-1338 [DOI] [PubMed] [Google Scholar]
- 27.Hempen PM, Zhang L, Bansal RK, Iacobuzio-Donahue CA, Murphy KM, Maitra A, Vogelstein B, Whitehead RH, Markowitz SD, Willson JK, Yeo CJ, Hruban RH, Kern SE: Evidence of selection for clones having genetic inactivation of the activin A type II receptor (ACVR2) gene in gastrointestinal cancers. Cancer Res 2003, 63:994-999 [PubMed] [Google Scholar]