

Abstract
Vascular endothelial growth factor (VEGF) expression is enhanced in ischemic skeletal muscle and is thought to play a key role in the angiogenic response to ischemia. However, it is still unknown whether, in addition to new blood vessel growth, VEGF modulates skeletal muscle cell function. In the present study immunohistochemical analysis showed that, in normoperfused mouse hindlimb, VEGF and its receptors Flk-1 and Flt-1 were expressed mostly in quiescent satellite cells. Unilateral hindlimb ischemia was induced by left femoral artery ligation. At day 3 and day 7 after the induction of ischemia, Flk-1 and Flt-1 were expressed in regenerating muscle fibers and VEGF expression by these fibers was markedly enhanced. Additional in vitro experiments showed that in growing medium both cultured satellite cells and myoblast cell line C2C12 expressed VEGF and its receptors. Under these conditions, Flk-1 receptor exhibited constitutive tyrosine phosphorylation that was increased by VEGF treatment. During myogenic differentiation Flk-1 and Flt-1 were down-regulated. In a modified Boyden Chamber assay, VEGF enhanced C2C12 myoblasts migration approximately fivefold. Moreover, VEGF administration to differentiating C2C12 myoblasts prevented apoptosis, while inhibition of VEGF signaling either with selective VEGF receptor inhibitors (SU1498 and CB676475) or a neutralizing Flk-1 antibody, enhanced cell death approximately 3.5-fold. Finally, adenovirus-mediated VEGF165 gene transfer inhibited ischemia-induced apoptosis in skeletal muscle. These results support a role for VEGF in myoblast migration and survival, and suggest a novel autocrine role of VEGF in skeletal muscle repair during ischemia.
Vascular endothelial growth factor-A (VEGF-A), also known as vascular permeability factor (VPF), plays a key role in mediating physiological and pathological angiogenesis. VEGF induces vasodilation, enhances vascular permeability and stimulates proliferation, migration, and survival of endothelial cells (ECs). 1 Further, VEGF has been shown to induce mobilization of endothelial precursors that home to ischemic tissue and differentiate into vascular cells. 2-4 At least five VEGF-A isoforms have been described, consisting of polypeptides with 121–145-165–189- and 206-amino acid residues; these isoforms differ in their ability to bind heparin, neuropilin-1, and −2, and in their solubility. 5,6 VEGF-A is known to exert its effects via two high-affinity receptors, the kinase insert domain-containing receptor (KDR/Flk-1; VEGF-R2) and Fms-like tyrosine kinase (Flt-1; VEGF-R1). The expression of VEGF and its receptors is not restricted to vascular ECs since their expression has been detected in vascular smooth muscle cells, 7 osteoblasts, 8 cardiac myocytes, 9 regenerating myotubes, 10 neurons, 11 and hematopoietic stem cells. 12 Although a role for Flk-1 and Flt-1 in non-ECs has not been clearly identified, recent studies suggest that these receptors and their ligand VEGF-A could have multiple functions. For example they are involved in chemotaxis and migration of vascular smooth muscle cells, 13 coordinate longitudinal bone growth and endochondral bone formation, 14 transduce survival signals in neuronal cells 15-17 and in hematopoietic stem cells. 12
VEGF production is enhanced by hypoxia both in vitro 18 and in vivo.19 In addition, it has been shown that in the ischemic limb, VEGF and its receptors are up-regulated several hours after the induction of ischemia. 10,20,21 Under these conditions VEGF is produced by skeletal myocytes, smooth muscle cells (SMC), ECs, and infiltrating cells and plays a key role in stimulating angiogenesis. 13,22 In certain pathological states such as diabetes 23 and hypercholesterolemia 24 as well as in aged 25,26 animals, the angiogenic response to ischemia is impaired and, under these conditions, VEGF expression is reduced. Several preclinical studies have established that VEGF administered as recombinant protein or by gene transfer can induce angiogenesis and enhance blood flow to ischemic tissues. 27,28 In addition to an effect on new blood vessel development it is possible that, in ischemic tissues, VEGF may also play other roles. Specifically, Flk-1 expression has been recently observed in regenerating muscle fibers, 10 but it is still unknown whether VEGF has an effect on skeletal muscle cell function.
Skeletal muscle regeneration after injury is characterized by the proliferation and differentiation of muscle precursor cells, known as satellite cells, followed by fusion with each other to form multinucleated myotubes. 29 This process has been largely investigated by using C2C12 myoblasts, a cell line derived from murine satellite cells, which provide a useful model system, to study skeletal muscle growth and differentiation in vitro. Various pathophysiological changes associated with skeletal muscle regeneration have been described. 30 Initially, damaged tissue is infiltrated by fibroblasts, inflammatory cells, and macrophages. This is followed by a removal of necrotic tissue, revascularization, and proliferation of muscle precursor cells. Here we report that VEGF receptors Flk-1 and Flt-1 are expressed in satellite cells and, during ischemia, in regenerating fibers. To avoid problems linked to the use of primary cell culture such as the low number of cells obtained and the difficulty to control their differentiation process in culture, most experiments are performed with C2C12 myoblasts. In these cells, Flk-1 and Flt-1 expression is down-regulated in concomitance with myogenic differentiation. VEGF administration enhances C2C12 myoblasts migration and survival and inhibits, in vivo, ischemia-induced skeletal muscle apoptosis. These results demonstrate that VEGF modulates skeletal myoblast function.
Materials and Methods
Cell Culture and Reagents
The murine myoblast C2C12 cell line 31 (a gift of Dr. A. Musaro, University of Rome “La Sapienza”) was cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 20% fetal bovine serum (FBS; Euroclone Inc., Milan, Italy), 20 mmol/L-glutamine, 100 U/ml penicillin and 100 mg/ml streptomycin (growth medium, GM) (Gibco BRL, Paisley, UK). To induce differentiation, cells at 70% to 80% confluence were shifted to DMEM supplemented with 1% bovine serum albumin (BSA) (differentiation medium, DM).
Primary satellite cells were isolated from freshly harvested mouse skeletal muscle as described. 32 Cells were cultured in DMEM with 20% horse serum (Sigma, St. Louis, MO) and 3% chick extract (Gibco).
Recombinant VEGF-A (VEGF165) and hepatocyte growth factor (HGF) (R&D Systems Inc., Minneapolis, MN) were diluted in PBS containing 0.1% BSA and stored at −80°C. To make cells hypoxic, culture dishes were placed in airtight modular incubator chambers (Forma Scientific, Mountain View, CA), infused for 20 minutes with 95% N2 plus 5% CO2, and incubated at 37°C.
(ε)-3(3,5-Diisopropyl-4-hydroxyphenyl)-2-[3-phenyl-n-propyl) amino-carbonyl] acrylonitrile (SU1498), 33 a potent and selective inhibitor of Flk-1 kinase, and 4-[94′-Chloro-2′fluoro-phenylamino]-6,7-dimethoxyquinazoline (CB676475), a potent and selective inhibitor of VEGFR1 and 2 tyrosine kinase activity, were purchased from Calbiochem (San Diego, CA). Anti-VEGF (C1), anti-Flk-1 (A3), monoclonal (mAb) antibodies, anti-Flt-1 (C17), and anti-M-cadherin (N19) polyclonal (PAb) antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Neutralizing mouse Flk-1 mAb (nFlk-1) was from R&D Systems. Anti-Desmin mAb was obtained from Sigma. Anti-α-tubulin mAb was purchased from Oncogene Science Inc. (Cambridge, MA). mAb anti-phosphotyrosine was from Upstate Biotechnology and anti α-myosin heavy chain (MF20) was a kind gift of M. Coletta (University of Rome, “La Sapienza”).
Animal Model of Hindlimb Ischemia
CD1 male mice (Charles River, Calco, Italy), 2 to 3 months of age and weighing 25 to 35 g, were used for all experiments. Animals were anesthetized with a mixture of 10 g of tribromoethyl alcohol in 10 ml of tert-amyl alcohol (Avertin, Sigma), diluted 1:50 and intraperitoneally injected 0.015 ml/g body weight. Unilateral hindlimb ischemia was induced by ligating the left femoral artery as described. 34
Laser Doppler Perfusion Imaging (LDPI; Lisca, Inc., North Brunswick, NJ) was used to record serial blood flow measurements over the course of 14 days postoperatively as previously described. 20 The animal study protocol was approved by the Institutional Animal Care and Use Committee.
Intramuscular Administration of Adenovirus Vectors
Replication-deficient recombinant Ad vector containing cDNA for VEGF165 has been previously described. 35 Two days before the induction of ischemia, mice were randomly assigned to receive intramuscular administration of AdCMV.VEGF165 or AdCMV.Null (1010 pfu/ml). The viral vectors were stored in dialysis buffer solution (3% sucrose, 10 mmol/L Tris-HCl pH 7.8, 150 mmol/L NaCl and 10 mmol/L MgCl2) at −70°C. Each solution for injection was prepared immediately before use and injected intramuscularly in three different sites in the thigh (3 μl/injection) along the projection of the femoral artery.
Immunohistochemistry
Animals from all experimental groups were anesthetized with 2.5% Avertin (100% Avertin: 10 g of 2,2,2-tribromoethyl alcohol and 10 ml of tert-amylalcohol; Sigma). Adductor muscles were removed, fixed in formalin for 48 hours and embedded in paraffin (Bio-plast special; melting point 52–54°C). Sections from each sample were cut at a thickness of 4 μm with the muscle fibers oriented transversely. For immunohistochemical analysis, sections were deparaffinized and incubated at room temperature for 20 minutes with a solution of methanol containing 0.03% H2O2. Serial sections were incubated for 1 hour with 3% goat serum and overnight at 4°C with the following antibodies: anti-VEGF (2 μg/ml; Santa Cruz), anti-Flk-1 (1 μg/ml; Santa Cruz), anti-Flt-1 (0.8 μg/ml, Santa Cruz), anti-M-Cadherin (0.7 μg/ml, Santa Cruz) and anti-desmin (1:30; Sigma). After a brief rinse, sections were incubated with biotinylated secondary antibodies (7.5 μg/ml, Vector Laboratories, Peterborough, UK), washed and incubated with avidin-biotinylated peroxidase complex (ABC Elite Kit; Vector Laboratories). The stain was visualized by treatment for 10 minutes in a 0.05% solution of 3-diaminobenzidine (DAB) and 0.01% H2O2 in 0.1 mol/L PBS. Sections were counterstained with hematoxylin to identify nuclei. Control reactions included the omission of the primary antibody, which was substituted by non-immune rabbit serum.
VEGF Immunoassays
VEGF levels were measured using an ELISA assay (R&D Systems Inc.) according to manufacturer’s instructions. C2C12 plated in 60 mm dishes (105 cells/dish) were cultured either in GM for 1 day or in DM for 1, 3, and 5 days. Conditioned medium (CM) was collected at the indicated time points and concentrated 10 times with centricon-3 microconcentrators (Millipore, Bedford, MA). Values were assayed in triplicate and corrected for the protein amounts calculated by the Bradford method (Bio-Rad, Hercules, CA).
Western Blotting and Immunoprecipitation
C2C12 cells cultured either in GM or in DM were lysed in RIPA buffer containing 10 mmol/L Tris-HCl (pH 7.4), 150 mmol/L NaCl, 1% NP40, 1% deoxycolic acid, 0.1% sodium dodecyl sulfate (SDS), 10% glycerol and protease inhibitors. Equal amounts of total cellular proteins (100 μg/lane) were resolved by 8% SDS-polyacrylamide gel electrophoresis and transferred to nitrocellulose membrane (Amersham Pharmacia Biotech, Little Chalfont, UK). Membranes were probed with specific antibodies (0.4 μg/ml anti Flk-1 mAb; 0.2 μg/ml anti Flt-1 Mab; 1:40 dilution MF20; 0.1 μg/ml anti α-tubulin mAb) followed by horseradish peroxidase-coupled secondary antibodies and developed by a chemiluminescence-based detection system (ECL; Amersham Biosciences). For immunoprecipitation experiments, 1 mg of whole cell extract was incubated at 4°C with 1 μg of the indicated antibodies for 2 hours followed by Protein G Sepharose (Amersham Biosciences) for 1 hour. The precipitates were analyzed as described above.
RT-PCR
C2C12 were grown in DM and RNA extraction was performed by use of TRIzol reagent (Invitrogen, Carlsbad, CA). Preamplification system was used to reverse transcribe total RNA (1 μg) into complementary DNA according to manufacturer’s instructions (Invitrogen). An aliquot (2 μl) of the reverse transcription reaction was subjected to 39 polymerase chain reaction (PCR) cycles: 1 minute at 94°C, 1 minute at 54°C, and 1 minute at 72°C, in the presence of 50 pmol of each primer, 1.5 mmol/L MgCl2, 200 mmol/L dATP, dCTP, dGTP, and dTTP, and 2.5 U of AmpliTaq polymerase (Invitrogen). Sequence of the primers were: Flk-1, 5′-GTGATCAGGGGTCCTGAA-3′ and 5′-GCAAACATAGTCGCCTTGGT-3′; Flt-1, 5′-GGCACAAAGACCCCAAAGAG-3′ and 5′-AACAGCAGGACTCCTTTCCC-3′. The expected product size was 288 bp for Flk-1 and 539 bp for Flt-1. The PCR products were electrophoresed on 2% agarose gel containing 0.5 μg/ml of ethidium bromide.
Chemotaxis Assays
Chemotaxis was performed in 48-microwell chemotaxis chambers (Neuroprobe, Cabin John, MD) using 8-μm pore-size polycarbonate filters (Costar Scientific Corporation, Cambridge, MA) coated with murine collagen type IV (BD Biosciences, Bedford, MA).
The lower compartment of each chamber was filled with 28 μl DMEM with 0.1% BSA. VEGF was added at the concentrations indicated in the Figure ▶ legends; DMEM with 0.1% BSA and GM were used as negative and positive controls for migration, respectively. Each well of the upper compartment was filled with 50 μl DMEM with 0.1% BSA, containing C2C12 cells (0.7 × 106 cells/ml). In some experiments either SU1498, CB676475, or nFlk-1 were added to the cell suspension. Each point was run in triplicate. After 4 hours incubation at 37°C in a 5% CO2 humidified atmosphere, the chemotaxis assay was stopped, cells on the filter were fixed and stained using Diff Quik (Dade AG, Dudingen, Switzerland). Cells on five random fields on the lower face of the filter were counted at ×40 magnification and migration index was calculated by dividing the number of migrated cells in the presence of chemoattractants by the cells migrated in response to DMEM with 0.1% BSA.
Figure 1.

Quantitative evaluation of blood flow recovery after hindlimb ischemia. LDPI was used to quantify both right and left hindlimb perfusion, preoperatively (C), immediately after femoral artery ligation (0), and at the indicated time points, postoperatively. Analysis was performed by calculating the average perfusion of each ischemic and non-ischemic foot and expressing it as a ratio of left (ischemic) to right (normoperfused) foot.
Apoptotis Assessment
Subconfluent C2C12 were incubated in DM with or without VEGF (20 ng/ml) for 3 days before analysis of DNA fragmentation by terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) assay according to manufacturer’s instructions (Roche Molecular Biochemical, Milian, Italy). Fluorescence and phase-contrast photomicrographs were taken at ×40 using the Axioplan 2 fluorescence microscope (Zeiss, Oberkochen, Germany). Apoptosis was also analyzed by cell death detection ELISA (Roche Diagnostics, Mannheim, Germany). Apoptotic nuclei, in the in vivo experiments, were identified by TUNEL (Roche).
Statistical Analysis
Data were expressed as means ± SD. Student’s two-tailed t-test was performed and a P ≤ 0.05 was considered statistically significant.
Results
Flk-1, Flt-1, and VEGF Expression in Vivo
To investigate VEGF receptors expression during skeletal muscle regeneration, hindlimb ischemia was induced by ligation of the femoral artery. LDPI was used to document changes in hindlimb blood flow at the indicated time points following the induction of ischemia. The marked decrease in blood flow immediately after femoral artery ligation was followed by a progressive recovery, which, under the experimental conditions of the present study, was complete by day 14 (Figure 1) ▶ . Flk-1 and Flt-1 expression was evaluated in normoperfused skeletal muscle. Serial muscle sections were stained with specific antibodies for Flk-1 and Flt-1 and it was found that both receptors were expressed in cells closely associated with skeletal muscle fibers (Figure 2A) ▶ as well as in vascular structures (Figure 2B) ▶ . Immunostaining with anti- M-cadherin antibody, which recognizes a cell adhesion molecule expressed in quiescent and activated satellite cells, identified the cells expressing Flk-1 and Flt-1 as satellite cells (Figure 2A) ▶ . These cells represent 2% to 5% of nuclei associated with fibers and reside juxtaposed to skeletal muscle fibers beneath the basal lamina. 36 Immunostaining for Flk-1 and Flt-1 performed at day 3 after ischemia showed Flk-1 and Flt-1 immunoreactivity in cells which also expressed the intermediate filament desmin, a marker of activated satellite cells 37 (Figure 2C) ▶ . This result indicates that Flk-1- and Flt-1-expressing cells were proliferating myogenic cells. One week after femoral artery dissection, regenerating skeletal muscle fibers were distinguished from normal fibers because of their small size and central nuclei (Figure 2D) ▶ . At this time point, regenerating fibers exhibited diffuse Flk-1 and Flt-1 labeling (Figure 2D) ▶ . In mature fibers, as well as in regenerated muscle at 14 days after ischemia, immunostaining for Flk-1 and Flt-1 returned to the basal level observed in normoperfused muscle (Figure 2E) ▶ . VEGF expression in skeletal muscle was also investigated. In normoperfused hindlimbs VEGF immunostaining was found in satellite cells (Figure 3A) ▶ . Both at day 3 and 7 after ischemia, staining for VEGF was detected in activated satellite cells and in regenerating fibers (Figure 3B and C) ▶ . Thereafter VEGF immunostaining decreased but it was still present at day 14 after the induction of ischemia (Figure 3D) ▶ .
Figure 2.
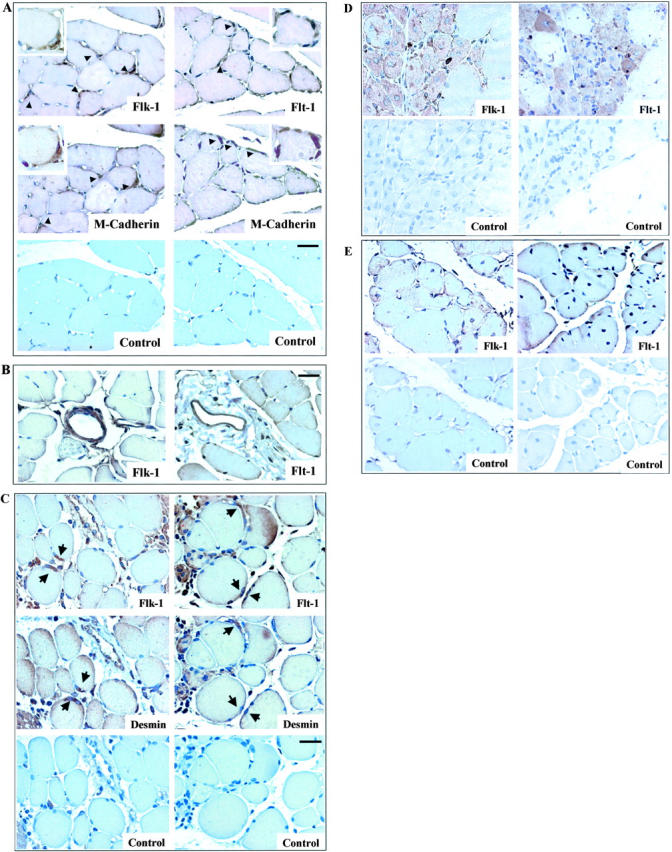
Expression of VEGF and its receptors in skeletal muscle cells in vivo. Flk-1 and Flt-1 expression in normoperfused mouse skeletal muscle (A) and in vascular structures (B). Serial muscle sections were immunostained for Flk-1 and Flt-1. Positive cells, indicated by arrowheads, were identified as satellite cells by their immunoreactivity with M-cadherin antibody. Insets show higher-power photomicrographs of satellite cell. Control immunostaining was performed by omitting the primary antibody. Magnification, ×40 (inset ×100); bar, 25 μm. Time-course of Flk-1 and Flt-1 expression (C to E). Serial sections from hindlimbs were obtained at 3 days (C), 7 days (D), and 14 days (E) following the induction of ischemia. Flk-1 and Flt-1 were expressed in activated satellite cells as identified by desmin labeling (C); 7 days after ischemia Flk-1 and Flt-1 were expressed in regenerating myotubes (D) and the expression of both receptors decreased at day 14 (E), when the regenerative process was nearly complete. Magnification, ×40; bar, 25 μm.
Figure 3.
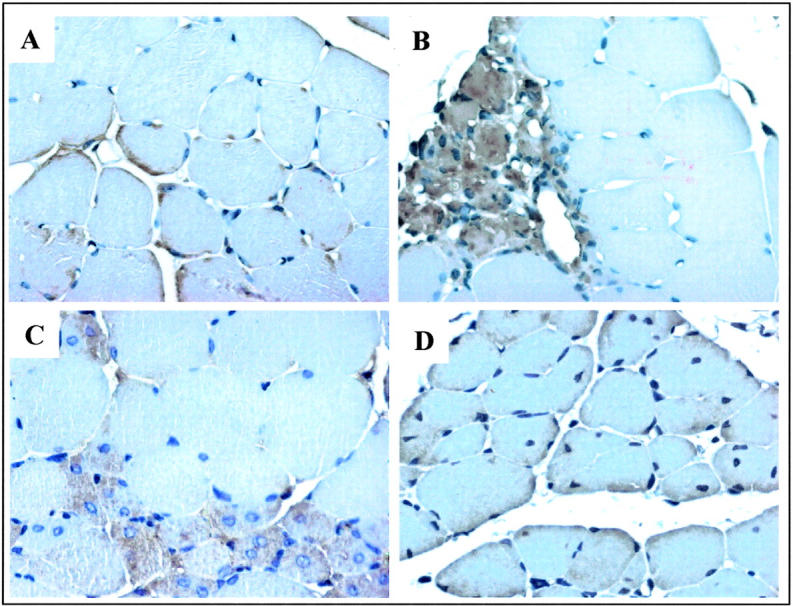
VEGF expression in skeletal muscle cells in vivo. Time-course of VEGF expression in mouse ischemic hindlimb. A: VEGF immunostaining was observed in satellite cells of normal skeletal muscle (A). VEGF protein was detected in satellite cells at day 3 (B) and in regenerating fibers at day 7 (C) after femoral artery ligation. The immunostaining decreased in regenerating fibers at 14 days after ischemic injury (D). Magnification, ×40; bar, 25 μm.
Flk-1 and Flt-1 Expression in Satellite Cells and in C2C12 Cell Line Cultured in Vitro
Flk-1 and Flt-1 expression was examined by RT-PCR in satellite cells and in C2C12 myoblasts cell line. Both satellite cells, isolated from adult mouse skeletal muscle, and C2C12 myoblasts were cultured in GM. Flk-1 and Flt-1 transcripts were readily detected in both cell types. RNA from total mouse heart was used as a positive control for Flk-1 and Flt-1 expression (Figure 4A) ▶ . Western blot analysis of total lysates from C2C12 and cultured satellite cells showed specific binding of anti-Flk-1 and Flt-1 antibodies to 200-kd bands. Similar bands were also present in HUVEC lysates, which were used as positive control (Figure 4B) ▶ . The highest bands detected with anti-Flk-1 antibody were the glycosylated form of Flk-1. 38 As expected, no bands were detected when isotype-matching immunoglobins were used in Western blot analysis (data not shown).
Figure 4.

Flk-1 and Flt-1 expression in myogenic cells in vitro. A: RT-PCR analysis of Flk-1 and Flt-1 expression in skeletal muscle cell culture. Total RNAs (1 μg) extracted from C2C12 cells, satellite cells, and newborn mice heart (positive control) were used for reverse transcription. PCR analysis was carried out using specific primers for Flk-1 and Flt-1. Negative control represents RT-PCR of C2C12 cells RNA without oligonucleotides. B: Western blot analysis showed the presence of Flk-1 and Flt-1 proteins from satellite cells and C2C12 cells in GM. Total extract from HUVEC was used as a positive control for the expression of both receptors. C: Flk-1 phosphorylation in C2C12 cells. Lysates from C2C12 untreated or treated either with VEGF165 (50 ng/ml) for 5 minutes or CB676475 (1 μmol/L) for 1 hour, were immunoprecipitated with anti-Flk-1 Mab or a preimmune serum (PI). Subsequently, immunoprecipitated proteins were subjected to Western blot analysis with anti-phosphotyrosine (top) and reprobed with antibody to Flk-1 (bottom).
To establish whether Flk-1 was activated, C2C12 cells were treated either with VEGF165 or CB676475, a broad-range VEGF receptor tyrosine kinase inhibitor. 39 Western blot analysis with an anti-phosphotyrosine Mab was performed on the immunoprecipitated Flk-1 protein. Phosphorylated Flk-1 was detected in C2C12 cells (Figure 4C) ▶ and in satellite cells (data not shown) but not in CB676475-treated cells (Figure 4C) ▶ . Moreover, VEGF165 stimulation enhanced Flk-1 phosphorylation (Figure 4C) ▶ . Using experimental conditions similar to those used for Flk-1 detection, there was no evidence of Flt-1 phosphorylation (data not shown).
VEGF, Flk-1, and Flt-1 Expression During in Vitro Myogenic Differentiation of C2C12 Cells
The sequence of events involved in muscle regeneration was reproduced in an in vitro model of differentiation. C2C12 myoblasts grow and divide when cultured in GM and, after 48–72 in DM, cells fuse to form multinucleated myotubes. In this experimental model, it was investigated whether Flk-1, Flt-1, and VEGF expression varied during differentiation as observed in in vivo during muscle regeneration (Figure 2) ▶ . Western blot analysis of C2C12 lysates showed that when myoblasts were induced to differentiate by changing from GM to DM both Flk-1 and Flt-1 proteins markedly decreased over a 5-day time period (Figure 5A) ▶ . However, Flt-1 but not Flk-1 was still detectable at day 5 of differentiation. These changes in VEGF receptor expression were paralleled by a progressive increase in myosin heavy chain expression (MyHC), consistent with the increase in differentiation of C2C12 cells (Figure 5A) ▶ . Further, after 5 days in DM, a large number of myotubes was observed in the culture dishes (not shown).
Figure 5.

Expression of VEGF and its receptors during myogenic differentiation. A: Western blot analysis of total C2C12 cell lysates shows that Flk-1 and Flt-1 proteins decreased progressively over a 5-day time period when cells in GM at day 0 (d0) were changed to DM. In agreement with the myogenic differentiation of these cells, MyHC expression increased progressively over the same time period. Western blot analysis with anti α-tubulin antibody was performed on the same membrane to confirm equal loading of the lanes. In these experiments myoblasts cultured in GM were 80% confluent when they were switched to DM. B: ELISA determination of VEGF production from proliferating and differentiating C2C12 cells. At the onset of differentiation VEGF level decreased and over a 5-day time period in DM was significantly higher to that found in GM. Culture medium was changed every 24 hours and VEGF levels in conditioned media were determined after 1 day of culture in GM and at day 1, 3, and 5 of culture in DM. Results represent mean ± SD of six experiments. The asterisk indicates a P ≤ 0.05 vs. GM.
In additional experiments it was determined whether VEGF was secreted from C2C12 cells and, if so, whether VEGF levels in the conditioned medium (CM) varied during differentiation. CM was collected every 24 hours from growing and differentiating C2C12 cells, and assayed for the presence of VEGF by ELISA. In GM, VEGF concentration was 550 pg/mg of protein/24 hours. After 1 day of culture in DM, VEGF level decreased to 270 pg/mg of protein/24 hours; thereafter it increased progressively and at day 5 in DM VEGF level was higher (760 pg/mg of protein/24 hours) than in GM (Figure 5B) ▶ . It is noteworthy that the culture medium (DMEM with 20% fetal calf serum) did not contain detectable VEGF level (<3pg/ml).
Flt-1 and Flk-1 Modulate Myoblast Migration
In these experiments it was characterized the functional role of Flk-1 and Flt-1 receptors in myoblasts. Specifically, it was examined whether these receptors modulated C2C12 cell migration in response to VEGF165 in a multiwell chemotaxis chamber. In this assay, cells in the upper chamber migrate through an extracellular matrix (ECM) protein-coated nucleopore filter to a lower chamber which contains the chemotactic agent. Under the experimental conditions of the present study, VEGF165 exhibited a dose-dependent chemotactic effect on C2C12 myoblasts. The chemotactic activity of 50 ng/ml VEGF165 was comparable to that induced by GM (Figure 6A) ▶ . VEGF-induced C2C12 cell migration was inhibited by CB676475 and SU1498, a potent and selective Flk-1 tyrosine kinase inhibitor 33 (Figure 6B) ▶ . Both drugs exhibited a dose-dependent effect to inhibit C2C12 migration in response to 20 ng/ml VEGF165. It is noteworthy that under the experimental conditions used in the chemotaxis assay neither VEGFR inhibitor had an effect on cell viability assessed by trypan blue exclusion (data not shown). Therefore, it is unlikely that the effect of these drugs was related to a toxic action. Further, a strong inhibition of VEGF-induced C2C12 cell migration was also obtained when a recombinant murine Flk-1 antibody was used to neutralize Flk-1 activity (Figure 6B) ▶ .
Figure 6.
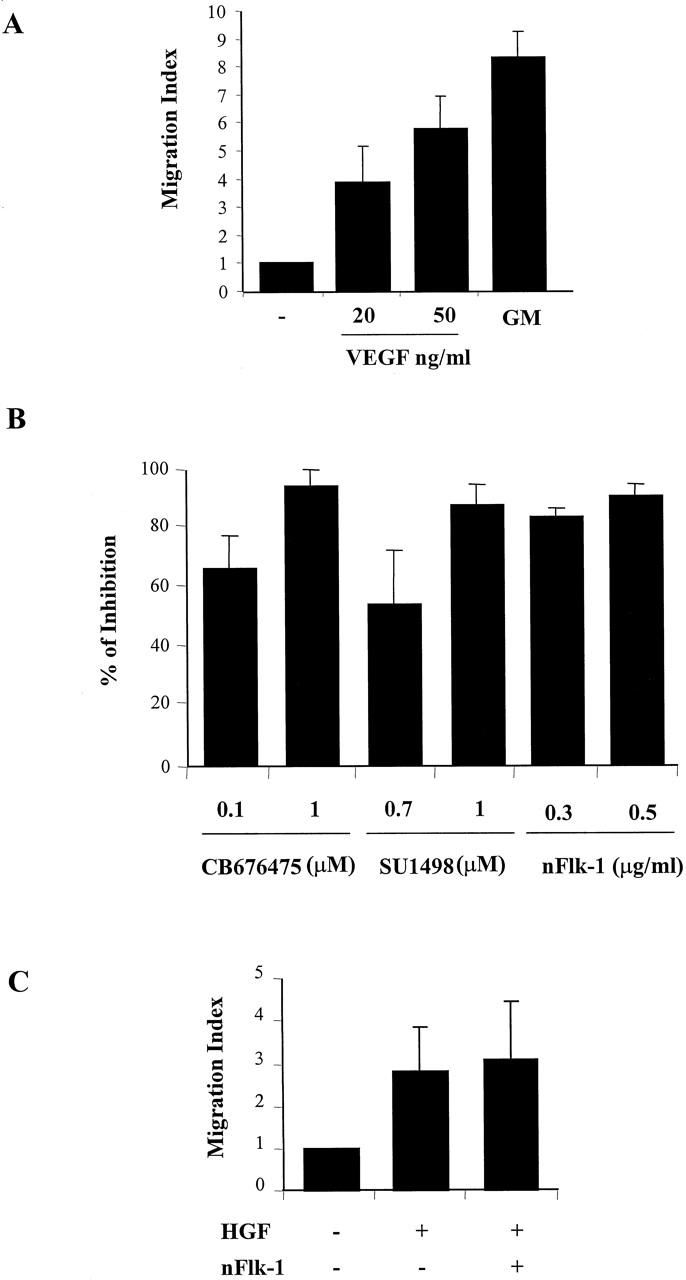
Chemotaxis of C2C12 myoblasts in response to VEGF. A: C2C12 (2 × 104) were placed in upper compartment of the modified Boyden chambers. VEGF165 at the indicated concentration was added to the lower compartment and incubated for 6 hours at 37°C. GM was used as a positive control. After staining with Giemsa solution, migrated cells were quantified by counting nuclei in five random microscope fields (×40). The data are expressed as the fold increase in the number of migrated cells relative to the number of migrated cells in the absence of factor (migration index) and are the means ± SD of at least four independent experiments performed in triplicate. B: Effect of Flk-1 and Flt-1 inhibitors on VEGF-mediated C2C12 migration. C2C12 cells were incubated with the indicated concentration of CB676475, SU1498, and nFlk-1 Mab and placed in the upper chamber. VEGF (20 ng/ml) was added to the lower chamber and quantification of migrated cells was performed as described in (A). The data are expressed as % inhibition of migration index. Results represent mean ± SD of three independent experiments performed in triplicate. C: Effect of Flk-1 inhibitors on HGF-induced C2C12 migration. C2C12 cells were incubated with 0.5 μg/ml of nFlk-1 in the upper chamber and HGF (15 ng/ml) was added to the lower chamber. Results represent the mean ± SD of three experiments.
Administration of both VEGFR inhibitors or a Flk-1 neutralizing antibody had no effect on migration induced by HGF (Figure 6C ▶ and data not shown) demonstrating the specificity of these molecules for VEGF receptors. Taken together these results indicate that VEGF165 is chemotactic for skeletal muscle precursors and that Flk-1 and Flt-1 receptors present in myoblasts are functional.
VEGF165 Protects Myoblasts from Cell Death
During in vitro myogenesis, some myoblasts undergo apoptosis, whereas others continue their differentiation program and form myotubes. After 3 days incubation in DM approximately 10% of C2C12 cells underwent apoptosis and no further increase in cell death was observed on longer incubation time. To analyze VEGF role in muscle cell viability, C2C12 cells cultured in DM were treated with 20 ng/ml VEGF165 and cell death was evaluated by TUNEL labeling. After 3 days culture in DM, VEGF decreased the number of apoptotic cells by 10.6% to 7% (Figure 7A) ▶ . Additional experiments performed by ELISA determination of histone-associated DNA fragments, revealed that cytoplasmic histone-associated DNA fragments in VEGF-treated C2C12 cells was 38.8% lower than in untreated cells confirming that VEGF decreased apoptosis (Figure 7B) ▶ . To determine whether VEGF affected myogenic differentiation, DM-cultured C2C12 cells were treated either with VEGF165 or VEGF receptor tyrosine kinase inhibitor CB676475. Both morphological and muscle-specific biochemical markers were evaluated. Morphologically, C2C12 myoblasts cultured in the presence of either VEGF or CB676475 retained their differentiative capacity and fused to form multinucleated myotubes (data not shown). Western blot analysis performed to detect MyHC accumulation after 1 and 3 days of culture showed no significant difference between untreated and VEGF- or CB676475-treated cells (Figure 7C) ▶ . Taken together, the results indicate that VEGF protects skeletal myoblasts from apoptosis without interfering with their differentiation process. It is noteworthy that VEGF had no effect on C2C12 cell number in a 48-hour assay in which cells were kept in DM supplemented with 50 μg/ml of VEGF165.
Figure 7.
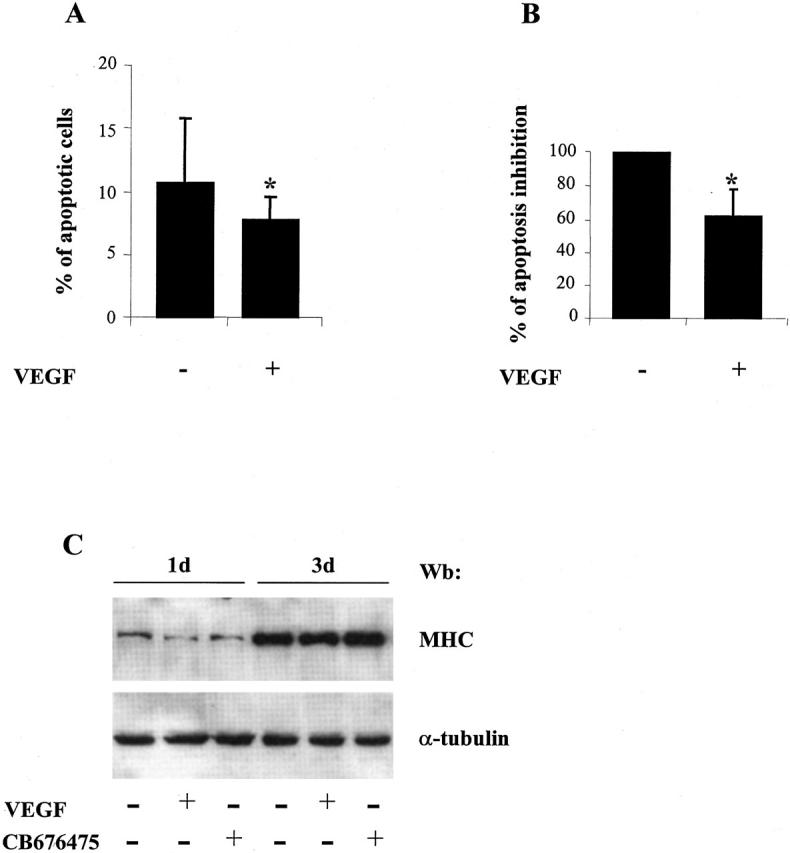
Effect of VEGF on differentiation-induced apoptosis of C2C12 myoblasts. C2C12 myoblasts were plated at 10 5 cells/60-mm diameter dish and cultured for 72 hours in DM without or with 20 ng/ml of VEGF165. A: TUNEL labeling was used to detect apoptotic myoblasts in the cultures. Apoptotic nuclei were counted in 20 random fields at ×40 magnification and expressed as a percentage of total nuclei. Results represent mean ± SD of five independent experiments. The asterisk indicates a P ≤ 0.01. B: ELISA quantification of histone-associated fragments in C2C12 cultures. Inhibition of apoptosis was reported as a % of optical density reduction between untreated and VEGF-treated C2C12 cells. Results represent mean ± SD of six independent experiments. The asterisk indicates a P ≤ 0.001. C: Effect of VEGF and CD676475 treatment on C2C12 myogenic differentiation. Western blot analysis of total extract from C2C12 cells treated for the indicated time-points either with VEGF or CB676475. Myogenic differentiation was assessed as MyHC expression with MF20 Mab. The same filter was probed with anti α-tubulin Mab to show equal protein concentration (lower panel).
Effect of Hypoxia on VEGF, Flk-1, and Flt-1 Expression in C2C12 Cells
Cell hypoxia is an environmental stress which occurs in many pathological conditions including ischemia. We sought to determine whether C2C12 cell exposure to hypoxia, modulated Flk-1 and Flt-1 expression as observed in vivo (Figure 2) ▶ . Western blot analysis performed on C2C12 culture lysates after 48 hours either in hypoxic or normoxic conditions, showed that Flk-1 and Flt-1 protein levels were unchanged in both experimental conditions (Figure 8A) ▶ . In contrast, VEGF levels in conditioned media increased approximately fivefold in response to 48-hour hypoxia (Figure 8B) ▶ . It is noteworthy that the C2C12 cell count per plate remained virtually constant 24 and 48 hours after exposure to hypoxia and only 2.8% of apoptotic cells were detected after 48 hours of culture in hypoxic medium (Figure 9, A and B) ▶ . To analyze whether the increased VEGF production observed in hypoxia was involved in hypoxia-mediated inhibition of apoptosis, neutralizing Flk-1 antibody (Figure 9A and B) ▶ or CB676475 (data not shown) were administered to C2C12 cells kept in hypoxic DM for 48 hours. In both instances, cultures containing either of these agents exhibited increased apoptosis as evaluated by TUNEL labeling (Figure 9, A and B) ▶ .
Figure 8.

Effect of hypoxia on the expression of VEGF and its receptors by C2C12 myoblasts. A: Cell lysates were prepared from C2C12 cultured in DM cells and kept either in normoxia or hypoxia for 48 hours and subjected to Western blot analysis using anti-Flk-1 and anti-Flt-1 Mabs. The same membrane was probed with anti α-tubulin antibody to confirm equal loading of the lanes. B: ELISA determination of VEGF production from normoxic and hypoxic C2C12 cells. CM from 1 day culture of C2C12 in normoxia and hypoxia conditions were collected. VEGF production was detected by ELISA as described in Materials and Methods. Results represent mean ± SD of four experiments. The asterisk indicates a P ≤ 0.05.
Figure 9.

Effect of Flk-1 and Flt-1 inactivation on hypoxia-mediated inhibition of C2C12 apoptosis. C2C12 myoblasts were plated in GM at 2 × 10 5 cells/60-mm diameter dish for 24 hours. Thereafter cells were switched to DM and cultured either in normoxic or hypoxic conditions for 48 hours. nFlk-1 (0.5 μg/ml) was added to the culture medium for the entire period of treatment. TUNEL labeling was used to detect apoptotic myoblasts. A: Micrographs: left panels illustrate the fluorescent TUNEL images from a representative experiment while right panels illustrate Hoechst staining of the same cells. B: Quantification of apoptotic cells obtained in the experimental conditions described for A. TUNEL-positive cells and total Hoechst-stained nuclei were counted on 20 fields for each experiment. Results represent mean ± SD of six independent experiments. The asterisk indicates a P ≤ 0.001.
Effect of AdCMV.VEGF165 in Ischemic Skeletal Muscle Cell Apoptosis
In these experiments, we evaluated the occurrence of apoptosis in muscle fibers following ischemia and the effect of adenovirus-mediated VEGF165 gene transfer in this phenomenon. AdCMV.VEGF165 was injected (see Methods) two days before surgery (n = 4) and animals treated with AdCMV.Null (n = 4) were used as controls. The efficiency of transduction was confirmed by immunohistochemical analysis for VEGF expression in muscle sections from both AdCMV.VEGF165 and AdCMV.Null injected muscles (data not shown). Only few TUNEL-positive nuclei were observed in normoperfused muscles. At 8 hours after ischemia, apoptotic nuclei were readily detected in muscle fibers of AdCMV.Null injected mice (Figure 10, A and D) ▶ . AdCMV.VEGF165 inhibited apoptosis in muscle cells (Figure 10, B and D) ▶ as well as in other cell types such as endothelial and smooth muscle cells (data not shown). Similar results were obtained 24 hours after ischemia, but quantification was difficult due to progressing tissue degeneration (not shown).
Figure 10.

In vivo effect of Ad.VEGF on ischemia-induced skeletal muscle apoptosis. Apoptosis was measured by TUNEL assay 8 hours after femoral artery ligation. Representative sections of ischemic adductor muscles treated with AdCMV.Null (A), AdCMV.VEGF165 (B), or DNAsi as a positive control (C). Arrowhead indicates apoptotic nuclei. Inset shows a higher-power photomicrograph of TUNEL-positive skeletal muscle nuclei indicated by the arrowhead. Magnification ×40; bar 25 μm. D: Bar graph of the mean TUNEL-positive skeletal muscle nuclei number/mm 2 ×10 6 cells from normoperfused and ischemic skeletal muscle injected either with Ad.CMV.Null or Ad.CMV.VEGF. The asterisk indicates a P < 0.05 vs. AdCMV.Null.
Discussion
The results of the present study show that VEGF receptors Flk-1 and Flt-1 are expressed by quiescent satellite cells in vivo and that their expression levels are modulated following acute ischemia, during satellite cell differentiation. Further, it is shown that VEGF increases Flk-1 phosphorylation and modulates skeletal myoblast function and survival in vitro and in vivo.
Skeletal muscle regeneration is a tightly regulated process involving multiple growth factors and cytokines. Although the role of VEGF and its receptors has been described in the regulation of blood vessel formation and hematopoiesis, the involvement of VEGF, Flk-1, and Flt-1 in muscle regeneration is still unknown. Prior reports examined the expression of VEGF, Flk-1, and Flt-1 in limb ischemia; however, the results have been controversial. Most studies in animal models have shown that VEGF mRNA and protein expression are either very low or absent in normoperfused limbs. 10,20 Following the induction of ischemia, both VEGF mRNA and protein increase predominantly in skeletal muscle cells and peak expression is achieved 1 to 2 days after surgery. Enhanced VEGF expression in skeletal muscle during both acute and chronic ischemia has also been described in human specimens. 10 In contrast to these studies, it has been reported that in a rabbit model of hindlimb ischemia, VEGF mRNA and protein did not increase in the ischemic quadriceps muscle during the first week following femoral artery ligation. 40 Fewer studies have addressed the effect of limb ischemia on VEGF receptors expression in skeletal muscle cells. Flk-1 increases in ischemic human and rabbit 10 as well as in dog 41 skeletal muscle while the effect of ischemia on Flt-1 expression in skeletal muscle has not been previously described. It is noteworthy that Flk-1 and Flt-1 mRNA levels have been examined in rabbit collateral arteries at different times following femoral artery ligation; the levels of both receptors transcripts were very low and did not vary after ischemia. 40
In the present study it is shown that both Flk-1 and Flt-1 were expressed in satellite cells of normoperfused adductor muscle. After the induction of ischemia, both receptors were identified in activated satellite cells and in regenerating skeletal muscle fibers. However, the expression of both receptors in mature muscle fibers was very low. The patterns of expression observed in vivo in undifferentiated and differentiating myoblasts, as well as in mature fibers, were also found in C2C12 cells cultured in growing medium and at different times during differentiation in vitro. In fact, the high levels of Flk-1 and Flt-1 protein found in undifferentiated C2C12 cells progressively decreased to very low levels as C2C12 cells differentiated. Therefore, Flk-1 and Flt-1 expression appeared subordinate to the proliferative state of myoblasts since a reduction of these receptors was observed after induction of differentiation. In contrast, as previously shown by others, 42,43 VEGF in the conditioned medium increased during C2C12 myoblast differentiation. This result apparently did not correlate with our in vivo observation showing a decrease of VEGF expression during skeletal muscle regeneration following femoral artery ligation. However, in light of the markedly different experimental conditions, VEGF secretion by differentiating C2C12 cells in vitro and VEGF expression by skeletal muscle fibers in response to ischemia cannot be compared.
Negative modulation of genes encoding other growth factor receptors has been observed in muscle cells when they enter the differentiation pathway. 44-46 This mechanism seems to contribute to the irreversible withdrawal from the cell cycle and, consequently, the stable expression of muscle-specific phenotype. Moreover, the results of the present study show that VEGF enhanced skeletal myoblast survival. This result is in agreement with the known effect of VEGF, to improve endothelial cell survival 47 by activating the serine-threonine protein kinase AKT. Exogenous VEGF decreased the number of apoptotic C2C12 cells during differentiation. Hypoxia increased VEGF secretion by C2C12 cells and reduced apoptosis following growth factor deprivation. It is noteworthy that under our experimental conditions the anti-apoptotic effect of VEGF played a dominant role over other anti-apoptotic factors potentially secreted by the cells. In fact, impairment of VEGF signaling led to massive apoptosis. The anti-apoptotic effect of VEGF did not interfere with the myogenic differentiation process since neither VEGF administration nor VEGF receptor inhibition modified the differentiative capacity of myogenic cells in vitro. Since apoptosis occurs during myogenesis and involves cells that do not withdraw from the cell cycle, it is possible that VEGF may exhibit its anti-apoptotic effect on these cells which fail to differentiate. Prior studies have shown that reperfusion injury occurs in skeletal muscle and it induces both apoptosis and necrosis. 48-50 However, the role of ischemia per se on skeletal muscle cell viability is still unknown. In the present study it was shown that hindlimb ischemia 8 hours following femoral artery ligation induced skeletal muscle cell apoptosis and that this effect was markedly inhibited in hindlimbs injected with AdCMV.VEGF165 48 hours prior the induction of ischemia. Taken together in vivo and in vitro results indicate that VEGF has a powerful anti-apoptotic action on skeletal muscle cells. Further, it is possible that VEGF could play an important role in preventing apoptosis in muscular dystrophy, in neuromuscular disorder 49 and possibly that it may coordinate the regulation of cell proliferation and death during embryonic development. 51
The agreement between the observations in vitro and in vivo described in the present study and the previously reported modulation of the expression of VEGF and Flk-1 by skeletal muscle cells in ischemic limbs 10 suggest that, in addition to an angiogenic effect, VEGF may also have a direct autocrine and paracrine action on skeletal muscle regeneration. A comparable direct action on muscle tissue may also be expected in response to therapeutic angiogenesis interventions in which VEGF gene transfer to the ischemic limb is used to improve blood flow. Accordingly, it is expected that the VEGF autocrine loop would become established only when satellite cells are induced to replicate and migrate to regions of muscle fiber damage. The initial release of VEGF into the local environment may prolong survival of cells that are not irreversibly damaged until angiogenesis is initiated. Further, since VEGF is locally produced in ischemic skeletal muscle by regenerating muscle cells, VEGF may attract satellite cells into muscle regenerating areas. Since homozygous deletion of both flk-1 and flt-1 resulted in mice death at embryonic day 8.5 52-54 for early defects in the development of hematopoietic and endothelial cells, we do not know whether VEGF plays a role in myoblast migration and survival during development. However it has been reported that VEGF is expressed by the somites of Xenopus and avian embryos and this expression modulates angioblast migration from the lateral plate of mesoderm, below the somites toward the midline of the embryo, where they organize into the dorsal aorta. 52,55 Although VEGF has never been shown to be a chemoattractant for myoblasts, it is possible that VEGF-mediated signaling modulates migration of ECs together with myogenic precursors inside developing muscle. Moreover, skeletal myogenic progenitors, expressing a number of endothelial and myogenic markers including Flk-1, have been identified in the embryonic dorsal aorta of mouse embryos 56 and have been shown to contribute to postnatal growth and regeneration of skeletal muscle. Therefore, it is conceivable that VEGF may also contribute to the recruitment of these precursors into the regenerating muscle. This could represent a coordinated process in which muscle regeneration and angiogenesis share a common molecular trigger.
Footnotes
Address reprint requests to Antonia Germani, Laboratorio di Biologia Vascolare e Terapia Genica, Centro Cardiologico Fondazione “I. Monzino,” Via Parea, 4, 20138 Milano, Italy. E-mail: a.germani@idi.it.
A. Germani and A. Di Carlo contributed equally to this work.
References
- 1.Matsumoto T, Claesson-Welsh L: VEGF receptor signal transduction. Sci STKE 2001, 112:1-17 [DOI] [PubMed] [Google Scholar]
- 2.Asahara T, Takahashi T, Masuda H, Kalka C, Chen D, Iwaguro H, Inai Y, Silver M, Isner JM: VEGF contributes to postnatal neovascularization by mobilizing bone marrow-derived endothelial progenitor cells. EMBO J 1999, 18:3964-3972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kalka C, Masuda H, Takahashi T, Gordon R, Tepper O, Gravereaux E, Pieczek A, Iwaguro H, Hayashi SI, Isner JM, Asahara T: Vascular endothelial growth factor(165) gene transfer augments circulating endothelial progenitor cells in human subjects. Circ Res 2000, 86:1198-1202 [DOI] [PubMed] [Google Scholar]
- 4.Kalka C, Tehrani H, Laudenberg B, Vale PR, Isner JM, Asahara T, Symes JF: VEGF gene transfer mobilizes endothelial progenitor cells in patients with inoperable coronary disease. Ann Thorac Surg 2000, 70:829-834 [DOI] [PubMed] [Google Scholar]
- 5.Tischer E, Mitchell R, Hartman T, Silva M, Gospodarowicz D, Fiddes JC, Abraham JA: The human gene for vascular endothelial growth factor: multiple protein forms are encoded through alternative exon splicing. J Biol Chem 1991, 266:11947-11954 [PubMed] [Google Scholar]
- 6.Ferrara N, Houck K, Jakeman L, Leung DW: Molecular and biological properties of the vascular endothelial growth factor family of proteins. Endocr Rev 1992, 13:18-32 [DOI] [PubMed] [Google Scholar]
- 7.Ishida A, Murray J, Saito Y, Kanthou C, Benzakour O, Shibuya M, Wijelath ES: Expression of vascular endothelial growth factor receptors in smooth muscle cells. J Cell Physiol 2001, 188:359-368 [DOI] [PubMed] [Google Scholar]
- 8.Deckers MM, Karperien M, van der Bent C, Yamashita T, Papapoulos SE, Lowik CW: Expression of vascular endothelial growth factors and their receptors during osteoblast differentiation. Endocrinology 2000, 141:1667-1674 [DOI] [PubMed] [Google Scholar]
- 9.Takahashi N, Seko Y, Noiri E, Tobe K, Kadowaki T, Sabe H, Yazaki Y: Vascular endothelial growth factor induces activation and subcellular translocation of focal adhesion kinase (p125FAK) in cultured rat cardiac myocytes. Circ Res 1999, 84:1194-1202 [DOI] [PubMed] [Google Scholar]
- 10.Rissanen TT, Vajanto I, Hiltunen MO, Rutanen J, Kettunen MI, Niemi M, Leppanen P, Turunen MP, Markkanen JE, Arve K, Alhava E, Kauppinen RA, Yla-Herttuala S: Expression of vascular endothelial growth factor and vascular endothelial growth factor receptor-2 (KDR/Flk-1) in ischemic skeletal muscle and its regeneration. Am J Pathol 2002, 160:1393-1403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sondell M, Lundborg G, Kanje M: Vascular endothelial growth factor has neurotrophic activity and stimulates axonal outgrowth, enhancing cell survival and Schwann cell proliferation in the peripheral nervous system. J Neurosci 1999, 19:5731-5740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gerber HP, Malik AK, Solar GP, Sherman D, Liang XH, Meng G, Hong K, Marsters JC, Ferrara N: VEGF regulates haematopoietic stem cell survival by an internal autocrine loop mechanism. Nature 2002, 417:954-958 [DOI] [PubMed] [Google Scholar]
- 13.Grosskreutz CL, Anand-Apte B, Duplaa C, Quinn TP, Terman BI, Zetter B, D’Amore PA: Vascular endothelial growth factor-induced migration of vascular smooth muscle cells in vitro. Microvasc Res 1999, 58:128-136 [DOI] [PubMed] [Google Scholar]
- 14.Gerber HP, Vu TH, Ryan AM, Kowalski J, Werb Z, Ferrara N: VEGF couples hypertrophic cartilage remodeling, ossification and angiogenesis during endochondral bone formation. Nat Med 1999, 5:623-628 [DOI] [PubMed] [Google Scholar]
- 15.Robinson GS, Ju M, Shih SC, Xu X, McMahon G, Caldwell RB, Smith LE: Nonvascular role for VEGF: vEGFR-1, 2 activity is critical for neural retinal development. EMBO J 2001, 15:1215-1217 [DOI] [PubMed] [Google Scholar]
- 16.Jin KL, Mao XO, Greenberg DA: Vascular endothelial growth factor: direct neuroprotective effect in in vitro ischemia. Proc Natl Acad Sci USA 2000, 97:10242-10247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ogunshola OO, Antic A, Donoghue MJ, Fan SY, Kim H, Stewart WB, Madri JA, Ment LR: Paracrine and autocrine functions of neuronal vascular endothelial growth factor (VEGF) in the central nervous system. J Biol Chem 2002, 277:11410-11415 [DOI] [PubMed] [Google Scholar]
- 18.Shweiki D, Itin A, Soffer D, Keshet E: Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature 1992, 359:843-845 [DOI] [PubMed] [Google Scholar]
- 19.Banai S, Shweiki D, Pinson A, Chandra M, Lazarovici G, Keshet E: Up-regulation of vascular endothelial growth factor expression induced by myocardial ischaemia: implications for coronary angiogenesis. Cardiovasc Res 1994, 28:1176-1179 [DOI] [PubMed] [Google Scholar]
- 20.Couffinhal T, Silver M, Zheng LP, Kearney M, Witzenbichler B, Isner JM: Mouse model of angiogenesis. Am J Pathol 1998, 152:1667-1679 [PMC free article] [PubMed] [Google Scholar]
- 21.Milkiewicz M, Brown MD, Egginton S, Hudlicka O: Association between shear stress, angiogenesis, and VEGF in skeletal muscles in vivo. Microcirculation 2001, 8:229-241 [DOI] [PubMed] [Google Scholar]
- 22.Waltenberger J, Claesson-Welsh L, Siegbahn A, Shibuya M, Heldin CH: Different signal transduction properties of KDR and Flt1, two receptors for vascular endothelial growth factor. J Biol Chem 1994, 269:26988-26995 [PubMed] [Google Scholar]
- 23.Couffinhal T, Silver M, Kearney M, Sullivan A, Witzenbichler B, Magner M, Annex B, Peters K, Isner JM: Impaired collateral vessel development associated with reduced expression of vascular endothelial growth factor in ApoE−/− mice. Circulation 1999, 99:3188-3198 [DOI] [PubMed] [Google Scholar]
- 24.Van Belle E, Rivard A, Chen D, Silver M, Bunting S, Ferrara N, Symes JF, Bauters C, Isner JM: Hypercholesterolemia attenuates angiogenesis but does not preclude augmentation by angiogenic cytokines. Circulation 1997, 96:2667-2674 [DOI] [PubMed] [Google Scholar]
- 25.Rivard A, Fabre JE, Silver M, Chen D, Murohara T, Kearney M, Magner M, Asahara T, Isner JM: Age-dependent impairment of angiogenesis. Circulation 1999, 99:111-120 [DOI] [PubMed] [Google Scholar]
- 26.Rivard A, Berthou-Soulie L, Principe N, Kearney M, Curry C, Branellec D, Semenza GL, Isner JM: Age-dependent defect in vascular endothelial growth factor expression is associated with reduced hypoxia-inducible factor 1 activity. J Biol Chem 2000, 275:29643-29647 [DOI] [PubMed] [Google Scholar]
- 27.Isner JM, Asahara T: Angiogenesis and vasculogenesis as therapeutic strategies for postnatal neovascularization. J Clin Invest 1999, 103:1231-1236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gowdak LH, Poliakova L, Wang X, Kovesdi I, Fishbein KW, Zacheo A, Palumbo R, Straino S, Emanueli C, Marrocco-Trischitta M, Lakatta EG, Anversa P, Spencer RG, Talan M, Capogrossi MC: Adenovirus-mediated VEGF(121) gene transfer stimulates angiogenesis in normoperfused skeletal muscle and preserves tissue perfusion after induction of ischemia. Circulation 2000, 102:565-571 [DOI] [PubMed] [Google Scholar]
- 29.Perry RL, Rudnick MA: Molecular mechanisms regulating myogenic determination and differentiation. Front Biosci 2000, 5:D750-D767 [DOI] [PubMed] [Google Scholar]
- 30.Hawke TJ, Garry DJ: Myogenic satellite cells: physiology to molecular biology. J Appl Physiol 2001, 91:534-551 [DOI] [PubMed] [Google Scholar]
- 31.Yaffe D, Saxel O: A myogenic cell line with altered serum requirements for differentiation. Differentiation 1977, 7:159-166 [DOI] [PubMed] [Google Scholar]
- 32.Rando TA, Blau HM: Primary mouse myoblast purification, characterization, and transplantation for cell-mediated gene therapy. J Cell Biol 1994, 125:1275-1287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Strawn LM, McMahon G, App H, Schreck R, Kuchler WR, Longhi MP, Hui TH, Tang C, Levitzki A, Gazit A, Chen I, Keri G, Orfi L, Risau W, Flamme I, Ullrich A, Hirth KP, Shawver LK: Flk-1 as a target for tumor growth inhibition. Cancer Res 1996, 56:3540-3545 [PubMed] [Google Scholar]
- 34.Turrini P, Gaetano C, Antonelli A, Capogrossi MC, Aloe L: Nerve growth factor induces angiogenic activity in a mouse model of hindlimb ischemia. Neurosci Lett 2002, 323:109-112 [DOI] [PubMed] [Google Scholar]
- 35.Muhlhauser J, Merrill MJ, Pili R, Maeda H, Bacic M, Bewig B, Passaniti A, Edwards NA, Crystal RG, Capogrossi MC: VEGF165 expressed by a replication-deficient recombinant adenovirus vector induces angiogenesis in vivo. Circ Res 1995, 77:1077-10786 [DOI] [PubMed] [Google Scholar]
- 36.Bischoff R: The satellite cell and muscle regeneration. Engel AG Franzini-Armstrong C eds. Myology. 1994:pp 97-118 McGraw-Hill New York
- 37.Kaufman SJ, Foster RF: Replicating myoblasts express a muscle-specific phenotype. Proc Natl Acad Sci USA 1988, 85:9606-9610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shen BQ, Lee DY, Gerber HP, Keyt BA, Ferrara N, Zioncheck TF: Homologous up-regulation of KDR/Flk-1 receptor expression by vascular endothelial growth factor in vitro. J Biol Chem 1998, 273:29979-29985 [DOI] [PubMed] [Google Scholar]
- 39.Hennequin LF, Thomas AP, Johnstone C, Stokes ES, Lohmann JJ, Ogilvie DJ, Dukes M, Wedge SR, Curwen JO, Kendrew J, Lambert-van der Brempt C: Design and structure-activity relationship of a new class of potent VEGF receptor tyrosine kinase inhibitors. J Med Chem 1999, 42:5369-5389 [DOI] [PubMed] [Google Scholar]
- 40.Deindl E, Buschmann I, Hoefer IE, Podzuweit T, Boengler K, Vogel S, van Royen N, Fernandez B, Schaper W: Role of ischemia and of hypoxia-inducible genes in arteriogenesis after femoral artery occlusion in the rabbit. Circ Res 2001, 89:779-786 [DOI] [PubMed] [Google Scholar]
- 41.Miraliakbari R, Francalancia NA, Lust RM, Gerardo JA, Ng PC, Sun YS, Chitwood WR, Jr: Differences in myocardial and peripheral VEGF and KDR levels after acute ischemia. Ann Thorac Surg 2000, 69:1750-1754 [DOI] [PubMed] [Google Scholar]
- 42.Claffey KP, Wilkison WO, Spiegelman BM: Vascular endothelial growth factor: regulation by cell differentiation and activated second messenger pathways. J Biol Chem 1992, 267:16317-16322 [PubMed] [Google Scholar]
- 43.Takahashi A, Kureishi Y, Yang J, Luo Z, Guo K, Mukhopadhyay D, Ivashchenko Y, Branellec D, Walsh K: Myogenic Akt signaling regulates blood vessel recruitment during myofiber growth. Mol Cell Biol 2002, 22:4803-4814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Florini JR, Magri KA: Effects of growth factors on myogenic differentiation. Am J Physiol 1989, 256:C701-C711 [DOI] [PubMed] [Google Scholar]
- 45.Florini JR, Ewton DZ, Magri KA: Hormones, growth factors, and myogenic differentiation. Annu Rev Physiol 1991, 53:201-216 [DOI] [PubMed] [Google Scholar]
- 46.Olson EN: Interplay between proliferation and differentiation within the myogenic lineage. Dev Biol 1992, 154:261-272 [DOI] [PubMed] [Google Scholar]
- 47.Gerber HP, McMurtrey A, Kowalski J, Yan M, Keyt BA, Dixit V, Ferrara N: Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3′-kinase/Akt signal transduction pathway: requirement for Flk-1/KDR activation. J Biol Chem 1998, 273:30336-30343 [DOI] [PubMed] [Google Scholar]
- 48.Cowled PA, Leonardos L, Millard SH, Fitridge RA: Apoptotic cell death makes a minor contribution to reperfusion injury in skeletal muscle in the rat. Eur J Vasc Endovasc Surg 2001, 21:28-34 [DOI] [PubMed] [Google Scholar]
- 49.Adams V, Gielen S, Hambrecht R, Schuler G: Apoptosis in skeletal muscle. Front Biosci 2001, 6:D1-D11 [DOI] [PubMed] [Google Scholar]
- 50.Hatoko M, Tanaka A, Kuwahara M, Yurugi S, Iioka H, Niitsuma K: Difference of molecular response to ischemia-reperfusion of rat skeletal muscle as a function of ischemic time: study of the expression of p53, p21(WAF-1), Bax protein, and apoptosis. Ann Plast Surg 2002, 48:68-74 [DOI] [PubMed] [Google Scholar]
- 51.Walsh K: Coordinate regulation of cell cycle and apoptosis during myogenesis. Prog Cell Cycle Res 1997, 3:53-58 [DOI] [PubMed] [Google Scholar]
- 52.Cleaver O, Krieg PA: VEGF mediates angioblast migration during development of the dorsal aorta in Xenopus. Development 1998, 125:3905-3914 [DOI] [PubMed] [Google Scholar]
- 53.Shalaby F, Rossant J, Yamaguchi TP, Gertsenstein M, Wu XF, Breitman ML, Schuh AC: Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature 1995, 376:62-66 [DOI] [PubMed] [Google Scholar]
- 54.Fong GH, Rossant J, Gertsenstein M, Breitman ML: Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature 1995, 376:66-70 [DOI] [PubMed] [Google Scholar]
- 55.Aitkenhead M, Christ B, Eichmann A, Feucht M, Wilson DJ, Wilting J: Paracrine and autocrine regulation of vascular endothelial growth factor during tissue differentiation in the quail. Dev Dyn 1998, 212:1-13 [DOI] [PubMed] [Google Scholar]
- 56.De Angelis L, Berghella L, Coletta M, Lattanzi L, Zanchi M, Cusella-De Angelis MG, Ponzetto C, Cossu G: Skeletal myogenic progenitors originating from embryonic dorsal aorta coexpress endothelial and myogenic markers and contribute to postnatal muscle growth and regeneration. J Cell Biol 1999, 147:869-878 [DOI] [PMC free article] [PubMed] [Google Scholar]