

Abstract
Genomic ablation of hepatocyte-specific fibroblast growth factor receptor (FGFR)4 in mice revealed a role of FGF signaling in cholesterol and bile acid metabolism and hepatolobular restoration in response to injury without effect on liver development or hepatocyte proliferation. Although the potential role of all 23 FGF polypeptides in the liver is still unclear, the most widely studied prototypes, FGF1 and FGF2, are present and have been implicated in liver cell growth and function in vitro. To determine whether FGF1 and FGF2 play a role in response to injury and fibrosis, we examined the impact of both acute and chronic exposure to carbon tetrachloride (CCl4) in the livers of FGF1- and FGF2-deficient mice. After acute CCl4 exposure, FGF1(−/−)FGF2(−/−) mice exhibited an accelerated release of serum alanine aminotransferase similar to FGFR4 deficiency, but no effect on overall hepatolobular restoration or bile acid metabolism. FGF1(−/−)FGF2(−/−) mice exhibited a normal increase in α-smooth muscle actin and desmin associated with activation and migration of hepatic stellate cells to damage, but a reduced level of hepatic stellate cell-derived matrix collagen α1(I) synthesis. Liver fibrosis resulting from chronic CCl4 exposure was markedly decreased in the livers of FGF1/FGF2-deficient mice. These results suggest an agonist role for FGF1 and FGF2 in specifically insult-induced liver matrix deposition and hepatic fibrogenesis and a potential target for the prevention of hepatic fibrosis.
The fibroblast growth factors (FGFs) comprise a family of 23 reported members that have varying affinities for variants of four different FGF receptor kinases (FGFR1 to FGFR4). 1-3 FGF1 and FGF2, the first two cloned members of the FGF family, 4,5 have received the most study, are widely expressed, and thus, predicted to be involved in tissue-specific functions and associated pathologies at their site of expression. 6,7 In vitro FGF1 affects cells of multiple origin whereas activity of FGF2 appears more limited to cells derived from mesenchyme and neuroectoderm. 8 FGF1 and FGF2 have been implicated in derivation of the liver from foregut endoderm. 9 However, mice lacking FGF1, FGF2, or both FGF1 and FGF2 are viable, fertile, and grossly indistinguishable from wild type (WT) except for modest defects in cardiovascular tissue, healing of skin wounds, and neuronal tissue. 3,10,11 This suggests that FGF1 and FGF2 alone are not essential for embryonic development or are compensated by other members of the extensive FGF ligand family. Consequently, FGF1- and FGF2-deficient animals are available for study of the role of the two factors in adult tissue homeostasis. Despite their ubiquity, little has emerged except for the modest effects of ablation of FGF1 and FGF2 in the cardiovascular, skin, and nervous systems.
Although the levels of mRNA transcripts are low, long-lived FGF1 and FGF2 polypeptides are present in the resting liver at significant levels. 12 This suggests that a significant reservoir of both ligands is present in the resting liver in an inactive state before activation and synthesis initiated by loss of liver mass or damage. Levels of mRNA and FGF1 and FGF2 polypeptides increase in the regenerating liver; 13,14 however, the pre-existent protein stores and the time frame suggests that the increase may be replenishment of stores or for support of long-term events in liver repair rather than triggering the hepatocyte proliferative response. Recently, we have shown that ablation of FGFR4, the only member of the FGFR family in hepatocytes, has little effect on the hepatocyte proliferative response to partial hepatectomy or insult, but plays a major role in homeostatic regulation of bile acid/cholesterol and potentially other metabolic functions and balanced hepatolobular restoration after carbon tetrachloride (CCl4) injury. 15 Dependent on cell-specific heparan sulfate, FGF1 and FGF2 are capable of binding to and activating FGFR4 in vitro. 16 Here, we show that the absence of FGF1 and FGF2 resulted in accelerated and increased hepatocyte necrosis, but without impairment of normal liver repair processes after acute CCl4 treatment. The absence of FGF1 and FGF2 caused a decrease in collagen α1(I) induction during the activation of hepatic stellate cells (HSCs) after acute CCl4 treatment and markedly decreased fibrosis that results from chronic CCl4 treatment. These results differ significantly from the impact of ablation of hepatocyte FGFR4 on the response of liver to CCl4 insult. They suggest a role of FGF1 and FGF2 in the response of nonparenchymal cells to insult and in indirect impact on hepatocytes rather than a role of activating ligands for hepatocyte FGFR4 functions.
Materials and Methods
Animals and Administration of CCl4
Generation of FGF1(−/−)/FGF2(−/−) double-knockout mice have been described. 10,11 Both FGF1(−/−)/FGF2(−/−) mice generated by crossing FGF1(−/−) and FGF2(−/−) and WT control mice homozygous for both FGF1 and FGF2 were 129 Sv:C57BL/6 mixes and bred in parallel for duration of the study. Single-knockout FGF1(−/−) and FGF2(−/−) mice for this study were identified by genotype after backcrossing the FGF1(−/−)/FGF2(−/−) mice with WT mice. Experiments were limited to 7- to 8-week-old males. Mice were maintained in 12-hour light/12-hour dark cycles with free access to food and water. Three to five mice were used for each experimental group as described in the text. For acute CCl4-induced liver injury, a single dose of 2.0 ml/kg of body weight (2:5 v/v in mineral oil) was administered by intraperitoneal injection. For chronic CCl4-induced liver injury, a dose of 2.0 ml/kg of body weight of CCl4 was administered intraperitoneally twice a week. Livers were excised for analysis after the mice were weighed, anesthetized, and exsanguinated. All procedures were performed in accordance with the Institutional Animal Care and Use Committee at the Institute of Biosciences and Technology, Texas A&M University System Health Science Center.
Partial Hepatectomy and Hepatocyte DNA Synthesis
A 70% hepatectomy, consisting of removal of the anterior and left lateral hepatic lobes, performed on male mice at 10:00 a.m. was performed as previously described. 17 Remnant livers were removed and weighed after 7 days.
Measurement of hepatocyte DNA synthesis was performed as described. 17 Briefly, 2 hours before sacrifice of the animals for analysis, 50 μg per g body weight of bromodeoxyuridine (BrdU) was administered intraperitoneally. The livers were removed and weighed at the times indicated in the text. BrdU incorporation in fixed liver sections was visualized with an anti-BrdU monoclonal antibody (no. 2531; Sigma, St. Louis, MO) and an alkaline phosphatase-conjugated second antibody. Positively stained nuclei among 1000 to 2000 nuclei per field in four to five fields (magnification, ×100) per tissue section for three different sections from each mouse was counted manually. BrdU incorporation was expressed as the percentage of positive-staining hepatocyte nuclei.
Blood Enzyme and Bile Acid Analysis
Blood plasma levels of alanine transaminase (aminotransferase) (ALT) activity were measured using the GP-transaminase kit (no. 505-P, Sigma). Fecal bile acid excretion was measured enzymatically using the Bile Acids kit (no. 450-A, Sigma) described previously. 17 Fecal bile acid excretion was expressed as μmol/day/100 g body weight. The total bile acid pool size was determined as bile acid content of the small intestine, the gallbladder, the liver, and their contents described previously. 17 The pool size was expressed as μmol/100 g body weight.
Histological Analysis
Liver tissues were fixed overnight in Histochoice Tissue Fixative MB (no. H120-4L; Amresco, Solon, OH), dehydrated through a series of ethanol treatments, and embedded in paraffin according to standard procedure. Liver sections were prepared and stained with hematoxylin and eosin and for collagen using Sirius Red (0.02%). For α-smooth muscle actin (SMA) immunostaining, liver sections were incubated with a horseradish peroxidase-conjugating anti-α-SMA antibody (no. U7033; DAKO, Glostrup, Denmark) followed by avidin-biotin-peroxidase complex (ABC) reagent (Amersham, Piscataway, NJ).
Collagen Determination
The total hepatic collagen in paraffin-embedded liver sections was determined according to the method of Lopez-De Leon and Rojkind. 18 Briefly, three 15-μm-thick liver sections from each mouse were deparaffinized and placed in 0.2 ml of a saturated solution of picric acid containing 0.1% Fast Green FCF and 0.1% Sirius Red. After washing with water, the dyes were eluted by 1 ml of 0.1 N NaOH in absolute methanol (1:1, v:v). The absorbance of eluted dyes at 540 and 605 nm was determined and used to calculate the amount of collagen that was expressed as μg of total collagen per mg of total protein.
Analysis of mRNA
Total RNA was isolated from livers with the Ultraspec RNA Isolation System (no. BL-10200; Biotecx Laboratories, Houston, TX), and specific mRNAs were measured by ribonuclease protection assay (RPA) using the HybSpeed RPA kit (no. 1412; Ambion, Austin, TX). Approximately 50 μg of liver RNA was hybridized with 1 × 105 cpm of [P32]-labeled specific anti-sense and β-actin riboprobes in the same reaction mixture. After treatment with ribonuclease, protected products were analyzed on 5% polyacrylamide sequencing gels, followed by autoradiography for a series of exposure times. The size of protection products was determined from the product of a DNA-sequencing reaction run parallel to the protection assays. The relative amount in arbitrary units of each radiographical product was assigned directly from the autoradiogram using a phosphorimager (Molecular Dynamics, Sunnyvale, CA). The relative intensity of experimental bands among samples was standardized by division of it by the intensity of the internal β-actin control band in each sample. Imager settings were at the minimal sensitivity compatible with adequate signal output to ensure detection of variations among β-actin standards among lanes. The level of expression in WT mice (treated or untreated as indicated) was assigned a value of 1. Images displayed in the text were captured at exposures designed to reveal differences in experimental bands rather than densitometric quantitation.
Murine collagen α1(I) cDNA was amplified by the reverse transcriptase-polymerase chain reaction from mouse liver using sense primer 5′-ACGTCCTGGTGAAGATGG-3′ and anti-sense primer 5′-GACCGCGTTCACCACTTG-3′. Murine β-actin cDNA has been described. 17 Murine transforming growth factor (TGF)-β1 cDNA was amplified by reverse transcriptase-polymerase chain reaction from mouse liver using sense primer 5′-GTCCAAACTAAGGCTCGC-3′ and anti-sense primer 5′-TATATACTGTGTGTGAGATGTC-3′. All products of reverse transcriptase-polymerase chain reaction were verified by sequencing.
Riboprobes complementary to part of the cDNAs described above that had been subcloned into pBluescript-SK were transcribed into [P32]-labeled anti-sense riboprobes by T3 or T7 RNA polymerase using the MAXIscript kit (no. 1326, Ambion). The size of probes and the predicted protected fragments were β-actin, 197 nucleotides and 139 nucleotides; collagen α1(I), 310 nucleotides and 236 nucleotides; and TGF-β1, 308 and 234 nucleotides, respectively.
Immunochemical Analyses
Livers were homogenized in phosphate-buffered saline containing 0.5% sodium deoxycholate and 0.1% sodium dodecyl sulfate and centrifuged. The protein concentration was determined using the BCA Protein Assay Reagent (no. 23225X; Pierce, Rockford, IL). A total of 25 μg of protein was subjected to 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, transferred to Hybond-P membrane (Amersham) that was incubated with a 1:200 dilution of mouse anti-human α-SMA monoclonal antibody (no. IMMH-2, Sigma) or mouse anti-desmin monoclonal antibody (no. D-1033, Sigma), washed, and then incubated with a 1:20,000 dilution of rabbit anti-mouse IgG conjugated to horseradish peroxidase (Bio-Rad, Hercules, CA). Bands were visualized by development with the Amersham ECL-Plus detection regents (Amersham).
Statistical Analyses
Values were expressed as the mean ± SD from the number of replicates described in the text. The statistical significance of differences between mean values (P < 0.05) was evaluated using the two-tailed Student’s t-test.
Results
No Effect of FGF1/FGF2 Deficiency on Bile Acid Metabolism and Partial Hepatectomy-Induced Liver Regeneration
Previously, we showed that mice lacking FGFR4, the only FGFR present in hepatocytes, exhibited elevated fecal bile acid excretion and bile acid pool size. 17 This implicated FGFR4 and its deficiency in cholesterol and bile acid metabolism in the liver. Because FGF1 and FGF2 are present at significant levels in the liver, we determined whether an FGF1/FGF2 deficiency would similarly affect bile acid and cholesterol metabolism by previously described methods. 17 The analysis revealed no differences in fecal bile acid excretion (7.3 ± 0.6 versus 7.5 ± 0.6 μmol/day/100 g body weight, n = 4 mice, P > 0.6) and bile acid pool size (58 ± 7 versus 55 ± 5 μmol/100 g body weight, n = 4, P > 0.5) between WT and FGF1(−/−)FGF2(−/−) mice, suggesting that in contrast to hepatocyte FGFR4, FGF1 and FGF2 do not play an essential uncompensated role in liver bile acid and cholesterol metabolism, 17 and, therefore, may not be essential activating ligands for hepatocyte FGFR4 in this metabolic domain. In addition, similar to mice with an FGFR4 deficiency, there was no difference in restoration of cellularity or liver mass after a 70% partial hepatectomy performed as described for the FGFR4 knockout mice (data not shown). 17 This suggested that similar to hepatocyte FGFR4, FGF1 and FGF2 do not play an essential uncompensated role in restoration of liver after loss of mass.
Accelerated Liver Injury Accompanied by Normal Hepatolobular Restoration in FGF1(−/−)FGF2(−/−) Mice after Acute CCl4 Administration
The ablation of FGFR4, the mature hepatocyte FGFR, results in accelerated damage indicated by release of liver alanine aminotransferase (ALT) accompanied by an accelerated schedule of DNA synthesis in response to the insult. 15 In addition, the FGFR4(−/−) livers exhibited a delay in hepatolobular restoration persisting long after DNA synthesis had ceased, accompanied by hypertrophy of the liver without an increase in cellularity. Despite mouse strain differences in the WT time curve, the overall time frame of lytic liver damage monitored by blood plasma liver ALT levels was accelerated in the livers of FGF1(−/−)FGF2(−/−) mice similar to the FGFR4(−/−) animals. At 24 and 38 hours after injection of CCl4, plasma ALT levels were 2.5 times that of WT mice at 24 and 38 hours after injection of CCl4 (Figure 1A) ▶ . In FGF1(−/−)FGF2(−/−) mice, plasma ALT levels peaked at 38 hours at ∼11,000 U/L ahead of the 48-hour peak in WT animals at ∼6000 U/L. By that time, ALT levels had fallen to 3000 U/L in the FGF1(−/−)FGF2(−/−) mice. In contrast to the FGFR4-deficient mice that exhibited an accelerated peak of DNA synthesis in response to CCl4 insult, 15 the FGF1(−/−)FGF2(−/−) mice exhibited a 12-hour delay in the peak hepatocyte DNA synthetic response (Figure 1B) ▶ . Although the FGF1(−/−)FGF2(−/−) mice exhibited altered peak ALT release and DNA synthesis after insult, the former similar to and the latter different from the FGFR4(−/−) mice, 15 the total extent of ALT release and DNA synthesis in response to insult was similar to WT when estimated by total area under the response curves. In addition, no difference could be observed in liver/body weight ratio after CCl4 administration in FGF1(−/−)FGF2(−/−) mice compared to WT controls (Figure 1C) ▶ . This was in marked contrast to the FGFR4(−/−) mice 15 that exhibited enlarged livers compared to WT controls.
Figure 1.

Accelerated injury in FGF1(−/−)FGF2(−/−) livers after acute CCl4 treatment. A: Plasma ALT levels after acute CCl4 treatment. ALT activity was measured using the GP-Transaminase kit (no. 505-P; Sigma, St. Louis, MO) as described. 15 Values were the mean ± SD (n = 4 mice). Significance of differences between FGF1(−/−)FGF2(−/−) and WT mice at 24, 38, and 48 hours was P < 0.05. B: Hepatocyte DNA synthesis after acute CCl4 treatment. DNA synthesis was assessed by BrdU incorporation as described in Materials and Methods. Values were the mean ± SD (n = 4 mice). Significance of differences between FGF1(−/−)FGF2(−/−) and WT mice at 38 and 48 hours was P < 0.02. C: Liver-to-total body weight ratio after acute CCl4 treatment. Livers were excised and weighed as described. 17 Values were the mean ± SD (n = 4 mice). Significance of the difference between FGF1(−/−)FGF2(−/−) and WT livers was P > 0.05 at all time points.
Consistent with the accelerated ALT release, histological examination of livers at the peak at 38 hours confirmed damage to livers of FGF1(−/−)FGF2(−/−) mice relative to WT controls (Figure 2) ▶ . FGF1(−/−)FGF2(−/−) livers showed severe centrizonal injury characterized by presence of scattered cell debris relative to the mild centrizonal necrosis observed in WT. However, at 48 hours after CCl4, a difference in the extent of hepatic necrosis between WT and the FGF1(−/−)FGF2(−/−) livers was not apparent (Figure 2) ▶ . At 168 hours, both FGF1(−/−)FGF2(−/−) and WT livers returned to normal and were indistinguishable. Despite the similarity to FGFR4-deficient mice indicated by the accelerated damage in the FGF1(−/−)FGF2(−/−) mice, complete recovery from the injury was unimpaired and similar to WT mice (Figure 2) ▶ . This was in marked contrast to observations in FGFR4-deficient animals in which hepatolobular restoration was severely impaired. 15
Figure 2.

Histology of livers from FGF1(−/−)FGF2(−/−) and WT mice after acute CCl4 treatment. Paraffin-embedded sections were prepared and stained with H&E as described in Materials and Methods. Note the severe centrizonal injury and cellular debris at 38 hours after CCl4 in FGF1(−/−)FGF2(−/−) livers relative to WT. Original magnifications, ×150.
Decreased Induction of Matrix Collagen α1(I) in FGF1(−/−)FGF2(−/−) Livers after Acute CCl4 Administration
The activation of nonparenchymal HSCs plays a key role in hepatic fibrogenesis in response to insult. 19 Based on results from in vitro experiments, FGF1 and FGF2 have been suggested as HSC activators. 20-22 A single acute dose of CCl4 administration can activate HSCs. 23 Activation of HSCs after acute CCl4 administration and hepatic injury 24 includes proliferation, 25 appearance of α-SMA, 26 and increased production of elements of the extracellular matrix including collagen α1(I). 27,28 Overexpression of collagen α1(I) and consequent formation of excessive and defective extracellular matrix is a hallmark of fibrotic abnormality in response to liver insult. 29,30 We investigated whether the lack of FGF1 and FGF2 affected the induction of hepatic collagen α1(I) in response to the acute CCl4 stimulus. Analysis of steady-state collagen α1(I) mRNA expression by RPA showed that in resting livers and those 24 hours after CCl4 insult, collagen α1(I) mRNA was equal in FGF1(−/−)FGF2(−/−) and WT animals. However, although collagen α1(I) mRNA expression increased ∼10-fold at 48 to 72 hours after CCl4 in WT livers, the increase was only approximately threefold in FGF1(−/−)FGF2(−/−) livers (Figure 3) ▶ . This suggested that the induction of collagen α1(I), presumably because of reduced activation of the source HSCs, was impaired in FGF1(−/−)FGF2(−/−) livers.
Figure 3.

Decreased induction of collagen α1(I) expression in FGF1(−/−)FGF2(−/−) livers after acute CCl4 treatment. Total liver RNA was isolated from three mice in each group at the indicated times after acute CCl4 treatment, pooled before assay by RNase protection, and relative intensity of the collagen α1(I) mRNA was quantitated and normalized as described in Materials and Methods. The signal in untreated WT livers was assigned a value of 1 and all other treatments expressed as a fold change over untreated wild type. P, labeled RNA probe. The displayed autoradiograph is representative of two independent reproductions.
To determine whether one or both FGFs contributed to the induction of collagen α1(I) after acute CCl4 insult, we compared the expression of collagen α1(I) mRNA expression in single-knockout mice FGF1 (−/−) and FGF2 (−/−) to the double-knockout FGF1(−/−)FGF2(−/−) and WT livers at 48 hours after CCl4 treatment. Although FGF1(−/−)FGF2(−/−) livers exhibited a reduced induction of collagen α1(I) mRNA of only ∼2.5-fold over untreated controls, the mice deficient in only FGF1 or FGF2 exhibited fivefold to sixfold increases more near the eightfold increase in WT livers (Figure 4) ▶ . Both untreated single- and double-knockout mice exhibited levels of α1(I) mRNA similar to that of untreated WT mice (WT-0) (not shown). This suggested that both FGF1 and FGF2 contribute additively to the induction of collagen α1(I) in livers after the acute CCl4 insult and that induction is significantly depressed in their combined absence.
Figure 4.
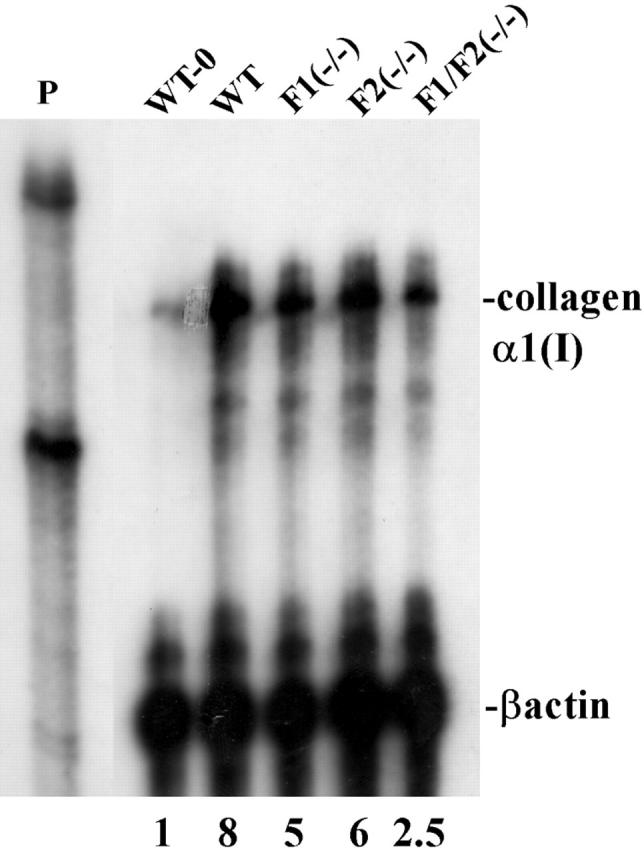
Both FGF1 and FGF2 contribute to induction of collagen α1(I) in livers after acute CCl4 administration. Collagen α1(I) mRNA was determined and quantitated as described in Figure 3 ▶ using 50 μg of pooled total RNA from three livers of mice with the indicated phenotype at 48 hours after CCl4 treatment, except the indicated untreated WT (WT-0) control. The displayed autoradiograph is representative of three independent reproductions.
Normal Activation of α-SMA and TGF-β1 Expression in FGF1(−/−)FGF2(−/−) Livers after Acute CCl4 Treatment
The expression of α-SMA is a marker of activated HSCs and of utility in tracking both their activation and presence. 24,26,31 We determined whether the activation of HSCs marked by α-SMA was reduced similar to collagen α1(I) gene expression in FGF1(−/−)FGF2(−/−) livers. Similar to expression of collagen α1(I) mRNA, α-SMA was apparent at 48 hours after the acute CCl4 treatment and declined somewhat by 96 hours (Figure 5A) ▶ . An identical time course and extent of α-SMA expression in FGF1(−/−)FGF2(−/−) livers as that of WT indicated that the deficiency of FGF1/FGF2 had no impact on activation of HSCs in general, but specifically affected the specific HSC matrix product collagen α1(I). The identical time course and extent of desmin expression in WT and FGF1(−/−)FGF2(−/−) livers further suggested that the increase in HSC number because of insult was not deficient in the FGF1(−/−)FGF2(−/−) mice (Figure 5A) ▶ . Appearance of desmin is an additional cytoskeletal marker associated with activated HSCs. 25,32,33 Immunohistochemical analysis at 72 hours after CCl4 liver sections further showed that α-SMA-staining HSCs accumulated in areas of damage both in FGF1(−/−)FGF2(−/−) and WT livers (Figure 5B) ▶ . In sum, neither the activation of α-SMA-staining HSCs nor the recruitment of the activated HSCs into damaged areas was impaired in FGF1(−/−)FGF2(−/−) livers. This indicated that neither FGF1 nor FGF2 are essential activators of these two processes but specific effectors of HSC collagen α1(I) production.
Figure 5.
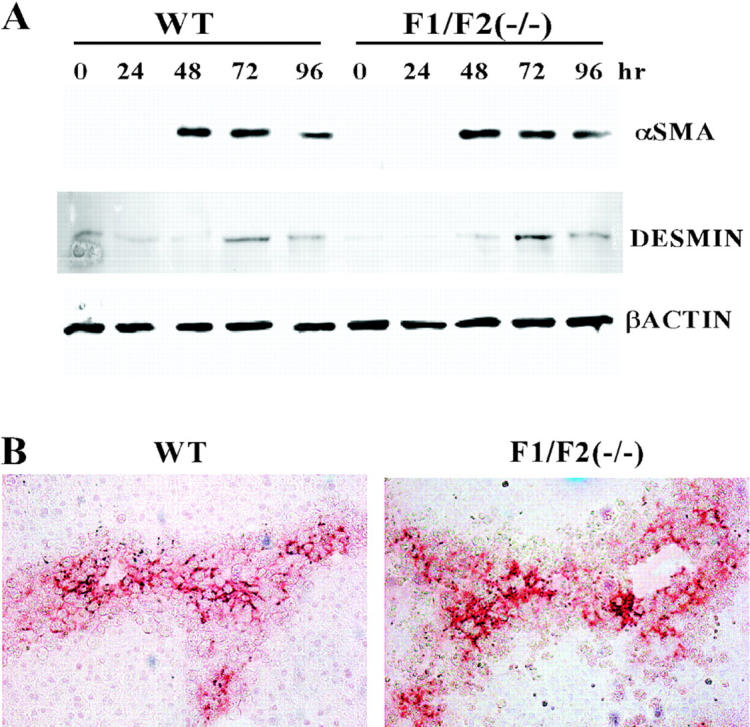
Lack of difference in α-SMA and desmin expression in FGF1(−/−)FGF2(−/−) and WT livers after acute CCl4 treatment. A: α-SMA and desmin expression in liver tissue extracts. Livers from mice of the indicated phenotype at the indicated times after acute CCl4 treatment were excised; the extract from three livers in each treatment group was pooled and analyzed by immunoblot (see Materials and Methods). The displayed autoradiograph is representative of three independent reproductions. B: Immunochemical analysis of activated HSCs expressing α-SMA in tissue sections. Livers at 72 hours after CCl4 treatment were processed and tissue sections prepared as described in Figure 2 ▶ . Original magnifications, ×100.
To date, TGF-β1 has been the single most-studied potent activator of HSCs that is induced by liver insult and has been implicated in liver fibrogenesis. 34 Similar to FGF1 and FGF2, genomic ablation of TGF-β1 depressed induction of hepatic matrix collagen α1(I) mRNA in response to injury. 23 To determine whether the induction of TGF-β1 was deficient in FGF1(−/−)FGF2(−/−) livers, we compared the expression of TGF-β1 mRNA in FGF1(−/−)FGF2(−/−) livers to WT after acute CCl4 administration. Figure 6 ▶ indicates that the time course and extent of induction of TGF-β1 mRNA was similar in both FGF1/FGF2-deficient and normal livers. This indicates that FGF1 and FGF2 are not acting through induction of TGF-β1 at the mRNA level.
Figure 6.

Normal activation of TGF-β1 expression in FGF1(−/−)FGF2(−/−) livers after acute CCl4 treatment. TGF-β1 mRNA was determined by RPA as described in Materials and Methods using 50 μg of pooled total RNA isolated from three livers. The intensity of TGF-β1 bands was quantitated as in Figure 3 ▶ and expressed relative to untreated WT assigned a value of 1. The displayed autoradiograph is representative of two independent reproductions.
Deficiency of FGF1 and FGF2 Reduces Liver Fibrosis after Chronic CCl4 Administration
Both FGF1(−/−)FGF2(−/−) and WT mice were administered 2.0 ml/kg/body weight of CCl4 twice a week after which paraffin-embedded sections of livers were examined after 3 weeks. Livers of the WT mice exhibited extensive centrilobular fibrosis indicated by clear areas that bridged from vein to vein (Figure 7A) ▶ . Staining with Sirius Red also confirmed the presence of extensive collagenous networks characteristic of fibrosis in the WT mouse livers. In contrast, the extent of centrizonal fibrosis and the collagenous matrix fibrils radiating from vein to vein were markedly reduced in the FGF1(−/−)FGF2(−/−) livers (Figure 7A) ▶ . Extraction and quantitation of total collagen in the sections confirmed the microscopic results that indicated that levels of collagen were significantly reduced (P < 0.02) in the FGF1(−/−)FGF2(−/−) mice after chronic CCl4 treatment for 21 days (Figure 7B) ▶ . These results indicate that the absence of FGF1 and FGF2 resulted in markedly decreased liver fibrosis after chronic CCl4 treatment, a phenotype that was opposite to that resulting from the absence of FGFR4. 15 Thus, intrinsic FGF1 and FGF2 play a major role in the chronic overproduction of abnormal matrix deposited by HSCs in response to chronic liver insult.
Figure 7.

Decreased fibrosis in FGF1(−/−)FGF2(−/−) livers after chronic CCl4 administration. A: The indicated type mice were subjected to chronic CCl4 treatment for 21 days as described in Materials and Methods. Livers were excised; tissue sections were prepared and stained with H&E (top) or Sirius Red (bottom). B: Quantitative analysis of collagen in liver sections. Total liver collagen in sections of WT or FGF1(−/−)FGF2(−/−) mice was quantitated as described in Materials and Methods. Values are the mean ± SD (n = 4 mice). Significance of differences between FGF1(−/−)FGF2(−/−) and WT mice at day 21 was P < 0.02. Original magnifications, ×60 (A).
Discussion
The presence of significant stores of both FGF1 and FGF2 in resting liver and induction of their mRNAs after perturbation 12 suggests the two factors as candidates for mediation of the immediate response of diverse liver cells expressing a FGFR to liver perturbation. This includes loss of mass, acute damaging insult, or chronic focal microenvironmental changes within the liver tissue matrix. 8 Here we show by genomic ablation that, similar to a variety of other tissues, 3,10,11 FGF1 and FGF2 play no apparent essential uncompensated role in embryonic development and compensatory growth of the adult liver. Of the four FGFR isotypes, only FGFR4 is expressed in mature hepatocytes. 16 Through gene targeting and genomic ablation, it has also been demonstrated that FGFR4 is not essential for embryonic development of the liver or for hepatocyte DNA synthesis and restoration of hepatocyte cell number in the adult. 17,35 However, hepatocyte FGFR4 plays a role in balanced bile acid/cholesterol metabolism, 17 limits metabolism of substrates with hepatocyte-damaging products in the hepatocyte, and is important for hepatolobular restoration after acute or chronic damaging insult in the adult liver. 15 The physiological activating FGF ligand(s) for FGFR4 in the liver has not been identified. FGFR4 binds and is activated specifically by FGF1 in isolated hepatocytes in vitro and can respond to FGF2 in other contexts dependent on cell-specific heparan sulfate. 16 The similar accelerated necrosis response to acute CCl4 insult in both FGF1(−/−)FGF2(−/−) and FGFR4(−/−) 15 suggests an activating ligand role for the FGF1/FGF2 pair for hepatocyte FGFR4 in its potential role in modulation of immediate damage to hepatocytes. After acute CCl4 treatment, FGF1 and FGF2 may be quickly activated by release from matrix or intracellular stores to stimulate hepatocyte FGFR4. This is consistent with the abundant stores of FGF1 and FGF2 in the liver available for an immediate protective response to perturbation or insult to the tissue matrix through hepatocyte FGFR4. However, all other aspects of the liver phenotype that we examined differed considerably between the FGF1(−/−)FGF2(−/−) and FGFR4(−/−) mice. FGF1(−/−)FGF2(−/−) mice do not exhibit the elevated cholesterol metabolism and bile acid synthesis that is evident in FGFR4-deficient mice, 17 making it unlikely that the two ligands are essential for activation of FGFR4 in modulation of that and related metabolic domains. The accelerated damage response to insult in FGF1/FGF2-deficient mice similar to the FGFR4 deficiency is consistent with ligand activity on FGFR4 in its role as a guardian of the immediate response against insult, but we observed a delay in the peak hepatocyte DNA synthesis response after CCl4 treatment in FGF1(−/−)FGF2(−/−) that is opposite to the effect of FGFR4 deficiency. This suggests that FGF1 and FGF2 may contribute to the normal schedule of hepatocyte proliferation in response to damage, but indirectly through nonparenchymal cells, because FGFR4 is the only FGFR present in mature hepatocytes. Moreover, overall hepatolobular restoration in FGF1(−/−)FGF2(−/−) livers with intact FGFR4 challenged with CCl4 insult appears normal in marked contrast to the FGFR4(−/−) livers that exhibit delayed hepatolobular repair for up to 3 weeks. 15 Together, these results suggest that other members of the FGF family either play the key roles or compensate for FGF1 and FGF2 in control of hepatocyte FGFR4 metabolic functions and their role in hepatolobular restoration after injury. Recently, FGF19 and FGF21 have emerged as candidate members of the FGF ligand family that have impact on the liver, although it is unclear whether the effect on hepatocytes is direct or indirect. Transgenic animals with FGF19 targeted to muscle exhibit an increased metabolic rate and decreased adiposity accompanied by a reduction in expression of liver acetyl CoA carboxylase 2 and triglyceride levels 36 and liver abnormalities associated with cancer in aging mice. 37 FGF21 appears to be specifically expressed in the liver 38 and elevated within both WT and FGF1(−/−)FGF2(−/−) livers after acute CCl4 treatment (data not shown). The role of these two FGF ligands in hepatocyte-specific FGFR4 functions and nonparenchymal cells of liver expressing other FGFR isotypes is under investigation.
Previously, we showed that the absence of FGFR4 results in profound liver fibrosis in response to chronic CCl4 insult, suggesting a role for hepatocyte FGFR4 in protection of liver against chronic long-term toxic injury. 15 Surprisingly, the deficiency of FGF1 and FGF2 resulted in essentially the opposite phenotype caused by the deficiency of FGFR4. Instead of the extensive fibrotic collagenous networks observed in the FGFR4(−/−) mouse livers after 3 weeks of chronic CCl4 insult, centrilobular fibrosis in the FGF1(−/−)FGF2(−/−) livers was reduced significantly below that observed in the WT mice. Instead of the limitations in fibrosis elicited by hepatocyte FGFR4, FGF1 and FGF2 apparently contribute to the development of fibrotic damage. This clearly eliminated FGF1 and FGF2 as activating ligands for hepatocyte FGFR4 in limiting fibrotic liver damage as a consequence of chronic toxic insult and prompted a search for nonparenchymal cell targets for the two ligands that could underlie the phenotype.
The chronic activation of nonparenchymal HSCs (also called Ito cells and fat-storing cells) has been established as the major contributor to liver fibrogenesis resulting from chronic toxic insult primarily through its production of extracellular matrix components and collagen α1(I), in particular. 29,39 Multiple cytokines have been implicated in HSC activation 29 including FGF1 and FGF2. 22,40 FGF1 is present in perisinusoidal HSCs in regenerating liver. 41 FGF2 elicits mitogenesis, chemotaxis, and chemoinvasion of HSCs in culture. 22,40 During acute CCl4 intoxication before formation of fibrotic lesions, FGF2 was observed in centrilobular clusters of mononuclear phagocytes that were surrounded by many FGF2-negative HSCs. 42 Thus, it was suggested that phagocyte-derived FGF2 may participate in the recruitment and activation of HSCs at the acute stage of CCl4 intoxication. 42 Here we showed that there was no apparent difference in recruitment of HSCs to areas of injury between FGF1/FGF2-deficient mice and WT livers after the acute CCl4 insult. Moreover, there was no effect because of the double FGF1 and FGF2 deficiency on the expression of liver desmin. Desmin has been used to estimate changes in total HSC number. 43 From these observations, we conclude that the absence of FGF1 and FGF2 impairs neither the total number of HSCs present nor their migration into areas of injury caused by acute CCl4 treatment. Therefore, a reduction of HSC number and their presence at the site of injury because of FGF1 or FGF2 deficiency cannot explain the reduction in extent of fibrosis after chronic insult to the livers of the FGF1(−/−)FGF2(−/−) animals. We conclude that FGF1 and FGF2 are not essential activating ligands for proliferation and migration of activated HSCs in vivo in either the early or chronic fibrotic phase of toxic insult to the liver.
Despite the apparent lack of requirement for FGF1 and FGF2 in HSC activation in respect to cell number and associated cytoskeletal markers, we clearly show an uncompensated role of both factors in induction of hepatic collagen α1(I) in response to insult that likely reflects normal homeostasis of liver matrix. However, chronic insults resulting in chronic mobilization of liver stores of FGF1 and FGF2 and induction of new synthesis to replenish stores results in the overproduction of HSC-derived matrix components and the irreversible scarring associated with hepatic fibrogenesis. It will be of interest to determine whether stimulation of HSC-derived matrix components by FGF1 and FGF2 is general or limited to collagen α1(I) or a related subset of matrix elements. TGF-β1 is considered to be the most potent profibrogenic cytokine in liver fibrosis through activation of HSCs that apparently includes stimulation of an increase in cell number, migratory behavior, and deposition of extracellular matrix components both in vitro and in vivo. 23,34,44,45 We showed here that it is unlikely that FGF1 or FGF2 exert their effect through limiting TGF-β expression at the mRNA level. However, FGF-mediated limits on activation of TGF-β protein and its access to and activation of the TGF-β receptor complex cannot be ruled out. Variations in FGF2 mRNA expression appear to mediate the mitogenic effects of TGF-β in cultured HSCs, but not its profibrogenic effects. 21 Clarification is needed as to whether TGF-β1 mediates the role of FGF1 and FGF2 in the deposition of extracellular matrix, or FGF1 and FGF2 mediate the TGF-β activity, or both factors play independent roles through convergent or different signaling pathways in vivo.
In summary, despite their nearly ubiquitous presence in vivo and wide range of reported biological effects in vitro, particularly cell growth and migratory behavior, the genomic ablation of FGF1 and FGF2 has remarkably little impact on gross organ phenotypes in development and the adult. 11 Our specific findings in the liver in vivo where FGF1 and FGF2 are relatively abundant in the tissue matrix in normal animals are generally consistent with these negative observations. However, they suggest an important role in the activation of HSCs in respect to control of collagen α1(I) synthesis, a prototype matrix element that may represent a more general role in deposition of extracellular matrix products from the HSCs. Our results may indicate a general physiological role of FGF1 and FGF2 as matrix-bound and cell-sequestered guardians for specific restoration of extracellular matrix components required on tissue perturbation and disruption rather than the compensatory cell growth factor activity so widely indicated in cell culture. On the one hand, chronic activation of FGF1 and FGF2 may underlie diseases of abnormal and excessive extracellular matrix as the hepatic fibrogenesis demonstrated here. On the other hand, a deficiency of FGF1 and FGF2 may result in defects in extracellular matrix production associated with diverse pathologies including wound healing, tissue remodeling, and deficiencies associated with aging. Conceivably, the neuronal defects and delay in healing of skin wounds observed in FGF2(−/−) and FGF1(−/−)FGF2(−/−) mice 10,11 are a consequence of defective matrix rather than restoration of cellularity. This suggests FGF1 and FGF2 and their presumed receptors FGFR1 and FGFR2 in HSCs 21 as potential therapeutic targets for prevention of liver fibrosis and potentially other disease of excess or deficient extracellular matrix. Our results suggest exercise of caution in excessive administration of FGF1 and FGF2 for presumed defects in restoration of cellularity based on its apparent mitogenic and anti-apoptotic effects that may result in fibrosis and scarring in various tissue contexts.
Footnotes
Address reprint requests to Wallace L. McKeehan, Institute of Biosciences and Technology, 2121 W. Holcombe Blvd., Houston, TX 77030-3303. E-mail: wmckeeha@ibt.tamu.edu.
Supported by Public Health Service grants DK35310 and DK47039 from the National Institute of Diabetes, Digestive, and Kidney Diseases; and grant CA59971 from the National Cancer Institute.
Current address of C. Y.: Department of Molecular and Cellular Biology, Baylor College of Medicine, One Baylor Plaza, Houston, TX 77030.
References
- 1.McKeehan WL, Wang F, Kan M: The heparan sulfate-fibroblast growth factor family: diversity of structure and function. Prog Nucleic Acid Res Mol Biol 1998, 59:135-176 [DOI] [PubMed] [Google Scholar]
- 2.Powers CJ, McLeskey SW, Wellstein A: Fibroblast growth factors, their receptors and signaling. Endocr Relat Cancer 2000, 7:165-197 [DOI] [PubMed] [Google Scholar]
- 3.Ornitz DM, Itoh N: Fibroblast growth factors. Genome Biol 2001, 2:3005.1-3005.12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abraham JA, Mergia A, Whang JL, Tumolo A, Friedman J, Hjerrild KA, Gospodarowicz D, Fiddes JC: Nucleotide sequence of a bovine clone encoding the angiogenic protein, basic fibroblast growth factor. Science 1986, 233:545-548 [DOI] [PubMed] [Google Scholar]
- 5.Jaye M, Howk R, Burgess W, Ricca GA, Chiu IM, Ravera MW, O’Brien SJ, Modi WS, Maciag T, Drohan WN: Human endothelial cell growth factor: cloning, nucleotide sequence, and chromosome localization. Science 1986, 233:541-545 [DOI] [PubMed] [Google Scholar]
- 6.Burgess WH, Maciag T: The heparin-binding (fibroblast) growth factor family of proteins. Annu Rev Biochem 1989, 58:575-606 [DOI] [PubMed] [Google Scholar]
- 7.Basilico C, Moscatelli D: The FGF family of growth factors and oncogenes. Adv Cancer Res 1992, 59:115-165 [DOI] [PubMed] [Google Scholar]
- 8.Kan M, McKeehan W: Liver Growth and Repair. 1998:pp 240-260 Chapman & Hall London
- 9.Jung J, Zheng M, Goldfarb M, Zaret KS: Initiation of mammalian liver development from endoderm by fibroblast growth factors. Science 1999, 284:1998-2003 [DOI] [PubMed] [Google Scholar]
- 10.Ortega S, Ittmann M, Tsang SH, Ehrlich M, Basilico C: Neuronal defects and delayed wound healing in mice lacking fibroblast growth factor 2. Proc Natl Acad Sci USA 1998, 95:5672-5677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miller DL, Ortega S, Bashayan O, Basch R, Basilico C: Compensation by fibroblast growth factor 1 (FGF1) does not account for the mild phenotypic defects observed in FGF2 null mice. Mol Cell Biol 2000, 20:2260-2268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nagasaki T, Lieberman MA: Liver contains heparin-binding growth factors as the major growth factor for cultured fibroblasts. Hepatology 1991, 13:6-14 [PubMed] [Google Scholar]
- 13.Kan M, Huang JS, Mansson PE, Yasumitsu H, Carr B, McKeehan WL: Heparin-binding growth factor type 1 (acidic fibroblast growth factor): a potential biphasic autocrine and paracrine regulator of hepatocyte regeneration. Proc Natl Acad Sci USA 1989, 86:7432-7436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Presta M, Statuto M, Rusnati M, Dell’Era P, Ragnotti G: Characterization of a Mr 25,000 basic fibroblast growth factor form in adult, regenerating, and fetal rat liver. Biochem Biophys Res Commun 1989, 164:1182-1189 [DOI] [PubMed] [Google Scholar]
- 15.Yu C, Wang F, Jin C, Wu X, Chan W, McKeehan WL: Increased carbon tetrachloride-induced liver injury and fibrosis in FGFR4-deficient mice. Am J Pathol 2002, 161:2003-2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kan M, Wu X, Wang F, McKeehan WL: Specificity for fibroblast growth factors determined by heparan sulfate in a binary complex with the receptor kinase. J Biol Chem 1999, 274:15947-15952 [DOI] [PubMed] [Google Scholar]
- 17.Yu C, Wang F, Kan M, Jin C, Jones RB, Weinstein M, Deng CX, McKeehan WL: Elevated cholesterol metabolism and bile acid synthesis in mice lacking membrane tyrosine kinase receptor FGFR4. J Biol Chem 2000, 275:15482-15489 [DOI] [PubMed] [Google Scholar]
- 18.Lopez-De Leon A, Rojkind M: A simple micromethod for collagen and total protein determination in formalin-fixed paraffin-embedded sections. J Histochem Cytochem 1985, 33:737-743 [DOI] [PubMed] [Google Scholar]
- 19.Reeves HL, Friedman SL: Activation of hepatic stellate cells—a key issue in liver fibrosis. Front Biosci 2002, 7:808-826 [DOI] [PubMed] [Google Scholar]
- 20.Pinzani M, Knauss TC, Pierce GF, Hsieh P, Kenney W, Dubyak GR, Abboud HE: Mitogenic signals for platelet-derived growth factor isoforms in liver fat-storing cells. Am J Physiol 1991, 260:C485-C491 [DOI] [PubMed] [Google Scholar]
- 21.Rosenbaum J, Blazejewski S, Preaux AM, Mallat A, Dhumeaux D, Mavier P: Fibroblast growth factor 2 and transforming growth factor beta 1 interactions in human liver myofibroblasts. Gastroenterology 1995, 109:1986-1996 [DOI] [PubMed] [Google Scholar]
- 22.Fibbi G, Pucci M, Grappone C, Pellegrini G, Salzano R, Casini A, Milani S, Del Rosso M: Functions of the fibrinolytic system in human Ito cells and its control by basic fibroblast and platelet-derived growth factor. Hepatology 1999, 29:868-878 [DOI] [PubMed] [Google Scholar]
- 23.Hellerbrand C, Stefanovic B, Giordano F, Burchardt ER, Brenner DA: The role of TGFbeta1 in initiating hepatic stellate cell activation in vivo. J Hepatol 1999, 30:77-87 [DOI] [PubMed] [Google Scholar]
- 24.Friedman SL: Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J Biol Chem 2000, 275:2247-2250 [DOI] [PubMed] [Google Scholar]
- 25.Geerts A, Lazou JM, De Bleser P, Wisse E: Tissue distribution, quantitation and proliferation kinetics of fat-storing cells in carbon tetrachloride-injured rat liver. Hepatology 1991, 13:1193-1202 [PubMed] [Google Scholar]
- 26.Ramadori G, Veit T, Schwogler S, Dienes HP, Knittel T, Rieder H, Meyer zum Buschenfelde KH: Expression of the gene of the alpha-smooth muscle-actin isoform in rat liver and in rat fat-storing (ITO) cells. Virchows Arch B Cell Pathol Incl Mol Pathol 1990, 59:349-357 [DOI] [PubMed] [Google Scholar]
- 27.Takahara T, Kojima T, Miyabayashi C, Inoue K, Sasaki H, Muragaki Y, Ooshima A: Collagen production in fat-storing cells after carbon tetrachloride intoxication in the rat. Immunoelectron microscopic observation of type I, type III collagens, and prolyl hydroxylase. Lab Invest 1988, 59:509-521 [PubMed] [Google Scholar]
- 28.Maher JJ, McGuire RF: Extracellular matrix gene expression increases preferentially in rat lipocytes and sinusoidal endothelial cells during hepatic fibrosis in vivo. J Clin Invest 1990, 86:1641-1648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu J, Zern MA: Hepatic stellate cells: a target for the treatment of liver fibrosis. J Gastroenterol 2000, 35:665-672 [DOI] [PubMed] [Google Scholar]
- 30.Bataller R, Brenner DA: Hepatic stellate cells as a target for the treatment of liver fibrosis. Semin Liver Dis 2001, 21:437-451 [DOI] [PubMed] [Google Scholar]
- 31.Rockey DC, Boyles JK, Gabbiani G, Friedman SL: Rat hepatic lipocytes express smooth muscle actin upon activation in vivo and in culture. J Submicrosc Cytol Pathol 1992, 24:193-203 [PubMed] [Google Scholar]
- 32.Burt AD, Robertson JL, Heir J, MacSween RN: Desmin-containing stellate cells in rat liver; distribution in normal animals and response to experimental acute liver injury. J Pathol 1986, 150:29-35 [DOI] [PubMed] [Google Scholar]
- 33.Yokoi Y, Namihisa T, Kuroda H, Komatsu I, Miyazaki A, Watanabe S, Usui K: Immunocytochemical detection of desmin in fat-storing cells (Ito cells). Hepatology 1984, 4:709-714 [DOI] [PubMed] [Google Scholar]
- 34.Gressner AM, Weiskirchen R, Breitkopf K, Dooley S: Roles of TGF-beta in hepatic fibrosis. Front Biosci 2002, 7:793-807 [DOI] [PubMed] [Google Scholar]
- 35.Weinstein M, Xu X, Ohyama K, Deng CX: FGFR-3 and FGFR-4 function cooperatively to direct alveogenesis in the murine lung. Development 1998, 125:3615-3623 [DOI] [PubMed] [Google Scholar]
- 36.Tomlinson E, Fu L, John L, Hultgren B, Huang X, Renz M, Stephan JP, Tsai SP, Powell-Braxton L, French D, Stewart TA: Transgenic mice expressing human fibroblast growth factor-19 display increased metabolic rate and decreased adiposity. Endocrinology 2002, 143:1741-1747 [DOI] [PubMed] [Google Scholar]
- 37.Nicholes K, Guillet S, Tomlinson E, Hillan K, Wright B, Frantz GD, Pham TA, Dillard-Telm L, Tsai SP, Stephan JP, Stinson J, Stewart T, French DM: A mouse model of hepatocellular carcinoma: ectopic expression of fibroblast growth factor 19 in skeletal muscle of transgenic mice. Am J Pathol 2002, 160:2295-2307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nishimura T, Nakatake Y, Konishi M, Itoh N: Identification of a novel FGF, FGF-21, preferentially expressed in the liver. Biochim Biophys Acta 2000, 1492:203-206 [DOI] [PubMed] [Google Scholar]
- 39.Gressner AM: The cell biology of liver fibrogenesis—an imbalance of proliferation, growth arrest and apoptosis of myofibroblasts. Cell Tissue Res 1998, 292:447-452 [DOI] [PubMed] [Google Scholar]
- 40.Pinzani M, Gesualdo L, Sabbah GM, Abboud HE: Effects of platelet-derived growth factor and other polypeptide mitogens on DNA synthesis and growth of cultured rat liver fat-storing cells. J Clin Invest 1989, 84:1786-1793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Marsden ER, Hu Z, Fujio K, Nakatsukasa H, Thorgeirsson SS, Evarts RP: Expression of acidic fibroblast growth factor in regenerating liver and during hepatic differentiation. Lab Invest 1992, 67:427-433 [PubMed] [Google Scholar]
- 42.Charlotte F, Win KM, Preaux AM, Mavier P, Dhumeaux D, Zafrani ES, Rosenbaum J: Immunolocalization of heparin-binding growth factors (HBGF) types 1 and 2 in rat liver. Selective hyperexpression of HBGF-2 in carbon tetrachloride-induced fibrosis. J Pathol 1993, 169:471-476 [DOI] [PubMed] [Google Scholar]
- 43.Schnabl B, Kweon YO, Frederick JP, Wang XF, Rippe RA, Brenner DA: The role of Smad3 in mediating mouse hepatic stellate cell activation. Hepatology 2001, 34:89-100 [DOI] [PubMed] [Google Scholar]
- 44.Win KM, Charlotte F, Mallat A, Cherqui D, Martin N, Mavier P, Preaux AM, Dhumeaux D, Rosenbaum J: Mitogenic effect of transforming growth factor-beta 1 on human Ito cells in culture: evidence for mediation by endogenous platelet-derived growth factor. Hepatology 1993, 18:137-145 [PubMed] [Google Scholar]
- 45.Fibbi G, Pucci M, D’Alessio S, Grappone C, Pellegrini G, Salzano R, Casini A, Milani S, Del Rosso M: Transforming growth factor beta-1 stimulates invasivity of hepatic stellate cells by engagement of the cell-associated fibrinolytic system. Growth Factors 2001, 19:87-100 [DOI] [PubMed] [Google Scholar]