

Abstract
Neonates are particularly susceptible to coxsackievirus infections of the central nervous system (CNS), which can cause meningitis, encephalitis, and long-term neurological deficits. However, viral tropism and mechanism of spread in the CNS have not been examined. Here we investigate coxsackievirus B3 (CVB3) tropism and pathology in the CNS of neonatal mice, using a recombinant virus expressing the enhanced green fluorescent protein (eGFP). Newborn pups were extremely vulnerable to coxsackievirus CNS infection, and this susceptibility decreased dramatically by 7 days of age. Twenty-four hours after intracranial infection of newborn mice, viral genomic RNA and viral protein expression were detected in the choroid plexus, the olfactory bulb, and in cells bordering the cerebral ventricles. Many of the infected cells bore the anatomical characteristics of type B stem cells, which can give rise to neurons and astrocytes, and expressed the intermediate filament protein nestin, a marker for progenitor cells. As the infection progressed, viral protein was identified in the brain parenchyma, first in cells expressing neuron-specific class III β-tubulin, an early marker of neuronal differentiation, and subsequently in cells expressing NeuN, a marker of mature neurons. At later time points, viral protein expression was restricted to neurons in specific regions of the brain, including the hippocampus, the entorhinal and temporal cortex, and the olfactory bulb. Extensive neuronal death was visible, and appeared to result from virus-induced apoptosis. We propose that the increased susceptibility of the neonatal CNS to CVB infection may be explained by the virus’ targeting neonatal stem cells; and that CVB is carried into the brain parenchyma by developing neurons, which continue to migrate and differentiate despite the infection. On full maturation, some or all of the infected neurons undergo apoptosis, and the resulting neuronal loss can explain the longer-term clinical picture.
Viral infection in the central nervous system (CNS) of newborns is an under-appreciated problem with potential long-term consequences that may include severe intellectual disabilities and scholastic performance deficiencies in children. 1 Enteroviruses, in particular, account for many cases of aseptic meningitis and encephalitis in newborn infants, who appear to be especially susceptible to these agents. 2,3 Enterovirus infections can occur not only in the immediate postpartum period, but also in utero; such fetal and congenital infections can have serious consequences, 4,5 including neurodevelopmental defects. 6,7 In addition to the acute pathology induced by neonatal CNS infection, there may be delayed effects; for example, some evidence links childhood enterovirus infection with adult-onset schizophrenia. 8 The enterovirus genus lies within the picornavirus family, and includes polioviruses, coxsackieviruses, and unclassified enteroviruses; since the advent of widespread poliovirus vaccination, coxsackievirus has emerged as the single most common cause of enteroviral CNS infection in neonates. Despite the frequency of coxsackievirus CNS infections, and the profound neuropathologies which may result, our understanding of coxsackievirus tropism in the neonatal CNS is very limited; the present study was initiated to partially redress this deficit.
Coxsackieviruses have been divided into subgroups A and B based on their pathogenicity in suckling mice. 9-11 Group A strains most commonly induce flaccid paralysis, as a result of extensive infection of skeletal musculature, while Group B strains are associated with spastic paralysis, indicative of upper motor neuron dysfunction resulting from meningoencephalitis. This CNS tropism of group B coxsackieviruses (CVB) is maintained in humans. CVB antigens have been detected in formalin-fixed, paraffin-embedded tissue sections from a patient with acute encephalitis, 12 infectious virus has been isolated from patients with meningo-encephalitis, 13 and CVB infection has resulted in encephalitis with widespread multi-focal areas of liquefactive necrosis in the cerebrum. 14 CVB-specific neutralizing antibodies have been identified in the ventricular fluid of newborns with severe congenital anatomical defects in the CNS, 15 suggesting that coxsackievirus infection may be an underlying factor in such diseases. Forebrain abnormalities, including hydranencephaly, have been seen after infection of newborn human infants, as well as in experimental infections of neonatal mice. 15,16 Coxsackievirus-induced encephalitis may cause long-term damage to the CNS, including to the basal ganglia, leading to Parkinson-like symptoms, and memory and learning disorders suggestive of subcortical dementia; 17 indeed, CVB can cause a syndrome very similar to encephalitis lethargica, 18 a disease long considered viral in origin.
To facilitate our investigation of CVB tropism in the CNS, we have used a recombinant CVB which expresses the enhanced green fluorescence protein (eGFP); 19 this virus (eGFP-CVB) was prepared using a method that we recently described for generating recombinant CVB. 20 eGFP was selected as the optimal marker protein because it is relatively small in size (enabling its incorporation into recombinant CVB), it can be used for live imaging of infected cells, 19 and the protein can be detected in thin, high-resolution sections of formaldehyde-fixed, paraffin-embedded tissue. 21,22 Furthermore, it is relatively resistant to photo-bleaching, and permits very straightforward studies to simultaneously compare viral protein expression with various cellular markers, identified using fluorescently tagged antibodies. Such dual-labeling studies, using eGFP as a surrogate marker for viral protein expression, in combination with various cell-specific markers in the CNS, have allowed us to identify the initial sites of CVB infection in the neonatal CNS, and to track the infection over a course of several days. We have identified the CNS sites infected at each stage, and the cellular tropism of the virus, which changes as the infection progresses. Our results reveal an enhanced susceptibility and increased pathology in newborn pups, compared to older mice, and suggest a possible mechanism for coxsackievirus-induced pathogenesis.
Materials and Methods
Recombinant eGFP-CVB Virus
The construction of our recombinant CVB3 expressing eGFP has been previously described. 19 Viral stocks were prepared on HeLa RW cells. Virus inoculations were done with concentrated stocks diluted in Dulbecco’s modified Eagle’s medium (DMEM; Gibco-BRL, Gaithersburg, MD).
Mice and Viral Inoculations
BALB/c mice obtained from the Scripps animal facilities were bred, and pups were infected with varying amounts eGFP-CVB intracranially (i.c.). All pups were infected within 24 hours of birth, unless otherwise indicated. Intracranial injections were carried out on pups anesthetized by hypothermia. 23 One pup at a time was placed in a paper-lined plastic dish resting on a bed of dry ice. After 1 to 3 minutes, general limb movement ceased, and the pup was removed and immediately inoculated i.c. with 25 μl of virus (viral dose indicated in the text) using a sleeved 30-gauge needle. Pups recovered within minutes, and were returned to their mothers after their breathing was regular, their limbs were moving normally, and their skin color had returned to normal. Control pups were mock infected by i.c. inoculation of 25 μl of DMEM. All pups recovered from hypothermia, regardless of their age. In control pups injected i.c. on the day of birth, a very low background mortality was seen (2 of 16 mice) in the days immediately following i.c. injection. This low procedural mortality did not occur in older pups.
Virus Titration
Pups were euthanized by hypothermia, followed by immediate decapitation. Organs were harvested, placed in dry ice, weighed, and homogenized in 1 ml complete DMEM for approximately 15 seconds using an Ultra-Turrax T 25 basic homogenizing apparatus (Ika Works Inc., Wilmington, NC). The samples were stored at −70°C, for subsequent measurement of virus titers by plaque assay, as described previously. 19 Briefly, six-well dishes were plated with 3.75 × 10 5 HeLa RW cells/well, and grown for 48 hours before infection. Ten-fold serial dilutions were prepared, and incubated with cell monolayers for 1 hour with rocking every 15 minutes. Cells were overlaid with 4 ml of 1X complete DMEM in 0.6% agar. After 40 to 48 hours incubation (37°C, 5% CO2), cells were fixed with 2 ml/well of methanol:acetic acid (3:1 v/v) for 10 minutes, after which the fixative and the agar plugs were removed. Cells were stained with 1 ml of crystal violet solution (0.5% crystal violet in 20% ethanol) for 1 hour, rinsed in tap water, and plaques were counted.
Detection of eGFP in Situ
Immediately following euthanasia, the brain was fixed by immersion in 10% neutral-buffered formalin for >4 hours, paraffin-embedded, and 3-μm sections were prepared. Unstained sections were overlaid with phosphate-buffered saline (PBS), and observed using either an Axiovert 200 inverted microscope (Carl Zeiss Light Microscopy, Goettingen, Germany), or a Nikon Optiphot fluorescent microscope. Images were acquired using a Spot RT camera (Diagnostic Instruments Inc., Sterling Heights, MI)) or an AxioCam HR digital camera (Axiovert).
Immunofluorescence Staining
Paraffin-embedded sections were deparaffinized with three washes in xylene and serial washes in 100%, 90%, 70% ethanol, followed by a final wash in PBS. Detection of activated caspase 3, nestin, and NeuN, required antigen unmasking; for these studies, deparaffinized sections were heat-treated by boiling for 25 minutes in 0.01 mol/L citrate buffer (pH 6.0) in an 1100W microwave oven set at full power, then were allowed to cool at room temperature for 30 minutes. All sections were blocked with 10% normal goat serum for 30 minutes, incubated overnight with primary antibody, and washed three times with PBS. Most of the primary antibodies used were from rabbit, including: rabbit anti-GFAP at 1:500, Sigma-Aldrich, Inc., St. Louis, MO; rabbit anti-class III β-tubulin at 1:500, Covance Research Products Inc., Cumberland, VA; and rabbit anti-human/mouse active caspase-3 at 1:100, R&D Systems, Inc., Minneapolis, MN. All antibody dilutions were made in 2% normal goat serum. The secondary antibody (goat biotinylated anti-rabbit IgG (H+L) at 1:500, Vector Laboratories Inc., Burlingame, CA) diluted in 2% normal goat serum was incubated on sections for 30 minutes. Two primary antibodies were derived from mouse (anti-NeuN at 1:1000, Chemicon International Inc., Temecula, CA; anti-nestin at 1:50, Chemicon International Inc.). To minimize background staining using these mouse antibodies on mouse tissues, the Mouse on Mouse (MOM) kit was used as described by the manufacturer (Vector Laboratories Inc.). Briefly, before addition of the primary antibodies, sections were blocked in MOM Mouse Ig Blocking Reagent for 1 hour, and then washed twice in PBS for 2 minutes. Primary mouse monoclonal antibodies in MOM diluent were added to sections and incubated for 30 minutes at room temperature, followed by two PBS washes for 2 minutes. The secondary antibody consisted of MOM biotinylated anti-mouse IgG reagent at 1:250 in MOM Diluent. After staining with secondary antibodies, all sections were washed twice with PBS, and incubated for 30 minutes with a streptavidin-rhodamine red complex (Jackson ImmunoResearch Laboratories Inc, West Grove, PA) diluted 1:500 in 2% normal goat serum. In sections stained to detect nestin, viral protein expression was detected using rabbit anti-GFP at 1:20 (Molecular Probes, Inc., Eugene, OR) incubated at room temperature for 1 hour, followed by a 30-minute incubation with anti-rabbit-FITC (Rockland Immunochemicals, Inc., Gilbertsville, PA) at 1:100. Specificity controls for immunostaining included sections stained in the absence of primary antibody, or in the presence of rabbit IgG control antibody at 0.1 μg/ml (Vector Laboratories, Inc). For detection of DNA/nuclei using DAPI (4′,6-diamidino-2-phenylindole), sections were incubated for 2 minutes in a 300 nmol/L DAPI dilactate stock solution (Molecular Probes Inc.) diluted in PBS, then washed once with PBS. Sections were observed by laser scanning confocal microscopy (Bio-Rad MRC1024) attached to a Zeiss Axiovert S100TV microscope, or by fluorescence microscopy (Zeiss Axiovert 200 inverted microscope) for eGFP (green), the indicated cellular marker (red), and DAPI nuclear staining (blue). Green, red, and blue channel images were merged using either Bio-Rad Confocal Assist software, or AxioVision software.
ApopTag Staining
Detection of cells undergoing apoptosis was evaluated using the ApopTag Red In Situ Apoptosis Detection Kit (Intergen, Purchase, NY), as specified by the manufacturer. Briefly, paraffin-embedded sections were deparaffinized and pre-treated with proteinase K (20 μg/ml) for 15 minutes. Equilibration buffer was added directly onto the specimen, after which terminal deoxynucleotidyl transferase (TdT) enzyme in reaction buffer was added for one hour at 37°C. Sections were washed in working strength Stop/Wash buffer for 10 minutes. Pre-warmed working strength anti-digoxigenin conjugate (rhodamine) was added to the sections and incubated at room temperature for 30 minutes. The samples were washed with PBS and observed by fluorescence microscopy, as described above.
In Situ Hybridization
A 33P-labeled anti-sense RNA (421 bases) probe for the 5′ untranslated region of CVB3 was generated using the MAXIscript In vitro Transcription Kit (Ambion Inc., Austin, TX), as described by the manufacturer. One microgram of plasmid (pBKS2 CVB3) containing the probe sequence was linearized with Asp718. The transcription reaction (20-μl final volume in nuclease-free water) contained the linearized DNA template, 0.5 μmol/L of each ribonucleotide, 1X Transcription buffer, T3 polymerase, and labeled UTP. The reaction mixture was incubated for 10 minutes at 37°C to produce high specific activity radiolabeling. The DNA template was degraded by addition of DNase I, and unincorporated nucleotides were removed by applying the transcription reaction to a Nuc Away Spin Column (Ambion Inc.). The efficiency of the transcription reaction was determined by measuring cpm/μl using a β-scintillation counter. In situ hybridization procedures were carried out using the mRNAlocater In Situ Hybridization Kit (Ambion Inc.), as described by the manufacturer. Paraffin-embedded sections were deparaffinized as described above, immersed in 1 μg/ml Proteinase K for 10 minutes at room temperature with agitation, and washed twice with nuclease-free water. Slides were incubated with 2X SSC for 2 minutes, dehydrated with increasing concentrations of ethanol, and dried under a hair dryer for 1 hour. The radiolabeled probe (107 cpm) was added to 100 μl of hybridization buffer, denatured by heating to 65°C for 5 minutes, and cooled on ice. The probe solution was applied to the section, and the sample was sealed in a humidified chamber and incubated at 46°C for 18 hours. Slides were washed twice in 4X SSC for 10 minutes at room temperature, then a pre-warmed solution of RNase A was added to the sections. After incubation for 30 minutes at 37°C, slides were washed in decreasing concentrations of SSC, dehydrated in graded ethanol solutions, and dried with a hair dryer for 30 minutes. Slides were immersed in photographic emulsion, held at 4°C for 6 days, then developed. Finally, slides were stained with hematoxylin (1 minute) and eosin (1.5 minutes), and mounted with Cytoseal (VWR, West Chester, PA).
Results
Coxsackievirus CNS Infection of Newborn Pups Leads to Dose-Related Mortality
The aim of this study was to establish the tropism of CVB in the neonatal CNS and, to this end, we first evaluated the outcomes of different routes of administration. eGFP-CVB was administered by the intraperitoneal, intraoral, intranasal, or intracranial routes, and only the latter route routinely resulted in CNS infection and lethal disease (data not shown). Therefore, for the remainder of this study, the i.c. route was used. Intracranial administration of a high dose (2 × 107 pfu) of eGFP-CVB was uniformly lethal within 2 days of infection of newborn pups (Figure 1A ▶ , green circles), and as few as 105 pfu of eGFP-CVB caused significant mortality (Figure 1A ▶ , red inverted triangles). These results illustrate the high susceptibility of neonatal mice to CVB3 infection with subsequent fatal consequences. As noted in the Materials and Methods section, a small number of mock-infected newborn pups died following the intracranial injection.
Figure 1.
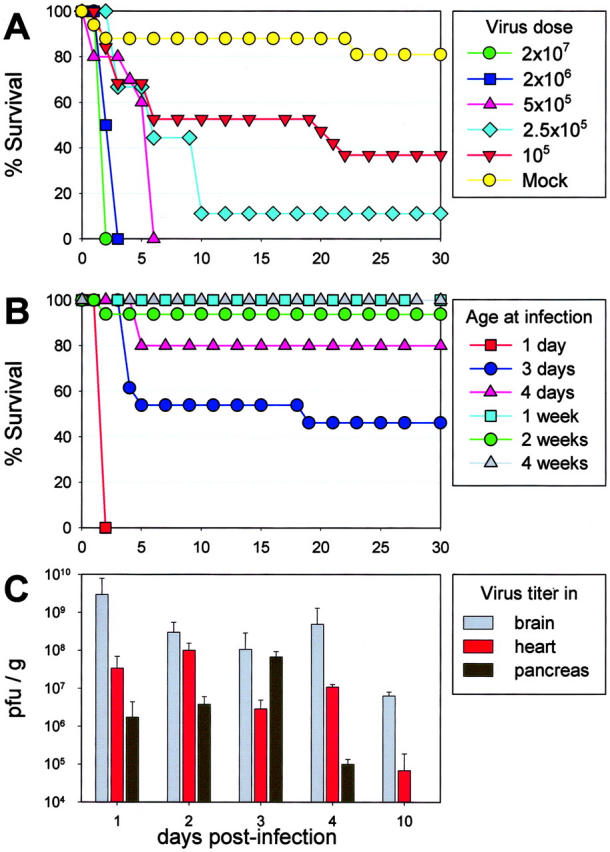
Outcome of eGFP-CVB CNS infection is dose-dependent, and changes dramatically within days of birth. A: Newborn BALB/c pups were inoculated i.c. with the indicated doses of eGFP-CVB, or with medium alone (mock), and their survival over a 30-day period is shown. B: Mice of the indicated ages were inoculated i.c. with eGFP-CVB (2 × 107 pfu) and observed for survival over a 30-day period. C: Newborn pups were infected with eGFP-CVB (2 × 105 pfu, i.c.) and, at the indicated time points post-infection, mice were sacrificed, the brains, hearts, and pancreata were harvested, and viral titers were determined.
Rapid, Age-Dependent Loss of Susceptibility to Intracranial CVB Infection
Susceptibility to virus-induced mortality decreased substantially within days after birth (Figure 1B) ▶ . Intracranial challenge with 2 × 107 pfu of eGFP-CVB was lethal to only ∼50% of pups inoculated at 3 days of age, and to only ∼20% of pups inoculated when 4 days old. Pups that were ≥ 1 week old when inoculated almost invariably survived (only 1 of 42 pups succumbed). In addition, no viral protein expression or pathology was seen in the brains of adult mice (8-week-old) that had been intracranially infected with eGFP-CVB (data not shown).
Viral Titers Peak Early after Intracranial Inoculation, and Infectious Virus Remains in the CNS at 10 Days Post-Infection
One-day old pups were infected i.c. with eGFP-CVB (2 × 105 pfu) and sacrificed at different time points after infection. Brain, heart, and pancreas were homogenized and titered, as described in Material and Methods. Following i.c. infection, virus titers in the brain rose rapidly, with maximum levels observed 24 hours post-infection, and slowly dropped over the course of 10 days (Figure 1C) ▶ . Somewhat delayed kinetics were observed for both the pancreas and heart from i.c. infected pups, suggesting that the CNS is the site of early infection, after which virus disseminates to other susceptible organs. However, the brain contained the highest titers of infectious virus at every time point analyzed. Infectious virus could no longer be detected in the brain by 30 days p.i., but viral RNA remained detectable at this time point (data not shown). Persistence of the viral RNA was also seen in the pancreas and heart tissues following i.c. inoculation. These results parallel numerous published reports which demonstrate the ability of coxsackievirus RNA to persist in tissues of adult mice, for many months after infection, and when no infectious virus can be detected. 24,25
CVB Infects Specific Regions of the Neonatal CNS
eGFP can be detected in very thin (3 μm) sections of formaldehyde-fixed, paraffin-embedded tissues, 22 and we have exploited this property to identify infected cells at very high resolution in situ. We first evaluated viral protein expression (using eGFP as a surrogate marker) at 1 day post-infection, and examination of several mice showed that eGFP expression was consistently restricted to specific areas of the neonatal brain. As indicated by the representative sections shown in Figure 2 ▶ , eGFP was expressed at extremely high levels in the choroid plexus, and also was readily detected in neighboring cells lining the lateral ventricle (Figure 2A) ▶ . Protein expression also was found in cells lining the roof of the third ventricle (Figure 2C) ▶ , and in the granular cell layer of the olfactory bulb (Figure 2E) ▶ . No viral protein expression or pathology was seen in the cerebellum of neonatal pups at any time point (data not shown), consistent with what has been observed previously in newborn human infants. We have been unable to detect eGFP expression within the CNS of older mice (>14 days old), even after i.c. inoculation of very high doses of recombinant virus (up to 2 × 107 pfu; data not shown); these findings are consistent with our observation (Figure 1B) ▶ that susceptibility to lethal infection dropped substantially within a few days after birth. Thus, specific regions of the neonatal CNS are exquisitely sensitive to CVB infection; and this susceptibility drops very dramatically within days of birth.
Figure 2.
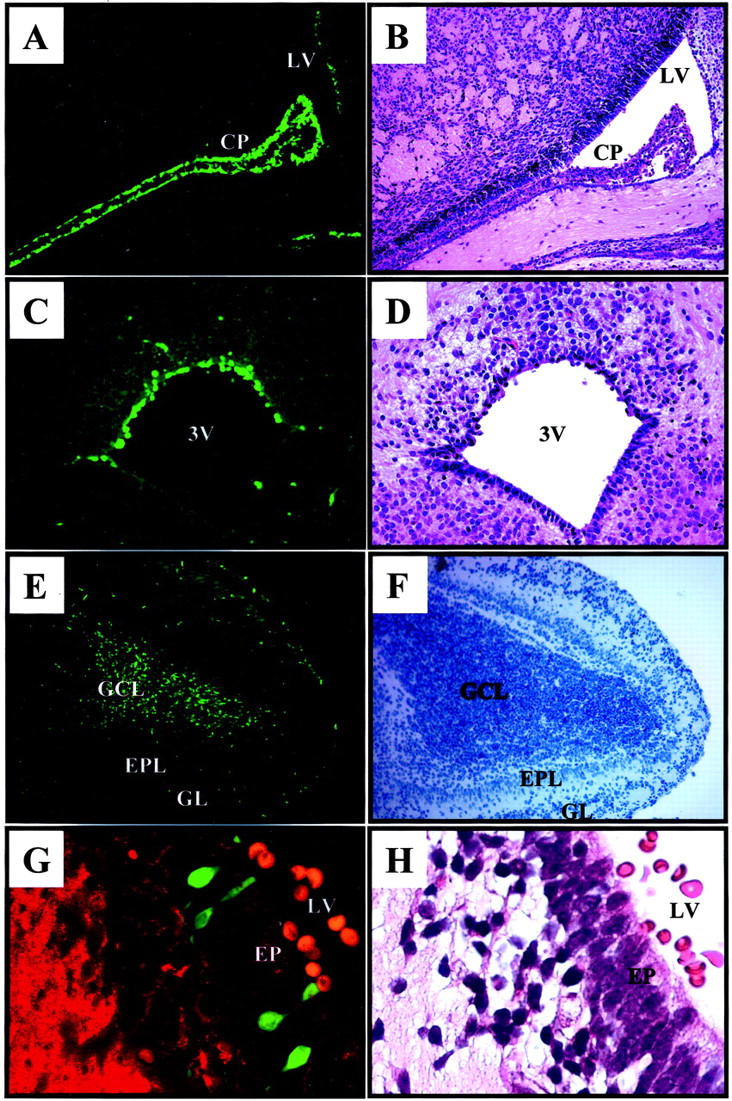
Viral protein expression within the neonatal CNS one day after infection with eGFP-CVB. Newborn pups were infected i.c. with eGFP-CVB (2 × 107 pfu) and sacrificed 1 day post-infection. Paraffin-embedded transverse sections of brain tissue were hydrated with PBS and observed by fluorescence microscopy. Fluorescent images are shown on the left; green represents eGFP, and red (G) represents β-tubulin. The corresponding H&E (B, D, H) or hematoxylin alone (F) images are shown on the right. Magnification, ×31 for images A, B, E and F; magnification, ×62 for images C and D. Magnification, ×63 for original images G and H, with a further ∼2-fold computer-generated magnification. CP, choroid plexus; LV, lateral ventricle; 3V, third ventricle; GCL, granular cell layer; EPL, external plexiform layer; GL, glomerular layer; EP, ependymal cells.
CVB Infects Progenitor Cells in the Neonatal CNS
The cerebroventricular system is lined by ependymal cells. These cuboidal cells have lengthy cilia, which assist in the circulation of cerebrospinal fluid, and such ciliated cuboidal cells can be clearly seen lining the floor of the third ventricle (Figure 2D) ▶ and in the lateral ventricle (Figure 2H) ▶ . It appears that most ependymal cells are not infected by CVB; the infected cells are limited to the roof of the third ventricle (Figure 2C) ▶ , and to morphologically distinctive cells, the apical portions of which appear to protrude through the ependymal layer of the lateral ventricle (Figure 2G) ▶ . Morphologically, these cells appear to be “type B” stem cells, 26 which can give rise both to neurons and astrocytes. 27 The existence of various types of CNS stem cells is now well-established, and stem cells have been identified in several anatomical areas, including the subventricular zone (SVZ), the hippocampus and the olfactory bulb. 26 To our knowledge, no marker is available that is unique to type B stem cells. However, these cells, and other progenitor cells in the CNS, express nestin, an intermediate filament protein whose expression is closely associated with rapidly proliferating progenitor cells during neurogenesis 28 and myogenesis. 29 Therefore, we evaluated nestin staining, and its relationship to viral protein expression, in 1-day-old mice that had been infected on the day of birth. As shown in Figure 3A ▶ , nestin (red staining) is absent from cells in the choroid plexus, but is expressed in cells adjacent to the ventricle, where progenitor cells are located; a higher-power photomicrograph of this region (Figure 3B) ▶ suggests that the majority of virus-infected cells also are nestin-positive. Co-localization of nestin and eGFP was observed in additional mice from the same experimental group (Figure 3, C and D) ▶ . Furthermore, evaluation of the subventricular region showed that nestin staining was filamentous, as previously reported, and was absent from the perinuclear areas in the cell bodies (Figure 3E ▶ , white arrows show “empty” perinuclear areas) These regions stained strongly for eGFP (Figure 3F) ▶ , and the co-localization is clearly visible in a merged image (Figure 3G) ▶ .
Figure 3.

Progenitor cells in the CNS are infected by CVB. Newborn pups were infected i.c. with eGFP-CVB (2 × 107 pfu) and sacrificed 1 day post-infection. Paraffin-embedded transverse sections of brain tissue were deparaffinized, antigen-unmasked (see Materials and Methods), and stained to detect nestin (red) and eGFP (green). A: A low-power merged nestin/eGFP image of the lateral ventricle, and the cyan box shows the region represented at higher magnification in B. C and D: Merged images from the ventricular regions of a different neonatal mouse. E–G: Further evidence of co-localization, showing respectively nestin, eGFP, and merged images. The white arrows show examples of “empty” perinuclear areas in nestin+ cells (see text).
Spread of Infection in the Brain Parenchyma, Identified by in Situ Hybridization and eGFP Expression
Next, we evaluated coxsackieviral spread in the CNS in the early phase post-infection, by tracking both viral RNA (detecting the viral genome by in situ hybridization) and viral protein expression (eGFP). Whole-brain distribution at days 1 and 2 p.i. are shown in Figure 4 ▶ . At both time points, there was an extremely high correlation between the viral RNA signal and eGFP expression, confirming the validity of using eGFP as an easily detected and accurate surrogate marker of viral protein expression. At day 1 p.i., viral genome was detected in or near the lateral ventricles (in both the anterior and posterior horns) and the olfactory bulb (Figure 4A) ▶ , and closely paralleled the distribution of eGFP (Figure 4B) ▶ . Higher-power photomicrographs of the lateral ventricles and hippocampus show that eGFP expression was restricted to cells lining the ventricles, and to the choroid plexus (Figure 4, C and D) ▶ confirming the findings shown in Figure 2 ▶ . Twenty-four hours later, the virus had begun to enter the brain parenchyma; viral genome and eGFP expression were more dispersed around the lateral ventricles, and could now be detected in the hippocampus (Figure 4, E and F) ▶ ; and higher-power fields show clearly that invasion of parenchymal tissue has taken place (Figure 4, G and H) ▶ .
Figure 4.
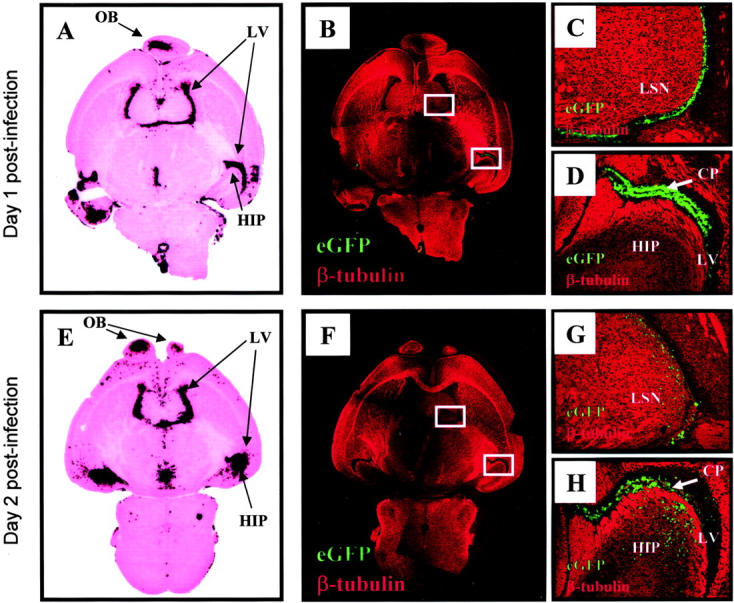
Distribution of infection over time within the CNS as determined by in situ hybridization for viral genome and by viral protein expression. Newborn pups were infected i.c. with eGFP-CVB (2 × 107 pfu) and harvested either 1 day (A–D) or 2 days (E–H) post-infection. A and E show sections of whole brain, probed by in situ hybridization to detect CVB genomic RNA, stained with H&E, then scanned using a Polaroid slide scanner. B and F: Identical fields of an adjacent section were captured at ×5 magnification using both the red channel (class III β-tubulin) and the green channel (eGFP); the two color channels were merged into a single image, and images of overlapping fields were assembled into the composites shown. Boxed areas in B and F are presented at higher magnification in C, D, G, and H. OB, olfactory bulb; LV, lateral ventricle; HIP, hippocampus; LSN, lateral septal nucleus; CP, choroid plexus.
In addition to identifying the distribution of viral materials, we sought also to determine the cell types involved in parenchymal spread. The neonatal CNS is a site of active neurogenesis and neuronal development, and several markers are available which detect neurons at various developmental stages. We first focused on class III β-tubulin, an early marker of neuronal differentiation. 30 Areas of active neurogenesis, which include the SVZ, are low in β-tubulin expression 31 but, as developing neurons move toward their final destinations, they express this marker protein. This developmental pattern is clearly demonstrated in Figure 2G ▶ , in which cells immediately adjacent to the lateral ventricle are negative for β-tubulin, while more distant cells express high levels of this protein (red). The distribution of this marker in the neonatal CNS is shown in more detail in Figure 4 ▶ (panels B to D and F to H). As expected for the neonatal brain, β-tubulin was widespread, and formed discrete tracts; the striated appearance is characteristic of β-tubulin, and reflects chains of migrating neurons. 30 At 1 day p.i., essentially all of the infected (eGFP+) cells were negative for β-tubulin (Figure 2G ▶ , and Figure 4, C and D ▶ ). However, by day 2 post-infection, apparent co-localization of eGFP and β-tubulin could be found in the lateral septal nucleus and the hippocampus (Figure 4, G and H) ▶ .
CVB Spread into the CNS Parenchyma Involves Immature Neurons
To confirm that coxsackievirus infected immature neurons, we carried out laser scanning confocal microscopy (Figure 5A) ▶ , which revealed a staining pattern similar to that obtained by fluorescence microscopy (Figure 5B) ▶ . A high-power photomicrograph of the lateral septal nucleus showed close apposition, but little overlap, of eGFP with β-tubulin (Figure 5C) ▶ , and analysis of β-tubulin alone (Figure 5D) ▶ revealed a honeycomb staining pattern. This appearance has been reported recently, 31,32 and results from the intracellular distribution of β-tubulin, which is cytoplasmic, with a concentration in extended axons/dendrites; the unstained areas represent the nucleus and perinuclear cytoplasm. To more precisely delineate eGFP expression relative to β-tubulin, sections were first counterstained with the nuclear stain DAPI, which showed that many of the nuclei were surrounded by a narrow corona of β-tubulin-negative cytoplasm (Figure 5E) ▶ ; and co-visualization of eGFP and DAPI showed that this perinuclear region contained the great majority of the viral protein (Figure 5F) ▶ . These results confirm that coxsackievirus infects immature neurons at day 2 post-infection. This could have occurred by de novo infection of β-tubulin+ immature neurons, or by the expression of β-tubulin by β-tubulin-negative cells which were infected on day 1.
Figure 5.
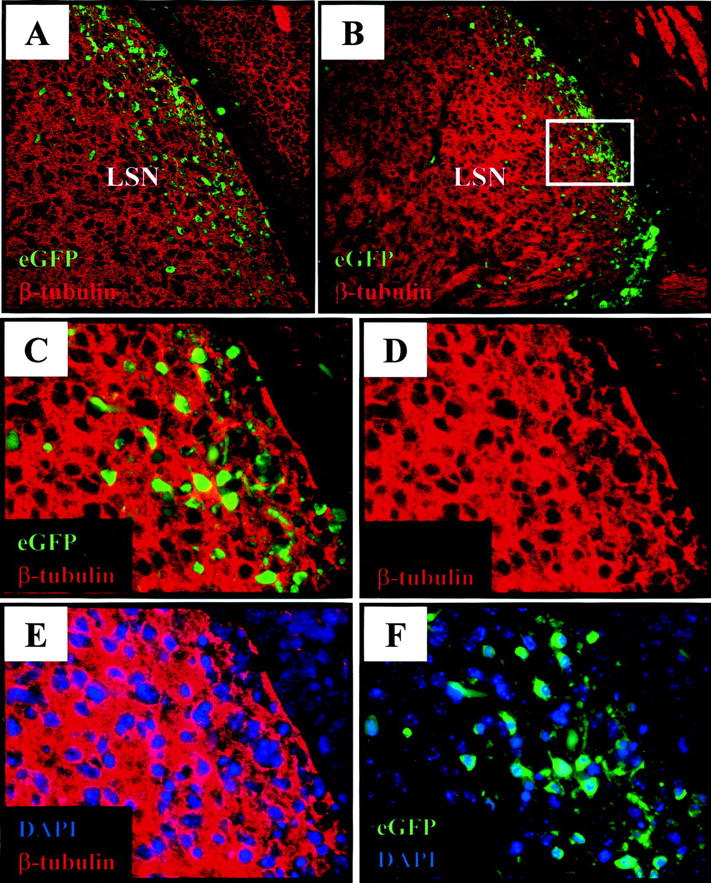
Immature neuronal cells within the lateral septal nucleus express viral protein by day 2 post-infection. Pups were infected i.c. with eGFP-CVB (2 × 107 pfu) and harvested 2 days post-infection. Paraffin-embedded sections were immunostained to detect neuron-specific class III β-tubulin (red), counterstained with DAPI (nuclear stain; blue), and observed by confocal or fluorescence microscopy. A: Confocal microscopy (Bio-Rad MRC1024, magnification, ×40) showing co-localization of neuron-specific class III β-tubulin expression with infected (eGFP+) cells in the lateral septal nucleus (LSN). B: Fluorescence microscopy (Axiovert 200 inverted microscope) of a parallel section (3-μm thickness, magnification, ×20) confirming co-localization of β-tubulin expression with infected cells. The boxed area in B indicates the ×63 magnified region depicted in C through F. C: Higher magnification shows little direct overlap between eGFP and β-tubulin, and the latter staining shows a honeycomb effect, seen even more clearly in D, in which β-tubulin appears to enclose empty spaces. Counterstaining with DAPI (E) shows that most of the honeycomb effect is attributable to the cytoplasmic distribution of β-tubulin, which leaves the large neuronal nuclei devoid of signal; however, some perinuclear regions in E are positive for neither β-tubulin nor DAPI, and it is mainly in those perinuclear areas that viral protein expression (eGFP) is seen, in a panel showing DAPI and eGFP (F).
As Infection Progresses, Viral Proteins Are Expressed in Mature Neurons, and Astrocytes Are Spared
A progressive increase in the number of infected cells within the hippocampus was observed over time. By day 4 post-infection, cells expressing viral proteins were found in the dentate gyrus and in Ammon’s horn, especially within the CA3 field of the hippocampus (Figure 6A) ▶ , and cells in the latter region had eGFP+ extended axonal processes, characteristic of pyramidal neurons of the hippocampus. At this time point, β-tubulin expression was reduced in the dentate gyrus and hippocampus (Figure 6, A to C) ▶ , consistent with neuronal maturation. To more directly assess the relationship between the ongoing coxsackievirus infection, and neuronal maturation, sections were stained for NeuN, a nuclear marker for mature neurons. 33 Intense NeuN staining was observed in the dentate gyrus and hippocampus (Figure 6, D to F) ▶ , and eGFP+ cells in these regions co-localized well with NeuN. Individual infected neurons can be identified both in the dentate gyrus (Figure 6E) ▶ and in the CA3 region of the hippocampus (Figure 6F) ▶ . Again, the ontogeny of the infected cells is unclear: eGFP+, NeuN+, β-tubulin− cells may have come from CVB-infected β-tubulin+ immature neuronal cells that, despite the infection, continued their migration, and underwent differentiation into mature neurons; or they may represent mature neurons which have been recently infected by virus. A more detailed analysis of BrdU-labeled pulsed cells followed over time in combination with coxsackievirus infection (eGFP expression) may help to clarify this issue. We also evaluated whether or not CVB3 infection of the CNS involved astrocytes, which can be identified by their expression of glial fibrillary acidic protein (GFAP). These cells, with their characteristic branched morphology, could be detected in the hippocampus of a 5-day-old mouse (Figure 6, G to I) ▶ , but we were unable to identify eGFP expression in any of these cells. Thus, 4 days after infection, coxsackievirus is located mostly in mature neurons.
Figure 6.
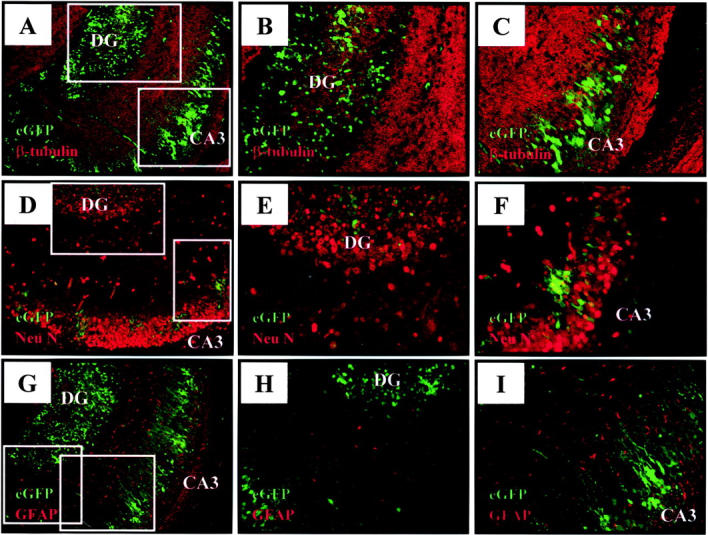
Mature hippocampal neurons express viral proteins by day 4 post-infection. Pups were infected i.c. with eGFP-CVB (2 × 106 pfu), and harvested 4 days post-infection. Paraffin-embedded sections were immunostained for neuron-specific class III β-tubulin (A–C), NeuN (mature neuronal marker, D–F), or GFAP (astrocytic marker, G–I), and observed by fluorescence microscopy. All pictures represent merged images for eGFP and the corresponding cell-type specific marker (red). Boxed areas in A, D, and G identify sites of magnification for images B and C; E and F; and H and I, respectively. A–C: Infected cells in the hippocampus express little, if any, β-tubulin. D–F: Mature hippocampal neurons expressing NeuN (red) co-localize with cells expressing high levels of viral protein (eGFP) within the hippocampus. G–I: Astrocytes (red) show a characteristic branched morphology, and are found in regions that are anatomically distinct from cells expressing viral protein (eGFP). Magnification, ×31 for images A, D, and G. All other images: magnification, ×62. DG, dentate gyrus; CA3, field CA3 of Ammon’s horn.
Severe Pathology Observed in the Olfactory Bulbs, Hippocampus, and Cortex Following Intracranial Infection with eGFP-CVB
The pathological consequences of the infection correlate strongly with the distribution of viral materials. By 5 days post-infection, widespread neuronal cell loss was evident in the granular cell layer of the olfactory bulb (Figure 7A) ▶ and in the dentate gyrus and hippocampus (Figure 7, C and E) ▶ . In a few pups infected with high doses of virus, areas of damage were also observed in the temporal and entorhinal cortex (Figure 7C) ▶ . Generally, the size of the hippocampal region was substantially smaller in infected pups 5 days post-infection, as compared to mock-infected controls. In addition, closure and fusion of the ependyma of the lateral ventricle 34 sometimes failed to occur and, in many cases, the lateral ventricle was distended. In a limited number of pups, areas of hemorrhage were observed in the olfactory bulb, the ventricles and parts of the temporal cortex. In contrast, mock infections produced no apparent pathology in these regions (Figure 7, B, D, and F) ▶ .
Figure 7.
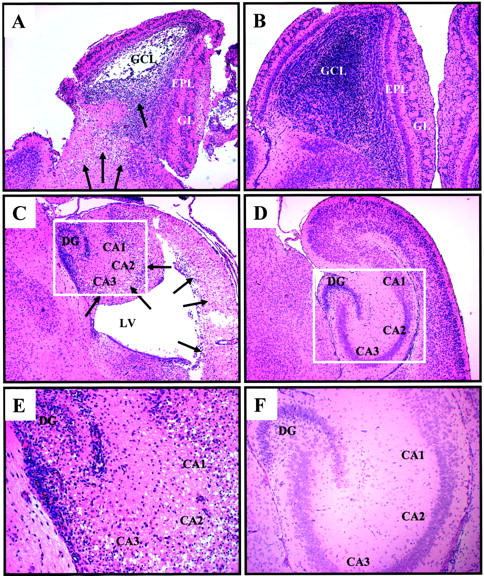
Coxsackievirus-induced damage observed in the olfactory bulb, hippocampus, and cortex after infection. One-day-old pups were infected intracranially with eGFP-CVB (2 × 106 pfu; A and C, ×8 E, ×20) or mock-infected (B and D, ×5; F, ×10) and sacrificed 5 days later. Brains were fixed in 10% neutral-buffered formalin, and paraffin-embedded transverse sections were stained by H&E. Extensive cellular damage was observed within the olfactory bulb (A), the temporal and entorhinal cortex (C), and the hippocampus (E) of infected mice. Arrows point to sites of cellular damage and condensed nuclei. Mock-infected pups revealed the normal morphology of the neonatal olfactory bulb, and hippocampus/cortex regions. Boxed regions in C and D are shown at higher magnification in E and F, respectively. Condensed chromatin was seen within cells in the hippocampus (E, ×20), but not in mock-infected controls (F, ×10). GCL, granular cell layer; EPL, external plexiform layer; GL, glomerular layer; DG, dentate gyrus; CA1, CA2, CA3, fields of Ammon’s horn; LV, lateral ventricle.
Regional Co-Localization of eGFP Expression and Apoptotic Staining Were Observed at Sites of CNS Damage
Next, we investigated the mechanism of neuronal cell death. Virus-induced apoptosis has been shown to play a role in myocarditis and pancreatitis in CVB3 infected adult mice, 35-37 and has recently been identified in rat cortical cultures following infection with CVB4. 38 Therefore, we used the TUNEL assay to detect fragmented DNA, indicative of cells undergoing apoptosis. Early after infection (day 1 post-infection), viral protein expression was observed in the absence of apoptosis (data not shown). Apoptosis was first observed by 2 days p.i. (not shown) and, by days 3 and 4 p.i. (Figure 8,A and D ▶ , respectively) was extensive near areas of infection in the hippocampus and dentate gyrus. Apoptosis also was detected in the temporal and entorhinal cortex, and in areas near the lateral ventricles (not shown). Higher magnification of the hippocampus demonstrated regional co-localization of infection and apoptosis (Figure 8, B, C, E, and F) ▶ ; and the morphological criteria of apoptosis (eg, nuclear condensation) were observed in sections stained with hematoxylin (Figure 8G) ▶ . Negative controls, in which adjacent tissue sections were incubated in the absence of terminal deoxytransferase, confirmed the specificity of the apoptotic signal (Figure 8H) ▶ and, as expected, mock-infected pups showed few signs of CNS apoptosis or pathology (Figure 8I) ▶ . eGFP signal, and TUNEL signal, were occasionally found to be immediately adjacent to one another (Figure 8C ▶ , arrowed); they did not directly overlap (which would have produced a yellow signal in the merged image), but this is not surprising, because eGFP is cytoplasmic, while the TUNEL signal is nuclear. However, it is clear that, in the majority of cases, the signals were in neighboring but separate cells (Figure 8F) ▶ ; many eGFP+ cells were negative by TUNEL assay, and vice versa. How might this disparity be explained? It is possible that coxsackievirus infection of neurons may lead to apoptosis of adjacent, uninfected cells, and such “bystander apoptosis” has been reported in other viral infections. 39,40 However, we favor an alternative explanation: that cells can remain eGFP+ for some time, before entering apoptosis (explaining the eGFP+, TUNEL− cells); and that eGFP is very rapidly degraded in cells as they enter apoptosis (explaining eGFP−, TUNEL+ cells). To investigate this, we next attempted to identify cells that were at an earlier stage of apoptosis, in the hope that such cells might contain residual eGFP.
Figure 8.

Viral protein expression co-localizes with ApopTag staining within the hippocampus. Newborn pups were infected with eGFP-CVB and 3 (A–C) or 4 (D–H) days later, brains were harvested, and transverse brain sections were examined for apoptotic cells using ApopTag staining as described in Materials and Methods. Merged ×31 images (eGFP-viral protein expression; red-ApopTag) in A and D demonstrate that apoptotic areas in the hippocampus regionally localized with areas in which viral proteins were expressed. The boxed regions in A are presented at higher magnification ×62 in B and C. Nuclei which appear positive for ApopTag, and may be related to eGFP+ cells, are arrowed. Increasing magnifications of the hippocampus at 4 days p.i. (D) are shown in E ×62 and F (×100); magnified regions are boxed in the preceding panel. Cells undergoing apoptosis are circled (F), and these cells showed nuclear condensation by hematoxylin staining (G). A control section was included (H), in which the TdT enzyme was omitted during staining, thus demonstrating that the red signal is specific for apoptotic cells; and a mock-infected control brain harvested at day 4 post-injection (I) showed staining for neither eGFP, nor for apoptosis. DG, dentate gyrus; CA1, CA2, CA3, fields of Ammon’s horn.
CVB Infection of Mature Neurons Can Lead to Their Apoptosis
To this end, we evaluated the expression of caspase-3, a cytosolic protein that is a key regulator of apoptosis. This protein is expressed as a proenzyme, and triggering of apoptosis is accompanied by cleavage of this precursor into the active enzyme, 41 for which a specific antibody is available. Activated caspase-3 was detected in all areas in which TUNEL signal was present, including the hippocampus. By day 3 p.i., direct co-localization of active caspase-3 expression (red) and viral protein expression (green) in the cytoplasm could be demonstrated by merged immunofluorescence images (yellow signal, Figure 9A ▶ ). No staining was seen in the hippocampus of a mock-infected mouse (Figure 9B) ▶ . Higher-power magnification of the boxed area of Figure 9A ▶ shows extensive eGFP signal (Figure 9C) ▶ , and somewhat more restricted active caspase-3 signal (Figure 9D) ▶ , and it is clear that almost all of the caspase signal overlaps with eGFP signal (Figure 9E) ▶ . H&E staining of the same section showed that the eGFP+, activated capsase-3+, cells had condensed nuclei (arrowed, Figure 9F ▶ ). Thus, coxsackievirus infection of mature neurons can result in their apoptosis.
Figure 9.
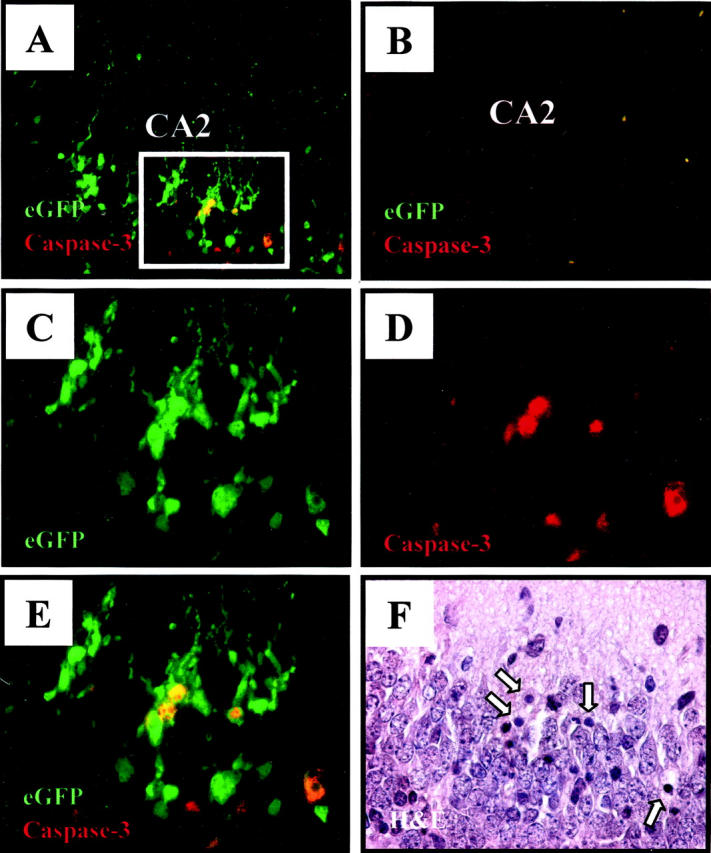
Viral protein expression co-localizes with activated caspase-3 staining in the hippocampus. Transverse brain sections from infected pups (2 × 106 pfu eGFP-CVB) harvested 4 days post-infection were examined for apoptotic activity by staining with an antibody against active caspase-3 (red signal). Magnification, ×40 for images A and B, magnification, ×63 for other images. A: Merged image demonstrating direct co-localization (yellow) of eGFP and active caspase 3 in infected hippocampal neurons in field CA3. No signal is detected in the hippocampus of a mock-infected mouse (B). The boxed region in A corresponds to higher magnification (×63) images shown in C to F. C: Single channel image representing infected neuronal cells (eGFP) within the hippocampus. D: Single channel image demonstrating active caspase-3 staining (red) within the hippocampus. E: Merged image of C and D, revealing direct co-localization of infected neurons expressing active caspase-3. The corresponding H&E section (F) showed condensed nuclei (arrowed) in the same cells in which active caspase-3 was detected. CA2, field CA2 of Ammon’s horn.
Discussion
Newborn infants are especially susceptible to coxsackievirus-induced diseases, including myocarditis, meningitis, and pancreatitis. Neonatal CVB infections have been associated with choriomeningitis and encephalitis, 42-44 and it has been suggested that CVB may play a role in sudden infant death syndrome. 45,46 In this study, we show (Figure 1) ▶ that newborn pups are inherently much more susceptible to coxsackievirus CNS infection than are older mice. This susceptibility dropped dramatically with age, and 7-day-old mice survived challenge with very high doses of CVB. Others have attributed the enhanced susceptibility of neonates to the immature status of the neonatal immune system, but few studies have investigated this claim; furthermore, recent findings, including some from our laboratory, have indicated that the neonatal immune system may be more competent than commonly assumed. 47-58 An alternative explanation for the enhanced susceptibility of neonates is that some feature of the neonatal CNS renders its cells more capable of supporting CVB infection. We have shown that the cell cycle status can play a role in determining cellular susceptibility to CVB infection in tissue culture, 19,59 and we hypothesize that the neonatal CNS may be better able to support CVB infection because many more cells are undergoing active division, migration, and differentiation. As a first step toward testing this hypothesis, we have chosen to carefully map the anatomical and cellular tropisms of CVB in the neonatal CNS.
We examined the tropism of eGFP-CVB over time within the CNS by co-localizing viral protein expression (eGFP) with neuronal and astrocytic markers. At early time points following infection, high levels of viral protein were identified in the choroid plexus, in cells lining the ventricles, and in the olfactory bulb. These locations reflect the anatomical regions in which CNS stem cells are found; primarily in the SVZ and in the olfactory bulb. 60 Intriguingly, one strain of the parvovirus minute virus of mice, known to target proliferative areas in vivo, also infects the SVZ, the olfactory bulb, and the dentate gyrus, 61 and infection of neonatal rats with LCMV also targets these regions of the CNS. 62 Although the anatomical locations of CNS stem cells are well-established, their precise cellular nature is somewhat controversial 63-65 and, as a result, we cannot confidently identify infected stem cells by immunostaining. However, as shown in Figure 2G ▶ , it appears that the infected cells in the SVZ are not ependymal cells, and may instead be type B stem cells, which have a stretched morphology and protrude through the ependymal cell layer. These intercalating type B stem cells can give rise to both astrocytes and neurons, 26,63 and we hypothesize that their particular metabolic status may render them susceptible to CVB infection. Nestin staining confirmed that CVB infection, at this early time point, was largely confined to CNS progenitor cells. Consistent with neuronal precursor cells being a major target for coxsackievirus infection within the CNS, by day 2 post-infection, high levels of viral protein were found in immature (β-tubulin+) neurons and, later in infection, the bulk of viral protein was found in cells expressing NeuN, a marker of mature neurons. This gradual spread of the infection into the CNS parenchyma, and its association with maturing neurons, raises an intriguing possibility. In the CNS of newborn rodents, there is very extensive neurogenesis; 66-68 indeed, the first week of birth represents the period of peak production of olfactory bulb interneurons in the neonatal rat. 69 During neurogenesis, stem cells give rise to immature neurons, which migrate to many areas of the brain, including the olfactory bulb and the cortex. It is tempting to speculate that the early infection of type B stem cells allows CVB to “hitchhike” in these migrating cells, which initially show the phenotype of immature neuronal cells, and subsequently express markers of neuronal maturation. We have previously shown that B lymphocytes can acts as “Trojan horses” to disseminate CVB in the blood; 70 perhaps type B cells, and their neuronal progeny, play a similar role in the CNS. If so, then the virus appears very selective in the progenitor cell in which it books passage; we found no sign of eGFP expression in astrocytes, which also can be derived from type B stem cells.
The sites of cell death closely paralleled those areas in which CVB was identified, either by in situ hybridization, or by viral protein expression. Cell death was observed in Ammon’s horn of the hippocampus, the dentate gyrus, the olfactory bulbs, and regions of the cortex, and many infected neurons contained condensed nuclei, characteristic of apoptotic cells. Coxsackievirus infection has been previously shown to induce apoptosis in mice, in pancreatic acinar cells and in myocardiocytes, 35,37,71 and CVB4 has recently been shown to induce apoptosis in rat cortical neuronal cultures. 38 Therefore, we investigated the role of apoptosis in neuronal death in vivo. The TUNEL assay confirmed the induction of apoptosis in infected hippocampal neurons as early as 3 days post-infection. Areas of apoptotic activity invariably co-localized with areas of infection, but the TUNEL assay rarely identified cells that scored positive for eGFP expression. We thought that this might result from the rapid disappearance of eGFP when a cell entered apoptosis, and therefore repeated the analysis, this time using an earlier marker of programmed cell death, activated caspase 3. As shown in Figure 9E ▶ , double-positive (yellow) cells were more readily detected, consistent with both of these markers being cytoplasmic. Nevertheless, even using this enzyme as an indicator of early apoptosis, a few active caspase-3+ cells showed no eGFP signal, perhaps because the enzyme’s protease activity targets many other cellular proteins, including eGFP, for cleavage in addition to its downstream target. 72 In addition, many eGFP+ cells failed to express detectable active caspase-3; it remains to be determined if all infected cells will eventually undergo apoptosis, or whether a population of infected cells remains protected from this fate. Such a scenario may explain how coxsackievirus is able to persist in vivo. Few inflammatory cells were observed during acute infection of the neonatal CNS, suggesting that the neuronal cell death and the pathology observed after acute infection are not significantly influenced by an adaptive immune response. This conclusion is strengthened by our evaluation of coxsackievirus CNS infection in RAG knockout pups, which exhibit pathologies identical to those seen in immunocompetent BALB/c pups (data not shown). The finding that coxsackievirus-induced neuronal cell death is apoptotic in origin opens the possibility of limiting CVB-induced CNS pathology by treating newborn infants with drugs designed to inhibit the apoptotic pathway. 73
Thus, we suggest that a major determinant of susceptibility in the neonatal CNS is the ability of specific cells, including type B stem cells, to support CVB infection; and we propose that the infection is disseminated through the CNS by neurons which continue their migration and maturation despite the infection. On full maturation, the infected neurons are triggered to initiate programmed cell death that may, perhaps, assist in the release of infectious virions. Although we find these hypotheses attractive, alternative explanations cannot be ignored. For example, the age-dependent susceptibility to infection could be related to the cellular distribution of the murine coxsackievirus and adenovirus receptor protein (mCAR). This molecule appears to be a major receptor for CVB3, 74,75 and we have shown that its expression in the adult pancreas correlates with the viral distribution. 36 mCAR mRNA expression in the developing murine CNS begins at ∼day E14, is maximal in the perinatal period, 76 and decreases rapidly after birth. 77 Expression has been observed in primary neurons, including the growth cones of the hippocampi, and it is thought that mCAR may function as an adhesion molecule that may be involved in neural-network formation in the developing nervous system. 77 Thus, it is possible that mCAR expression determines the distribution of CVB in the neonatal CNS; we are currently investigating this question. Furthermore, spread into the brain parenchyma, in the days following infection, could result from the release of infectious virus, and subsequent infection of susceptible cells; this may contribute to the extensive cell death seen by 5 days post-infection. However, this interpretation is rendered less likely by our observation that susceptibility to i.c. infection decreases markedly within 3 days of birth (Figure 1B) ▶ . We suggest that, as an infected newborn mouse ages, its cells rapidly become less susceptible to infection; as a result, any virus released from infected cells after the mouse has reached ∼3 days of age will encounter an inhospitable population of uninfected cells. Experiments also are underway to address this issue. Finally, it is possible that some infected cells do not undergo apoptosis, and instead survive, perhaps retaining viral RNA for months or years after the infection has apparently been eradicated. The ability of CVB RNA to persist for months or years following the initial infection is well-established both in humans, 78 and in experimental models, 79,80 and we have shown that viral RNA can persist in tissue culture, in the absence of infectious virus. 19
The possibility of CVB RNA persistence suggests a mechanism for chronic or delayed neuropathology and, although most enteroviral diseases are acute in nature, some symptoms can emerge many years after the original infection; for example, coxsackievirus infections also have been associated with the development of delayed neuropathologies, including amyotrophic lateral sclerosis, 81 schizophrenia, 8 and encephalitis lethargica (EL). The latter disease, named by the Austrian physician Constantin von Economo (and sometimes referred to as von Economo’s disease), was first identified in 1915 in Romania, and rapidly reached epidemic proportions, affecting at least 250,000 individuals, and eventually subsiding in the 1930s. It was usually biphasic; in the acute phase, patients developed symptoms consistent with infectious encephalitis; headache, fever, and diplopia, sometimes progressing to delirium. Some cases were fatal, but many of those who recovered subsequently, sometimes years later, developed chronic disease, characterized by lethargy, near-catatonia, and Parkinson-like symptoms. The cause of this epidemic has never been satisfactorily resolved. It is generally assumed that the disease had an infectious origin, and its temporal coincidence to the 1918–1919 worldwide pandemic of “Spanish flu” has often been cited to support the popular notion that influenza virus was the primary cause of EL. However, this hypothesis has been challenged, and recent PCR analyses of archived clinical samples from both acute and chronic EL patients failed to detect influenza virus RNA. 82 Although EL is often considered a disease of the past, cases are still reported, 18,83,84 sometimes in children, 85 and the biochemical changes in the cerebrospinal fluid, which include the identification of oligoclonal immunoglobulin banding, are consistent with a viral etiology. 83,84 Attempts at viral isolation are not always successful 84 but, in some cases, it is clear that the clinical syndrome can be caused by CVB. 18 Therefore, given that influenza virus is unlikely to have caused this epidemic, enteroviruses, which very commonly infect the CNS, should be considered as candidates in EL. The delayed symptoms might reflect a gradual deterioration in neuronal function, but also could be explained by the pathogenic consequences of persistent viral materials. In our own studies, a number of pups have demonstrated instances of paralysis and hydrocephalus at later time points during development (data not shown). Therefore, we are currently evaluating the ability of coxsackievirus to induce chronic diseases of the CNS.
Acknowledgments
We thank Annette Lord for excellent secretarial support and the histology laboratory of the Scripps Research Institute for skilled technical assistance. This is manuscript number 15584-NP from the Scripps Research Institute.
Footnotes
Address reprint requests to Lindsay Whitton, Department of Neuropharmacology, CVN-9, The Scripps Research Institute, 10550 N. Torrey Pines Rd., La Jolla, CA 92037. E-mail: lwhitton@scripps.edu.
Supported by National Institutes of Health grants R-01 AI-42314 (to J.L.W.) and F32 HL-10490 (to R.F.).
Current address for I.M. is Institut Pasteur, Paris, France.
References
- 1.Chamberlain RN, Christie PN, Holt KS, Huntley RM, Pollard R, Roche MC: A study of school children who had identified virus infections of the central nervous system during infancy. Child Care Health Dev 1983, 9:29-47 [DOI] [PubMed] [Google Scholar]
- 2.Hsueh C, Jung SM, Shih SR, Kuo TT, Shieh WJ, Zaki S, Lin TY, Chang LY, Ning HC, Yen DC: Acute encephalomyelitis during an outbreak of enterovirus type 71 infection in Taiwan: report of an autopsy case with pathologic, immunofluorescence, and molecular studies. Mod Pathol 2000, 13:1200-1205 [DOI] [PubMed] [Google Scholar]
- 3.Ratzan KR: Viral meningitis. Med Clin North Am 1985, 69:399-413 [DOI] [PubMed] [Google Scholar]
- 4.Sauerbrei A, Gluck B, Jung K, Bittrich H, Wutzler P: Congenital skin lesions caused by intrauterine infection with coxsackievirus B3. Infection 2000, 28:326-328 [DOI] [PubMed] [Google Scholar]
- 5.Daley AJ, Isaacs D, Dwyer DE, Gilbert GL: A cluster of cases of neonatal coxsackievirus B meningitis and myocarditis. J Paediatr Child Health 1998, 34:196-198 [DOI] [PubMed] [Google Scholar]
- 6.Gear JH, Measroch V: Coxsackievirus infections of the newborn. Prog Med Virol 1973, 15:42-62 [PubMed] [Google Scholar]
- 7.Euscher E, Davis J, Holzman I, Nuovo GJ: Coxsackie virus infection of the placenta associated with neurodevelopmental delays in the newborn. Obstet Gynecol 2001, 98:1019-1026 [DOI] [PubMed] [Google Scholar]
- 8.Rantakallio P, Jones P, Moring J, Von Wendt L: Association between central nervous system infections during childhood and adult onset schizophrenia and other psychoses: a 28-year follow-up. Int J Epidemiol 1997, 26:837-843 [DOI] [PubMed] [Google Scholar]
- 9.Dalldorf G, Melnick JL: Coxsackieviruses. Horsfall FL Tamm I eds. Viral and Rickettsial Infections of Man. 1965:pp 472-512 JB Lippincott Philadelphia
- 10.Hyypia T, Kallajoki M, Maaronen M, Stanway G, Kandolf R, Auvinen P, Kalimo H: Pathogenetic differences between coxsackie A and B virus infections in newborn mice. Virus Res 1993, 27:71-78 [DOI] [PubMed] [Google Scholar]
- 11.Harvala H, Kalimo H, Dahllund L, Santti J, Hughes P, Hyypia T, Stanway G: Mapping of tissue tropism determinants in coxsackievirus genomes. J Gen Virol 2002, 83:1697-1706 [DOI] [PubMed] [Google Scholar]
- 12.Kamei S, Hersch SM, Kurata T, Takei Y: Coxsackie B antigen in the central nervous system of a patient with fatal acute encephalitis: immunohistochemical studies of formalin-fixed, paraffin-embedded tissue. Acta Neuropathol (Berl) 1990, 80:216-221 [DOI] [PubMed] [Google Scholar]
- 13.de la Fuente G, Palacios O, Villagra E, Villanueva ME: Isolation of Coxsackieviruses B5 in a fatal case of meningoencephalitis. Rev Med Chile 1995, 123:1510-1513 [PubMed] [Google Scholar]
- 14.Estes ML, Rorke LB: Liquefactive necrosis in Coxsackie B encephalitis. Arch Pathol Lab Med 1986, 110:1090-1092 [PubMed] [Google Scholar]
- 15.Gauntt CJ, Gudvangen RJ, Brans YW, Marlin AE: Coxsackievirus group B antibodies in the ventricular fluid of infants with severe anatomic defects in the central nervous system. Pediatrics 1985, 76:64-68 [PubMed] [Google Scholar]
- 16.Gauntt CJ, Jones DC, Huntington HW, Arizpe HM, Gudvangen RJ, DeShambo RM: Murine forebrain anomalies induced by coxsackievirus B3 variants. J Med Virol 1984, 14:341-355 [DOI] [PubMed] [Google Scholar]
- 17.Peatfield RC: Basal ganglia damage and subcortical dementia after possible insidious Coxsackie virus encephalitis. Acta Neurol Scand 1987, 76:340-345 [DOI] [PubMed] [Google Scholar]
- 18.Cree BC, Bernardini GL, Hays AP, Lowe G: A fatal case of coxsackievirus B4 meningoencephalitis. Arch Neurol 2003, 60:107-112 [DOI] [PubMed] [Google Scholar]
- 19.Feuer R, Mena I, Pagarigan RR, Slifka MK, Whitton JL: Cell cycle status affects coxsackievirus replication, persistence, and reactivation in vitro. J Virol 2002, 76:4430-4440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Slifka MK, Pagarigan RR, Mena I, Feuer R, Whitton JL: Using recombinant coxsackievirus B3 to evaluate the induction and protective efficacy of CD8+ T cells in controlling picornaviral infection. J Virol 2001, 75:2377-2387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Henry SC, Schmader K, Brown TT, Miller SE, Howell DN, Daley GG, Hamilton JD: Enhanced green fluorescent protein as a marker for localizing murine cytomegalovirus in acute and latent infection. J Virol Methods 2000, 89:61-73 [DOI] [PubMed] [Google Scholar]
- 22.Walter I, Fleischmann M, Klein D, Muller M, Salmons B, Gunzburg WH, Renner M, Gelbman W: Rapid and sensitive detection of enhanced green fluorescent protein expression in paraffin sections by confocal laser scanning microscopy. Histochem J 2000, 32:99-103 [DOI] [PubMed] [Google Scholar]
- 23.Sjokin K, Dalmasso AP, Smith JM, Martinez C: Thymectomy in newborn and adult mice. Transplantation 1963, 1:521-525 [DOI] [PubMed] [Google Scholar]
- 24.Kandolf R, Sauter M, Aepinus C, Schnorr JJ, Selinka HC, Klingel K: Mechanisms and consequences of enterovirus persistence in cardiac myocytes and cells of the immune system. Virus Res 1999, 62:149-158 [DOI] [PubMed] [Google Scholar]
- 25.Reetoo KN, Osman SA, Illavia SJ, Cameron-Wilson CL, Banatvala JE, Muir P: Quantitative analysis of viral RNA kinetics in coxsackievirus B3-induced murine myocarditis: biphasic pattern of clearance following acute infection, with persistence of residual viral RNA throughout and beyond the inflammatory phase of disease. J Gen Virol 2000, 81:2755-2762 [DOI] [PubMed] [Google Scholar]
- 26.Temple S: The development of neural stem cells. Nature 2001, 414:112-117 [DOI] [PubMed] [Google Scholar]
- 27.Doetsch F, Garcia-Verdugo JM, Alvarez-Buylla A: Cellular composition and three-dimensional organization of the subventricular germinal zone in the adult mammalian brain. J Neurosci 1997, 17:5046-5061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lendahl U, Zimmerman LB, McKay RD: CNS stem cells express a new class of intermediate filament protein. Cell 1990, 60:585-595 [DOI] [PubMed] [Google Scholar]
- 29.Sejersen T, Lendahl U: Transient expression of the intermediate filament nestin during skeletal muscle development. J Cell Sci 1993, 106:1291-1300 [DOI] [PubMed] [Google Scholar]
- 30.Doetsch F, Alvarez-Buylla A: Network of tangential pathways for neuronal migration in adult mammalian brain. Proc Natl Acad Sci USA 1996, 93:14895-14900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pencea V, Bingaman KD, Freedman LJ, Luskin MB: Neurogenesis in the subventricular zone and rostral migratory stream of the neonatal and adult primate forebrain. Exp Neurol 2001, 172:1-16 [DOI] [PubMed] [Google Scholar]
- 32.Brazelton TR, Rossi FM, Keshet GI, Blau HM: From marrow to brain: expression of neuronal phenotypes in adult mice. Science 2000, 290:1775-1779 [DOI] [PubMed] [Google Scholar]
- 33.van Praag H, Schinder AF, Christie BR, Toni N, Palmer TD, Gage FH: Functional neurogenesis in the adult hippocampus. Nature 2002, 415:1030-1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kawamata S, Stumpf WE, Bidmon HJ: Adhesion and fusion of ependyma in rat brain. Acta Anat (Basel) 1995, 152:205-214 [DOI] [PubMed] [Google Scholar]
- 35.Gebhard JR, Perry CM, Harkins S, Lane T, Mena I, Asensio VC, Campbell IL, Whitton JL: Coxsackievirus B3-induced myocarditis: perforin exacerbates disease, but plays no detectable role in virus clearance. Am J Pathol 1998, 153:417-428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mena I, Fischer C, Gebhard JR, Perry CM, Harkins S, Whitton JL: Coxsackievirus infection of the pancreas: evaluation of receptor expression, pathogenesis, and immunopathology. Virology 2000, 271:276-288 [DOI] [PubMed] [Google Scholar]
- 37.Henke A, Launhardt H, Klement K, Stelzner A, Zell R, Munder T: Apoptosis in coxsackievirus B3-caused diseases: interaction between the capsid protein VP2 and the proapoptotic protein siva. J Virol 2000, 74:4284-4290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Joo CH, Kim YK, Lee H, Hong H, Yoon SY, Kim D: Coxsackievirus B4-induced neuronal apoptosis in rat cortical cultures. Neurosci Lett 2002, 326:175-178 [DOI] [PubMed] [Google Scholar]
- 39.Geleziunas R, Xu W, Takeda K, Ichijo H, Greene WC: HIV-1 Nef inhibits ASK1-dependent death signalling providing a potential mechanism for protecting the infected host cell. Nature 2001, 410:834-838 [DOI] [PubMed] [Google Scholar]
- 40.Zhang M, Atherton SS: Apoptosis in the retina during MCMV retinitis in immunosuppressed BALB/c mice. J Clin Virol 2002, 25(Suppl 2):S137-S147 [DOI] [PubMed] [Google Scholar]
- 41.Cohen GM: Caspases: the executioners of apoptosis. Biochem J 1997, 326:1-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chalhub EG, Devivo DC, Siegel BA, Gado MH, Feigin RD: Coxsackie A9 focal encephalitis associated with acute infantile hemiplegia and porencephaly. Neurology 1977, 27:574-579 [DOI] [PubMed] [Google Scholar]
- 43.Draganescu N, Nereantiu F, Girjabu E: Coxsackie B2 virus isolation from a case of postnatal meningoencephalitis. Virologie 1980, 31:9-12 [PubMed] [Google Scholar]
- 44.Price RA, Garcia JH, Rightsel WA: Choriomeningitis and myocarditis in an adolescent with isolation of coxsackie B-5 virus. Am J Clin Pathol 1970, 53:825-831 [DOI] [PubMed] [Google Scholar]
- 45.Dettmeyer R, Baasner A, Schlamann M, Haag C, Madea B: Coxsackie B3 myocarditis in 4 cases of suspected sudden infant death syndrome: diagnosis by immunohistochemical and molecular-pathologic investigations. Pathol Res Pract 2002, 198:689-696 [DOI] [PubMed] [Google Scholar]
- 46.Cioc AM, Nuovo GJ: Histologic and in situ viral findings in the myocardium in cases of sudden, unexpected death. Mod Pathol 2002, 15:914-922 [DOI] [PubMed] [Google Scholar]
- 47.Ridge JP, Fuchs EJ, Matzinger P: Neonatal tolerance revisited: turning on newborn T cells with dendritic cells. Science 1996, 271:1723-1726 [DOI] [PubMed] [Google Scholar]
- 48.Hassett DE, Zhang J, Whitton JL: Neonatal DNA immunization with an internal viral protein is effective in the presence of maternal antibodies and protects against subsequent viral challenge. J Virol 1997, 71:7881-7888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Manickan E, Yu Z, Rouse BT: DNA immunization of neonates induces immunity despite the presence of maternal antibody. J Clin Invest 1997, 100:2371-2375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Martinez X, Brandt C, Saddallah F, Tougne C, Barrios C, Wild F, Dougan G, Lambert PH, Siegrist CA: DNA immunization circumvents deficient induction of T helper type 1 and cytotoxic T lymphocyte responses in neonates and during early life. Proc Natl Acad Sci USA 1997, 94:8726-8731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Prince AM, Whalen R, Brotman B: Successful nucleic acid based immunization of newborn chimpanzees against hepatitis B virus. Vaccine 1997, 15:916-919 [DOI] [PubMed] [Google Scholar]
- 52.Wang Y, Xiang Z, Pasquini S, Ertl HC: Immune response to neonatal genetic immunization. Virology 1997, 228:278-284 [DOI] [PubMed] [Google Scholar]
- 53.Adkins B: T-cell function in newborn mice and humans. Immunol Today 1999, 20:330-335 [DOI] [PubMed] [Google Scholar]
- 54.Bot A, Shearer M, Bot S, Woods C, Limmer J, Kennedy R, Casares S, Bona C: Induction of antibody response by DNA immunization of newborn baboons against influenza virus. Viral Immunol 1999, 12:91-96 [DOI] [PubMed] [Google Scholar]
- 55.Watts AM, Stanley JR, Shearer MH, Hefty PS, Kennedy RC: Fetal immunization of baboons induces a fetal-specific antibody response. Nat Med 1999, 5:427-430 [DOI] [PubMed] [Google Scholar]
- 56.Hassett DE, Zhang J, Slifka MK, Whitton JL: Immune responses following neonatal DNA vaccination are long-lived, abundant, and qualitatively similar to those induced by conventional immunization. J Virol 2000, 74:2620-2627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Marshall-Clarke I, Reen I, Tasker I, Hassan I: Neonatal immunity: how well has it grown up? Immunol Today 2000, 21:35-41 [DOI] [PubMed] [Google Scholar]
- 58.Zhang J, Silvestri N, Whitton JL, Hassett DE: Neonates mount robust and protective adult-like CD8+ T cell responses to DNA vaccines. J Virol 2002, 76:11911-11919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Feuer R, Mena I, Pagarigan RR, Hassett DE, Whitton JL: Coxsackievirus replication and the cell cycle: a potential regulatory mechanism for viral persistence/latency. Med Microbiol Immunol (Berl), in press [DOI] [PubMed]
- 60.Gritti A, Bonfanti L, Doetsch F, Caille I, Alvarez-Buylla A, Lim DA, Galli R, Verdugo JM, Herrera DG, Vescovi AL: Multipotent neural stem cells reside into the rostral extension and olfactory bulb of adult rodents. J Neurosci 2002, 22:437-445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ramirez JC, Fairen A, Almendral JM: Parvovirus minute virus of mice strain i multiplication and pathogenesis in the newborn mouse brain are restricted to proliferative areas and to migratory cerebellar young neurons. J Virol 1996, 70:8109-8116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bonthius DJ, Mahoney J, Buchmeier MJ, Karacay B, Taggard D: Critical role for glial cells in the propagation and spread of lymphocytic choriomeningitis virus in the developing rat brain. J Virol 2002, 76:6618-6635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Doetsch F, Caille I, Lim DA, Garcia-Verdugo JM, Alvarez-Buylla A: Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell 1999, 97:703-716 [DOI] [PubMed] [Google Scholar]
- 64.Johansson CB, Momma S, Clarke DL, Risling M, Lendahl U, Frisen J: Identification of a neural stem cell in the adult mammalian central nervous system. Cell 1999, 96:25-34 [DOI] [PubMed] [Google Scholar]
- 65.Kornblum HI, Geschwind DH: Molecular markers in CNS stem cell research: hitting a moving target. Nat Rev Neurosci 2001, 2:843-846 [DOI] [PubMed] [Google Scholar]
- 66.Menezes JR, Smith CM, Nelson KC, Luskin MB: The division of neuronal progenitor cells during migration in the neonatal mammalian forebrain. Mol Cell Neurosci 1995, 6:496-508 [DOI] [PubMed] [Google Scholar]
- 67.Smith CM, Luskin MB: Cell cycle length of olfactory bulb neuronal progenitors in the rostral migratory stream. Dev Dyn 1998, 213:220-227 [DOI] [PubMed] [Google Scholar]
- 68.Coskun V, Luskin MB: Intrinsic and extrinsic regulation of the proliferation and differentiation of cells in the rodent rostral migratory stream. J Neurosci Res 2002, 69:795-802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Law AK, Pencea V, Buck CR, Luskin MB: Neurogenesis and neuronal migration in the neonatal rat forebrain anterior subventricular zone do not require GFAP-positive astrocytes. Dev Biol 1999, 216:622-634 [DOI] [PubMed] [Google Scholar]
- 70.Mena I, Perry CM, Harkins S, Rodriguez F, Gebhard JR, Whitton JL: The role of B lymphocytes in coxsackievirus B3 infection. Am J Pathol 1999, 155:1205-1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Huber SA, Budd RC, Rossner K, Newell MK: Apoptosis in coxsackievirus B3-induced myocarditis and dilated cardiomyopathy. Ann NY Acad Sci 1999, 887:181-190 [DOI] [PubMed] [Google Scholar]
- 72.Back SH, Shin S, Jang SK: Polypyrimidine tract-binding proteins are cleaved by caspase-3 during apoptosis. J Biol Chem 2002, 277:27200-27209 [DOI] [PubMed] [Google Scholar]
- 73.DeBiasi RL, Edelstein CL, Sherry B, Tyler KL: Calpain inhibition protects against virus-induced apoptotic myocardial injury. J Virol 2001, 75:351-361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bergelson JM, Krithivas A, Celi L, Droguett G, Horwitz MS, Wickham T, Crowell RL, Finberg RW: The murine CAR homolog is a receptor for coxsackie B viruses and adenoviruses. J Virol 1998, 72:415-419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bergelson JM, Cunningham JA, Droguett G, Kurt-Jones EA, Krithivas A, Hong JS, Horwitz MS, Crowell RL, Finberg RW: Isolation of a common receptor for coxsackie B viruses and adenoviruses 2 and 5. Science 1997, 275:1320-1323 [DOI] [PubMed] [Google Scholar]
- 76.Xu R, Crowell RL: Expression and distribution of the receptors for coxsackievirus B3 during fetal development of the Balb/c mouse and of their brain cells in culture. Virus Res 1996, 46:157-170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Honda T, Saitoh H, Masuko M, Katagiri-Abe T, Tominaga K, Kozakai I, Kobayashi K, Kumanishi T, Watanabe YG, Odani S, Kuwano R: The coxsackievirus-adenovirus receptor protein as a cell adhesion molecule in the developing mouse brain. Brain Res Mol Brain Res 2000, 77:19-28 [DOI] [PubMed] [Google Scholar]
- 78.Bowles NE, Rose ML, Taylor P, Banner NR, Morgan-Capner P, Cunningham L, Archard LC, Yacoub MH: End-stage dilated cardiomyopathy: persistence of enterovirus RNA in myocardium at cardiac transplantation and lack of immune response. Circulation 1989, 80:1128-1136 [DOI] [PubMed] [Google Scholar]
- 79.Tam PE, Messner RP: Molecular mechanisms of coxsackievirus persistence in chronic inflammatory myopathy: viral RNA persists through formation of a double-stranded complex without associated genomic mutations or evolution. J Virol 1999, 73:10113-10121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tam PE, Schmidt AM, Ytterberg SR, Messner RP: Viral persistence during the developmental phase of Coxsackievirus B1-induced murine polymyositis. J Virol 1991, 65:6654-6660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Woodall CJ, Riding MH, Graham DI, Clements GB: Sequences specific for enterovirus detected in spinal cord from patients with motor neurone disease. Br Med J 1994, 308:1541-1543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.McCall S, Henry JM, Reid AH, Taubenberger JK: Influenza RNA not detected in archival brain tissues from acute encephalitis lethargica cases or in postencephalitic Parkinson cases. J Neuropathol Exp Neurol 2001, 60:696-704 [DOI] [PubMed] [Google Scholar]
- 83.Howard RS, Lees AJ: Encephalitis lethargica: a report of four recent cases. Brain 1987, 110:19-33 [DOI] [PubMed] [Google Scholar]
- 84.Kiley M, Esiri MM: A contemporary case of encephalitis lethargica. Clin Neuropathol 2001, 20:2-7 [PubMed] [Google Scholar]
- 85.Mellon AF, Appleton RE, Gardner-Medwin D, Aynsley-Green A: Encephalitis lethargica-like illness in a five-year-old. Dev Med Child Neurol 1991, 33:158-161 [DOI] [PubMed] [Google Scholar]