

Abstract
The activity of endothelial nitric oxide synthase (eNOS) is subject to complex transcriptional and post-translational regulation including the association with several proteins and variations in subcellular distribution. In the present study we describe a transgenic mouse model expressing eNOS fused to green fluorescent protein (GFP), which allows the study of localization and regulation of eNOS expression. We tested the functionality of eNOS in the eNOS-GFP mice. Expression of eNOS was restricted to the endothelial lining of blood vessels in various tissues tested, without appreciable expression in non-endothelial cells. Activity of the enzyme was confirmed by assaying the conversion of L-arginine to L-citrulline. NO production in isolated vessels was increased in transgenic mice when compared to non-transgenic control animals (4.88 ± 0.59 and 2.48 ± 0.47 μmol/L NO, respectively, P < 0.005). Both the mean aortic pressure and the pulmonary artery pressure were reduced in eNOS-GFP mice (both ∼30%, P < 0.05). Plasma cholesterol levels were also slightly reduced (∼20%, P < 0.05). In conclusion, eNOS-GFP mice express functional eNOS and provide a unique model to study regulation of eNOS activity or eNOS-mediated vascular events, including response to ischemia, response to differences in shear stress, angiogenesis and vasculogenesis, and to study the subcellular distribution in relation with functional responses to these events.
In endothelial cells, nitric oxide (NO) is generated by the enzyme endothelial nitric oxide synthase (eNOS) via the conversion of L-arginine to L-citrulline. NO produced by eNOS affects a number of biological processes in the vessel wall. It is important for the regulation of blood pressure and plays an important role in the aggregation of blood platelets, adhesion of leukocytes to the vessel wall, and migration of vascular smooth muscle cells. 1 A decrease in NO availability is one of the hallmarks of endothelial dysfunction, which can occur in a number of cardiovascular disorders, including hypertension, heart failure, diabetes, and atherosclerosis. 2 It has been shown in animal models that a decrease in eNOS activity results in accelerated atherosclerosis. 3,4 Conversely, stimulation of eNOS activity by statin treatment has been implicated in the protective actions of these drugs. 5-7
For these reasons, the regulation of eNOS activity is considered to be of major physiological and pathophysiological importance. Although eNOS was originally termed endothelial constitutive NOS (ecNOS), 8-10 several laboratories have found that its protein expression and enzymatic activity is under tight regulation. 10-13 In addition to regulation at the transcriptional level, protein activity is controlled in several ways. Regulatory processes include N-myristoylation and cysteine palmitoylation, serine/threonine phosphorylation, and protein-protein interactions. These events affect the intracellular localization of eNOS which might influence its enzymatic activity. Normally, eNOS is localized in two subcellular compartments: the Golgi complex and the plasma membrane. 14 Although it has been suggested that translocation between these two compartments is important in the regulation of the enzymatic activity, it has been demonstrated that eNOS in both pools can be phosphorylated and activated. 14 In vitro studies of cells transfected with eNOS cDNA fused to DNA encoding green fluorescent protein (GFP) have provided valuable information of eNOS localization and regulation. However, to date such studies have not been performed in vivo. Consequently, we developed a transgenic mouse model in which an eNOS-GFP fusion protein is expressed. The results indicate that the eNOS fusion protein is functionally intact. These eNOS-GFPtg mice can be used to study vascular reactions in which eNOS is involved, including angiogenesis or response to variations in shear stress.
Materials and Methods
Generation of eNOS-GFP Transgenic Mice
A genomic DNA fragment was isolated from a home-made human cosmid library. It included 6 kb of 5′ sequence, the complete eNOS gene, and 3 kb of 3′ sequence. At the STOP codon of the eNOS gene, a linker was introduced that allowed the in-frame insertion of a BamHI-NotI DNA fragment encoding eGFP which was derived from the pEGFP-N1 plasmid (BD Biosciences Clontech, Palo Alto, CA). Fertilized oocytes from FVB mice were microinjected with a solution of 1–2 μg/ml in 8 mmol/L Tris-HCl and 0.1 mmol/L ethylenediaminetetraacetate (EDTA) and transplanted into the oviducts of pseudopregnant B10xCBA mice. Founder mice and offspring were genotyped by PCR on DNA isolated from tail biopsies. Primers used were: 5′-GTCCTGCAGACCGTGCAGC-3′ (sense) and 5′-GGCTGTTGGTGTCTGAGCCG-3′ (antisense). Mice were back-crossed to C57Bl6 for at least five generations (>96% C57Bl6). Transgenic mice used in the present study were hemizygous. All animal experiments were performed in compliance with institutional and national guidelines.
Immunohistochemistry, Fluorescence Microscopy, Confocal Microscopy
Immunohistochemistry experiments were performed according to Bakker et al 15 on 7-μm paraffin sections. The antibodies used were raised against the carboxy terminus of eNOS (Santa Cruz Biotechnology Inc., Santa Cruz, CA; catalog number sc-654). For confocal microscopy, mice were sacrificed using an overdose of isoflurane (1-chloro-2,2,2-trifluoroethyl-difluoromethyl-ether). Subsequently, in situ perfusion fixation was performed by flushing 20 ml PBS through a cardiac puncture followed by 20 ml 4% (v/v) paraformaldehyde in PBS. The common carotid arteries were then harvested, fixed in 4% paraformaldehyde in PBS at 4°C, and mounted in Vectashield (Vector Laboratories, Inc., Burlingame, CA) between glass slides. Samples were examined under a Zeiss LSM inverted laser-scanning confocal fluorescence microscope. Images of GFP fluorescence were acquired after excitation with a 488-nm argon laser and after filtering the fluorescence with a 500- to 550-nm bandpass barrier filter. The thickness of the optical slice images was 0.6 μm.
Western Blotting
Western blotting experiments were performed asdescribed previously. 16 The antibodies used were directed against human eNOS or human eNOS phosphorylated at Ser-1177, both obtained from Santa Cruz Biotechnology, Inc. (catalog numbers sc-654 and sc-12972, respectively).
eNOS Enzyme Activity and Nitric Oxide Measurements
eNOS activity was measured in the L-arginine to L-citrulline conversion assay using a nitric oxide synthase assay kit (Calbiochem, La Jolla, CA; catalog number 482700) as described previously. 16 For NO measurements, mice were anesthetized with 0.2 ml i.p. sodium pentobarbital (Apharmo BV, Arnhem, The Netherlands) and intracardially perfused with 10 ml Krebs Henseleit buffer solution (118 mmol/L NaCl, 4.7 mmol/L KCl, 3.0 mmol/L CaCl2, 1.2 mmol/L KH2PO4, 1.2 mmol/L MgSO4, 25 mmol/L NaHCO3, 0.5 mmol/L EDTA, 10 mmol/L D-glucose, and 0.1 mmol/L L-arginine, equilibrated with 95% air and 5% CO2 at room temperature resulting in a pH of 7.4). The aorta was dissected and in situ cannulated with two metal cannulas, proximal at the level of aortic arch and distal at the level of the diaphragm. The cannulas were connected to a home-made device keeping the vessel at its original length. Next, the vessel was transported to the set-up, where the cannulas were fixed to two holders. The two holders were placed in a chamber filled with Krebs buffer. The cannula at the proximal end of the vessel was connected to Tygon tubing coming out of a reservoir with Krebs buffer. Between the reservoir and the cannula, a flow pump (Watson Marlow, Falmouth, Cornwall, UK) and an infusion pump (Perfusor VI; B.Braun, Melsungen, Germany) were positioned. The settings of the pump were adjusted to perfuse the aorta with a constant flow of 10 ml/min, measured by an electromagnetic flow sensor. This value was based on measurements of the cardiac output of our mice with a clinical Doppler device (Prosound 4000, Aloka, Japan) or by a transit-time flow probe (see below). Next, the aorta vessel was equilibrated by perfusing it for 30 minutes. During that period the experimental set-up was switched to its calibration state and the NO sensor was calibrated by infusing known NO concentrations through a tube mounted in parallel to the vessel (0.1 to 30 μmol/L). Then, L-arginine (0.1 mol/L) in Krebs buffer was added to the perfusate via the infusion pump. The NO-sensitive electrode was positioned at the distal end of the aorta, to measure the NO production by the vessel. The electrode was connected to an NO measurement system (Iso-NO mark II; World Precision Instruments, Sarasota, FL). All data, including temperature, pressure drop, flow and NO measurements were stored in a PC equipped with an analog-digital converter and analysis software (Advanced Technology CODAS; Dataq Instruments, Akron, OH).
Hemodynamics and Heart Weights
Hemodynamic measurements were performed as described. 16 Shortly, blood pressure measurements were performed on mice anesthetized with ketamine (100 mg/kg ip) and xylazine (20 mg/kg ip), intubated and ventilated with a mixture of O2 and N2 (1/2 v/v). A flame stretched PE 50 polyethylene catheter was inserted into the right carotid artery and advanced into the aortic arch for measurement of aortic pressure and connected to a pressure transducer (B.Braun). For infusion of L-NAME, a second catheter was introduced into the right external jugular vein and advanced into the superior caval vein. After thoracotomy through the second right intercostal space, the ascending aorta was exposed and a transit-time flow probe (ID 1.5 mm; Transonics systems T206, Ithaca, NY) was placed around the aorta for measuring aorta flow. Ten minutes after a second intraperitoneal bolus of 100 mg/kg ketamine and 20 mg/kg xylazine, baseline recordings were obtained. Then, a continuous 10-minute intravenous infusion of L-NAME (100 mg/kg) was started and measurements were continued until 10 minutes after completion of the infusion. For the measurements of pulmonary artery pressure, the second left intercostal space was opened and a 30G needle connected to a PE 10 polyethylene catheter was directly inserted into the pulmonary artery. Hemodynamic data were recorded and digitized using an on-line 4 channel data acquisition program (Advanced Technology CODAS; Dataq Instruments). Ten consecutive beats were selected for determination of heart rate, aortic and pulmonary artery pressures, and aortic blood flow. For determination of ventricular weights the heart was removed and the ventricles were separated from the atria, the aorta, and the pulmonary artery. The right ventricle (free wall) was carefully separated from the left ventricle (including septum) and the right and left ventricle were weighed on a microbalance (Sartorius AG Göttingen, Germany). For the measurements of dry weights, tissue fluids were removed by lyophilization.
Cholesterol and Lipoprotein Analysis
Blood was collected by orbital puncture after an overnight fast. Plasma from 6 to 10 mice was pooled and subjected to gel filtration analysis on two HR10/30 FPLC columns in tandem (Superdex 200 and Superose 6, both prepgrade; Pharmacia AB, Uppsala, Sweden). 17 Cholesterol concentrations were measured with the free cholesterol C kit (WAKO, Neuss, Germany) after hydrolysis of cholesterol esters with cholesterol esterase from Candida cylindracea.
Data Analysis
Analysis of data were performed using two-way or one-way analysis of variance followed by Scheffé’s test, as appropriate. Statistical significance was accepted when P < 0.05 (two-tailed). Data are presented as mean ± SEM.
Results
eNOS-GFP Transgenic Mice
We aimed to create mice transgenic for human eNOS, in which the expression resembled that of the endogenous gene as much as possible, both with respect to the expression pattern as well as the regulation. Therefore, we used a DNA fragment to generate transgenic mice that comprised the complete human eNOS genomic sequence, including the natural promoter. This fragment was isolated from a human cosmid library. In addition to the eNOS gene, the DNA fragment contained ∼6 kb of 5′ sequence, including the endothelial enhancer sequence identified by Laumonnier et al, 18 and ∼3 kb of 3′ sequence. At the STOP codon of the eNOS gene, a DNA fragment encoding enhanced GFP was inserted in frame with the eNOS gene, to obtain a DNA construct encoding an eNOS-GFP fusion protein. This fragment (Figure 1a) ▶ was used for the generation of eNOS-GFP transgenic (eNOS-GFPtg) mice. Figure 1b ▶ shows sections from mouse aorta, taken from eNOS-GFPtg mice or wild-type littermates (controls). Immunohistochemistry studies on these sections using an antibody directed against human eNOS revealed a strong staining of the endothelial layer of the aorta in the eNOS-GFPtg mice, while there was no staining in the media of the aorta (Figure 1b ▶ , lower left panel). Sections from non-transgenic controls showed virtually no staining (Figure 1b ▶ , upper left panel). Inspection with fluorescence microscopy showed a strong fluorescent signal in the endothelial layer of the aorta from eNOS-GFPtg mice (Figure 1b ▶ , lower right panel), which is much higher than the autofluorescence from the elastic lamellae that is present in both eNOS-GFPtg mice and in wild-type controls (Figure 1b ▶ , upper right panel). The subcellular localization was studied by confocal microscopy in the carotid artery (Figure 1c) ▶ . The expression pattern exactly matches the sites where eNOS is known to be primarily located: in the Golgi complex and at the plasma membrane. We performed Western blotting experiments with aorta and lung tissue from eNOS-GFPtg mice and non-transgenic controls (Figure 1d) ▶ . Using an antibody directed against human eNOS, both the endogenous murine eNOS is detected, caused by cross-reactivity of the antibody, as well as the eNOS-GFP fusion protein, which has a slightly higher molecular mass caused by the GFP moiety (approximately 130 kd and 155 kd, respectively; Figure 1d ▶ , left panels). In addition, we used an antibody that recognizes eNOS that is phosphorylated on serine 1177 (P-eNOS), which is the activated form of the enzyme. P-eNOS-GFP is present in both aorta and lung tissue from eNOS-GFPtg mice (Figure 1d ▶ , right panels). To determine whether the expressed eNOS-GFP fusion protein is catalytically active, enzymatic activity was measured in aorta and lung tissue from eNOS-GFPtg mice and non-transgenic controls. As shown in Figure 1e ▶ , the transgenic mice show an increase in eNOS activity of approximately 35-fold in aorta and 27-fold in lung tissue. In addition, NO production was measured in isolated vessels ex vivo, using an NO sensor. Aortas from eNOS-GFP transgenic mice showed an increased NO production compared with aortas from control mice (Figure 1f) ▶ . The data could be described by a first order kinetic reaction, which was confirmed by a Lineweaver-Burke transformation (data not shown).
Figure 1.

Transgenic mice expressing eNOS-GFP. a: The DNA construct used for the generation of eNOS-GFPtg mice consisted of the complete eNOS gene in which an eGFP encoding cassette was cloned in frame at the position of the eNOS STOP codon. The natural flanking sequences of the eNOS gene were left intact. b: Sections of aorta from control mice or eNOS-GFP transgenic mice, inspected with immunohistochemistry using an antibody directed against human eNOS (left panels) or by fluorescent microscopy (right panels). Original magnifications, ×630. c: Green fluorescence in endothelial cells from an intact carotid artery inspected with confocal microscopy. Double arrowheads indicate fluorescence at the position of the plasma membrane; the filled arrow indicates fluorescence at the position of the Golgi complex. The open arrow indicates the cell nucleus. Scale bar, 10 μm. d: Immunoblotting of aorta (upper panels) or lung (lower panels) homogenates with antibodies directed against human eNOS (left panels), which show cross-reactivity with endogenous mouse eNOS, or antibodies directed against phosphorylated Ser-1177 from human eNOS (right panels). e: eNOS activity measured in aorta or lung homogenates by the L-arginine to L-citrulline conversion assay. n = 9. *, P < 0.001. f: NO production in isolated aortas measured with an NO-sensitive electrode. n = 11 (C), n = 8 (Tg). *, P < 0.005. C, control mice; Tg, eNOS-GFP transgenic mice.
Localization of eNOS-GFP
To investigate the cell specificity of the expression of the transgene, sections from organs of eNOS-GFPtg mice were inspected by immunohistochemistry using an antibody directed against human eNOS (Figure 2) ▶ . eNOS immunoreactivity was detected in small blood vessels in the heart (Figure 2a) ▶ , in the sinusoids in the liver (Figure 2b) ▶ , in the peritubular capillaries, and in the endothelial cells in the glomeruli in the kidney (Figure 2c) ▶ and in the capillary sinusoids in the adrenal (Figure 2d) ▶ . eNOS protein was not found in the parenchymal cells of these organs. Sections from non-transgenic controls showed no immunoreactivity (not shown). This expression pattern was confirmed by fluorescence microscopy. Figure 3 ▶ shows micrographs from heart (Figure 3, a and b) ▶ , liver (Figure 3, c and d) ▶ , kidney (Figure 3, e and f) ▶ , and adrenal gland (Figure 3, g and h) ▶ . The lining of the larger vessels was clearly fluorescent (Figure 3c) ▶ . A dense capillary network was fluorescent in the heart, surrounding the cardiomyocytes, and in the adrenal. The sinusoids in the liver, as well as the peritubular capillaries in the kidney showed a positive signal, just like the capillaries of the glomeruli in the kidney. The parenchymal cells of none of these organs showed appreciable expression. In sections from non-transgenic controls, no fluorescent signal was detected (not shown).
Figure 2.
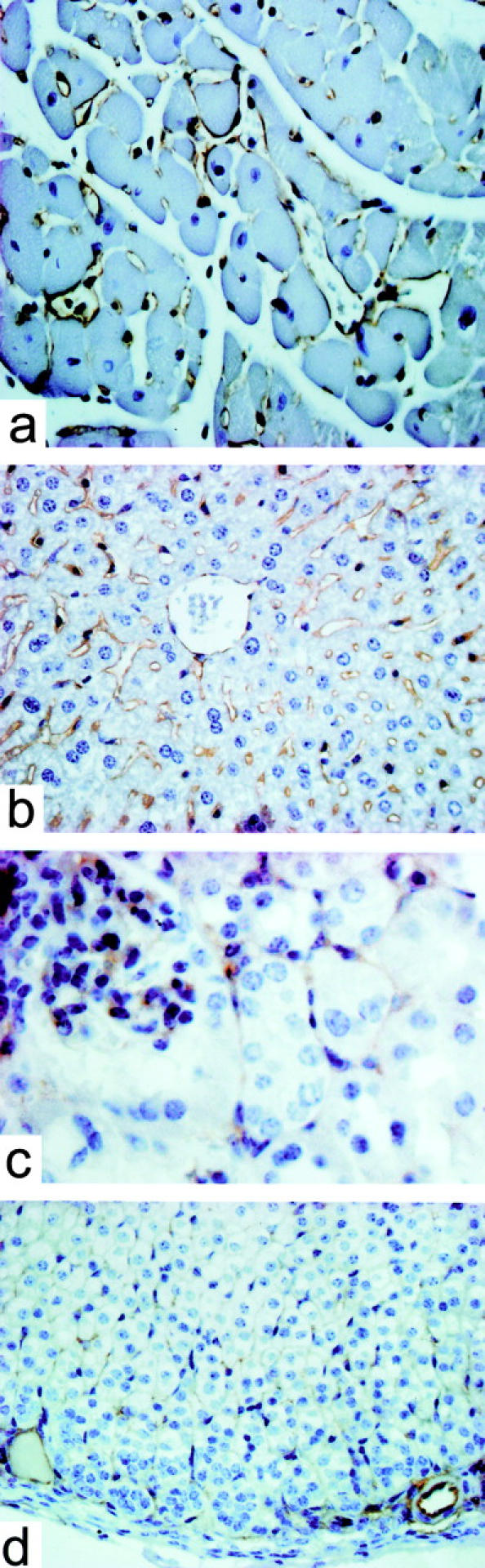
Localization of eNOS-GFP in various organs from eNOS-GFPtg mice visualized by immunohistochemistry using an antibody directed against human eNOS. a: Heart. b: Liver. c: Kidney. d: Adrenal. Original magnifications, ×400.
Figure 3.
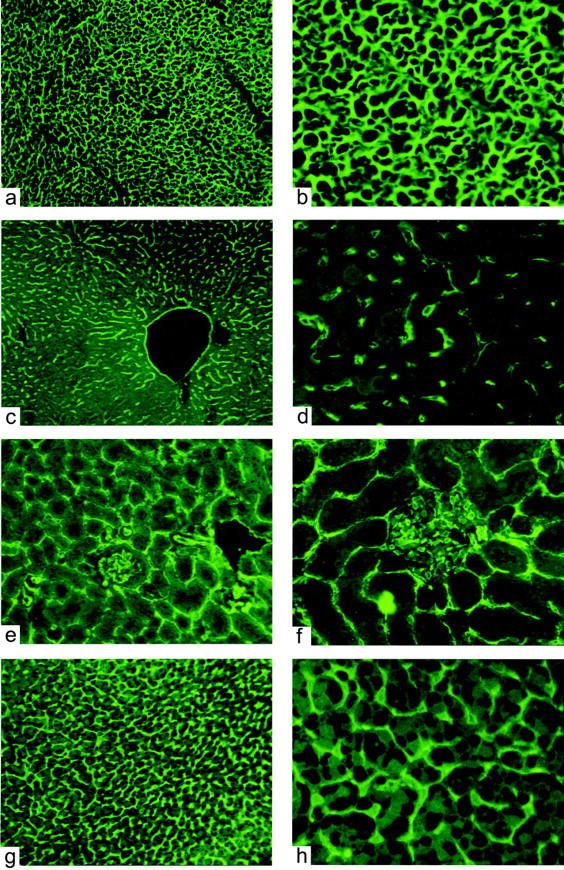
Localization of eNOS-GFP in various organs from eNOS-GFPtg mice visualized by fluorescence microscopy. a and b: Heart. c and d: Liver. e and f: Kidney. g and h: Adrenal. Original magnifications: ×100 (a,c,e,g) and ×400 (b,d,f,h).
Hemodynamic Properties of eNOS-GFPtg Mice
We performed hemodynamic measurements in eNOS-GFPtg mice compared to non-transgenic control mice. This was done to establish the functional activity of the eNOS-GFP fusion protein, as eNOS-derived NO is an important modulator of vascular tone. As shown in Figure 4a ▶ , the mean aortic pressure (MAP) was decreased in eNOS-GFP-expressing mice compared to wild-type littermates by approximately 29%. Treatment with the NO synthase-inhibitor NG nitro-L-arginine methyl ester (L-NAME) resulted in an increase of the MAP and abolished the difference between transgenic mice and wild-type littermates. The increase in the MAP (ΔMAP) caused by L-NAME treatment was greater in transgenic mice than in non-transgenic controls. There were no differences found in heart rate (Figure 4b) ▶ or mean aortic blood flow (Figure 4c) ▶ between transgenic or control mice, with or without treatment with L-NAME. Consequently, the difference in MAP can be attributed to a difference in systemic vascular resistance (Figure 4d) ▶ . In addition to a lower pressure in the systemic bed, we also observed a lower mean pulmonary artery pressure in eNOS-GFPtg mice of 30% (Figure 4e) ▶ . We also analyzed the ventricular weights (normalized to body weight) of the transgenic mice in comparison with controls. No differences were found (Figure 4, f and g) ▶ .
Figure 4.
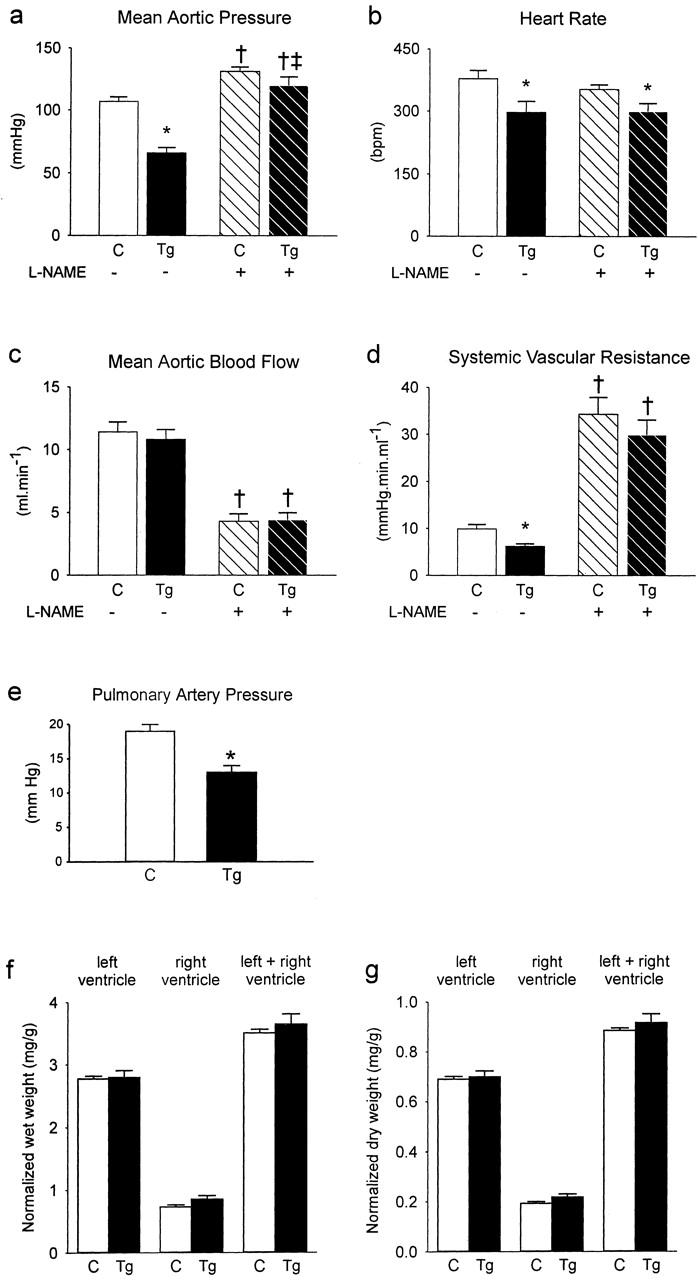
Hemodynamics and ventricular weights were measured in control mice (C) or eNOS-GFPtg mice (Tg) before and after addition of L-NAME. a: Mean aortic pressure. b: Heart rate. c: Mean aortic blood flow. d: Systemic vascular resistance. e: Pulmonary artery pressure. f: Ventricular wet weights. g: Ventricular dry weights. a to d: n = 10 (C), n = 9 (Tg). e: n = 8 (C), n = 6 (Tg). f to g: n = 20 (C), n = 12 (Tg). *, P < 0.05 versus corresponding controls; †, P < 0.05 versus corresponding baseline before addition of L-NAME; ‡, P < 0.05 Δ in C versus Δ in Tg (Δ produced by addition of L-NAME).
Plasma Cholesterol Levels in eNOS-GFPtg Mice
Finally, we measured levels of plasma cholesterol in eNOS-GFPtg mice with different genetic backgrounds as models for variations in plasma cholesterol levels. We compared wild-type background, heterozygous low-density lipoprotein receptor-deficient background, which results in a condition of slight hypercholesterolemia, and apolipoprotein-E-deficient background, which results in a condition of more severe hypercholesterolemia. We found a slight decrease in cholesterol levels in all backgrounds when comparing eNOS-GFP transgenic animals with animals without this transgene (Figure 5a) ▶ . Next, we analyzed the lipoprotein profile in two of these backgrounds. In wild-type background, plasma cholesterol is predominantly found in high-density lipoproteins (HDL). 19 Thus, the cholesterol-lowering observed in eNOS-GFP transgenic animals is found in HDL (Figure 5b) ▶ . In apolipoprotein-E-deficient background, however, plasma cholesterol is present in the atherogenic lipoproteins, very low-density lipoproteins (VLDL) and low-density lipoproteins (LDL). 16 In this background, expression of eNOS-GFP results in a decrease in both VLDL+LDL and HDL (Figure 5c) ▶ .
Figure 5.
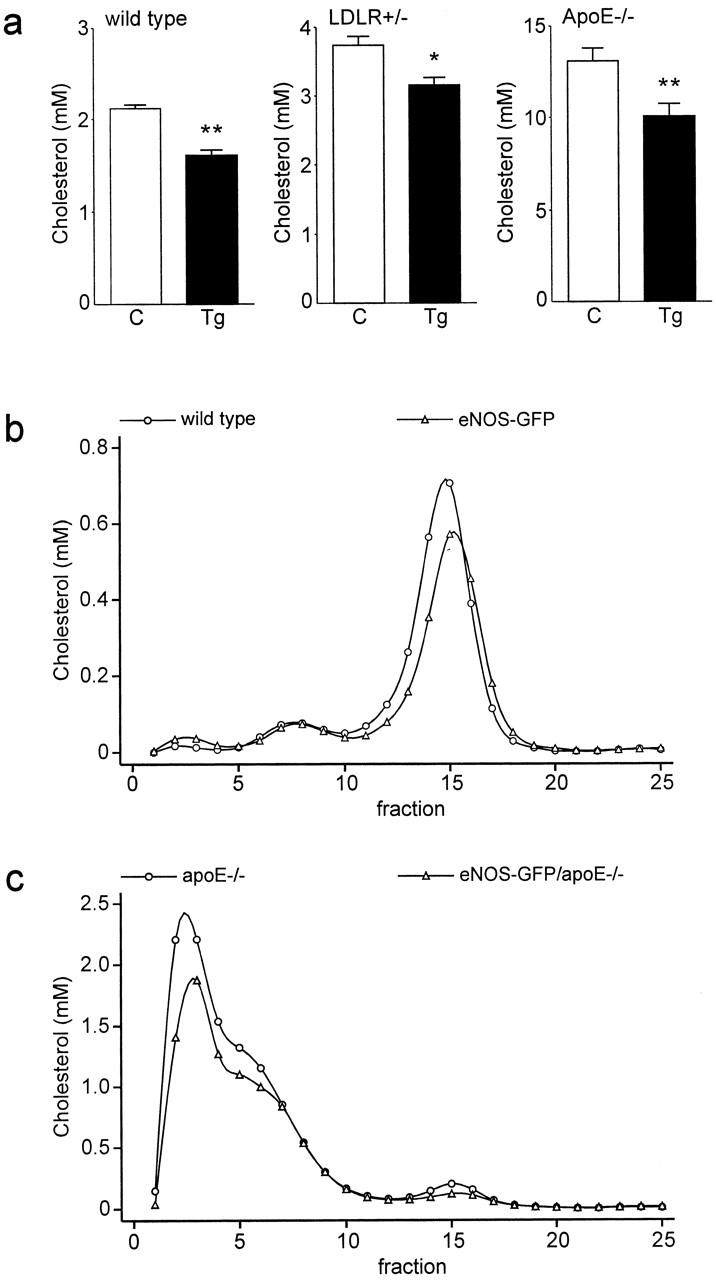
a: Plasma cholesterol concentrations of wild-type, LDLR heterozygous knockout (LDLR+/−) and apolipoprotein E homozygous knockout (ApoE−/−) mice with (Tg) or without (C) the eNOS-GFP transgene. *, P < 0.01 versus controls; **, P < 0.001 versus controls. b and c: Fractionation of lipoproteins by gel filtration of plasma from mice with wild-type background (b) or apolipoprotein E knockout background (c). Fraction 1–5 contain VLDL, 6–11 contain LDL, and 12–20 contain HDL.
Discussion
We generated transgenic mice that express human eNOS in fusion with GFP. To preserve transcriptional regulation, we used a genomic construct which contains the intronic sequences as well as the natural flanking sequences, including the previously identified endothelial enhancer located 4.9 kb upstream of the transcription initiation site. 18 The expression pattern was visualized either indirectly via immunohistochemistry using antibodies directed against eNOS, or directly via fluorescence microscopy. Both techniques showed virtually the same distribution of the eNOS-GFP transgene in a variety of organs: endothelial cells of smaller and larger vessels in various organs are positive, while the parenchymal cells are not. These results indicate that the DNA construct used for microinjections resulted in expression of the transgene predominantly, if not exclusively, in endothelial cells.
The functionality of the eNOS-GFP fusion protein was demonstrated in two different ways. Using the L-arginine to L-citrulline conversion assay, it was shown that eNOS enzyme activity is ∼30-fold higher in eNOS-GFP transgenic animals compared with non-transgenic controls. In ex vivo measurements with a NO sensor, NO production by endothelial cells in isolated blood vessels was shown to be 1.8-fold higher when vessels from eNOS-GFP transgenic mice were used.
The apparent discrepancy between these measurements can be explained by the difference between the methods that were used. In the L-arginine to L-citrulline conversion assay, a homogenate of aortic tissue was used. Cofactors and reagents are added to the assay to optimize the conditions for the enzymatic reaction. Therefore, the actual measurement represents the maximal eNOS activity, thereby reflecting the total amount of enzyme present. In contrast, the ex vivo measurements with the NO sensor were performed in intact blood vessels. As a consequence, the cellular regulation of the activity of the enzyme is still intact. In this system, the actual NO production is measured. This is much lower than the maximal enzymatic activity, probably because post-translational control of eNOS activity is intact.
In addition, blood pressure measurements showed that the mean aortic pressure was lower in eNOS-GFP transgenic animals compared with non-transgenic controls. This is in agreement with findings in other transgenic models of eNOS overexpression. 16,20 In the latter models, pulmonary pressures have not been measured. However, these are expected to be decreased as well, as the pulmonary vasodilator response following NO inhalation in patients is well known. 21,22 Indeed, we found lower pulmonary pressure in eNOS-GFP transgenic animals compared with non-transgenic controls.
GFP expression has been used in various transgenic mouse models and in numerous in vitro studies using cultured cells. It is generally assumed that the GFP protein itself does not interfere with any biological function of other proteins studied. However, specific expression in cardiomyocytes has been reported to result in hypertrophy. 23 Although the transgene in our mouse model is expressed in endothelial cells and not in cardiomyocytes, we measured the ventricular weights of transgenic and control mice. No differences were found, demonstrating that hypertrophy probably does not occur in eNOS-GFPtg mice.
Previously, we generated and studied transgenic mice overexpressing eNOS without GFP. 16 In addition to a lower blood pressure, these animals displayed a decrease in plasma levels of cholesterol. Thus, we measured plasma cholesterol in eNOS-GFP transgenic mice and found a decrease of ∼20% compared with non-transgenic mice (Figure 5a) ▶ . Plasma cholesterol in mice is for the larger part in HDL, which is generally believed to be atheroprotective. 19 Therefore, atherosclerosis in mouse models is usually studied in a genetic background with increased cholesterol in atherogenic very low-density lipoproteins (VLDL) and low-density lipoproteins (LDL). We crossbred eNOS-GFPtg mice with two of these models. First, we crossbred them to low-density lipoprotein receptor-deficient mice, resulting in a background of heterozygous deficiency. This genetic background causes mild hypercholesterolemia. 24 In addition, we crossbred the mice in an apolipoprotein E deficient background, which causes more severe hypercholesterolemia. 16 In both of these backgrounds, we found that expression of eNOS-GFP results in lower levels of plasma cholesterol (Figure 5a) ▶ . While in wild-type background a reduction in HDL-cholesterol was found (Figure 5b) ▶ , the effect in apolipoprotein E-deficient mice occurred primarily in the VLDL+LDL cholesterol fraction (Figure 5c) ▶ . This is in agreement with our previous work, in which it was tentatively concluded that this effect contributed to the anti-atherogenic properties of elevated eNOS expression. 16
The mouse model described in the present study can be used to study the unusually complex regulation of eNOS. 11,13 Regulation takes place at the transcriptional level, 25-27 and is responsive to variations in shear stress. 28,29 In addition, extensive regulation of eNOS activity at the post-translational level exists. These pathways are probably also active in the eNOS-GFPtg mice, as these are most likely responsible for the remarkable discrepancy between the level of protein overexpression and the physiological level of NO production (compare Figure 1, d and e with f ▶ ). The post-translational regulatory mechanisms include protein-protein interactions, some of which affect the subcellular localization of the protein. eNOS is primarily located in the Golgi complex and in the caveolae in the plasma membrane, which were shown to be two pools of active enzyme, 14 although others have suggested that active eNOS is largely restricted to caveolae. 30-32 A number of proteins have been shown to interact with eNOS and affect its activity. Caveolin, the structural protein of the caveolae, forms an inhibitory complex with eNOS, 32 for which further evidence was provided by studies in caveolin-deficient mice. 33 On activation by calcium, calmodulin can activate eNOS by displacing it from caveolin. 34,35 Another protein that binds to eNOS and subsequently activates it is heat shock protein 90. 36 The latter protein appears to play a role in the caveolin-calmodulin regulatory complex. 37 Recently identified proteins that interact with eNOS have been termed NOSIP and NOSTRIN. 38,39 The functions of these proteins have not been established yet. Still another way of post-translational regulation of eNOS activity is protein modification, like palmitoylation and myristoylation, 40,41 or phosphorylation, especially of the serine at position 1177 (human eNOS). 42,43
The eNOS-GFPtg mice described in the present study are particularly suitable for studying various aspects of the complex regulation of eNOS activity. Subcellular localization under various conditions or dynamic changes in this pattern on various stimuli can be studied, as well as interactions with other proteins or with the cytoskeleton. 44 Valuable information has been achieved by studying expression of eNOS-GFP cDNA in transfected cells. 45,46 Our model also allows ex vivo and in vivo studies, including real-time imaging by intravital microscopy. These type of studies will be useful to further assess the impact of eNOS activity in processes like reactivity to variations in shear stress or angiogenesis under conditions of ischemia or in tumor growth.
Acknowledgments
We thank Dr. G. van Cappellen for help with confocal microscopy, T. de Vries Lentsch and R. Koppenol for help with artwork, and Dr. J.F. Hamming and Dr. H. van Urk for continuous support.
Footnotes
Address reprint requests to Rini de Crom, Ph.D., Department of Cell Biology and Genetics, Erasmus University Medical Center, P.O. Box 1738, 3000 DR Rotterdam, The Netherlands. E-mail: m.decrom@erasmusmc.nl.
D. J. D and R. K. are established investigators of the Netherlands Heart Foundation (2000D038 and 2002T45, respectively)
References
- 1.Li H, Forstermann U: Nitric oxide in the pathogenesis of vascular disease. J Pathol 2000, 190:244-254 [DOI] [PubMed] [Google Scholar]
- 2.Harrison DG: Cellular and molecular mechanisms of endothelial cell dysfunction. J Clin Invest 1997, 100:2153-2157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Knowles JW, Reddick RL, Jennette JC, Shesely EG, Smithies O, Maeda N: Enhanced atherosclerosis and kidney dysfunction in eNOS(−/−)Apoe(−/−) mice are ameliorated by enalapril treatment. J Clin Invest 2000, 105:451-458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kuhlencordt PJ, Gyurko R, Han F, Scherrer-Crosbie M, Aretz TH, Hajjar R, Picard MH, Huang PL: Accelerated atherosclerosis, aortic aneurysm formation, and ischemic heart disease in apolipoprotein E/endothelial nitric oxide synthase double-knockout mice. Circulation 2001, 104:448-454 [DOI] [PubMed] [Google Scholar]
- 5.Laufs U, La Fata V, Plutzky J, Liao JK: Upregulation of endothelial nitric oxide synthase by HMG CoA reductase inhibitors. Circulation 1998, 97:1129-1135 [DOI] [PubMed] [Google Scholar]
- 6.Kano H, Hayashi T, Sumi D, Esaki T, Asai Y, Thakur NK, Jayachandran M, Iguchi A: A HMG-CoA reductase inhibitor improved regression of atherosclerosis in the rabbit aorta without affecting serum lipid levels: possible relevance of up-regulation of endothelial NO synthase mRNA. Biochem Biophys Res Commun 1999, 259:414-419 [DOI] [PubMed] [Google Scholar]
- 7.Sessa WC: Can modulation of endothelial nitric oxide synthase explain the vasculoprotective actions of statins? Trends Mol Med 2001, 7:189-191 [DOI] [PubMed] [Google Scholar]
- 8.Nishida K, Harrison DG, Navas JP, Fisher AA, Dockery SP, Uematsu M, Nerem RM, Alexander RW, Murphy TJ: Molecular cloning and characterization of the constitutive bovine aortic endothelial cell nitric oxide synthase. J Clin Invest 1992, 90:2092-2096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marsden PA, Heng HH, Scherer SW, Stewart RJ, Hall AV, Shi XM, Tsui LC, Schappert KT: Structure and chromosomal localization of the human constitutive endothelial nitric oxide synthase gene. J Biol Chem 1993, 268:17478-17488 [PubMed] [Google Scholar]
- 10.Forstermann U, Boissel JP, Kleinert H: Expressional control of the ‘constitutive’ isoforms of nitric oxide synthase (NOS I and NOS III). EMBO J 1998, 12:773-790 [PubMed] [Google Scholar]
- 11.Fulton D, Gratton JP, Sessa WC: Post-translational control of endothelial nitric oxide synthase: why isn’t calcium/calmodulin enough? J Pharmacol Exp Ther 2001, 299:818-824 [PubMed] [Google Scholar]
- 12.Govers R, Rabelink TJ: Cellular regulation of endothelial nitric oxide synthase. Am J Physiol Renal Physiol 2001, 280:F193-F206 [DOI] [PubMed] [Google Scholar]
- 13.Marletta MA: Another activation switch for endothelial nitric oxide synthase: why does it have to be so complicated? Trends Biochem Sci 2001, 26:519-521 [DOI] [PubMed] [Google Scholar]
- 14.Fulton D, Fontana J, Sowa G, Gratton JP, Lin M, Li KX, Michell B, Kemp BE, Rodman D, Sessa WC: Localization of endothelial nitric-oxide synthase phosphorylated on serine 1179 and nitric oxide in Golgi and plasma membrane defines the existence of two pools of active enzyme. J Biol Chem 2002, 277:4277-4284 [DOI] [PubMed] [Google Scholar]
- 15.Bakker CE, de Diego Otero Y, Bontekoe C, Raghoe P, Luteijn T, Hoogeveen AT, Oostra BA, Willemsen R: Immunocytochemical and biochemical characterization of FMRP, FXR1P, and FXR2P in the mouse. Exp Cell Res 2000, 258:162-170 [DOI] [PubMed] [Google Scholar]
- 16.Van Haperen R, De Waard M, Van Deel E, Mees B, Kutryk M, Van Aken T, Hamming J, Grosveld F, Duncker DJ, De Crom R: Reduction of blood pressure, plasma cholesterol, and atherosclerosis by elevated endothelial nitric oxide. J Biol Chem 2002, 277:48803-48807 [DOI] [PubMed] [Google Scholar]
- 17.Lie J, de Crom R, van Gent T, van Haperen R, Scheek L, Lankhuizen I, van Tol A: Elevation of plasma phospholipid transfer protein in transgenic mice increases VLDL secretion. J Lipid Res 2002, 43:1875-1880 [DOI] [PubMed] [Google Scholar]
- 18.Laumonnier Y, Nadaud S, Agrapart M, Soubrier F: Characterization of an upstream enhancer region in the promoter of the human endothelial nitric-oxide synthase gene. J Biol Chem 2000, 275:40732-40741 [DOI] [PubMed] [Google Scholar]
- 19.van Haperen R, van Tol A, Vermeulen P, Jauhiainen M, van Gent T, van den Berg P, Ehnholm S, Grosveld F, van der Kamp A, de Crom R: Human plasma phospholipid transfer protein increases the antiatherogenic potential of high-density lipoproteins in transgenic mice. Arterioscler Thromb Vasc Biol 2000, 20:1082-1088 [DOI] [PubMed] [Google Scholar]
- 20.Ohashi Y, Kawashima S, Hirata K, Yamashita T, Ishida T, Inoue N, Sakoda T, Kurihara H, Yazaki Y, Yokoyama M: Hypotension and reduced nitric oxide-elicited vasorelaxation in transgenic mice overexpressing endothelial nitric oxide synthase. J Clin Invest 1998, 102:2061-2071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hurford WE: Inhaled nitric oxide. Respir Care Clin N Am 2002, 8:261-279 [DOI] [PubMed] [Google Scholar]
- 22.Sitbon O, Humbert M, Simonneau G: Primary pulmonary hypertension: current therapy. Prog Cardiovasc Dis 2002, 45:115-128 [DOI] [PubMed] [Google Scholar]
- 23.Huang WY, Aramburu J, Douglas PS, Izumo S: Transgenic expression of green fluorescence protein can cause dilated cardiomyopathy. Nat Med 2000, 6:482-483 [DOI] [PubMed] [Google Scholar]
- 24.van Haperen R, van Tol A, van Gent T, Scheek L, Visser P, van der Kamp A, Grosveld F, de Crom R: Increased risk of atherosclerosis by elevated plasma levels of phospholipid transfer protein. J Biol Chem 2002, 277:48938-48943 [DOI] [PubMed] [Google Scholar]
- 25.Zhang R, Min W, Sessa WC: Functional analysis of the human endothelial nitric oxide synthase promoter. Sp1 and GATA factors are necessary for basal transcription in endothelial cells. J Biol Chem 1995, 270:15320-15326 [DOI] [PubMed] [Google Scholar]
- 26.Karantzoulis-Fegaras F, Antoniou H, Lai SL, Kulkarni G, D’Abreo C, Wong GK, Miller TL, Chan Y, Atkins J, Wang Y, Marsden PA: Characterization of the human endothelial nitric-oxide synthase promoter. J Biol Chem 1999, 274:3076-3093 [DOI] [PubMed] [Google Scholar]
- 27.Wu KK: Regulation of endothelial nitric oxide synthase activity and gene expression. Ann NY Acad Sci 2002, 962:122-130 [DOI] [PubMed] [Google Scholar]
- 28.Topper JN, Cai J, Falb D, Gimbrone MA, Jr: Identification of vascular endothelial genes differentially responsive to fluid mechanical stimuli: cyclooxygenase-2, manganese superoxide dismutase, and endothelial cell nitric oxide synthase are selectively up-regulated by steady laminar shear stress. Proc Natl Acad Sci USA 1996, 93:10417-10422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Go YM, Boo YC, Park H, Maland MC, Patel R, Pritchard KA, Jr, Fujio Y, Walsh K, Darley-Usmar V, Jo H: Protein kinase B/Akt activates c-Jun NH(2)-terminal kinase by increasing NO production in response to shear stress. J Appl Physiol 2001, 91:1574-1581 [DOI] [PubMed] [Google Scholar]
- 30.Blair A, Shaul PW, Yuhanna IS, Conrad PA, Smart EJ: Oxidized low-density lipoprotein displaces endothelial nitric-oxide synthase (eNOS) from plasmalemmal caveolae and impairs eNOS activation. J Biol Chem 1999, 274:32512-32519 [DOI] [PubMed] [Google Scholar]
- 31.Uittenbogaard A, Shaul PW, Yuhanna IS, Blair A, Smart EJ: High-density lipoprotein prevents oxidized low density lipoprotein-induced inhibition of endothelial nitric-oxide synthase localization and activation in caveolae. J Biol Chem 2000, 275:11278-11283 [DOI] [PubMed] [Google Scholar]
- 32.Everson WV, Smart EJ: Influence of caveolin, cholesterol, and lipoproteins on nitric oxide synthase: implications for vascular disease. Trends Cardiovasc Med 2001, 11:246-250 [DOI] [PubMed] [Google Scholar]
- 33.Schubert W, Frank PG, Woodman SE, Hyogo H, Cohen DE, Chow CW, Lisanti MP: Microvascular hyperpermeability in caveolin-1 (−/−) knockout mice: treatment with a specific nitric-oxide synthase inhibitor, L-name, restores normal microvascular permeability in Cav-1 null mice. J Biol Chem 2002, 277:40091-40098 [DOI] [PubMed] [Google Scholar]
- 34.Michel JB, Feron O, Sase K, Prabhakar P, Michel T: Caveolin versus calmodulin: counterbalancing allosteric modulators of endothelial nitric oxide synthase. J Biol Chem 1997, 272:25907-25912 [DOI] [PubMed] [Google Scholar]
- 35.Michel T, Feron O: Nitric oxide synthases: which, where, how, and why? J Clin Invest 1997, 100:2146-2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Garcia-Cardena G, Fan R, Shah V, Sorrentino R, Cirino G, Papapetropoulos A, Sessa WC: Dynamic activation of endothelial nitric oxide synthase by Hsp90. Nature 1998, 392:821-824 [DOI] [PubMed] [Google Scholar]
- 37.Gratton JP, Fontana J, O’Connor DS, Garcia-Cardena G, McCabe TJ, Sessa WC: Reconstitution of an endothelial nitric-oxide synthase (eNOS), hsp90, and caveolin-1 complex in vitro: evidence that hsp90 facilitates calmodulin stimulated displacement of eNOS from caveolin-1. J Biol Chem 2000, 275:22268-22272 [DOI] [PubMed] [Google Scholar]
- 38.Dedio J, Konig P, Wohlfart P, Schroeder C, Kummer W, Muller-Esterl W: NOSIP, a novel modulator of endothelial nitric oxide synthase activity. EMBO J 2001, 15:79-89 [DOI] [PubMed] [Google Scholar]
- 39.Zimmermann K, Opitz N, Dedio J, Renne C, Muller-Esterl W, Oess S: NOSTRIN: a protein modulating nitric oxide release and subcellular distribution of endothelial nitric oxide synthase. Proc Natl Acad Sci USA 2002, 99:17167-17172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Robinson LJ, Michel T: Mutagenesis of palmitoylation sites in endothelial nitric oxide synthase identifies a novel motif for dual acylation and subcellular targeting. Proc Natl Acad Sci USA 1995, 92:11776-11780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sakoda T, Hirata K, Kuroda R, Miki N, Suematsu M, Kawashima S, Yokoyama M: Myristoylation of endothelial cell nitric oxide synthase is important for extracellular release of nitric oxide. Mol Cell Biochem 1995, 152:143-148 [DOI] [PubMed] [Google Scholar]
- 42.Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh K, Franke TF, Papapetropoulos A, Sessa WC: Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature 1999, 399:597-601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM: Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature 1999, 399:601-605 [DOI] [PubMed] [Google Scholar]
- 44.Venema VJ, Marrero MB, Venema RC: Bradykinin-stimulated protein tyrosine phosphorylation promotes endothelial nitric oxide synthase translocation to the cytoskeleton. Biochem Biophys Res Commun 1996, 226:703-710 [DOI] [PubMed] [Google Scholar]
- 45.Liu J, Hughes TE, Sessa WC: The first 35 amino acids and fatty acylation sites determine the molecular targeting of endothelial nitric oxide synthase into the Golgi region of cells: a green fluorescent protein study. J Cell Biol 1997, 137:1525-1535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sowa G, Liu J, Papapetropoulos A, Rex-Haffner M, Hughes TE, Sessa WC: Trafficking of endothelial nitric-oxide synthase in living cells: quantitative evidence supporting the role of palmitoylation as a kinetic trapping mechanism limiting membrane diffusion. J Biol Chem 1999, 274:22524-22531 [DOI] [PubMed] [Google Scholar]