

Abstract
Toll-like receptors (TLRs) have been thought to mediate the antibody-enhancing effects of adjuvants. However, we find here that Myd88−/−; TrifLps2/Lps2 mice, which cannot transmit signals through TLRs, mount robust antibody responses to T cell dependent antigen given in four typical adjuvants: alum, complete Freund’s adjuvant (CFA), incomplete Freund’s adjuvant (IFA), and “Ribi” adjuvant. We therefore conclude that TLR signaling does not account for the action of classical adjuvants, nor does it fully explain the action of a strong adjuvant that contains a TLR ligand. This suggests that TLR ligands, and their attendant side effects, may be excluded from vaccine adjuvants.
Adjuvants are vaccine additives that enhance the elicited levels of antibodies and T lymphocytes. These effects improve immunological protection from infection and, hence, enhance public health. Adjuvants promote inflammatory responses of leukocytes in ways that presumably mimic natural infection. Toll-like receptor (TLR) mediated recognition of microbial signature molecules is one of the cues normally used by leukocytes to react to real microbial challenges (1, 2). Each of the 10 different functional TLRs in humans (12 in mice) have apparently evolved to recognize a specific set of evolutionarily conserved molecules, including components of bacterial cell walls, and endocytosed nucleic acids such as dsRNA, ssDNA, and unmethylated CpG DNA (1, 2). TLR recognition leads to the activation of transcription factors that drive cytokine expression, proliferation, survival, and inflammatory mediator expression. TLR signaling is initiated by four adapters MyD88, TRIF, TIRAP/Mal and TRAM, which associate with the cytoplasmic TIR domains of TLRs (1, 2). MyD88 associates with all TLRs except TLR3, while TRIF associates with TLR3 and TLR4. TIRAP/Mal and TRAM appear to function as bridging adapters for MyD88 and TRIF, respectively (1, 2). TIRAP/Mal and TRAM are essential for signaling by TLR4, with TIRAP/Mal also required for TLR2 function. MyD88 also contributes to signaling in B cells, and is required for maximal B cell responses to foreign proteins, when present in the context of TLR ligands (3). Nevertheless, there is debate about whether such signals are necessary for this class of response (4, 5). Mice genetically deficient in both MyD88 and TRIF (Myd88−/−; TrifLps2/Lps2 mice) have a complete lack of known TLR signaling (6–8), thus allowing an assessment of the TLR dependence of antibody responses. We took advantage of this to explore more precisely the role of TLR signaling in antibody responses to immunization, and the augmenting roles of adjuvants in this response.
Myd88−/−; TrifLps2/Lps2 and control C57BL/6 mice were immunized with the T cell dependent antigen TNP-hemocyanin (TNP-Hy) given in CFA and the induced anti-TNP titers in the serum determined (9). In these analyses, antibody responses of Myd88−/−; TrifLps2/Lps2 mice were entirely comparable to those of C57BL/6 mice, indicating that signals transmitted by TRIF and MyD88 made no appreciable contribution to the antibody response (Fig 1). This experiment included initial immunization, followed by a boost with TNP-Hy in phosphate buffered saline (PBS) on day 21, and showed no significant defect in sera at any time point for IgM, IgG1, IgG2b, IgG2c, IgG3 and IgE anti-TNP responses. Furthermore, when the TNP-Hy challenge was given with the adjuvant alum, a frequently used adjuvant in human vaccines, antibody responses of Myd88−/−; TrifLps2/Lps2 mice were also comparable to those of C57BL/6 mice (5 and Suppl. Fig 1).
Figure 1.
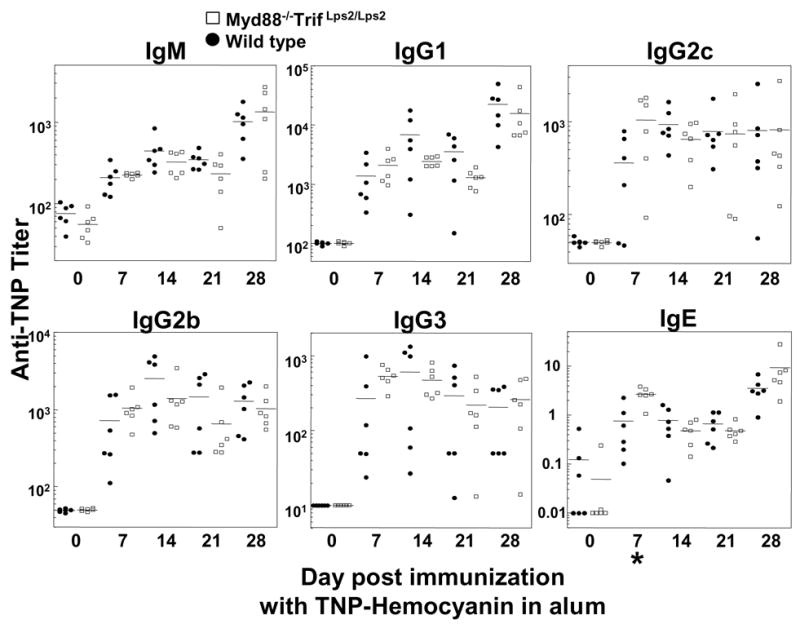
T dependent antibody responses of Myd88−/−; TrifLps2/Lps2 and control mice using CFA. Two month old mice were immunized with TNP-Hy in CFA on day 0 and boosted with antigen in PBS on day 21. Each point represents the serum anti-TNP titer of an individual mouse. IgE titers represent ng/ml, whereas other measurements represent reciprocal serum dilution yielding half maximal signal. Closed circles, wild type C57BL/6; open squares, Myd88−/−; TrifLps2/Lps2 double knockout mice. Statistically significant differences between mutant and control groups are as indicated with asterisks; *, p>.05; **, p>.01. For methods, see (9).
To reevaluate the augmenting effects of adjuvant on antibody production and its suggested dependence on MyD88 and TRIF, additional immunizations of C57BL/6 and of Myd88−/−; TrifLps2/Lps2 mice were carried out, using a second antigen, TNP-KLH, in which the adjuvants CFA, IFA, and monophosphoryl-lipid A/trehalose dicorynomycolate (“Ribi” adjuvant) were compared to responses in the absence of adjuvant. TNP specific antibody responses were assessed 7 and 14 days later (Fig 2). Ribi adjuvant contains the TLR4 ligand monophosphoryl-lipid A, whereas IFA is not known to contain any TLR ligand. Both C57BL/6 and Myd88−/−; TrifLps2/Lps2 mice responded strongly to TNP-KLH only when given in adjuvant (Fig 2). (The IgG1 responses to TNP-KLH given in PBS, though low, were significantly above preimmune background [titer<10]). Responses of Myd88−/−; TrifLps2/Lps2 mice to antigen given in CFA or IFA were unimpaired, whereas responses to TNP-KLH given in Ribi adjuvant were reduced relative to those of wild type mice at day 14 (Fig 2). However, the reduction in antibody titers elicited in Myd88−/−; TrifLps2/Lps2 mice was modest and observed mainly in the IgG2c and IgG2b responses (Fig 2). We conclude that, under these experimental conditions, TLR signaling is not required for T cell dependent antibody responses and makes a relatively insignificant contribution to responses to antigen given in either alum or CFA.
Figure 2.

Comparison of serum antibody responses of C57BL/6 and Myd88−/−; TrifLps2/Lps2 mice given immunogen in different adjuvants. Groups of four mice were immunized with 100μg TNP-KLH in PBS in the indicated adjuvants and anti-TNP titers of the indicated immunoglobulin isotypes determined on day 7 and 14.
It was notable that even in mice challenged with Ribi adjuvant, which contains a potent TLR ligand, most of the antibody-augmenting adjuvant effect was independent of MyD88/TRIF (Fig 2). Consistent with this, Ribi, but not IFA or CFA, stimulated peritoneal macrophages to produce type I interferon and TNF, when used to treat cells at concentrations approximating those achieved in vivo (Fig S2). As expected, isolated splenic B cells from Myd88−/−; TrifLps2/Lps2 mice failed to proliferate in response to the TLR ligand LPS (Fig S3B). By contrast, responses of Myd88−/−; TrifLps2/Lps2 B cells to BCR ligation were indistinguishable from those of normal cells (Fig S3A,C). Nevertheless, it was apparent that the observed adjuvant effect on antibody responses in vivo failed to correlate with their potential to activate myeloid cells or B cells in vitro.
Myd88−/−; TrifLps2/Lps2 mice were next tested for their responses to the T cell independent type 2 (TI-2) antigen TNP-Ficoll. Overall antibody responses did not generally differ significantly from normal controls (Fig S4). These results show for the first time that the antibody response to a typical TI-2 antigen is independent of TLR signaling.
Preimmune serum levels of immunoglobulins (Ig) in Myd88−/−; TrifLps2/Lps2 mice were next analyzed and found to have a complex pattern, with some isotypes reduced in amount and others increased, relative to C57BL/6 levels (Fig 3). Relative to control, IgG2b and IgG2c levels were lower in Myd88−/−; TrifLps2/Lps2 mice, by 50% and 70%, respectively; a statistically significant, but modest reduction (Fig 3). Also, the serum levels of one Ig subclass- IgG3- in Myd88−/−; TrifLps2/Lps2 mice was strikingly lower. By contrast, Myd88−/−; TrifLps2/Lps2 mice displayed a 3-fold elevation in levels of IgG1 (p>.001) and 20 times the level of IgE (p>.0001). Collectively, such differences suggest that TLR signaling may control the class rather than the magnitude of Ig levels in naive mice, at least under specific pathogen free conditions, such as found in our facility.
Figure 3.
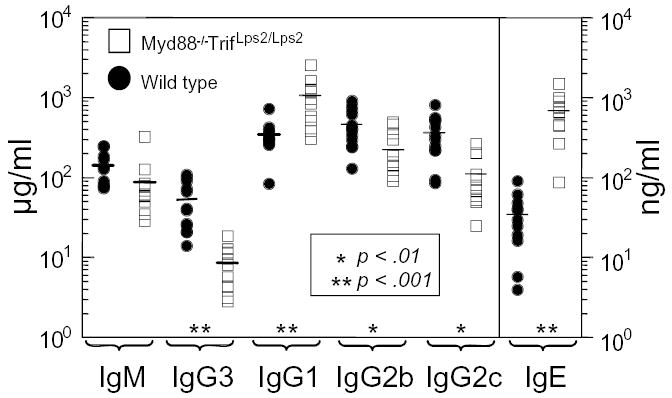
Preimmune serum immunoglobulin levels in Myd88−/−; TrifLps2/Lps2 compared to wild type C57BL/6 controls as measured by ELISA. Each point represents data from a different mouse. Closed circles, wild type, C57BL/6; open squares, Myd88−/−; TrifLps2/Lps2 mice.
Consistent with the robust preimmune Ig levels and antibody responses of Myd88−/−; TrifLps2/Lps2 mice, nearly normal B-1, marginal zone, and follicular B cell numbers were found in these animals. Compared to wild type mice, the only consistent differences found in Myd88−/−; TrifLps2/Lps2 mice tested at 2 and 6 months of age were an approximately two-fold higher B cell surface level of CD23 (Fig S5) and a slight reduction in splenic transitional type 1 B cells (T1) (Tables S1,S2). We conclude that TLR signaling is not required for preimmune B cell development, but may partly affect the abundance and CD23 levels of B cell subsets. As levels of CD23, IgE, and IgG1 are upregulated by interleukin 4, we speculate that environmental stimuli transduced by TLRs in a MyD88- and/or TRIF-dependent manner suppress the expression of, or response to, this cytokine.
Our findings reveal that TLR signaling is not required for robust antibody responses to antigen, when given in four commonly used adjuvants, in particular CFA, which is widely thought to depend upon TLR signaling for its adjuvant effect (10). However, ligands that signal through the MyD88 and TRIF pathways can costimulate these responses, and affect the antibody class of the response, as has been known for many years (3) and is indicated by the modest boost seen in C57BL/6 mice immunized with Ribi adjuvant (Fig 2). By exclusion, our data suggest the likelihood that non-TLR mediated “innate” signals may be involved in the augmentation of adaptive antibody responses. It is also formally possible that there exist MyD88/TRIF independent modes of TLR signaling yet to be discovered, possibly through TRAM and TIRAP, although so far there is no evidence for this.
Our results extend and clarify previous studies. A recent paper involving MyD88 deficient mice asserted that T-dependent antibody responses require activation of TLRs in B cells (4). However, that inference was based upon use of LPS as an adjuvant rather than more commonly used adjuvants, such as complete Freund’s adjuvant or alum. Indeed, an earlier study found that though MyD88 deficient mice failed to make an IgG2a response to ovalbumin given in CFA, they still made good IgG1 and IgE antibody responses, and antigen given in alum could promote an IgE response (11). However, in those studies, the remaining antibody responses in MyD88-deficient mice could conceivably have involved TLR signaling through TRIF dependent pathways, a caveat that does not apply to our data. We clearly find that IgG2c (also known as IgG2ab) responses to protein antigen given in alum or CFA are robust in Myd88−/−; TrifLps2/Lps2 mice, in apparent contradiction to both studies of MyD88-deficient mice mentioned (4, 11) and other studies implicating a requirement of MyD88 or TLR signaling in the generation of IgG2a antibodies. It may be that TLR ligand-driven suppression of IgG2c/a production occurs in mice with intact TRIF signaling that lack MyD88; for example, through cytokine driven polarization of the T helper response (11, 12). The IgG2c/a response is promoted by IFN-α/β or IFN-γ (13, 14). In viral infection, the IgG2c/a response is lost in mice lacking both IFN-α/βR and IFN-γ R (15). As we found good IgG2c responses upon immunization of Myd88−/−; TrifLps2/Lps2 mice, our results may indicate that these immunizations can stimulate TLR-independent interferon production.
The antibody response to the T independent type II antigen TNP-Ficoll was relatively normal in Myd88−/−; TrifLps2/Lps2 mice, though it was slightly lower at later time points. Interestingly, the IgG3 component of the antigen specific response of Myd88−/−; TrifLps2/Lps2 mice was not significantly lower than normal despite their reduced preimmune total serum IgG3 levels. It has been argued that the class switch to IgG3 requires cytokines produced by accessory cells (16). If this is so, it is clear from these results that production of the cytokines in question does not require TLR stimulation of B cells or accessory cells.
That B cells can respond to autologous DNA and RNA through TLR signaling (17, 18) raised the possibility that TLRs could affect preimmune development or maintenance of B cell subsets, particularly the marginal zone and B-1 compartments. However, our findings that Myd88−/−; TrifLps2/Lps2 mice generate abundant B-1 and marginal zone B cells appear to rule out definitively a required role for TLR signaling in their development.
Our data do not contest the long-held understanding that TLR ligands can augment antibody responses. However, it is surprising, given the recent emphasis on the importance of TLRs in the initiation of the adaptive immune response, that we fail to find a deficit in the early antibody responses of Myd88−/−;TrifLps2/Lps2 mice using conventional antigens and immunization regimens. Our data are more consistent with a model in which TLRs play roles in early microbial suppression, regulation of the antibody class, and sustaining antibody secretion at late times after immunization, rather than as an essential component of the self/non-self discrimination of the adaptive immune response. These data have implications for vaccine design as they indicate that robust antibody responses to moderate doses of antigens can be achieved when given in the total absence of TLR ligands. As TLR mediated signals can be toxic, our findings raise the possibility that unwanted side effects of adjuvants may be avoided by excluding TLR ligands from adjuvants, rather than including them.
Supplementary Material
Footnotes
This manuscript has been accepted for publication in Science. This version has not undergone final editing. Please refer to the complete version of record at http://www.sciencemag.org/. This manuscript may not be reproduced or used in any manner that does not fall within the fair use provisions of the Copyright Act without the prior, written permission of AAAS
One sentence summary: This paper analyzes for the first time antibody responses in mice lacking all toll-like receptor signaling and shows that their antibody responses to antigens given in a wide range of common adjuvants are robust.
References
- 1.Takeda K, Akira S. Int Immunol. 2005;17:1. doi: 10.1093/intimm/dxh186. [DOI] [PubMed] [Google Scholar]
- 2.Beutler B, et al. Annu Rev Immunol. 2006;24:353. doi: 10.1146/annurev.immunol.24.021605.090552. [DOI] [PubMed] [Google Scholar]
- 3.Andersson J, Sjoberg O, Moller G. Transplant Rev. 1972;11:131. doi: 10.1111/j.1600-065x.1972.tb00048.x. [DOI] [PubMed] [Google Scholar]
- 4.Pasare C, Medzhitov R. Nature. 2005;438:364. doi: 10.1038/nature04267. [DOI] [PubMed] [Google Scholar]
- 5.Nemazee D, Gavin A, Hoebe K, Beutler B. Nature. 2006;441:E4. doi: 10.1038/nature04875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoebe K, et al. Nature. 2003;424:743. doi: 10.1038/nature01889. [DOI] [PubMed] [Google Scholar]
- 7.Yamamoto M, et al. Science. 2003;301:640. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- 8.Janssen E, et al. Immunity. 2006;24:787. doi: 10.1016/j.immuni.2006.03.024. [DOI] [PubMed] [Google Scholar]
- 9.Materials and methods are available as supporting material on Science Online.
- 10.Pasare C, Medzhitov R. Curr Opin Immunol. 2003;15:677. doi: 10.1016/j.coi.2003.09.002. [DOI] [PubMed] [Google Scholar]
- 11.Schnare M, et al. Nat Immunol. 2001;2:947. doi: 10.1038/ni712. [DOI] [PubMed] [Google Scholar]
- 12.Kaisho T, et al. Int Immunol. 2002;14:695. doi: 10.1093/intimm/dxf039. [DOI] [PubMed] [Google Scholar]
- 13.Snapper CM, Paul WE. Science. 1987;236:944. doi: 10.1126/science.3107127. [DOI] [PubMed] [Google Scholar]
- 14.Finkelman FD, et al. J Exp Med. 1991;174:1179. doi: 10.1084/jem.174.5.1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van den Broek MF, Muller U, Huang S, Aguet M, Zinkernagel RM. J Virol. 1995;69:4792. doi: 10.1128/jvi.69.8.4792-4796.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Snapper CM, et al. J Exp Med. 1992;175:1367. doi: 10.1084/jem.175.5.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leadbetter EA, et al. Nature. 2002;416:603. doi: 10.1038/416603a. [DOI] [PubMed] [Google Scholar]
- 18.Lau CM, et al. J Exp Med. 2005;202:1171. doi: 10.1084/jem.20050630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.We thank Drs. Kono and Theofilopoulos for comments on the manuscript, Dr. Rolink for FGK4.5 antibody, and Drs. Nemerow and Sarvetnick for use of instruments. Supported by NIH grants RO1GM44809 to D.N., AI050241 to B.B., and T32AI07606 to B.D..
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.