

Abstract
In human methylenetetrahydrofolate reductase (MTHFR) the Ala222Val (677C → T) polymorphism encodes a heat-labile gene product that is associated with elevated levels of homocysteine and possibly with risk for cardiovascular disease. Generation of the equivalent Ala to Val mutation in Escherichia coli MTHFR, which is 30% identical to the catalytic domain of the human enzyme, creates a protein with enhanced thermolability. In both human and E. coli MTHFR, the A → V mutation increases the rate of dissociation of FAD, and in both enzymes, loss of FAD is linked to changes in quaternary structure [Yamada, K., Chen, Z., Rozen, R., and Matthews, R. G. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 14853–14858; Guenther, B. D., Sheppard, C. A., Tran, P., Rozen, R., Matthews, R. G., and Ludwig, M. L. (1999) Nat. Struct. Biol. 6, 359–365]. Folates have been shown to protect both human and bacterial enzymes from loss of FAD. Despite its effect on affinity for FAD, the A → V mutation is located at the bottom of the (βα)8 barrel of the catalytic domain in a position that does not contact the bound FAD prosthetic group. Here we report the structures of the Ala177Val mutant of E. coli MTHFR and of its complex with the 5,10-dideazafolate analogue, LY309887, and suggest mechanisms by which the mutation may perturb FAD binding. Helix α5, which immediately precedes the loop bearing the mutation, carries several residues that interact with FAD, including Asn168, Arg171, and Lys172. In the structures of the mutant enzyme this helix is displaced, perturbing protein–FAD interactions. In the complex with LY309887, the pterin-like ring of the analogue stacks against the si face of the flavin and is secured by hydrogen bonds to residues Gln183 and Asp120 that adjoin this face. The direct interactions of bound folate with the cofactor provide one mechanism for linkage between binding of FAD and folate binding that could account in part for the protective action of folates. Conformation changes induced by folate binding may also suppress dissociation of FAD.
Methylenetetrahydrofolate reductase (MTHFR1) catalyzes the only reaction that forms methyltetrahydrofolate (CH3-H4folate) and lies at an important metabolic switch point where the flow of one-carbon units from methylenetetrahydrofolate is directed either to the methionine/adenosylmethionine (AdoMet) cycle or to synthesis of thymidylate (Figure 1). In the mammalian enzyme, which is a dimer of identical ~70 kDa chains containing catalytic and regulatory modules (1, 2), AdoMet acts as an allosteric inhibitor. Its effects are suppressed by AdoHcy, which competes for the AdoMet binding site (3, 4).
Figure 1.
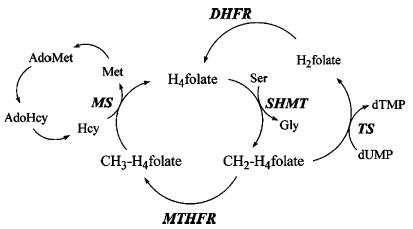
Segments of the pathways of one-carbon metabolism showing the fates of methylenetetrahydrofolate. In the AdoMet cycle on the left, CH2-H4folate is used to form methionine by the dual action of methylenetetrahydrofolate reductase (MTHFR) and methionine synthase (MS); in the thymidylate synthase (TS) cycle on the right, the methylene group is utilized for thymidylate biosynthesis. Dihydrofolate reductase (DHFR) catalyzes the conversion of dihydrofolate (H2folate) to tetrahydrofolate (H4folate), and CH2-H4folate is regenerated by the enzyme serine hydroxymethyl transferase (SHMT). The conversion of homocysteine to methionine catalyzed by betaine-homocysteine methyltransferase is not shown here.
MTHFR has been a focus of extensive clinical research because of its role in homocysteine homeostasis. Very high homocysteine (Hcy) levels in subjects with impaired cystathionine β-synthase first drew attention to a connection between Hcy and cardiovascular disease (5, 6). Subsequently, the polymorphism Ala222Val in MTHFR has been associated with risks for cardiovascular disease (7–9) and neural tube defects (10, 11). The Ala222Val polymorphism was initially characterized on the basis of its thermolability as measured by assays in lymphocyte extracts (12, 13). This mutation leads to modest increases in plasma Hcy (14, 15) by lowering the levels of CH3-H4folate, the cosubstrate required for conversion of Hcy to methionine by methionine synthase (MS). The latest epidemiological studies indicate that the Ala222Val mutation is a significant risk factor for coronary heart disease in populations where diets are not supplemented with folates (16–18).
To obtain a structure for the catalytic domain of MTHFR, and to examine the properties of the A → V mutation, we have employed Escherichia coli MTHFR as a model system. This bacterial MTHFR, with only 296 residues, is much smaller than mammalian MTHFRs. It is not regulated by AdoMet, and aggregates to form a tetramer rather than a dimer. Sheppard (19, 20) characterized an E. coli MTHFR carrying the mutation Ala177Val that corresponds to the Ala222Val polymorphism in the human enzyme. She showed that the mutation did not affect the kinetic properties of the enzyme but instead inactivated MTHFR by enhancing the loss of the essential FAD cofactor, and further demonstrated in vitro that addition of folates decreased the rate of dissociation of FAD in dilute solutions of the enzyme. Loss of the flavin occurs in concert with dissociation of the tetrameric enzyme to dimers (19). In parallel experiments Rozen and co-workers compared extracts of the wild type and mutant human enzymes and established a similar phenotype for the mutant human enzyme (2, 19). Loss of FAD and activity from both the thermolabile mutant enzymes and the wild type enzymes could be suppressed by addition of folates and/or FAD (19). These effects of folates have helped to provide the rationale for dietary supplementation with the vitamin (21–23).
Structure analysis of E. coli MTHFR (19) has established that the bacterial enzyme is a (βα)8 barrel in which the entire sequence constitutes the catalytic domain. The initial structure of E. coli MTHFR, determined in the absence of substrates (19), located the mutation site at the bottom of the (βα)8 barrel near the N-terminal end of strand β6, away from direct contact with any atoms of the bound FAD cofactor. Although this remote position was consistent with the observation that catalytic activity (per flavin) is not impaired in the Ala177Val mutant, it complicated elucidation of the mechanism by which the mutation prompts dissociation of the FAD. Because the Ala177 side chain resides in a tight loop between α5 and β6, it was suggested that steric overlap of the valine with helix α5 might displace this helix, disturbing some of the FAD-binding residues that are contributed by α5 (19). It also seemed possible that shifts of this same helix could affect interchain interactions responsible for holding the tetramer together.
This paper describes structures of the Ala177Val mutant of E. coli MTHFR. A His6 tag was introduced at the C-terminus to facilitate purification of this relatively unstable enzyme (19). We were able to obtain crystals in the presence of a stable 5,10-dideaza analogue (24) of the folate substrate and were also able to crystallize the mutant enzyme in the absence of substrates. The structure determinations allow a description of structural changes associated with the mutation at position 177 as well as the assignment of the binding determinants for the folate analogue. The dideazafolate proves to be an ideal mimic of the product, CH3-H4folate, occupying the same site as product bound to the Glu28Gln mutant enzyme (25). The several structures of free enzyme and of substrate complexes, determined earlier (19, 25), help to distinguish changes induced by ligand binding at the flavin from those associated with the Ala → Val mutation. Structural changes induced by folates may contribute to the effects of folate on FAD binding.
MATERIALS AND METHODS
Enzyme Purification and Crystallization
The mutant enzyme was prepared as described by Sheppard et al. (26, 27). The antifolate LY309887 is a stable 5,10-dideaza analogue of tetrahydrofolate with the p-aminobenzoyl (PABA) ring replaced by thiophene (C19H20N5O6S1). This compound, 6R-2′,5′-thienyl-5,10-dideazatetrahydrofolate (monoglutamate) (24), was a gift from Eli Lilly & Co.
His-tagged protein in 10% glycerol, 50 mM KPi pH 7.2, 0.2 mM EDTA, and 125 μM excess FAD was concentrated to about 15 mg/mL (~450 μM) by centrifugation using Microcon30 filters (Amicon). For hanging drop crystallization 2 μL of protein was mixed with 2 μL of reservoir solution (0.2 M Mg acetate, 0.1 M cacodylate pH 6.5, 14% PEG 8K, 5% glycerol) and 1 μL of LY309887 (5 mM). Crystals appeared in a day at 22 °C and grew in the same C2 space group as the wild type enzyme (19) but with the slightly different cell dimensions, a = 101.56 Å, b = 128.64 Å, c = 96.74 Å, β = 120.87°. In this crystal form three chains constitute the asymmetric unit; these chains are arranged around the crystallographic dyad to form a hexamer from which we can choose the tetrameric unit that exists in solution. Substrate-free mutant enzyme was crystallized by adding 2 μL of reservoir solution (14% PEG 4K, 225 mM Li2SO4, 100 mM sodium cacodylate pH 6.5, 5% ethanol) to 2μL of protein, with nucleation promoted by seeding.
Cryoprotection
Before flash-cooling, crystals of the LY309887 complex were transferred in two steps to a holding solution that was 14% PEG 8K, 10% MPD, 7% glycerol, 100 μM excess FAD, and 1 mM LY309887, with cacodylate and Mg acetate at the same concentrations as in the reservoir solution. Substrate-free crystals were transferred to a holding solution (15% PEG 4K, 225 mM Li2SO4, 100 mM sodium cacodylate pH 6.5, 5% ethanol) containing 5% methylpentanediol (MPD) for 5 min. They were subsequently transferred to a solution containing 10% methylpentanediol and flash-frozen.
Data Collection and Structure Determination
Datasets for both the substrate-free and LY309887 complexes were collected on an RAXIS IV detector. Initial molecular replacement searches with the coordinates for wild type MTHFR (1B5T.pdb) were necessary to position the chains comprising the asymmetric unit. The first refinements were conducted in X-PLOR (28); later calculations used CNS v1.1 (29). Rounds of refinement in CNS included simulated annealing in torsional space, coordinate minimization, and restrained individual B-factor adjustment with maximum-likelihood targets. The program PRODRG (30) was used to construct restraints for refinement of LY309887, and ligand occupancies were fixed at unity. Because of differences in the conformations of loop regions, NCS restraints were not applied to the three chains constituting the asymmetric unit. Data/parameter ratios for independent refinement of the chains were as follows: 2FMN, 2.17, and 2FMO, 1.94. The three chains differ from one another in their average temperature factors (25). Residues 123–128 are not visible in the electron density of the A → V mutant structure or in the wild type structures. The N-terminal helix of the C chain is disordered in all structures, and cannot be modeled completely in the A and B chains of the substrate-free structure of Ala177Val.
Quality indices for the model geometry and agreement with structure factors were evaluated with the programs PROCHECK and SFCHECK, respectively. The mutant LY309887 complex has 92.3% of the residues in the most favored region of the Ramachandran plot, with 7.1% in additional allowed regions. The substrate-free mutant has 91.5% of the residues in the most favored regions and 8.1% in additional allowed areas.
Pairwise comparisons of A, B, or C chains from different structures were carried out in LSQKAB (32) using backbone atoms of a core of residues judged not to be affected by mutation, ligation, or pH. Sequences used for these structural alignments were 22–59, 68–90, 96–117,131–160,185–200, and 254–294. Average RMS differences between the cores established a background deviation (σ) against which to measure putative structural changes. Having three independently refined chains in six structures of bacterial MTHFR (this work and ref 25) allows good measurement of deviations, and the relatively large radius of convergence of simulated annealing should sample experimental variations in fitting models to the X-ray data.
Dynamic Light Scattering
Measurements of light scattering (31) were performed on a Protein Solutions Dyna Pro instrument. Histidine-tagged wild type MTHFR was studied at a concentration of 1 mg/mL in 0.1 M sodium cacodylate pH 6.0.
RESULTS AND DISCUSSION
The Structure of the Monomer and the FAD Binding Site
A drawing of the MTHFR monomer, designating the secondary structure elements of the (βα)8 barrel, is shown in Figure 2A. The chain of E. coli MTHFR begins with an extra-barrel helix at its N-terminus. The largest insertion in the main barrel follows strand β7 and includes two extra-barrel helices denoted α7a and α7b. In barrels that bind FMN, the phosphate of the cofactor is associated with the N-terminus of an extra-barrel helix, but binding of FAD to MTHFR does not follow this pattern. The FAD side chain adopts an unusual kinked conformation and appears to be held in place by a salt bridge between Arg118 and Glu158. Residues that contribute to FAD binding have been described earlier (19) and are noted in Figure 2B. The FAD–protein interactions that are important for analysis of the Ala177Val mutant are depicted in Figure 2C.
Figure 2.
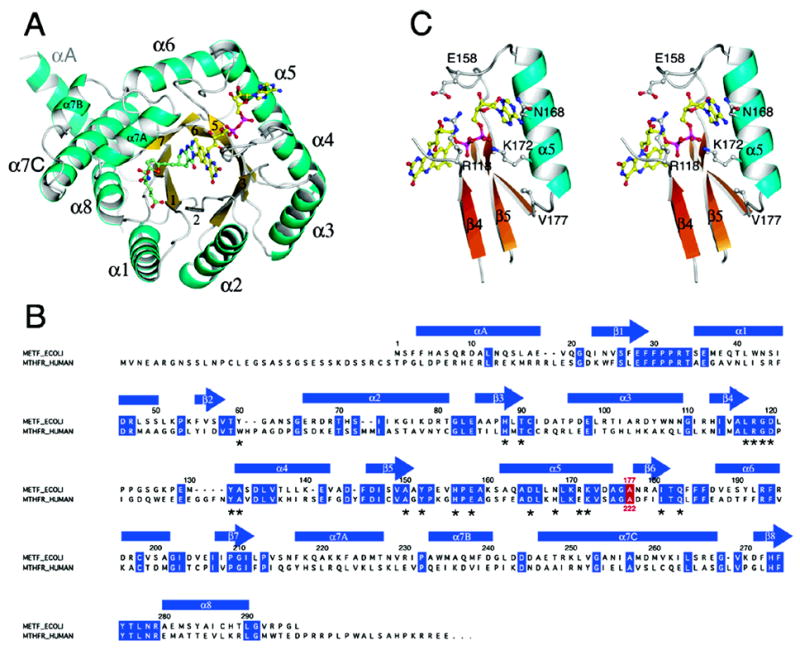
(A) The monomer of MTHFR with FAD and LY309887 shown in ball-and-stick mode. The view is along the axis of the (βα)8 barrel. Loops at the C-termini of barrel strands are referenced to the preceding strand, e.g. L4 connects β4 to α4. The groove that accommodates the folate analogue is formed by the shortened β8 strand and L8, and lies next to helix α7a. (B) Alignment of the sequence of E. coli MTHFR with the N-terminal region of human MTHFR. Secondary structures in the bacterial enzyme are displayed above the sequences, and residues that are conserved in the bacterial and human sequences are highlighted in blue. The location of Ala177/Ala222 is marked in red. Residues that interact with the FAD cofactor are marked by asterisks. (C) A stereoview of features of the binding site for FAD in the Ala177Val mutant showing Val177, helix α5, and some of the residues that interact with FAD.
Structural Changes Induced by the Ala177Val Mutation and Effects on Protein–FAD Interactions
To examine the structural perturbations resulting from the mutation, the structures of the folate-free A → V mutant and of the A → V mutant complexed with LY309887, both determined at pH 6.5 (Tables 1 and 2), have been compared with structures of the wild type enzyme and its complexes with the product, CH3-H4folate, or the substrate, NADH (25). The initial structure analysis of wild type E. coli MTHFR at 2.5 Å resolution employed untagged protein and crystals grown at pH 6.1 (19). We subsequently obtained higher resolution (1.85 Å) in an analysis of substrate-free His-tagged protein, using crystals equilibrated at pH 7.4 (25). Comparisons with the A → V mutant are made with this higher resolution wild type structure, but the patterns of changes resulting from the mutation (Figure 3) are the same regardless of the choice of substrate-free wild type structure.
Table 1.
Data Collection Statistics
| EoxA177V·LY309887 (pH 6.5) | EoxA177V (pH 6.5) | |
|---|---|---|
| resolution (Å) | 35–2.05 | 20–2.25 |
| reflectionsa | 222652 | 121116 |
| unique reflectionsa | 64861 | 50833 |
| completeness (%)b | 96.8 (92.6) | 99.7 (98.7) |
| Rsyma,b,c | 0.048 (0.286) | 0.048 (0.297) |
| I/σ (I)a | 22.0 (4.35) | 16.6 (3.25) |
Statistics calculated using the programs DENZO and SCALEPACK (48).
Statistics for highest resolution shell are enclosed in parentheses.
Rsym = ∑|Iobs − Iav|/∑Iobs.
Table 2.
Refinement Statistics
| EredA177V·LY309887 (pH 6.5) | EoxA177V (pH 6.5) | |
|---|---|---|
| resolution (Å) | 35–2.05 | 20–2.25 |
| average B-factor | 34.6 | 32.2 |
| Rcrysta/Rfreea,b | 0.216/0.249 | 0. 219/0.247 |
| (highest resolution shell) | 0.292/0.316 | 0.273/0.300 |
| correlation coefficientc | 0.923 | 0.912 |
| RMSD bonds (Å) | 0.0081 | 0. 0089 |
| RMSD angles (deg) | 1.538 | 1.485 |
| Ramachandran plot | ||
| most favored (%) | 92.3 | 91.5 |
| additional allowed (%) | 7.1 | 8.1 |
Rcryst = ∑|| Fobs| − |Fcalc||/∑|Fobs|, where |Fobs| and |Fcalc| are the observed and calculated structure factor amplitudes, respectively.
Rfree was calculated using a random test set of reflections (5%) which were omitted throughout all stages of refinement.
Calculated using the CCP4 program SFCHECK: correlation coefficient = 〈FobsFcalc〉 − 〈Fobs 〉〈Fcalc〉)/[(〈 F2obs〉 − 〈Fobs〉2)(〈F2calc〉 − 〈Fcalc〉2)]½.
Figure 3.
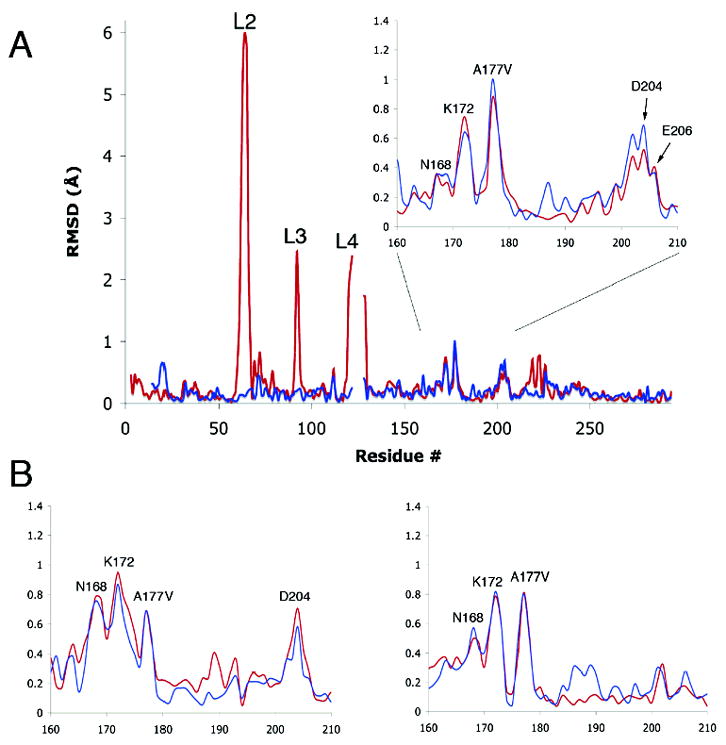
(A) RMS differences in positions of backbone Cα atoms in the B chains of wild type MTHFR (pH 7.4) and the A177V mutant, with or without bound LY309887 (pH 6.5). Differences between A177V·LY309887 and wild type are shown in red, and differences between A177V and wild type are in blue. The Ala177Val mutation primarily affects residues 165–180. Large differences are observed in loops L2, L3, and L4, which undergo pH- and ligand-induced conformational changes that are not caused by the substitution at position 177; smaller differences near 223 and 243 are attributed to folate binding. Chains were aligned as described in Materials and Methods. The inset magnifies the region in which changes are ascribed to the A → V mutation. (B) Inset regions showing the RMS difference values for the A (left) and C (right) chains.
RMS differences of atomic positions were obtained after pairwise alignments of the backbone cores of the models using LSQKAB (32) (see Materials and Methods). Background (σ) values for the matches were taken as the average RMSDs in the core regions, and vary from 0.15 to 0.23 Å in comparisons between wild type MTHFR and the A → V mutant or its complex with LY309887 (Figure 3). Significant differences from wild type that are attributed to the A → V mutation occur in α5 and in the α5–β6 connector that carries the mutation site. In this region, differences between the mutant and wild type structures are larger in chains A and C, where the side chain of Asn168 in helix α5 fully adopts a new conformation (see below). Segments of helix α6 and its C-terminus also appear to be perturbed in the A and B chains of the mutant (Figure 3). Structural differences in other regions are attributed to pH- and/or ligand-dependent changes in conformation (Figure 3).
The prediction that steric effects at the site of the mutation might initiate structural perturbations is borne out by the changes observed in the loop that connects α5 to β6 (Figure 4). Introduction of valine leads to overlaps of the Val177 side chain with backbone oxygens at 172 and 179 and with the Cα of 173 that are relieved by spreading of the loop and displacement of helix α5. As seen in Figure 4B and Table 3 the helix backbone is displaced by 0.75 to 0.95 Å at Lys172. Smaller backbone shifts are propagated as far as Asn168.
Figure 4.
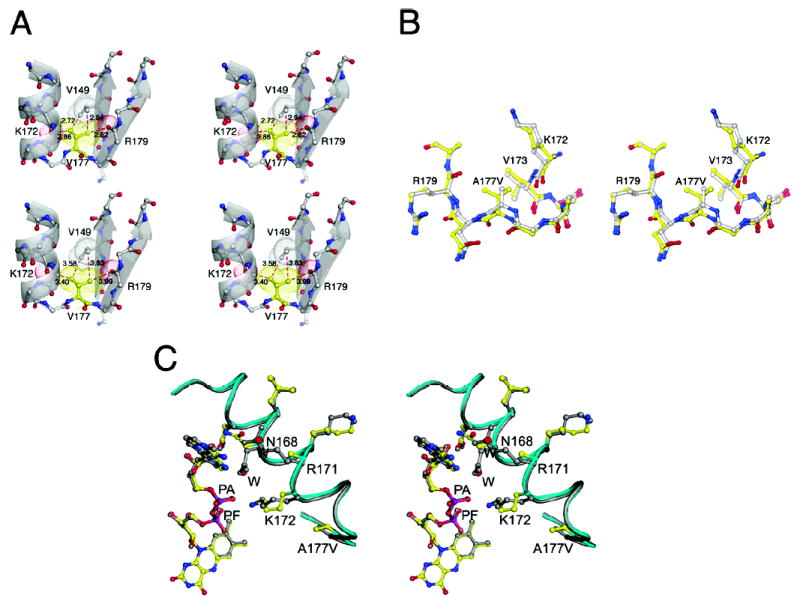
(A) Perturbations at the mutation site and their propagation to helix α5. The upper panel shows the crowding and steric overlap that result when valine is substituted for alanine without allowing any relaxation of the structure. The lower panel displays the actual structure of the C chain from the LY309887 complex of the Ala → Val mutant. (B) Structural changes in the α5–β6 loop bearing the Ala177Val mutation. A stereoview of the superposition of wild type (gray and atom coloring) and A177V mutant (yellow and atom coloring) showing expansion of the α–β loop bearing the mutation. (C) A stereoview centered at Asn168 of the α5 helix, showing the effect of mutation on the interactions of the protein with FAD. Wild type enzyme (gray) is superposed onto the A177V mutant (yellow and atom coloring). In the A177V mutant, Asn168, which forms an interaction with the adenine phosphate (PA) in wild type enzyme, undergoes a rearrangement which allows for a water to intervene between PA and the neighboring helix. Arg171, which interacts with adenine, becomes disordered in the A177V mutant and is replaced by solvent. The two solvents that are introduced in the A → V mutant are in red and labeled W. The interaction of Lys172 with PA is maintained despite the displacement of the backbone.
Table 3.
RMS Displacements (Å) of Cα Atoms of Residues Perturbed by the Ala177Val Mutationa
| A177V · LY309887
|
A177V
|
|||||
|---|---|---|---|---|---|---|
| chain | Val177 | Lys172 | Asn168 | Val177 | Lys172 | Asn168 |
| A | 0.6(9) | 0.9(5) | 0.7(9) | 0.6(9) | 0.8(7) | 0.7(6) |
| B | 0.8(7) | 0.7(5) | 0.2(8) | 1.0(0) | 0.6(4) | 0.3(5) |
| C | 0.8(1) | 0.7(9) | 0.5(0) | 0.8(1) | 0.8(2) | 0.5(7) |
In the B chains, displacements are calculated for the dominant conformer of Asn168, which maintains a hydrogen bond to the adenine phosphate of FAD.
Despite the displacement of the helix backbone, the amino group of Lys172 maintains its interaction with the adenine phosphate of FAD (Figure 4C). However, the conformations and interactions of the Asn168 side chain, in the previous turn of α5, are completely altered in the A and C chains of the tetramer (Figure 4C, Table 3). Rearrangement of Asn168 breaks a hydrogen bond with the adenine phosphate, but generates a new hydrogen bond to the 2′ O of the adenine ribose. The displacement of Asn168 appears to be coordinated with movement and increased mobility of Arg171 and is accompanied by the introduction of a solvent that interacts with the adenine phosphate (Figure 4C). Arg171 swings out toward bulk solvent to accommodate the displacement of Asn168, and the adenine ring of FAD shifts, as it is no longer constrained by a contact of C8 with Asn168. The displacement of Arg171 breaks an interaction with N7 that is replaced by water. The changes in FAD–protein interactions and the introduction of solvents at the adenine phosphate and N7 are consistent with weaker binding of FAD in the mutant.
In chain B the mutation is accommodated with subtly different structural adjustments. The α–β connector carrying Val177 is displaced a bit more than in the other chains, helix α5 is correspondingly less perturbed, and the dominant conformer of Asn168 remains close to the adenine phosphate. However, significant electron density corresponding to the alternate conformation of Asn168B is observed in omit maps for the A → V mutant and its complex with LY309887.
Rearrangement of Asn168 is a characteristic feature of the A → V mutant structures. Examination of the density around Asn168 in simulated annealing omit maps of the twelve chains in the four high-resolution wild type structures that we have determined (25) fails to reveal density corresponding to the alternative conformation of Asn168. In all of the chains from structures of wild type MTHFR, Asn168 forms a hydrogen bond to the adenine phosphate oxygen.
Effects of the A → V Mutation on Dissociation of MTHFR to Dimers
Evidence that E. coli MTHFR is a tetramer in solution comes from gel filtration experiments (20) and from dynamic light scattering measurements that yield a weight-average molecular weight of 143.1 kDa (this work). The tetramer of MTHFR is unusual; it does not display the full 222 symmetry found in most tetramers, but instead shows only 2-fold point symmetry (Figure 5). This lower symmetry is not an artifact of crystallization but is an inherent property of the molecule. In the MTHFR tetramer there are two possible ways of dissociating chains to form dimers: these modes of dissociation disrupt different interfaces and hence involve different energies. Using the notation of Figure 5, the resulting dimers could be either type A–C (B–B′ by molecular symmetry) or A–B′ (B–C by symmetry). Earlier calculations of the areas of the two different interfaces found in MTHFR suggested that the plane of dissociation would be vertical as viewed in Figure 5, so that interfaces not far from the site of the Ala177Val mutation would be broken on disaggregation to give A–C dimers (19). Inclusion of the N-terminal helices in chains designated A and B (Figure 5), helices which could not be built in the initial structure determination (19), has increased the estimates of the interface areas. They are now ~1350 Å2 per monomer face (2700 Å2 buried per dimer for formation of the tetramer from dimers) for the horizontal interfaces and 570 Å2 for the vertical interfaces, rather than the smaller values of 800 and 500 Å2 cited earlier (19). Thus the more complete model still predicts that dissociation will produce identical dimers of A and C (or B and B′) chains. These dimers are held together by interactions between helices αA, α7b, α7c, and α8 and their equivalents that are related by 2-fold symmetry (Figure 5). The observation that the N-terminal αA helix could not be modeled in the substrate-free structures determined at pH 6.1 (19) or 6.5 (this work) indicates that the stability or mobility of these N-terminal helices varies with pH.
Figure 5.
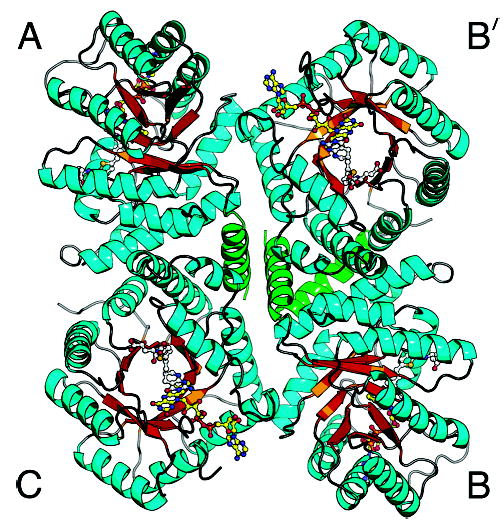
The MTHFR tetramer with bound LY309887 viewed down the local 2-fold axis. As in Figure 2, sheet strands are gold and helices are cyan, with the exception of the N-terminal helix (green). The N-terminal helix, which could not be built in the initial structure, fills in the center of the tetramer. Chains are designated differently from those of our previous drawing (19).
Sheppard and Matthews (19, 20) showed that loss of FAD from the Ala177Val mutant or from wild type enzyme is linked to dissociation of the tetramer to dimers, and that the mutant enzyme loses FAD much more rapidly than does wild type MTHFR (see introductory comments). Their data are best fit to a model in which FAD dissociates from the dimer rather than from the tetramer. These results do not distinguish between effects of the mutation on the tetramer–dimer equilibrium and effects on the affinity of the dimeric protein for FAD.
Although comparisons of the FAD binding sites in wild type and mutant enzymes, described above, suggested that the A → V mutation alters the affinity of each chain for FAD, we also looked for mutation-linked changes at the interchain contacts that might affect the tetramer–dimer equilibrium. Superimposing the tetramers of wild type and A177V MTHFR did not reveal any significant differences in the relative orientation of the chains in the mutant protein. We then examined the structures for evidence of more local changes at the A–B′ and B–C interfaces. Residues in the α5–β5 connector that carries the mutation do not contact neighboring chains, but in all three of the unique chains, residues 173 and 176–180 at the site of the mutation contact residues 204–206 in the α6–β7 region of the same chain. As a result small shifts in the vicinity of the mutation can be transmitted to the α6–β 7 connector, which does form part of the A–B′ (or B–C) interface (Figure 5). The shifts observed at 204–206 are greater than the background RMSD values in chains A and B. Interactions of Glu206B with the backbone nitrogens of C chain residues 162–163 at the B–C interface are slightly altered in the A → V mutant. However these contacts are not replicated in the corresponding A–B′ interface of the tetramer, and these and other features of the interfaces are not systematically different in the wild type and mutant structures. Thus while we can describe pathways tracing structural perturbations from the mutation site in chain B to the B–C interface, there is no definitive evidence for loosening of the interchain contacts or for transmission of structural perturbations to the neighboring chain. In particular, the environment of Gln163C, which occupies a pocket surrounded by residues from chain B, seems undisturbed by the mutation. Although the LY309887 complex was obtained by cocrystallization rather than by adding the ligand to preformed crystals, it remains possible that changes in interchain interactions are suppressed in the crystal structure.
The Conformation and Interactions of the Bound Folate Analogue LY309887
A fundamental question is how binding of folates can protect MTHFR from loss of its essential FAD. Protection against inactivation is of considerable clinical importance. To study the interactions of a folate with the A177V mutant enzyme we cocrystallized the mutant protein with LY309887, an unreactive analogue of the reaction product, CH3-H4folate. This strategy is an alternative to investigating the binding of CH3-H4folate, which is feasible only with the inactive mutant enzyme Glu28Gln (25) and would therefore require introduction of a second mutation. In the complex, the deazapterin ring of LY309887 stacks against the si face of the isoalloxazine ring of FAD, with the C6 side chain of the S-isomer pointing into a central groove that is carved into the top of the barrel (Figures 2A, 6). The overall shape of the ligand is determined by the pucker at the tetrahedral C6 atom, which is displaced from the plane of the remaining atoms of the deazapterin ring. The conserved Gln183 hydrogen bonds to pterin nitrogens that establish the orientation of the Gln side chain. The oxygens of Asp120 are close to N3 and the 2-exo-NH2 group of the deazapterin, but the carboxylate of Asp120 is not perfectly in-plane with the pterin. Tyr275 acts as cushion for the C9 and C10 atoms of the dideazafolate analogue. The thiophene ring is enclosed in a conserved hydrophobic box with sides provided by Phe223, Leu212, Leu277, and Phe184. Phe223 swings away from its position in the substrate-free enzyme in order to stack over the thiophene ring.
Figure 6.
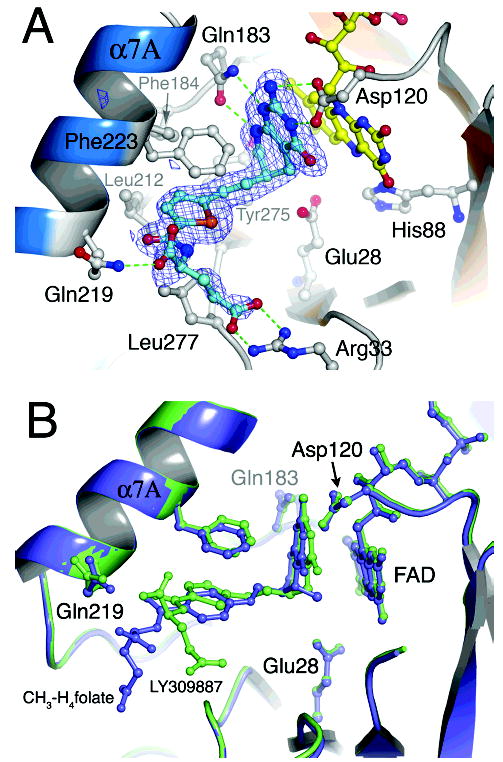
(A) A larger perspective of interactions made by the 5,10-dideazafolate, LY309887. Omit difference density, calculated after simulated annealing, is overlaid onto the final model for LY309887. The 5-deazapterin ring is oriented by hydrogen bonding interactions with Asp120 and Gln183, which are conserved among both bacterial and mammalian MTHFRs. The isoalloxazine and pterin planes are in van der Waals contact. The thiophene is sandwiched between Phe223 and Leu277, and hydrogen bonds from Gln219 and Arg33 position the α- and γ-carboxylates of the glutamate tail of deazafolate. (B) Comparison of bound LY309887 with bound methyltetrahydrofolate. The LY309887 complex of the A177V mutant (lime) is superposed on the structure of CH3-H4-folate bound to the Glu28Gln mutant of E. coli MTHFR (purple) (25). The binding modes of LY309887 and CH3-H4folate are very similar: the deazapterin of LY309887 and the pterin of CH3-H4- folate are oriented by similar interactions with Asp120 and Gln183, and the thiophene of LY309887 occupies the same hydrophobic pocket as the PABA ring (25).
The glutamate tail of the folate analogue extends along the groove whose base is formed by strand β8 and the following loop L8 (Figure 6). The α-COO− binds to the backbone and side chain of Gln219, and the γ-COO− makes a bidentate interaction with Arg33, a semiconserved residue that is sometimes lysine and sometimes arginine. Physiological folates are polyglutamylated, and the Km and Ki depend on the length of the glutamylated side chain (33, 34); in E. coli the triglutamates are the major form of folates. With a polyglutamylated substrate or inhibitor, the interaction with Arg33 would be modified to accommodate the second γ-linked glutamate.
The interactions made by LY309887 are very similar to those described in the Glu28Gln complex with the product, CH3-H4folate (25) (Figure 6B). In particular, Gln183 and Asp120 make interactions with the pterin and deazapterin rings, while the hydrophobic quartet (Phe223, Leu212, Leu277, and Phe184) surrounds the thiophene ring in the complex with LY309887 and the PABA ring in the product complex. Because the deazapterin ring of LY309887 lacks a methyl substituent at position 5, it is more tightly stacked against the flavin isoalloxazine ring. In both structures the α-COO− of the glutamate tail interacts with Gln219. However the γ-COO− of CH3-H4folate interacts with Arg279 rather than Arg33. A mutation of the residue equivalent to Arg279 is among the variants observed as severe in patient populations (35). Therefore the interaction of CH3-H4folate with Arg279 (not shown) is more likely to be physiologically relevant than the apparently opportunistic contact of LY309887 with Arg33.
As expected for a mimic of one of the substrates, LY309887 is an inhibitor of MTHFR. Preliminary measurements of inhibition, in assays that monitored the oxidation of NADH by menadione (36), are consistent with competitive inhibition by LY309887 with a Ki of approximately 20 μM.
How Do Folates Affect the Affinity of MTHFR for FAD?
Direct measurements of Kd for the binding of FAD to the E. coli enzyme have been precluded by the inability to find conditions for reversible resolution and reconstitution of the holoenzyme. Instead, the rates of dissociation of FAD have been used to estimate the relative affinities for FAD. Several folates, including CH3-H4folate, dihydrofolate, 5,10-dideazafolate (Glu5), and CH2-H4folate, protect MTHFR against dissociation of FAD and the consequent loss of activity (19). These observations can be explained either by kinetic effects, in which folates act simply to impede dissociation, or by positive cooperativity between the binding of FAD and the binding of folates.
Structures of the complexes of A177V with LY309887 and of CH3-H4folate with the E28Q mutant suggest mechanisms to account for interaction between FAD binding and folate binding. One likely contribution to tighter binding of FAD in the presence of folates comes from the stacking of the pterin and isoalloxazine rings, which presumably provides substantial favorable free energy of interaction. There is ample evidence that stacking interactions are preferred by the flavin ring. Free FAD is folded in solution (37, 38), and stacking of the isoalloxazine ring with protein side chains (39, 40) and substrates or other ligands (41) is very common in flavoproteins. Stacking interactions of protein-bound pterin rings have been documented for several enzymes, including nitric oxide synthase (42, 43) and phenylalanine hydroxylase (44).
Folates may also affect FAD binding indirectly via conformation changes that are induced by folate binding. Alternate “open” and “closed” conformations are adopted by the loops L2 (61–67) and L4 (118–131), depending on pH and the presence of ligands. These states have been described elsewhere (25) and are illustrated in Figure 7. The closed conformers of loops L2 and L4 form protein–flavin hydrogen bonds that cannot occur in the open conformations: in the closed form of L2, NH 62 interacts with the flavin O2, and in the closed form of L4, NH 120 interacts with O2. The affinity for FAD may therefore be modulated by opening and closing of these loops.
Figure 7.
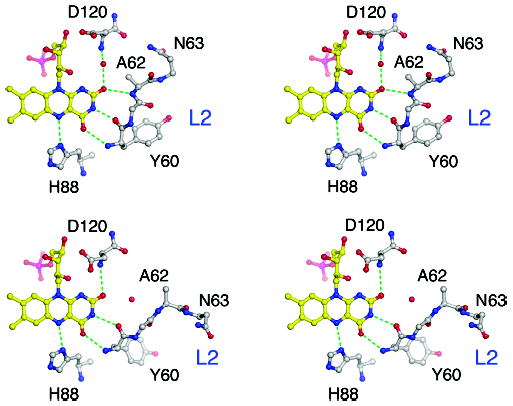
The conformations of loops L2 and L4 and their interactions with the flavin ring. (A) Stereoview of the flavin ring with L2 closed and L4 open. NH of Ala 62 from L2 is hydrogen bonded to the flavin O2, but NH of Asp120 interacts through an intervening water molecule. (B) Stereoview of the flavin ring with L2 open and L4 closed. The hydrogen bond between NH 62 and O2 is lost, but NH of Asp120 interacts directly with O2. In the complex of Glu28Gln MTHFR with CH3-H4folate, both loops are closed.
Analysis of the structures of the A177V mutant in the presence and absence of inhibitor shows that binding of LY309887 at pH 6.5 induces closure of loop L4, which moves to allow Asp120 to interact with both the deazapterin and flavin rings. Comparison of substrate-free MTHFR with the complex MTHFR (Glu28Gln) · CH3-H4folate (25) at pH 7.4 similarly shows that binding of CH3-H4folate induces closure of the L4 loop. Thus it appears that folates uniformly shift the distribution of L4 conformers toward the closed state (Table 4).
Table 4.
Conformations of L2 and L4 Loops
The effects of LY309887 and CH3-H4folate on the conformation of L2 are more complicated. In the A → V mutant at pH 6.5, binding of inhibitor is accompanied by opening of L2. However in structures determined at pH 7.4, L2 is closed in the presence or absence of CH3-H4folate (Table 4). Thus the equilibria in which conformation changes are coupled to ligand binding vary with pH, with the open form of L2 observed only in the inhibitor complex at the nonphysiological acid pH. At physiological pH, L2 and L4 are simultaneously closed when CH3-H4folate binds, so that the protein makes more direct interactions with FAD in the presence of CH3-H4folate than in the absence of substrates.2
A complete analysis of the energetics of folate binding should include contributions from the conformation changes that are induced in loops L2 and L4 (45). For example, conversion of L2 to the closed conformation unravels the start of helix α2 and substitutes an energetically less favorable type II turn, and the simultaneous closure of L2 and L4 requires disordering of residues downstream of Pro122 in the L4 loop (25).
Opening and closing of L4 might also affect the stability of the Arg118–Glu158 “belt” that surrounds the ribityl side chain of FAD (Figure 2C). The salt bridge between Arg118 and Glu158 must open to release or admit FAD (19). The functional importance of this bridge is supported by the dysfunction of the Arg118Gln mutant in E. coli (Campbell, H., unpublished results) and by the devastating effect of the corresponding R → Q mutation in humans (46). Although the populations of the open and closed forms of L4 are shifted by folate binding, the salt bridge itself is not disrupted in the Ala177Val structures or in any of the other structures that we have determined.
Conclusions
Relevance of the Bacterial Model for the Human A222V Polymorphism
In both human and E. coli MTHFR, the A → V mutation enhances the rate of dissociation of FAD, and in both enzymes loss of FAD is linked to changes in quaternary structure (2, 19). The present study reveals local structural perturbations that may account for weaker binding of FAD by the mutant E. coli enzyme. It also suggests mechanisms by which folate binding can protect the enzyme against loss of FAD, but it does not explain how the mutation affects the tetramer–dimer equilibrium.
The perturbations in the FAD binding site that are propagated from the site of the mutation are modest but reproducible in the structures of the bacterial enzyme; backbone displacements of similar magnitude occur in all three chains of both the folate-bound and substrate-free mutant structures (Table 3). Rearrangement of Asn168, which breaks an interaction with the adenine phosphate of FAD, and alters other–FAD interactions in the structures of the mutant protein, is observed only in the A → V mutant. Twelve independent images of this region in wild type MTHFR and its complexes with ligands all show Asn168 interacting with the adenine phosphate. To reduce bias that may arise from fitting a single model to the electron densities (49), we have examined three independently refined chains in each of two mutant structures and have sampled four different wild type structures for comparison with the mutant (see Materials and Methods).
Perturbations of the FAD binding site that may occur in the human A222V polymorphism are likely to resemble the local changes that are observed in the mutant bacterial enzyme. Conservation of the sequences within the catalytic domains (Figure 2B) strongly suggests that the human enzyme employs similar mechanisms to transmit structural distortions from Val222 to the FAD binding site. Direct interactions between folates and the flavin ring are also likely to be similar in the bacterial and mammalian enzymes. To the extent that these interactions account for the protective effects of folate binding, then the present structures of folate complexes also provide good models for the phenomenon of protection in mammalian enzymes. However the differences in length and sequence of the loops L2 and L4 suggest that the contributions of loop rearrangements induced by folate binding might not be identical in bacterial and human MTHFRs.
In contrast, the mechanisms by which changes at the site of mutation affect enzyme oligomerization are expected to be specific for the bacterial enzyme and may not be relevant to the human protein. The structure of the dimeric human MTHFR has so far resisted determination by X-ray crystallography. Although dimeric mammalian MTHFR is a planar object with four lobes (47) that superficially resembles the tetrameric enzyme from E. coli, the domain–domain contacts are expected to be different from those found in the bacterial protein.
Acknowledgments
The authors wish to dedicate this paper to the late Professor Vincent Massey. They thank Dr. Victor Chen of Eli Lilly & Co. for samples of the folate analogue, LY309887, and Cristal Sheppard for preparations of the mutant protein used in initial studies. They are also pleased to acknowledge many helpful discussions with Dr. David Ballou and Dr. Elizabeth Trimmer.
Abbreviations
- MTHFR
methylenetetrahydrofolate reductase
- CH3-H4folate
N5-methyl-5,6,7,8-tetrahydrofolate
- CH2-H4folate
N5,N10-methylene-5,6,7,8-tetrahydrofolate
- FAD
flavin adenine dinucleotide
- Hcy
homocysteine
- MS
methionine synthase
- NAD(P)H
nicotinamide adenine dinucleotide (phosphate)
- AdoHcy
S-adenosylhomocysteine
- AdoMet
S-adenosylmethionine
Footnotes
Financial support was received from the National Institutes of Health (GM16429 to M.L.L., GM24908 to R.G.M, and GM08720 to R.P.).
Atomic coordinates and structure factors (codes 2FMN and 2FMO) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ.
In chain C, the L2 loop adopts an alternative conformation that is stabilized by local crystal packing contacts and not influenced by a shift in pH or presence of substrate.
References
- 1.Kutzbach C, Stokstad ELR. Mammalian methylenetetrahydrofolate reductase: partial purification, properties, and inhibition by S-adenosylmethionine. Biochim Biophys Acta. 1971;250:459–477. doi: 10.1016/0005-2744(71)90247-6. [DOI] [PubMed] [Google Scholar]
- 2.Yamada K, Chen Z, Rozen R, Matthews RG. Effects of common polymorphisms on the properties of recombinant human methylenetetrahydrofolate reductase. Proc Natl Acad Sci USA. 2001;98:14853–14858. doi: 10.1073/pnas.261469998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jencks DA, Matthews RG. Allosteric inhibition of methylenetetrahydrofolate reductase by adenosylmethionine: Effects of adenosylmethionine and NADPH on the equilibrium between active and inactive forms of the enzyme and on the kinetics of approach to equilibrium. J Biol Chem. 1987;262:2485–2493. [PubMed] [Google Scholar]
- 4.Matthews RG, Daubner SC. Modulation of methylenetetrahydrofolate reductase activity by S-adenosylmethionine and by dihydrofolate and its polyglutamate analogues. In: Weber G, editor. Advances in Enzyme Regulation. Pergamon Press; Oxford, New York: 1982. pp. 123–131. [DOI] [PubMed] [Google Scholar]
- 5.McCully KS. Vascular pathology of homocysteinemia: implications for the pathogenesis of arteriosclerosis. Am J Pathol. 1969;56:111–128. [PMC free article] [PubMed] [Google Scholar]
- 6.Mudd SH, Uhlendorf BW, Freeman JM, Finkelstein JD, Shih VE. Homocystinuria associated with decreased methylenetetrahydrofolate reductase activity. Biochem Biophys Res Commun. 1972;46:905–912. doi: 10.1016/s0006-291x(72)80227-4. [DOI] [PubMed] [Google Scholar]
- 7.Frosst P, Blom HJ, Milos R, Goyette P, Sheppard CA, Matthews RG, Boers GJ, den Heijer M, Kluijtmans LA, van den Heuvel LP, et al. A candidate genetic risk factor for vascular disease: a common mutation in methylenetetrahydrofolate reductase. Nat Genet. 1995;10:111–113. doi: 10.1038/ng0595-111. [DOI] [PubMed] [Google Scholar]
- 8.Kluijtmans LA, van den Heuvel LP, Boers GH, Frosst P, Stevens EM, van Oost BA, den Heijer M, Trijbels FJ, Rozen R, Blom HJ. Molecular genetic analysis in mild hyperhomocysteinemia: a common mutation in the methylenetetrahydrofolate reductase gene is a genetic risk factor for cardiovascular disease. Am J Hum Genet. 1996;58:35–41. [PMC free article] [PubMed] [Google Scholar]
- 9.Refsum H, Ueland PM, Nygard O, Vollset SE. Homocysteine and cardiovascular disease. Annu Rev Med. 1998;49:31–62. doi: 10.1146/annurev.med.49.1.31. [DOI] [PubMed] [Google Scholar]
- 10.de Franchis R, Sebastio G, Mandato C, Andria G, Mastroiacovo P. Spina bifida, 677T → C mutation, and role of folate. Lancet. 1995;346:1703. doi: 10.1016/s0140-6736(95)92865-0. [DOI] [PubMed] [Google Scholar]
- 11.Shields DC, Kirke PN, Milols JL, Ramsbottom D, Molloy AMB, Weir H, Scott DGJM, Whitehead AS. The “thermolabile” variant of methylenetetrahydrofolate reductase and neural tube defects: an evalution of gnetic risk and the relative importance of the genotypes of the embryo and the mother. Am J Hum Genet. 1999;64:1045–1055. doi: 10.1086/302310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kang SS, Wong PWK, Susmano A, Sora J, Norusis M, Ruggie N. Thermolabile methylenetetrahydrofolate reductase: An inherited risk factor for coronary artery disease. Am J Hum Genet. 1991;48:536–545. [PMC free article] [PubMed] [Google Scholar]
- 13.Kang S-S, Wong PWK, Zhou J, Sora J, Lessick M, Ruggie N, Grcevich G. Thermolabile methylenetetrahydrofolate reductase in patients with coronary artery disease. Metabolism. 1988;37:611–613. doi: 10.1016/0026-0495(88)90076-5. [DOI] [PubMed] [Google Scholar]
- 14.Engbertsen AMT, Franken DG, Boers GHJ, Stevens EMB, Trijbels FJM, Blom HJ. Thermolabile 5,10-methylenetetrahydrofolate reductase as a cause of mild hyperhomocysteinemia. Am J Hum Genet. 1995;56:142–150. [PMC free article] [PubMed] [Google Scholar]
- 15.Jacques PF, Bostom AG, Williams RR, Ellison RC, Eckfeldt JH, Rosenberg IH, Selhub J, Rozen R. Relation between folate status, a common mutation in methylenetetrahydrofolate reductase, and plasma homoycsteine concentrations. Circulation. 1996;93:7–9. doi: 10.1161/01.cir.93.1.7. [DOI] [PubMed] [Google Scholar]
- 16.McNulty H, McKinley MC, Wilson B, McPartlin J, Strain JJ, Wier DG, Scott JM. Impaired functioning of thermolabile methylenetetrahydrofolate reductase is dependent on riboflavin status: implications for riboflavin requirements. Am J Clin Nutr. 2002;76:436–431. doi: 10.1093/ajcn/76.2.436. [DOI] [PubMed] [Google Scholar]
- 17.Klerk M, Verhoef P, Clarke R, Blom HJ, Kok FJ, Shouten EG. MTHFR 677C → T polymorphism and risk of coronary heart disease. J Am Med Assoc. 2002;288:2023–2032. doi: 10.1001/jama.288.16.2023. [DOI] [PubMed] [Google Scholar]
- 18.Meleady R, Ueland PM, Blom H, Whitehead AS, Refsum H, Daly LE, Vollset SE, Donohue C, Giesendorf B, Graham IM, Ulvik A, Zhang Y, Bjorke Monsen AL. Thermolabile methylenetetrahydrofolate reductase, homocysteine, and cardiovascular disease risk: the European Concerted Action Project. Am J Clin Nutr. 2003;77:63–70. doi: 10.1093/ajcn/77.1.63. [DOI] [PubMed] [Google Scholar]
- 19.Guenther BD, Sheppard CA, Tran P, Rozen R, Matthews RG, Ludwig ML. The structure and properties of methylenetetrahydrofolate reductase from Escherichia coli suggest how folate ameliorates human hyperhomocysteinemia. Nat Struct Biol. 1999;6:359–365. doi: 10.1038/7594. [DOI] [PubMed] [Google Scholar]
- 20.Sheppard CA. PhD Dissertation. University of Michigan; 1998. [Google Scholar]
- 21.Homocysteine Lowering Trialists’ Collaboration. Dose-dependent effects of folic acid on blood concentrations of homocysteine: a meta-analysis of the randomized trials. Am J Clin Nutr. 2005;82:806–812. doi: 10.1093/ajcn/82.4.806. [DOI] [PubMed] [Google Scholar]
- 22.Malinow MR, Nieto FJ, Kruger WD, Duell PB, Hess DL, Gluckman RA, Block PC, Holzgang CR, Anderson PH, Seltzer D, Upson B, Lin QR. The effects of folic acid supplementation on plasma total homocysteine are modulated by multivitamin use and methylenetetrahydrofolate genotypes. Arterioscler, Thromb, Vasc Biol. 1997;17:1157–1162. doi: 10.1161/01.atv.17.6.1157. [DOI] [PubMed] [Google Scholar]
- 23.Nelen WL, Blom HJ, Thomas CM, Steegers EA, Boers GH, Eskes TK. Methylenetetrahydrofolate reductase polymorphism affects the change in homocysteine and folate concentrations resulting from low dose folic acid supplementation in women with unexplained recurrent miscarriages. J Nutr. 1998;128:1336–1341. doi: 10.1093/jn/128.8.1336. [DOI] [PubMed] [Google Scholar]
- 24.Budman DR, Johnson R, Barile B, Bowsher RR, Vinciguerra V, Allen SL, Kolitz J, Ernst SEI, Kries W, Zervos P, Walling J. Phase I and pharmacokinetic study of LY309887: a specific inhibitor of purine biosynthesis. Caner Chemother Pharmacol. 2001;47:525–531. doi: 10.1007/s002800000272. [DOI] [PubMed] [Google Scholar]
- 25.Pejchal R, Sargeant R, Ludwig ML. Structures of NADH and CH3-H4folate complexes of E. coli methylenetetrahydrofolate reductase reveal a Spartan strategy for a ping-pong reaction. Biochemistry. 2005; 44:11447–11457. doi: 10.1021/bi050533q. [DOI] [PubMed] [Google Scholar]
- 26.Matthews RG, Sheppard C, Goulding C. Methylenetetrahydrofolate reductase and methionine synthase: biochemistry and molecular biology. Eur J Pediatr. 1998;157(Suppl 2):S54–9. doi: 10.1007/pl00014305. [DOI] [PubMed] [Google Scholar]
- 27.Sheppard CA, Trimmer EE, Matthews RG. Purification and properties of NADH-dependent 5,10-methylenetetrahydrofolate reductase (MetF) from Escherichia coli. J Bacteriol. 1999;181:718–725. doi: 10.1128/jb.181.3.718-725.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brünger AT. X-PLOR: A System for X-ray Crystallography and NMR. Yale University Press; New Haven, CT: 1992. [Google Scholar]
- 29.Brünger AT, Adams PA, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RQ, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 30.vanAalten DMF, Bywater R, Findlay JBC, Hendlich M, Hooft RWW, Vriend G. PRODRG, a program for generating molecular topologies and unique molecular descriptors from coordinates of small molecules. J Comput-Aided Mol Des. 1996;10:255–262. doi: 10.1007/BF00355047. [DOI] [PubMed] [Google Scholar]
- 31.Ferre-Damare AR, Burley SK. Dynamic light scattering in evaluating crystallizability of macromolecules. Methods Enzymol. 1997;276:157–166. doi: 10.1016/S0076-6879(97)76056-7. [DOI] [PubMed] [Google Scholar]
- 32.Kabsch W. A solution for the best rotation to relate two sets of vectors. Acta Crystallogr. 1976;32:922–923. [Google Scholar]
- 33.Matthews RG, Baugh CM. Interactions of pig liver methylenetetrahydrofolate reductase with methylenetetrahydropteroylpolyglutamate substrates and dihydropteroylpoly-glutamate inhibitors. Biochemistry. 1980;19:2040–2045. doi: 10.1021/bi00551a005. [DOI] [PubMed] [Google Scholar]
- 34.Matthews RG, Ross J, Baugh CM, Cook JD, Davis L, Goldman ID, Chabner J, Bertino JR. Folyl- and Antifolylpolyglutamates. Plenum Press; New York: 1983. The role of folylpolyglutamates in the regulation of folate metabolism; pp. 35–44. [DOI] [PubMed] [Google Scholar]
- 35.Goyette P, Frosst P, Rosenblatt DS, Rozen R. Seven novel mutations in the methylenetetrahydrofolate reductase gene and genotype/phenotype correlations in severe methylenetetrahydrofolate reductase deficiency. Am J Hum Genet. 1995;56:1052–1059. [PMC free article] [PubMed] [Google Scholar]
- 36.Matthews RG. Methylenetetrahydrofolate reductase from pig liver. Methods Enzymol. 1986;122:372–381. doi: 10.1016/0076-6879(86)22196-5. [DOI] [PubMed] [Google Scholar]
- 37.Weber G. Intramolecular complexes of flavins. In: Slater EC, editor. Flavins and Flavoproteins. Elsevier; Amsterdam: 1966. pp. 15–21. [Google Scholar]
- 38.Spencer RD, Weber G. Thermodynamics and kinetics of the intramolecular complex in flavin-adenine dinucleotide. In: Akeson A, Ehrenberg A, editors. Structure and Function of Oxidation-Reduction Enzymes. Pergamon; Oxford and New York: 1972. pp. 393–399. [Google Scholar]
- 39.Wang M, Roberts DL, Paschke R, Shea TM, Masters BS, Kim JJ. Three-dimensional structure of NADPH-cytochrome P450 reductase: prototype for FMN- and FAD-containing enzymes. Proc Natl Acad Sci USA. 1997;94:8411–8416. doi: 10.1073/pnas.94.16.8411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hoover DM, Ludwig ML. A flavodoxin that is required for enzyme activation: the structure of oxidized flavodoxin from Escherichia coli at 1.8 Å resolution. Protein Sci. 1997;6:2525–2537. doi: 10.1002/pro.5560061205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Deng Z, Aliverti A, Zanetti G, Arakaki AK, Ottado J, Orellano EG, Calcaterra NB, Ceccarelli EA, Carillo N, Karplus PA. A productive NADP+ binding mode of ferredoxin-NADP+ reductase revealed by protein engineering and crystallographic studies. Nat Struct Biol. 1999;6:847–853. doi: 10.1038/12307. [DOI] [PubMed] [Google Scholar]
- 42.Crane BR, Rosenfeld RJ, Arvai AS, Ghosh DK, Ghosh S, Tainer JA, Stuehr DJ, Getzoff ED. N-terminal domain swapping and metal ion binding in nitric oxide synthase dimerization. EMBO J. 1999;18:6271–6281. doi: 10.1093/emboj/18.22.6271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pant K, Bilwes AM, Adak S, Stuehr DJ, Crane BR. Structure of a nitric oxide synthase heme protein from Bacillus subtilis. Biochemistry. 2002;41:11071–11079. doi: 10.1021/bi0263715. [DOI] [PubMed] [Google Scholar]
- 44.Andersen OA, Flatmark T, Hough E. High resolution crystal structures of the catalytic domain of human phenylalanine hydroxylase in its catalytically active Fe(II) form and binary complex with tetrahydrobiopterin. J Mol Biol. 2001;314:279–291. doi: 10.1006/jmbi.2001.5061. [DOI] [PubMed] [Google Scholar]
- 45.Ludwig ML, Pattridge KA, Metzger AL, Dixon MM, Eren M, Feng Y, Swenson RP. Control of oxidation–reduction potentials in flavodoxin from Clostridium beijerinckii: the role of conformation changes. Biochemistry. 1997;36:1259–1280. doi: 10.1021/bi962180o. [DOI] [PubMed] [Google Scholar]
- 46.Goyette P, Sumner JS, Milos R, Duncan AM, Rosenblatt DS, Matthews RG, Rozen R. Human methylenetetrahydrofolate reductase: isolation of cDNA, mapping and mutation identification [published erratum appears in Nat. Genet. (1994) Aug; 7 (4), 551] Nat Genet. 1994;7:195–200. doi: 10.1038/ng0694-195. [DOI] [PubMed] [Google Scholar]
- 47.Matthews RG, Vanoni MA, Hainfeld JF, Wall J. Methylenetetrahydrofolate reductase: Evidence for spatially distinct subunit domains obtained by scanning transmission electron microscopy and limited proteolysis. J Biol Chem. 1984;259:11647–11650. [PubMed] [Google Scholar]
- 48.Otwinowski Z, Minor W. Processing of x-ray diffraction data collected in the oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 49.DePristo MA, Bakker PIW, Blundell TL. Heterogeneity and inaccuracy in protein structures solved by X-ray crystallography. Structure. 2004;12:831–838. BI052294C. doi: 10.1016/j.str.2004.02.031. [DOI] [PubMed] [Google Scholar]