

Abstract
The l-menthone-derived TADDOL phosphite 6b catalyzes highly enantioselective conjugate additions of acyl silanes to α,β-unsaturated amides. p-Methoxybenzoyl cyclohexyldimethylsilane adds to a variety of N,N-dimethyl acrylamide derivatives in the presence of the lithium salt of 6b. In many instances the α-silyl-γ-ketoamide product undergoes facile enantioenrichment (to 97–99% ee) upon recrystallization. Desilylation with HF·pyr affords the formal Stetter addition products. Baeyer–Villiger oxidation of the desilylated γ-ketoamides affords useful ester products. An X-ray diffraction study of 6b reveals that the isopropyl group of the menthone ketal influences the position of the syn-pseudoaxial phenyl group in the TADDOL structure. Through a crossover experiment, the silicon migration step in the reaction mechanism is shown to be strictly intramolecular.
Introduction
The conjugate addition of acyl anion equivalents to α,β-unsaturated carbonyls is a useful and direct approach to the synthesis of 1,4-dicarbonyl compounds that can simultaneously introduce two new stereocenters.1 The prototypical catalytic reaction type involves the use of cyanide or heterazolium carbenes as umpolung catalysts.2–4 Employing scalemic versions of the latter, the asymmetric intramolecular Stetter reaction was recently realized as a useful transformation by Enders and coworkers.5,6 The transformation has undergone some significant improvements in enantioselectivity and scope in a body of work by Rovis and co-workers that now stands as the benchmark in this family of reactions.7–12 Their optimized process lends itself to a number of α,β-unsaturated ester and ketone acceptors delivering the annulated products in high yield and enantioselectivity; however, extension to asymmetric intermolecular alkene acylation has generally proven challenging. Enders was the first, to our knowledge, to successfully investigate this variant, achieving the addition of butanal to chalcone in 4–29% yield and 30–39% enantiomeric excess;13 these results represent the current state-of-the-art.
In parallel with the development of aldehydes as acyl donors for conjugate additions, acyl silanes have shown promise as suitable pronucleophiles in conjugate addition reactions. As in the classic Stetter reaction, cyanide and heterazolium carbenes can trigger the needed umpolung reactivity: −CN-catalyzed addition of acyl silanes to cyclic enones and acyclic acrylates was demonstrated by Degl’Innocenti,14 while carbene catalysis was shown by Scheidt to be effective for the acylation of α,β-unsaturated esters and ketones.15,16 Since the initiating steps in these catalytic processes involving acyl silanes are presumed to be carbonyl addition and [1,2]-Brook rearrangement,17,18 a pressing question related to catalysis was whether other entities might trigger these events. Indeed, Reich and Takeda had already shown that lithium phosphites add to acyl silanes and cause silicon migration.19,20 The latter study was particularly relevant since a pendant α,β-unsaturated ester was employed to trap the nascent (siloxy)phosphonate anion.21 The work of Reich and Takeda was instrumental to our development of chiral metallophosphites as nucleophilic catalysts for aldehyde and alkene acylation.22,23 We recently demonstrated a preliminary asymmetric variant of the alkene acylation reaction that proceeded in 67% yield and 50% enantiomeric excess using the Enders (R,R)-TADDOL-phosphite 1-Ph.24,25 From that platform we hypothesized that more highly enantioenriched 1,4-dicarbonyl products could be obtained through tuning of the phosphite catalyst and reagent functional groups. This article details the development of an effective asymmetric metallo-phosphite-catalyzed intermolecular addition of acyl silanes to α,β-unsaturated amides that can be achieved under mild reaction conditions in good yield and high enantioselectivity (eq 1).

Results and Discussion
I. Optimization Studies
Phosphite Structure
The asymmetric cross silyl benzoin reaction developed in our laboratory22 exhibited significant enantioselectivity enhancement through varying the aromatic groups on the (R,R)-TADDOL-phosphite (1-Ar). Due to this positive effect, our first instinct was to examine those electronic modifications on the phosphite in the 1,4-addition of benzoyl dimethylphenylsilane 2 to morpholinocinnamide 3 (Scheme 1). Electron-withdrawing substituents on 1 gave very low conversion, but in all reactions, neither electron-withdrawing nor electron-donating aromatic groups gave a significant boost in enantioselectivity.
Scheme 1.
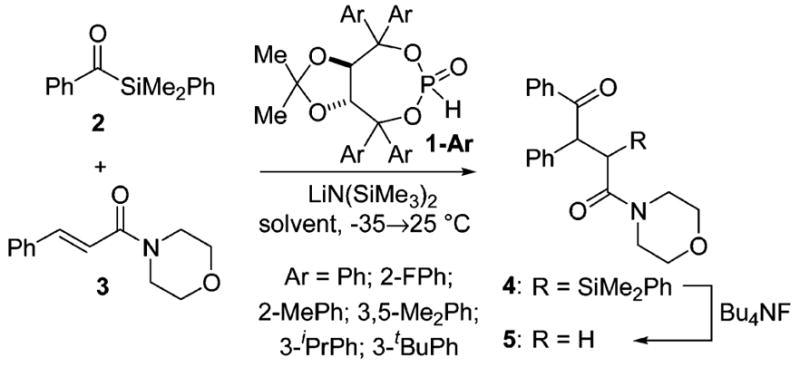
Alkene Acylation Catalyzed by Phosphites 1-Ar
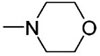
Another candidate for site modification in the catalyst architecture was the ketal backbone. In previous studies, we had observed minimal effect in changes at this site;22 however, we were pleased to find that replacement of acetone with l-menthone in the initial ketalization using either L- or D-diethyl tartrate afforded, after Grignard addition and phosphinylation, diastereomeric phosphite catalysts 6a and 6b (Figure 1) that exhibited notably higher levels of enantiocontrol in the addition of 2 to 3. Although the menthone tartrate has been reported by Seebach,26 to the best of our knowledge, our preparation of 6a and 6b constitutes the first synthesis of a menthone-based TADDOL.25 The therapeutic effects of this modification were noted in not only the enantiomeric excess of 5 (−60% ee using 6b, Table 1, entry 2) but also the improved anti/syn selectivity for the intermediate α-silyl-γ-ketoamide 4 (6b: anti/syn = 20: 1; 1-Ph: anti/syn = 10:1). The latter point impacts the former since we had previously demonstrated that anti and syn diastereomers possess the opposite absolute configuration at the phenyl-bearing stereocenter and that desilylation of the mixture results in erosion in the enantiomeric excess of 5.23
Figure 1.
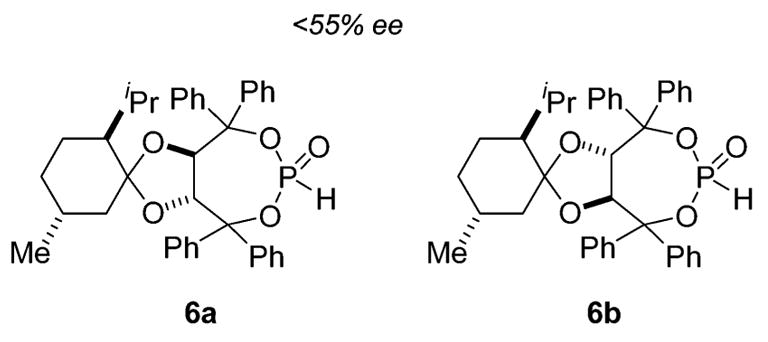
TADDOL-phosphites containing the l-menthone ketal.
Table 1.
Optimization Summary for Asymmetric Alkene Acylationa
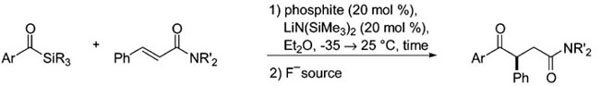
| entry | Ar | SiR3 | NR′2 | phosphite | time (h) | F− source | yield (%) | %ee |
|---|---|---|---|---|---|---|---|---|
| 1 | Ph | SiMe2Ph |
|
1-Ph | 1.0 | TBAF | 67 | 50 |
| 2 | Ph | SiMe2Ph |
|
6b | 1.0 | TBAF | 57 | −60 |
| 3 | Ph | SiMe2Ph | NMe2 | 6b | 2.5 | TBAF | 40 | −71 |
| 4 | Ph | SiEt3 | NMe2 | 6b | 2.0 | TBAF | 44 | −88 |
| 5 | p-MeOPh | SiEt3 | NMe2 | 6b | 0.75 | TBAF | 85 | −90 |
| 6 | p-MeOPh | SiCyMe2 | NMe2 | 6b | 0.25 | TBAF | 78 | −87 |
| 7 | p-MeOPh | SiCyMe2 | NMe2 | 1-Ph | 1.0 | TBAF | 54 | 81 |
| 8 | p-MeOPh | SiCyMe2 | NMe2 | 6b | 0.25 | HF·pyr | 73 | −89 |
| 9 | p-MeOPh | SiCyMe2 | NMe2 | 6a | 1.0 | HF·pyr | 82 | 88 |
ArC(O)SiR3 (1.0 equiv), PhCH=CHC(O)NR′2 (1.5 equiv), phosphite (0.2 equiv.), and LiN(SiMe3)2 (0.2 equiv) in Et2O from −35 → 25 °C for 0.25–2.0 h after slow addition of alkene and acyl silane.
Amide
The impact of the amide structure on enantioselec-tivity and reactivity was probed through an examination of eleven cinnamides; a complete summary of this study may be found in the Supporting Information. Pyrrole, pyrazole, and Weinreb cinnamides failed to provide any acylation product. In fact, of the amides surveyed, only N,N-dimethylcinnamide (7) exhibited significant improvement relative to the morpholinocinnamide 3 in both conferred enantiocontrol (71% ee using phosphite 6b) and diastereoselectivity for the derived α-silyl-γ-ketoamide 9 (Scheme 2). The reaction proceeded at a somewhat slower rate and product yield was lower (40%, Table 1, entry 3), but only a single diastereomer of the α-silyl-γ-ketoamide 9 could be distinguished by 1H NMR spectroscopy (anti/syn > 30:1).
Scheme 2.
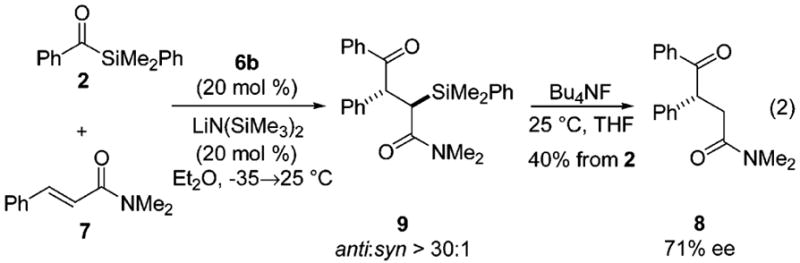
Enantioselective Acylation of N,N-Dimethylcinnamide
Acyl Silane
Attention was then directed at improving the enantiomeric excess and yield for the alkene acylation through modification of the acyl donor. Our experiments in this vein revealed that the identity of the silyl moiety was crucial for maximizing enantiocontrol and that the aryl group of the acyl silane was the key in modulating reactivity. The addition of benzoyl triethylsilane to N,N-dimethylcinnamide (7) catalyzed by 6b gave the product α-silyl-γ-ketoamide as one distinguishable diastereomer by 1H NMR spectroscopy and the desilylated γ-ketoamide product 8 in 88% enantiomeric excess (Table 1, entry 4). Further increases in the steric demand of the silyl group (benzoyl triisopropylsilane, benzoyl trihexylsilane, benzoyl tert-butyldimethylsilane) resulted in dramatic decreases in reactivity. The use of benzoyl cyclohexyldimethylsilane27 furnished 8 in comparable enantioselectivity to PhC(O)SiEt3 but conferred the added bonus of crystallinity to the intermediate α-silyl-γ-ketoamide. This characteristic was found to be relatively common (vide infra) and was used advantageously in purification and upgrades in product enantiomeric excess. Although the boost in enantioselectivity using PhC(O)SiEt3 and PhC(O)-SiCyMe2 was encouraging, product yields were still unacceptably low. In probing this issue, we were fortunate to discover that substitution of the phenyl group of the acyl silane with a para-anisyl group reduced reaction times, provided the needed increase in yield, and had little effect on the enantioselectivity. Specifically, phosphite 6b catalyzed the efficient conjugate addition of para-methoxybenzoyl cyclohexyldimethylsilane 10 to cinnamide 7; desilylation of an aliquot of α-silyl-γ-ketoamide 11 indicated that the kinetic enantioselectivity for the initial addition was 95:5 (Scheme 3). The remainder of amide 11 was recrystallized from hot hexanes and upon desilylation afforded 12a in 99% ee and 68% yield.
Scheme 3.
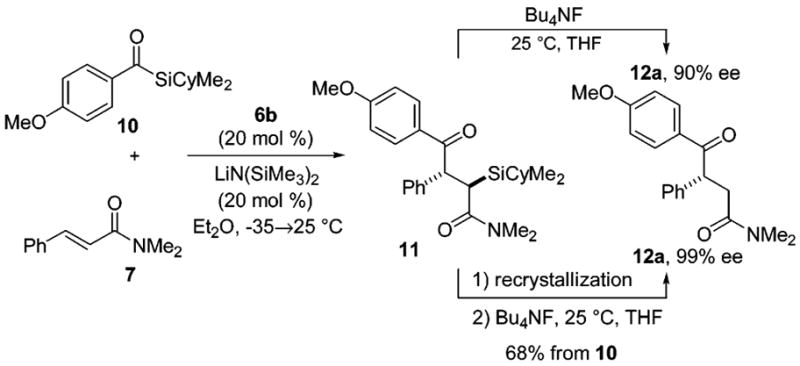
Recrystallization and Enantiomeric Excess Upgrade of α-Silyl-γ-ketoamide 11
The key experiments in the reaction evolution are summarized in Table 1, and a more extensive tabulation can be found in the Supporting Information. All reactions were run on a 100 mg scale in a glovebox using 20 mol % of the phosphite catalyst and base, lithium hexamethyldisilazide. The optimized reagents were also tested using catalyst 1-Ph (entry 7) for a direct comparison to the menthone phosphite 6b, and phosphite 6a (entry 9) was employed to demonstrate that both product enantiomers can be obtained. A final operational modification was made involving the deprotection step: the use of tetra-N-butylammonium fluoride (TBAF) was occasionally found to cause erosion in product enantioselectivity depending on contact time with the substrate. Desilylation using HF·pyridine in acetonitrile gave comparable yields and consistent enantiose-lectivities (entries 8,9).
After an optimized reaction protocol had been established for the asymmetric conjugate addition of acylsilanes to α,β-unsaturated amides in the glovebox, it was necessary from a practicality standpoint to achieve those yields and enantioselectivities outside of the glovebox. Our initial efforts resulted in inconsistent results with incomplete conversion arising as a common problem even under nominally anhydrous and anerobic conditions. Separation and isolation of materials from these reactions and identification of them by 31P spectroscopy and mass spectrometry revealed the presence of (1) the conjugate addition product between the phosphite and unsaturated amide24 and (2) the (siloxy)phosphonate resulting from quenching of the Brook rearrangement product. The precursors to these byproducts are understandably sensitive to traces of proton sources. The use of excess base was sufficient in our cross benzoin studies to achieve a simple reaction protocol and avoid this problem, but conditions employing excess base gave nearly racemic product in the current study. To achieve full conversion outside of the glovebox, we found it optimal to conduct reactions on at least a 200 mg scale using 30 mol % of phosphite and 30mol % of LiN(SiMe3)2 at room temperature. This gave a simple and reproducible protocol and also simplified recrystallization of the silylated product.
II. Substrate Scope and Limitations
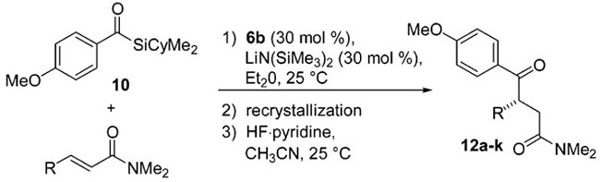
The reaction scope with respect to the Michael acceptor was then analyzed employing the aforementioned reaction conditions. An aliquot from each reaction was removed and desilylated to determine the kinetic enantioselectivity for the addition. When possible, the remaining α-silyl-γ-ketoamide was recrystallized. Deprotection with HF·pyridine in acetonitrile afforded γ-ketoamides 12a–k. Yields and enantioselectivities of the final products are listed in Table 2.
Table 2.
Substrate Scope of Asymmetric Metallophosphite-Catalyzed Alkene Acylationa
| entry | R | % yield | % ee (aliquot) | % ee (12) |
|---|---|---|---|---|
| 1 | Ph (a) | 68 | 90 | 99 |
| 2 | p-MeOPh (b) | 63 | 92 | b |
| 3 | p-MePh (c) | 78 | 90 | b |
| 4 | m-MePh (d) | 67 | 93 | 99 |
| 5 | 2-furyl (e) | 15 | 24 | c |
| 6 | p-ClPh (f) | 66 | 95 | 98 |
| 7 | N-tosylindol-3-yl (g) | 60 | 97 | 97 |
| 8 | p-CF3Ph (h)d | 80 | 90 | b |
| 9 | 2-naphthyl (i) | 66 | 89 | 97e |
| 10 | Me (j)d | 56f | 86f | g |
| 11 | Et (k)d | 82 | 71 | b |
Slow addition of p-MeOPhC(O)SiCyMe2 (1.0 equiv) and RCH=CHC(O)NMe2 (1.5 equiv) to the phosphite (0.3 equiv) and LiN-(SiMe3)2 (0.3 equiv) in Et2O at 25 °C; reaction times = 0.25–3.0 h.
Attempts at recrystallization of the α-silyl-γ-ketoamide or final product (12) were unsuccessful.
Yield was too low to attempt recrystallization.
1.1 equiv of RCH=CHC(O)NMe2 was employed and was added with the acyl silane to the metallophosphite mixture.
The major enantiomer was found in the mother liquor after recrystallization of the final product.
Yield and % ee after separation of the major diastereomer.
Recrystallization yielded little improvement in enantioselectivity.
The alkene acylation is applicable to α,β-unsaturated amides containing both electron-donating (entries 2–4) groups and electron-withdrawing groups (entries 6–8) in the β-position of the Michael acceptor affording the product in reasonable yields and high enantioselectivities except for the strongly electron-donating furyl substituent (entry 5). β-Alkyl amides also undergo productive coupling with somewhat lower product enantioselectivities (entries 10–11). While the scope is good for the Michael acceptor, a notable limitation of this acylation protocol in its current form is its inability to successfully couple alkyl acyl silanes (RC(O)SiR′3, R = alkyl) with unsaturated amides in yields > 10%. Since the metallophosphite addition and Brook rearrangement steps have been demonstrated for alkyl acyl silanes,22 this lack of reactivity is apparently due to the inability of the (silyloxy)phosphonate anion to participate in conjugate addition.
The optimized reaction conditions described above work well on a reasonable laboratory scale. On a 5-g scale, the addition of 10 to N,N-dimethylcinnamide catalyzed by 6b gives the α-silyl-γ-ketoamide 11 in >99% enantiomeric excess after crystallization. Deprotection with HF·pyridine and recrystallization give the enantiopure γ-ketoamide 12a in 61% yield for the two steps.
III. Product Manipulation and Stereochemistry
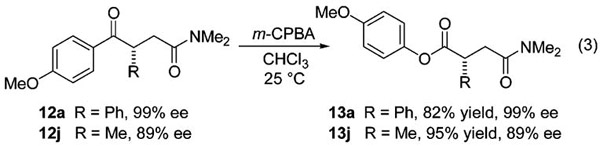
Table 1 suggests, and experiments with other acyl silanes confirm, that the p-anisyl substitutent in 10 is needed for high yield and enantioselectivity. We viewed this apparent limitation as an opportunity to develop useful second-stage reactions involving the acylation products. In particular, we considered that oxidation of the Caryl–Ccarbonyl bond would provide a broader selection of 1,4-dicarbonyl compounds with more flexibility for subsequent manipulation. Such Baeyer–Villiger reactions of (methoxyphenyl)ketones in conjunction with asymmetric synthesis are well-documented.28–32 Indeed, exposure of 12a and 12j to m-chloroperbenzoic acid (m-CPBA) in chloroform resulted in oxidation of the ketones to the derived p-methoxyphenyl (PMP) esters 13a and 13j, respectively (eq 3). As expected, no loss in optical purity was observed for the ester products. The difference in the yield for 13a relative to 13j arises from a regioisomeric Baeyer–Villiger product in the former case.
The PMP esters are useful compounds that permit differentiation of the two carbonyl groups and provide access to other useful building blocks (Scheme 4). Transesterification of 13a with methanol occurs to afford the corresponding methyl ester 14 with little racemization. Ester 13a undergoes chemoselective reduction with NaBH4 to give γ-hydroxyamide 15 in >95% yield with negligible loss in enantiopurity.
Scheme 4.
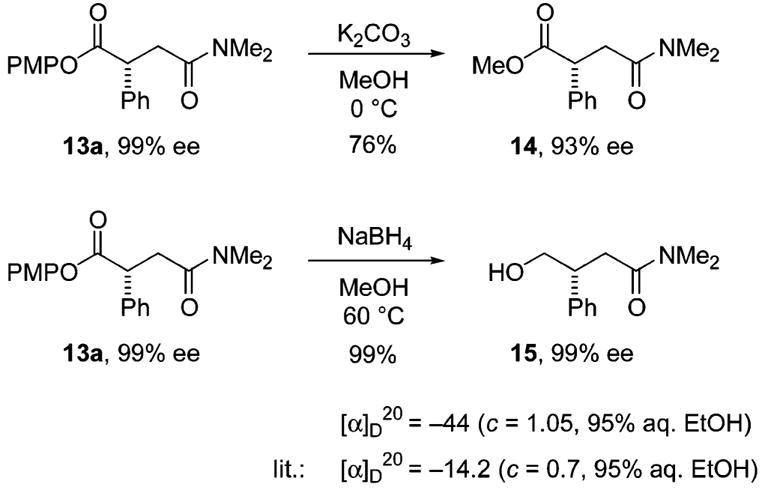
Synthetic Operations Involving 13a
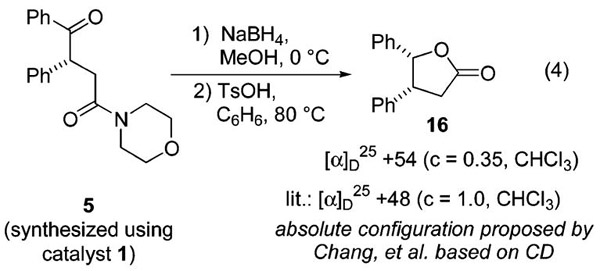
The absolute configuration of 15 (and thence 13a and 12a) was established through comparison of its optical rotation to that reported in the literature for the 3(R) isomer.33 This assignment was cross-verified through an X-ray diffraction study of enantiomerically pure 12f; this amide also possessed the (R) configuration.34 These stereochemical proofs are at odds with the one provided in our original communication for compound 5. That analysis involved the conversion of γ-ketoamide 5 to the γ-lactone 16, whose absolute stereostructure had been previously proposed on the basis of CD studies on related compounds (eq 4).35 The discrepancy between that assignment and the stereochemical assignments established in this article through both chemical correlation and X-ray crystallography suggests either (1) the stereochemical assignment made by Chang et al. (and by corollary our assignment of 5) is incorrect or (2) there is a turnover in absolute stereochemical preference for acyl silane/amide combination 2/3 versus 7/10. Difficulty in obtaining 5 in high enantiomeric excess has so far precluded the use of derivatization and X-ray crystallography to resolve this issue. The absolute configuration of 5 must therefore be considered tentative. In practice, however, the most synthetically useful and highly enantioenriched adducts described in this article (i.e., 12) are those whose absolute stereostructures have been unambiguously determined. It is noteworthy that the topicity preference for the tartrate-based (silyloxy)phosphonate anion intermediate is the same for both aldehyde and alkene electrophiles, a fact that would seem to augur well for extension to other electrophiles.
IV. Reaction Characteristics. Catalyst Structure
To better understand the structural features of the l-menthone-modified catalyst that result in enhanced enantiocontrol, we undertook X-ray diffraction studies of phosphites 1-Ph and 6b (Figure 2).34 The structure of 6b reveals that the isopropyl group of the menthone moiety is disposed approximately on the pseudo-C2 axis of the catalyst tartrate framework. Further inspection indicates that the presence of the isopropyl group results in nonbonded compression of the nearby pseudoaxial phenyl group into the reaction site (i.e., toward the phosphorus atom). This is most clearly manifested in the distance between the ketal carbon and the para carbon of the pseudoaxial phenyl group syn to the isopropyl group. For 6b this distance is 5.482 Å, while the analogous measurement for 1-Ph gives a distance of 5.192 Å. For comparison, the distance from the ketal carbon to the para carbon of the pseudoaxial phenyl group anti to the isopropyl group in 6b is 5.110 Å. These observations may reveal a structural basis for the enhanced enantioselectivity, since the isopropyl group appears to force one of the phenyl groups in closer proximity to the obligatory transition structure. Such transmission effects have been observed crystallographically in other nonacetonide TADDOL derivatives.25 The fact that 6a and 6b provide opposite but equal enantioselectivity suggests that the absolute configuration of the menthone is not important, a supposition that is congruent with the position of the iso-propyl group on the pseudo-C2 axis.
Figure 2.

X-ray structures of phosphites 1-Ph and 6b.
Test for Silyl Group Transfer Pathway
A crossover experiment was designed (Scheme 5) to distinguish whether the silyl transfer in the title reaction was occurring via an intramolecular or intermolecular pathway. Phosphite 6b was used to catalyze the reaction of para-methoxybenzoyl cyclohexyldim-ethylsilane (10) and benzoyl triethylsilane (17) with a single α,β-unsaturated amide, N,N-dimethylcinnamide (7), under conditions stated in the Supporting Information. By 1H NMR spectroscopy, the α-silyl-γ-ketoamides 11 and 18 were each obtained in >80% yield (versus internal standard) and to the exclusion of any intermolecular Si-transfer products.
Scheme 5.
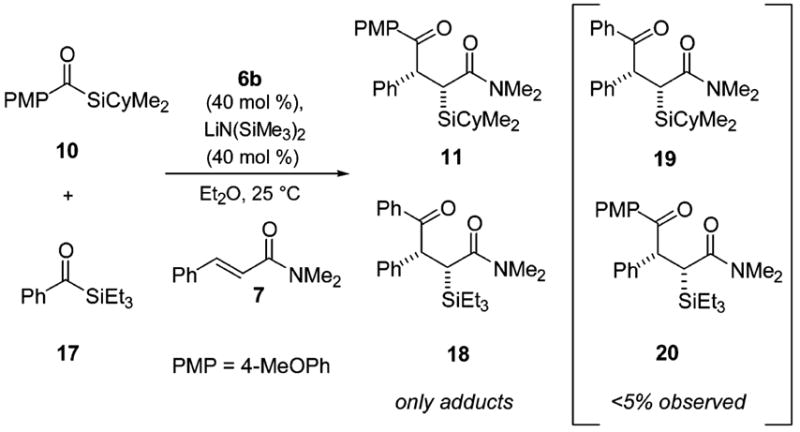
Crossover Experiment to Determine Silyl Group Transfer Pathway
With the confirmation that the silicon migration is intramolecular, we propose that the high anti diastereoselectivity10 can be explained through consideration of some simple conformational issues. In the catalyzed alkene acylation, the reactive conformer of the acceptor is presumed to be s-cis.36 Conjugate addition of the (siloxy)phosphonate anion21 should provide the nascent lithium enolate in the favored (Z)-geometry. In this conformation, intramolecular retro-[1,4] Brook rearrangement37 is expected to be strongly favored from the rear face of the enolate due to associated A1,3 constraints (as depicted in Figure 3).
Figure 3.

Proposed model for anti diastereoselectivity.
Conclusion
High enantioselectivities can now be achieved in the catalytic intermolecular conjugate addition of acyl silanes to α,β-unsaturated amides. The reactions take place at room temperature in less than 2 h for a variety of aryl and alkyl substrates. In many cases the α-silyl-γ-ketoamides formed after the nucleophilic addition are stable, white solids that may be purified by recrystallization. This simple purification leads to useful enantioselectivity upgrades in several cases. While the catalyst loading is not optimal in the current iteration, the phosphites are trivial to prepare from cheap starting materials that are available in either antipode. The levels of enantiocontrol realized for the title reaction are the highest for any intermolecular Stetter-type reaction to date, and the products may be transformed to terminally differentiated chiral succinates.
Supplementary Material
Acknowledgments
Funding was provided by the National Institutes of Health (National Institute of General Medical Sciences, GM068443). Research support from Eli Lilly, 3M, Amgen, and GSK is gratefully acknowledged.
Footnotes
Supporting Information Available: Experimental procedures, compound characterization data, and crystallographic information files. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Stetter H, Kuhlmann H. Org React (N Y) 1991;40:407–496. [Google Scholar]
- 2.Seebach D. Angew Chem, Int Ed Engl. 1979;18:239–258. [Google Scholar]
- 3.Johnson JS. Angew Chem, Int Ed. 2004;43:1326–1328. doi: 10.1002/anie.200301702. [DOI] [PubMed] [Google Scholar]
- 4.For an exception using PBu3, see Gong JH, Im YJ, Lee KY, Kim JN. Tetrahedron Lett. 2002;43:1247–1251.
- 5.Enders D, Breuer K, Runsink J, Teles JH. Helv Chim Acta. 1996;79:1899–1902. [Google Scholar]
- 6.Enders D, Breuer K. In: Comprehensive Asymmetric Catalysis. Jacobsen EN, Pfaltz A, Yamamoto H, editors. Vol. 3. Springer; New York: 1999. pp. 1093–1102. [Google Scholar]
- 7.Kerr MS, Read de Alaniz J, Rovis T. J Am Chem Soc. 2002;124:10298–10299. doi: 10.1021/ja027411v. [DOI] [PubMed] [Google Scholar]
- 8.Kerr MS, Rovis T. Synlett. 2003:1934–1936. [Google Scholar]
- 9.Kerr MS, Rovis T. J Am Chem Soc. 2004;126:8876–8877. doi: 10.1021/ja047644h. [DOI] [PubMed] [Google Scholar]
- 10.Read de Alaniz J, Rovis T. J Am Chem Soc. 2005;127:6284–6289. doi: 10.1021/ja0425132. [DOI] [PubMed] [Google Scholar]
- 11.Mennen SM, Blank JT, Tran-Dube MB, Imbriglio JE, Miller SJ. Chem Commun. 2005:195–197. doi: 10.1039/b414574g. [DOI] [PubMed] [Google Scholar]
- 12.Christmann M. Angew Chem, Int Ed. 2005;44:2632–2634. doi: 10.1002/anie.200500761. [DOI] [PubMed] [Google Scholar]
- 13.Enders D, Balensiefer T. Acc Chem Res. 2004;37:534–541. doi: 10.1021/ar030050j. [DOI] [PubMed] [Google Scholar]
- 14.Degl’Innocenti A, Ricci A, Mordini A, Reginato G, Colotta V. Gazz Chim Ital. 1987;117:645–648. [Google Scholar]
- 15.Mattson AE, Bharadwaj AR, Scheidt KA. J Am Chem Soc. 2004;126:2314–2315. doi: 10.1021/ja0318380. [DOI] [PubMed] [Google Scholar]
- 16.Bharadwaj AR, Scheidt KA. Org Lett. 2004;6:2465–2468. doi: 10.1021/ol049044t. [DOI] [PubMed] [Google Scholar]
- 17.Brook AG. Acc Chem Res. 1974;7:77–84. [Google Scholar]
- 18.Moser WH. Tetrahedron. 2001;57:2065–2084. [Google Scholar]
- 19.Reich HJ, Holtan RC, Bolm C. J Am Chem Soc. 1990;112:5609–5617. [Google Scholar]
- 20.Takeda K, Tanaka T. Synlett. 1999:705–708. [Google Scholar]
- 21.Koenigkramer RE, Zimmer H. J Org Chem. 1980;45:3994–3998. [Google Scholar]
- 22.Linghu X, Potnick JR, Johnson JS. J Am Chem Soc. 2004;126:3070–3071. doi: 10.1021/ja0496468. [DOI] [PubMed] [Google Scholar]
- 23.Nahm MR, Linghu X, Potnick JR, Yates CM, White PS, Johnson JS. Angew Chem, Int Ed. 2005;44:2377–2379. doi: 10.1002/anie.200462795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Enders D, Tedeschi L, Bats JW. Angew Chem, Int Ed. 2000;39:4605–4607. [PubMed] [Google Scholar]
- 25.Seebach D, Beck AK, Heckel A. Angew Chem, Int Ed. 2001;40:92–138. [PubMed] [Google Scholar]
- 26.Seebach D, Beck AK, Imwinkelried R, Roggo S, Wonnacott A. Helv Chim Acta. 1987;70:954–974. [Google Scholar]
- 27.Dimethylcyclohexylsilyl chloride is commerically available and is comparable in price to triethylsilyl chloride.
- 28.Evans PA, Lawler MJ. J Am Chem Soc. 2004;126:8642–8643. doi: 10.1021/ja049080n. [DOI] [PubMed] [Google Scholar]
- 29.Yoshikawa N, Suzuki T, Shibasaki M. J Org Chem. 2002;67:2556–2565. doi: 10.1021/jo0162538. [DOI] [PubMed] [Google Scholar]
- 30.Boyes SA, Hewson AT. J Chem Soc, Perkin Trans 1. 2000:2759–2765. [Google Scholar]
- 31.Reissig HU, Schumacher R, Ferse D. Liebigs Ann/Recl. 1997:2119–2124. [Google Scholar]
- 32.Baures PW, Eggleston DS, Flisak JR, Gombatz K, Lantos I, Mendelson W, Remich JJ. Tetrahedron Lett. 1990;31:6501–6504. [Google Scholar]
- 33.Braun M, Unger C, Opdenbusch K. Eur J Org Chem. 1998;2389:9–2396. [Google Scholar]
- 34.CCDC 282470 (1-Ph), CCDC 282471 (6b), and CCDC 293708 (12f) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif
- 35.Chang CJ, Fang JM, Liao LF. J Org Chem. 1993;58:1754–1761. [Google Scholar]
- 36.Montaudo G, Librando V, Caccamese S, Maravigna P. J Am Chem Soc. 1973;95:6365–6370. [Google Scholar]
- 37.Gibson C, Buck T, Noltemeyer M, Brückner R. Tetrahedron Lett. 1997;38:2933–2936. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.