

Abstract
We have shown previously that ulcerogenic (type I) strains of Helicobacter pylori (Hp) retard their entry into macrophages. However, the signaling pathways that regulate Hp phagocytosis are largely undefined. We show here that Hp strongly activated class IA phosphoinositide3-kinases (PI3Ks) in macrophages, coincident with phagocytosis, and endogenous p85 and active protein kinase Bα accumulated on forming phagosomes. PI3K inhibitors, wortmannin and LY294002, inhibited phagocytosis of Hp in a dose-dependent manner, and blockade of engulfment correlated directly with loss of 3′-phosphoinositides in the membrane subjacent to attached bacteria. During uptake of large immunoglobulin G (IgG)-coated particles, PI3Ks regulate pseudopod extension and phagosome closure. In marked contrast, we show here that 3′-phosphoinositides regulated actin polymerization at sites of Hp uptake. Moreover, Hp and IgG beads activated distinct PI3K isoforms. Phagosomes containing IgG-coated particles accumulated 3′-phosphatase and tensin homologue deleted on chromosome 10 and Src homology 2 domain-containing inositol 5′-phosphatase, yet Hp phagosomes did not. Finally, rapid uptake of IgG-opsonized Hp or a less-virulent type II Hp was PI3K-independent. We conclude that Hp and IgG beads are ingested by distinct mechanisms and that PI3Ks regulate the actin cytoskeleton during slow phagocytosis of ulcerogenic Hp.
Keywords: macrophage, signal transduction, phagosome
INTRODUCTION
Helicobacter pylori (Hp) is a highly successful pathogen that has colonized the gastric epithelium of ~50% of humans [1]. These Gram-negative, microaerophilic organisms cause a broad spectrum of disease that includes gastritis, peptic ulceration, or more rarely, gastric adenocarcinoma. In general, strains of Hp have been divided into two groups [2, 3]. Type II organisms induce moderate inflammation and are common in persons with asymptomatic gastritis. In contrast, ulcerogenic (type I) Hp induce substantial inflammation and tissue damage that is accompanied by a prominent influx of phagocytes to the gastric mucosa. The virulence of type I Hp is mediated, in part, by secretion of a potent vacuolating cytotoxin and the presence of a type IV secretion system encoded by the cag pathogenicity island. These virulence factors act on the gastric epithelium to induce cell death and trigger phagocyte influx, respectively.
Several research groups have demonstrated that a subset of Hp strains avoids killing after they are ingested by macrophages or neutrophils in vitro (reviewed in ref. [3]). In vivo, the ability of Hp to survive inside epithelial cells and macrophages contributes to organism persistence and treatment failure [4, 5]. We have shown previously that type I Hp evade phagocytic killing and actively modulate their entry into macrophages, whereas the less-virulent type II strains do not [6]. Delayed phagocytosis requires bacterial protein synthesis and is specific for live, type I organisms [6, 7]. In contrast, type II Hp are ingested rapidly and killed [6, 8]. As delayed phagocytosis is unique to type I Hp, we hypothesized that these organisms may define a distinct phagocytic pathway. Nevertheless, receptors for Hp remain obscure, and the signaling pathways that control bacterial engulfment are only beginning to be explored.
Class IA phosphoinositide3-kinases (PI3Ks) are heterodimers composed of a regulatory subunit and a catalytic subunit (p110α, p110β, or p110δ) [9, 10]. PI3Kδ is expressed primarily in leukocytes, whereas PI3Kα and PI3Kβ are widely distributed [11]. The p85 regulatory subunit targets these enzymes to membranes by interaction of its Src homology 2 (SH2) domains with phosphotyrosine residues or by interaction of its SH3 domain with proline-rich sequences [12]. At the membrane, the catalytic subunit phosphorylates the D3 position of the inositol ring of phosphatidylinositol-4,5-bisphosphate and to a lesser extent, phosphatidylinositol 4-phosphate [12]. In vitro, phosphoinositide can also be used as a substrate [10]. Activation of PI3K downstream of tyrosine kinases is required for Fc receptor for immunoglobulin G (IgG; FcγR)-mediated phagocytosis in macrophages and neutrophils [13–15]. In this context, 3′-phophoinositides regulate pseudopod extension and closure of phagosomes containing large IgG-coated particles but are dispensable for local actin polymerization [14, 15]. To our knowledge, PI3K activity has not been measured following engagement of other phagocytic receptors. However, PI3K inhibitors wortmannin (WTM) [16, 17] and/or LY294002 [18] prevent phagocytosis of zymosan [19], αvβ3-mediated uptake of apoptotic cells [20], and in some cases, particle ingestion via CD11b/CD18 [19, 21–23]. Conversely, WTM and LY294002 do not prevent ingestion of yeast, Escherichia coli, Yersinia enterocolitica, Legionella pneumophilia, Salmonella, or Brucella by macrophages [22–26]. PI3K is also dispensable for phagocytosis in Dictyostelium discoideum [27], and depletion of PI3Kα does not prevent engulfment of Mycobacterium tuberculosis [28]. Collectively, these data suggest that PI3Ks regulate only a subset of phagocytic processes.
Herein, we used biochemical approaches and confocal microscopy to assess the role of PI3K in Hp uptake by macrophages. We now show that class IA PI3Ks are activated strongly by ulcerogenic Hp and that accumulation of phospha-tidylinositol-3,4,5-trisphosphate [PI(3,4,5)P3] on forming phagosomes is essential for bacterial engulfment. In addition we demonstrate that PI3K activity regulates the actin cytoskeleton in Hp-infected cells, and as such, the results of this study define a new function for class IA PI3Ks in macrophages.
MATERIALS AND METHODS
Materials
Trypticase soy agar was obtained from Difco Laboratories (Detroit, MI). Pyrogen-free phosphate-buffered saline (PBS), Hepes-RPMI, minimum essential medium-α (MEMα), L-glutamine, and penicillin-streptomycin were from Bio-Whittaker (Walkersville, MD). Fetal bovine serum (FBS) was from Gibco (Grand Island, NY). Zymosan-opsonizing reagent, rhodamine-phalloidin, and rabbit antibody to PI(3,4,5)P3 were from Molecular Probes (Eugene, OR). IgG beads were from Dynal (Brown Deer, WI). Polyclonal antibodies (pAb) to Hp were from Accurate (Westbury, NY). Monoclonal antibodies (mAb), specific for Hp lipopolysaccharide (LPS) were from Biogenesis Limited (Kingston, NH). WTM, mouse anti-protein kinase Bα (PKBα) hybridoma supernatant [29], rabbit anti-p85 antiserum, and purified anti-p85 IgG [30] were the generous gift of Dr. Marcus Thelen (Institute for Research in Biomedicine, Bellinzona, Switzerland). Rabbit anti-p110α, -p110β, and -p110δ pAb and mouse mAb to phosphatase and tensin homologue deleted on chromosome 10 (PTEN) were from Santa Cruz Biotechnology (CA). Mouse mAb to SH2 domain-containing inositol 5′-phosphatase (SHIP) and class II PI3K (PI3KC2α) were from BD Biosciences (San Jose, CA). Mouse mAb to phosphorylated Ser473 of PKB and rabbit pAb to phosphorylated Thr308 of PKB were from Upstate Biotechnology (Charlottsville, VA) and Biosource (Camarillo, CA), respectively. Leupeptin was obtained from Boehringer-Mannheim (Indianapolis, IN). Affinity-purified fluorescein isothiocyaante (FITC)- and rhodamine-conjugated secondary antibodies were from Jackson ImmunoResearch Laboratories (West Grove, PA). PolyScreen polyvinylidene difluoride (PVDF) transfer membranes were from New England Nuclear (Boston, MA). Horseradish peroxidase (HRP)-conjugated secondary antibodies were from Amersham (Piscataway, NJ). Latrunculin B was from Biomol (Plymouth Meeting, PA). LY294002 was from Calbiochem (San Diego, CA). Coomassie Plus and bicinchoninic acid (BCA) protein assay kits and enhanced chemiluminescence (ECL) reagents were from Pierce (Rockford, IL). Additional reagents were obtained from Sigma Chemical Co. (St. Louis, MO).
Bacterial strains and culture
Hp were obtained from the American Type Culture Collection (Manassas, VA). Strains 11637, 11916, and DT61A contain the cag pathogenicity island and secrete an active form of VacA [31–33] and as such, are ulcerogenic (type I) organisms [2]. Strains Tx30a and MC123 are cag-negative, produce an inactive form of VacA [34, 35], and are classified as type II Hp [2]. All Hp were grown on pH 6 trypticase soy agar containing 5% sheep blood (Remel, Lenexa, KS) under microaerophilic conditions (5% O2, 10% CO2, 85% N2) as described previously [6]. Where indicated, Hp were opsonized with anti-Hp pAb or anti-LPS mAb for 30 min at 37°C, washed, and resuspended in MEMα [36].
Macrophage isolation and culture
Resident peritoneal macrophages (PMφ) and marrow for cultivation of bone marrow-derived macrophages (BMMs) were obtained from female CD-1 mice (Charles River Laboratories, Wilmington, MA) using procedures approved by the University Animal Care and Use Committee of the University of Iowa (Iowa City). BMMs were maintained in Hepes-RPMI containing 15% heat-inactivated (HI) FBS, 1% L-glutamine, 100 U/ml penicillin G, 100 μg/ml streptomycin, and 20% L cell-conditioned medium as a source of colony-stimulating factor (CSF)-1 and used after 7–16 days in culture. For each experiment, BMMs were scraped off petri dishes and replated on coverslips or in tissue-culture dishes as indicated. BMMs were switched to MEMα containing 10% HI FBS and 1% L-glutamine (without antibiotics or CSF-1) and were incubated overnight at 37°C prior to infection. Similarly, PMφ were plated on glass coverslips or tissue-culture dishes in MEMα containing 10% HI FBS and 1% L-glutamine, washed after 2 h to remove lymphocytes, and incubated overnight at 37°C prior to infection.
Drug treatments
To prevent actin rearrangements and phagocytosis, macrophages were treated with 2 μg/ml cytochalasin B or 63 nM latrunculin B for 30 min at 37°C prior to addition of Hp. To inhibit PI3K activity, macrophages were treated with 0 –100 nM WTM or 0 –100 μM LY294002 for 1 h at 37°C in serum-free MEMα prior to infection. Drugs were maintained in the medium for the duration of each experiment
Synchronized phagocytosis
Washed Hp, IgG beads, or IgG-opsonized zymosan (IgG-Z) were dispersed in a tissue-culture medium to achieve a ratio of five to 15 bacteria and three to five particles per macrophage (for microscopy experiments) or ~25 bacteria and 10 –12 particles per macrophage (for biochemistry experiments). In all cases, phagocytosis was synchronized using centrifugation as described previously [6, 37].
Immunofluorescence microscopy
An established, a differential staining procedure was used to distinguish intracellular and extracellular macrophage-associated Hp [36]. Fluorescence was visualized with a Zeiss Axioplan2 microscope (Carl Zeiss, Inc., Thorn-wood, NY). For each experiment Hp associated with at least 100 infected macrophages were counted on triplicate coverslips. By this assay, ≥80% of cell-associated Hp were ingested after 15 min at 37°C, and phagocytosis was blocked by ~95% in the presence of the actin filament-destabilizing agents cytochalasin or latrunculin B [36]. Ingestion of IgG-Z was quantified in a similar manner using rhodamine or FITC-conjugated secondary antibodies directed against the opsonin.
To assess the association of F-actin and marker proteins with forming phagosomes, cells were fixed in formalin, permeabilized with −20°C acetone, blocked, and then stained with rhodamine phalloidin and anti-Hp antibodies as described [6, 38]. Reagents were diluted as follows: rhodamine phalloidin (1:200); rabbit anti-Hp pAb (1:2000); mouse mAb to Hp LPS (1:400); rabbit anti-p85 IgG (1:50); mouse hybridoma supernatant to PKBα (neat); mouse mAb to phospho-PKB (1:200); mouse anti-PTEN mAb (1:20); mouse anti-SHIP mAb (1:30); mouse anti-PI3KC2 mAb (1:30); secondary antibodies (1:200). Fluorescence was viewed using an Axioplan2 photomicroscope or an LSM-510 laser-scanning confocal microscope (both from Carl Zeiss Inc.). Composite images were generated using Photoshop 6.0 (Adobe Systems Inc., Mountain View, CA).
Subcellular fractionation, sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and immunoblotting
BMMs (1 × 106/35 mm dish), which had ingested Hp or IgG-opsonized particles for 0 –15 min at 37°C, were disrupted by nitrogen cavitation, and post-nuclear supernatants were fractionated into total membranes and cytosol as described previously [36, 38]. Each sample (20 μg) was resolved by 8% SDS-PAGE and then transferred to PVDF membranes. Blocked membranes were probed overnight at 4°C with primary antibodies, washed with PBS-Tween, and then incubated with secondary antibody conjugated to HRP. Bands were developed using ECL reagents and quantified using an IS-1000 digital imaging system (Alpha Innotech, San Leandro, CA).
In vitro PI3K assay
To measure total class IA PI3K activity, BMMs (2 × 106 cells/60 mm dish) were treated with 0 –100 nM WTM and then infected with Hp or IgG particles, as described above. Thereafter, cells were washed twice with ice-cold PBS containing 1 mM Na3VO4 and scraped into lysis buffer (1% Triton X-100, 20 mM Tris-HCl, pH 8, 137 mM NaCl, 10% glycerol, 2 mM EDTA, 1 mM Na3VO4, 5 mM NaF, 1 mM phenylmethylsulfonyl fluoride, 50 μg/ml leupeptin, 10 μg/ml pepstatin, and 0.09 tyrpsin inhibitor unit aprotinin). Samples were normalized for protein content and then incubated at 4°C with anti-p85 antiserum. Immune complexes were collected on protein A sepharose beads and washed twice with lysis buffer and three times with 10 mM Tris-HCl, pH 7.4. PI3K activity was measured using an in vitro kinase assay that detects conversion of phosphatidylinositol into phosphatidylinositol 3-phosphate [PI(3)P] in the presence of [γ-32P]adenosine 5′-triphosphate (ATP) [39]. Extracted lipids were resolved by thin-layer chromatography on oxalate-impregnated silica gel plates using CHCl3:MeOH:H2O:NH4OH (18:14:3:1, v/v). PI(3)P was detected by autoradiography and quantified by liquid scintillation counting.
To quantify PI3K activity at the membrane during phagocytosis, BMMs were infected with Hp or IgG particles for 0 –15 min at 37°C, washed with cold PBS-Na3VO4, and disrupted by nitrogen cavitation, and postnuclear supernatants were fractionated to obtain membranes and cytosol. Isolated membranes were solubilized with lysis buffer, and PI3K activity associated with p85 immune complexes was measured as described above.
Antisense treatment
Phosphorothioate-modified sense (5′-atg-agt-gct-gag-ggg-tac-ca-3′) and anti-sense (5′-tgg-tac-ccc-tca-gca-ctc-at-3′) oligonucleotides directed against the transcription start site of murine p85 [40] were synthesized, and high-pressure liquid chromatography was purified by Sigma Genosys (Haverhill, UK). BMMs were left untreated or exposed to 25 μM sense or antisense oligos for 48 h at 37°C. Thereafter, Hp phagocytosis was assessed by differential staining, and macrophage p85 content was quantified by immunoblotting of cell lysates.
Other methods
Protein concentrations were determined using Coomassie-Plus or BCA kits (Pierce). IgG-Z particles were prepared as directed by Molecular Probes. Statistical significance was determined by Student’s t-test for paired samples or by ANOVA. P < 0.05 was considered significant.
RESULTS
Active class IA PI3K accumulate on type I Hp phagosomes
We and others [6, 7, 36] have shown that ulcerogenic type I Hp actively retard their entry into macrophages. Accordingly, local actin polymerization begins ~4 min after Hp attachment, and complete engulfment occurs after 15–20 min [6, 36]. To assess whether Hp uptake was accompanied by activation of class IA PI3Ks in primary murine macrophages, we determined first whether infection triggered PI3K translocation to the membrane. Using pAb to the PI3K regulatory subunit p85, we found that 4.4 ± 1.6% of class IA PI3K was associated with the membrane in resting BMMs, and infection with Hp 11637 increased membrane p85 levels 4.8 ±1.2-fold (n=3, P=0.032; Fig. 1A). Moreover, membrane p85 in Hp-infected cells exhibited slightly slower electrophoretic mobility compared with the soluble protein (Fig. 1A, arrowheads).
Fig. 1.
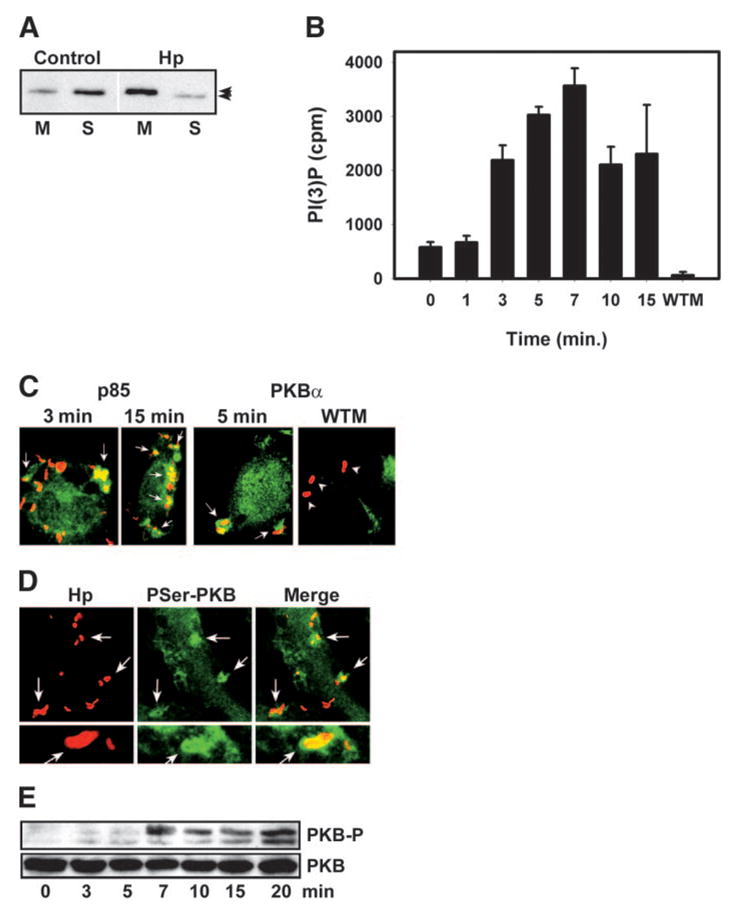
Class IA PI3K is activated at sites of Hp uptake. (A and B) BMMs were infected with Hp 11637 for 0 –15 min, and postnuclear supernatants were fractionated into membranes and cytosol. (A) p85 immunoblots of membrane (M) and cytosol (S) fractions from resting (Control) and Hp-infected BMMs. Arrowheads indicate mobility shift of membrane p85 in Hp-infected cells. (B) PI3K activity at the membrane, as judged by in vitro kinase assays of p85 immune complexes. PI(3)P synthesized is shown as average counts per min (cpm) ±SD (n = 3). Pretreatment with 100 nM WTM abrogated PI3K activity (7 min infection). (C) Confocal sections of PMφ infected with Hp 11637 for 3–15 min show Hp (red) and p85 or PKBα (green). Arrows indicate enrichment of p85 and PKBα on forming phagosomes. Pretreatment with 100 nM WTM prevented translocation of PKBα to the membrane beneath Hp (far right, arrowheads). (D) Confocal sections of BMMs infected with Hp 11916 for 7 min show active P-Ser473 PKB (green) and Hp (red). Arrows indicate phagosomes. (E) BMMs were infected with Hp 11637 for 0 –20 min. Immunoblots of cell lysates show total PKB and PKB phosphorylated on Thre308 (PKB-P).
Translocation to the membrane positions PI3K in close proximity to substrate, and in this locale, enzyme activity is regulated by allosteric mechanisms. We measured PI3K activity directly at the membrane of Hp-infected BMMs using an established in vitro kinase assay in which p85 immune complexes are incubated with [γ-32P]ATP and phosphatidylinositol [39]. By this assay, PI3K activity increased significantly 3 min after Hp binding, peaked at 7 min, and remained elevated for at least 15 min (Fig. 1B). It is important that the 6.1 ± 0.2-fold increase in 3′-phosphoinositide synthesis observed at 7 min was ablated by pretreatment of BMMs with 100 nM WTM (Fig. 1B). Comparable data were obtained for BMMs infected with Hp strains 11916 and DT61A (not illustrated). These data demonstrate that class IA PI3Ks are activated coincident with Hp internalization.
Next, we used confocal microscopy to localize PI3K and its lipid products in infected macrophages. Endogenous PI3K was detected using affinity-purified anti-p85 IgG [30]. As shown in Figure 1C, p85 was distributed diffusely throughout the cytoplasm and accumulated in the membrane specifically at sites of Hp uptake. PKB (also called Akt) is a PI3K effector that is recruited to membranes via the affinity of its pleckstrin homology (PH) domain for PI(3,4,5)P3 and phosphatidylinositol-3,4-bisphosphate [41, 42]. Therefore, local accumulation of PKB or a tagged PH domain construct is used to define sites of class IA PI3K activity in whole cells [43– 45]. During Hp infection, we found that endogenous PKBα accumulated on forming phagosomes in control cells but not in macrophages pretreated with 100 nM WTM (Fig. 1C). Thus, PKBα was recruited to sites of Hp engulfment in a PI3K-dependent manner.
After binding, PI(3,4,5)P3 PKB is activated by phosphorylation. Specifically, Thr308 is phosphorylated by 3-phospho-inositide-dependent protein kinase-1, and Ser473 is phosphorylated by DNA-dependent protein kinase [46, 47]. Using mAb specific for phospho-Ser473 and confocal microscopy, we found that active PKB was enriched on forming Hp phagosomes (Fig. 1D). At the same time, immunoblotting demonstrated that phosphorylation of Thr308 increased markedly~7 min after Hp binding and continued to increase for at least 20 min (Fig. 1E). Our finding that PKB phosphorylation was delayed slightly relative to PI3K activation (compare Fig. 1, B and E) is consistent with the fact that PKB and 3-phosphoino-sitide-dependent protein kinase-1 are PI3K effectors. Altogether, the biochemical and microscopy data demonstrate that PI3K is activated specifically at sites of Hp engulfment and indicate that PI(3,4,5)P3 is generated in amounts sufficient to induce membrane translocation and activation of PKBα.
Class IA PI3Ks are required for Hp phagocytosis
Having established that PI3K was activated by Hp, we next asked whether synthesis of 3′-phosphoinositides was essential for bacterial engulfment. BMMs were treated with increasing concentrations of WTM or LY294002, infected with Hp for 30 min, and ingestion of cell-associated bacteria was quantified by differential staining [36]. As shown in Figure 2A, WTM blocked Hp internalization in a dose-dependent manner. Bacterial engulfment was significantly inhibited in the presence of 10 nM WTM (67.8 ± 12.3% of control, P = 0.032, n = 4), profoundly impaired by 50 nM WTM (30.7 ± 6.4% of control, P = 0.001, n = 4), and nearly ablated by 100 nM WTM (5.6 ± 2.9% of control, P < 0.001, n = 4). As 20 –50 nM concentrations of WTM are highly specific for blockade of PI3K in intact cells [9], our data suggest that PI3K is a key regulator of Hp phagocytosis. In support of this notion, the structurally and mechanistically distinct PI3K inhibitor LY294002 [9, 18] also prevented Hp entry into macrophages (Fig. 2B).
Fig. 2.
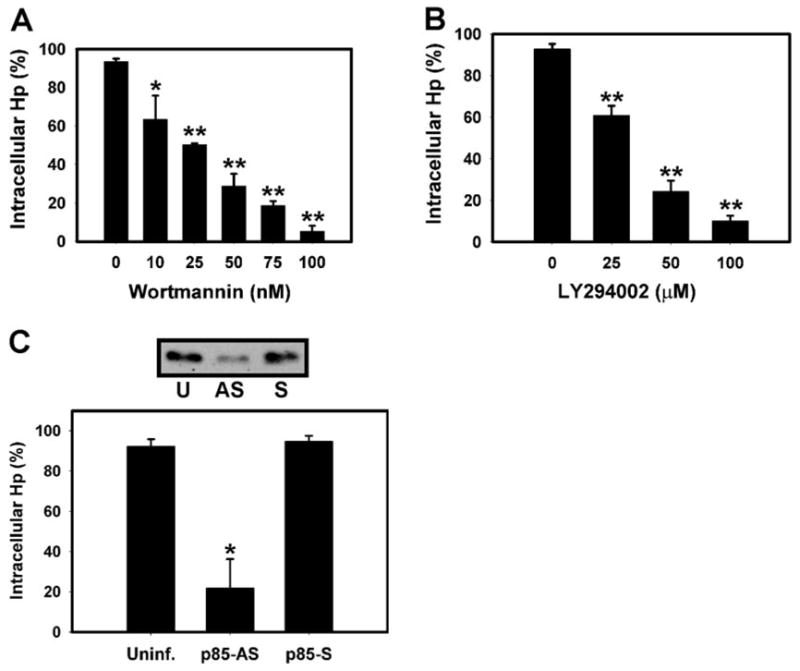
Class IA PI3Ks are essential for Hp phagocytosis. (A) BMMs pretreated with 0 –100 nM WTM were infected with Hp 11637 for 30 min, and Hp internalization was quantified by differential staining. Data are the average ± SD of three determinations performed in triplicate. *, P = 0.032, and **, P < 0.010, versus no drug control. (B) BMMs pretreated with 0 –100 μM LY294002 were infected with Hp, and phagocytosis was quantified as in A. Data are the average ± SD of three experiments performed in triplicate. (C, upper panel) Immunoblot, demonstrating the effect of sense or antisense p85 oligos on BMM p85 content. Data shown are representative of three independent experiments. U, Untreated BMMs; AS, antisense oligo-treated; S, sense oligo-treated. (Lower) Internalization of Hp 11637 by control (Uninf.) or antisense (p85-AS)- or sense (p85-S)-treated BMMs. Data shown are the average ± SD of three independent experiments. *, P = 0.008, versus untreated or p85-S controls.
As WTM and LY294002 inhibit class IA, class IB, and class III PI3Ks with equal potency [9], we tested directly the requirement for class IA PI3Ks in Hp phagocytosis by depleting cells of p85α using antisense oligonucleotides [40]. As shown in Figure 2C, antisense treatment reduced p85α levels ~70% and inhibited Hp phagocytosis by 76.6 ± 15.9% (n = 3, P = 0.008). In contrast, sense oligonuceotides were without effect. These data indicate that class IA PI3Ks are essential for Hp phagocytosis.
Blockade of Hp uptake by WTM parallels loss of phagosomal PKB
To assess more precisely the role of PI3K in Hp entry into macrophages, we compared the ability of increasing concentrations of WTM to inhibit PI3K activity, impair recruitment of PKBα to the membrane, and prevent phagocytosis. Cells stimulated with IgG-Z were used as a positive control. As expected, WTM inhibited BMM class IA PI3K activity in a dose-dependent manner (Fig. 3, A and B). In macrophages incubated with IgG-Z, particle engulfment and local accumulation of PKBα fell in parallel with PI3K activity [WTM inhibitory concentration of 50% (IC50) = 7 nM, 9 nM, and 11 nM, respectively; Fig. 3A]. Specific effects of WTM on PKBα recruitment to forming IgG-Z phagosomes are illustrated in Figure 3C. In control BMMs, PKBα was present throughout phagocytic cups. As WTM concentrations increased, fewer IgG-Z recruited PKBα (Fig. 3A), and where PKB was present, its appearance changed from full cups (0 –10 nM WTM) to pedestals (25 nM WTM) and then to small foci (50 –100 nM WTM; Fig. 3C).
Fig. 3.

Effect of WTM on PI3K activity, phagocytosis, and local accumulation of PKBα. (A and B) Macrophages were treated with 0 –100 nM WTM and then infected with IgG-Z (A) or Hp 11637 (B). Thereafter, cells were lysed, and total class IA PI3K activity was quantified by in vitro kinase assays of p85 immunoprecipitates (PI3K act.;●). Particle engulfment (Phago.;○;) was assessed by differential staining, and recruitment of PKBα; to forming phagosomes (PKB+; ▼) was scored using confocal microscopy. PI3K activity is normalized to cells infected in the absence of WTM. Phagocytosis and PKBα recruitment are shown as a percentage of cell-associated particles. In all cases, the data shown are the average ± SD of three to four independent experiments. *, P < 0.010, for Hp phagocytosis and PKBα recruitment versus PI3K activity at 25 nM WTM. (C and D) Effect of WTM on PKBα association with IgG-Z (C) and Hp (D) phagosomes. PMφ were treated with 0 –100 nM WTM and then infected with IgG-Z (C) or Hp (D). Arrowheads in each confocal section indicate areas of PKBα accumulation.
Surprisingly, we observed something somewhat different in Hp-infected cells (Fig. 3, B and D). In support of the notion that Hp entry required 3′-phosphoinositides, bacterial engulfment coincided precisely with local accumulation of PKBα (Fig. 3B). However, in the presence of low concentrations of WTM, Hp phagocytosis and PKBα translocation declined more slowly than PI3K activity (IC50 = 27 nM, 28 nM, and 11 nM, respectively). This discordance was most pronounced in BMMs treated with 25 nM WTM (Fig. 3B, arrow), wherein ~26% of total macrophage class IA PI3K activity supported ingestion of ~53% of Hp in PKBα-positive compartments (Fig. 3, B and D). It is remarkable that the few Hp that were ingested by BMM in the presence of 50 –75 nM WTM also recruited PKBα to forming phagosomes (Fig. 3, B and D). These data demonstrate that local accumulation of PI(3,4,5)P3 is required for phagocytosis of Hp and IgG-Z. However, under conditions where PI3K activity is limiting, forming Hp phagosomes accumulate 3′-phosphoinositides more efficiently than IgG-Z compartments.
Hp phagosomes do not acquire PI3KC2α, PTEN, or SHIP
PI3KC2α is at least tenfold less sensitive to inhibition by WTM than other PI3K isoforms [48, 49]. Our finding that 25 nM WTM did not ablate Hp phagocytosis suggested that PI3KC2αs might contribute to 3′-phosphoinositide synthesis at sites of bacterial uptake. Therefore, we localized this enzyme in BMMs using confocal microscopy. Unexpectedly, we found that PI3KC2α accumulated on phagosomes containing IgG-Z yet was undetectable on phagosomes containing Hp (Fig. 4, A and D).
Fig. 4.

PI3KC2, PTEN, and SHIP do not accumulate on Hp phagosomes. (A) Forming IgG-Z phagosomes recruit PI3KC2 (arrowhead, left panel), but Hp phagosomes do not (right panel, Hp, red; PI3KC2, green). (B) Confocal sections of PMφ show PTEN or SHIP (green) and Hp (red). Arrowheads, Rare SHIP-positive Hp phagosomes. (C) Time course of PTEN and SHIP accumulation on IgG-Z phagosomes (arrowheads). (D) Differential recruitment of PI3KC2, PTEN, and SHIP to IgG-Z and Hp phagosomes. Data are the average ± SD of three independent experiments performed in triplicate. *, P ≤ 0.003, for Hp versus IgG-Z. p’somes, Phagosomes.
Alternatively, slow phosphoinositide catabolism may allow Hp phagosomes to accumulate or retain PI(3,4,5)P3 and PKBα. PI3K-dependent signaling is terminated by the combined actions of the 3′-phosphoinositide phosphatase PTEN and the 5′-phosphoinositide phosphatase SHIP [9]. In macrophages, overexpression of either enzyme impairs internalization of IgG-opsonized particles [21, 50]. Conversely, phagocytosis is enhanced in macrophages from PTEN or SHIP null mice [21, 51]. In support of these data, we found that PTEN and SHIP associated with phagosomes containing IgG-Z (Fig. 4, C and D). PTEN was recruited to forming (1–3 min) phagosomes but was no longer detectable at the membrane, once particle ingestion was complete (Fig. 4C, 10 min). SHIP also accumulated on IgG-Z phagosomes (Fig. 4C); however, SHIP differed from PTEN in one respect. As particles were drawn into the macrophage, SHIP immunoreactivity accumulated at the phagosome apex before disappearing completely from the membrane.
In marked contrast to IgG-Z, PTEN was detected on less than 5% of Hp phagosomes (P < 0.001, n = 3), and only a small fraction of these structures accumulated SHIP [13.7 ± 2.2% (n = 3), P = 0.003; Fig. 4, B and D]. Taken together, the data demonstrate significant differences in the composition of Hp and IgG-Z phagosomes. Moreover, our findings suggest a model in which the paucity of SHIP and PTEN at the membrane may allow Hp phagosomes to accumulate the lipid products of PI3K more efficiently under basal conditions and in cells treated with small amounts of WTM.
PI3K regulates Hp-induced actin polymerization
During FcγR-mediated phagocytosis, PI3K activity is required for particle engulfment but not the formation of F-actin-rich phagocytic cups [14, 15, 44]. To assess whether WTM blocked Hp phagocytosis at a similar stage, BMMs were infected with IgG-Z or Hp in the presence and absence of 100 nM WTM, stained with rhodamine-phalloidin to detect F-actin, and examined by confocal microscopy. In good agreement with the results of previous studies [14, 15, 44], F-actin was enriched on forming phagocytic cups containing IgG-Z in control and WTM-treated BMMs (Fig. 5, A and B). By contrast, WTM prevented actin polymerization in the vicinity of bound Hp [88.6 ± 9.4% inhibition (n = 4), P = 0.014; Fig. 5, A and B]. These data demonstrate that PI3K regulates actin rearrangements at sites of Hp uptake.
Fig. 5.

PI3K regulates actin polymerization at sites of Hp uptake. Control Mφ or cells treated with 100 nM WTM were infected with IgG-Z or Hp. F-actin was detected in fixed and permeabilized cells using rhodamine-phalloidin and confocal microscopy. (A) Unlike IgG-Z, forming Hp phagosomes accumulate F-actin in control macrophages but not in cells pretreated with WTM. Hp are green, and F-actin is red or gray. Arrows and arrowheads indicate actin-positive and actin-negative particles, respectively. (B) Differential effect of WTM on actin polymerization triggered by Hp and IgG-Z. Data shown are the average ± SD of three independent experiments performed in triplicate. *, P = 0.014, versus IgG-Z. Con, Control.
Hp and IgG beads activate distinct class IA PI3K isoforms
Our finding that Hp and IgG-coated particles differed in their ability to recruit PTEN and SHIP to forming phagosomes and the ability of WTM to block Hp-induced actin polymerization suggested that these two stimuli may activate distinct class IA PI3K isoforms. To test this hypothesis, we incubated BMMs with Hp or IgG-coated beads and used subcellular fractionation and immunoblotting to assess translocation of p110α, p110β, and p110δ to the membrane. In unstimulated BMMs, ~20% of p110α was associated with the membrane fraction, whereas p110β and p110δ were exclusively cytosolic (Fig. 6A and data not shown). Phagocytosis of IgG beads was accompanied by a significant increase in membrane p110α and p110β yet had no effect on p110δ; (Fig. 6A). These data lend support to the notion that PI3Kα regulates cell adhesion [43, 52, 53] and are consistent with the findings of Leverrier et al. [54], which indicate that PI3Kβ regulates FcγR-mediated phagocytosis in macrophages. Conversely, infection of BMMs with Hp triggered membrane translocation of p110δ as well as p110α and p110β (Fig. 6A). In addition, a faster-migrating band was reproducibly detected in p110α immunoblots of membranes prepared from Hp-infected cells (Fig. 6A, arrow). Whether this band is an artifact or reflects Hp-induced degradation of PI3Kα is unknown.
Fig. 6.
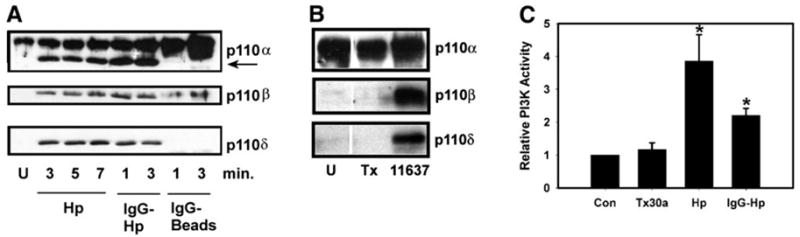
PI3Kδ translocates in response to type I Hp but not IgG beads. (A) Membrane translocation of class IA PI3K isoforms. BMMs were left uninfected (U) or incubated with type I Hp strain DT61A (Hp), IgG-opsonized DT61A (IgG-Hp), or IgG beads for the indicated times. Immunoblots of isolated membranes were probed with pAb to p110α, p110β, or p110δ as indicated. Data shown are from one experiment that is representative of four independent determinations. Note the faster-migrating band reactive with p110α pAb in Hp-infected cells (arrow). (B) Unlike type I Hp (strain 11637), type II Hp (strain Tx30a) does not trigger membrane translocation of p110β or p110δ. U, Uninfected BMM. Immunoblots are from one experiment representative of three. (C) Relative effect of type II Hp (Tx30a), type I Hp 11637 (Hp), and IgG-opsonized Hp 11637 (IgG-Hp) on class IA PI3K activity in BMM. Data are normalized to uninfected BMMs (516 ± 94 cpm) and are the average ± SD (n = 3– 4). *, P < 0.010, versus uninfected BMM. Con, Control.
Effect of opsonins on Hp-induced PI3K activity
It is well documented that the requirement for PI3K activity during FcγR-mediated phagocytosis depends on particle size but not particle load [15, 21, 55]. Consequently, WTM prevents ingestion of 4.5– 6 μm IgG beads but not internalization of smaller beads that approximate the size of most bacteria. Hp is ~0.5 μm × 2.5 μm, and we have shown previously that opsonization with IgG increases the efficiency of phagocytosis, such that 85 ± 4% of IgG-Hp are ingested within 3 min [36]. In accordance with their small size, rapid uptake of opsonized Hp is not prevented by pretreatment of macrophages with 100 nM WTM or 100 μM LY294002 [36]. Although ingestion of IgG-Hp is PI3K-independent, whether opsonins affect PI3K activation is unknown. We now show that coating Hp with IgG did not prevent membrane translocation of p110δ (Fig. 6A) or PI3K activation (Fig. 6C).
Rapid ingestion of type II Hp neither activates nor requires PI3K
Unlike type I Hp, less-virulent type II organisms are ingested quickly by macrophages in the absence of opsonins [6]. Interestingly, we found that infection of BMM with type II Hp (strain Tx30a) did not stimulate robust translocation of PI3K to the membrane or markedly enhance PI3K activity (Fig. 6, B and C). Total membrane p85 increased by only 12.8 ± 7.1% (n = 3, P = 0.548; not illustrated), and 3′-phosphoinositide synthesis increased by only 19.1 ± 11.2% (n = 3, P = 0.493; Fig. 6C). Indeed, Tx30a-infected cells resembled resting BMMs, as p110α was detected in the membrane fraction, but p110β and p110δ were not (Fig. 6B). Comparable data were obtained using the type II Hp strain MC123 (data not shown). Consistent with these findings, phagocytosis of type II Hp was not prevented by 100 nM WTM or 100 μM LY294002 [83.9 ± 1.3 and 841 ± 4.5% of control, respectively (n = 3), P ≥ 0.151]. Although we cannot exclude the possibility that type II Hp activated a form of PI3K that was not assayed here, the fact that delayed phagocytosis of type I Hp required class IA PI3Ks, whereas rapid uptake of type II Hp did not, suggests that these organisms were ingested by distinct mechanisms.
DISCUSSION
We and others [6, 7, 36] have shown previously that ulcerogenic type I Hp actively retard their entry into macrophages. Delayed phagocytosis has not been described for other microbes and is of interest, as the mechanism of entry has a profound effect on microbe fate [56, 57]. Accordingly, slowly ingested type I Hp resist intracellular killing, whereas rapidly ingested IgG-Hp and type II Hp do not [6, 36]. Herein, we explored the role of class IA PI3Ks in Hp phagocytosis, and our findings are significant in several respects. First, we show that class IA PI3Ks are activated specifically by type I Hp and demonstrate that the enzyme and its lipid products accumulate on forming phagosomes. Inhibitor treatments and antisense depletion studies indicate a requirement for class IA PI3Ks in phagocytosis of ulcerogenic bacteria. At the same time, we document important differences between Hp and IgG-opsonized particles with respect to PI3K isoform activation and effector recruitment. Most importantly, we show that PI3Ks serve distinct functions in these two systems. Downstream of FcγRs, 3′-phosphoinositides regulate phagosome closure but not local actin polymerization. Conversely, class IA PI3Ks regulate the actin cytoskeleton at sites of Hp engulfment. Thus, the results of this study document for the first time a new role for PI3K in macrophages and characterize further the unusual mechanism of phagocytosis that is defined by ulcerogenic Hp.
By combining subcellular fractionation, in vitro kinase assays, and confocal microscopy, we show here for the first time that class IA PI3Ks are activated sixfold by ulcergenic Hp and that the enzyme and its lipid products accumulate specifically on forming phagosomes. The time course of PI3K activation correlates precisely with the delayed and slow time course of Hp entry, which we described previously [36]. More importantly, our findings indicate a critical role for class IA PI3K in Hp engulfment. The PI3K inhibitors WTM and LY294002 have distinct structures and mechanisms of action [16 –18]. WTM is an irreversible inhibitor that forms an enamine with a key lysine residue, whereas LY294002 is a competitive inhibitor of ATP binding. LY294002 is more stable than WTM, but is 500- to 1000-fold less potent in vitro, and its efficacy in intact cells is reduced further by high intracellular concentrations of ATP. Although no pharmacological agent is absolutely specific for one target, at the doses used in this study, WTM and LY294002 are highly specific inhibitors of PI3K in intact cells [9]. Therefore, our finding that the effects of both agents were dose-dependent together with the fact that Hp internalization was reduced profoundly (~70% inhibition) in BMMs treated with 50 nM WTM or 50 7mu;M LY294002 indicate that PI3K activity is essential for Hp engulfment. In addition, a specific role for class IA PI3Ks in Hp phagocytosis is suggested by our finding that depletion of p85 prevented Hp uptake. Most importantly, our microscopy analyses demonstrate that blockade of phagocytosis correlated directly with loss of 3′-phosphoinositides in the membrane. Conversely, class IA PI3Ks were activated only slightly by type II Hp, and neither WTM nor LY294002 prevented rapid phagocytosis of these less-virulent organisms.
Prior to this study, a requirement for PI3K activity during phagocytosis has been described exclusively for large particles (≥4.5 μm diameter) such as IgG- or C3bi-opsonized particles, unopsonized zymosan, and apoptotic cells [14, 15, 19 –21, 54, 58]. As Hp are significantly smaller than these stimuli, we compared directly the ability of WTM to inhibit PI3K activity, local accumulation of PKBα, and phagocytosis. In so doing, we found that Hp had the unusual ability to accumulate 3′-phosphoinositides in BMM treated with low concentrations of WTM, but IgG-Z did not. As a result, slightly higher concentrations of WTM were required to inhibit Hp uptake. Specifically, PI3K activity was reduced by ~75% in BMM treated with 25 nM WTM. Under these conditions, 53–55% of Hp accumulated PI(3,4,5)P3 in the subjacent membrane and were ingested compared with only 25–33% of IgG-Z (P = 0.031). These data are reminiscent of studies in human neutrophils, which indicate that 20 nM WTM is sufficient to ablate the respiratory burst, whereas 75–100 nM WTM is required to inhibit degranulation [59]. The small size of Hp cannot, in and of itself, explain why forming Hp phagosomes accumulated the lipid products of PI3K under conditions where IgG-Z did not. Conversely, confocal analysis demonstrated that phagocytic cups containing IgG-Z (but not Hp compartments) acquired PI3KC2α, PTEN, and SHIP. The absence of PI3KC2α on Hp phagosomes indicates that WTM-resistant synthesis also cannot account for the ability of Hp to accumulate 3′-phosphoi-nositides. Rather, our data suggest that PI3K signaling on forming Hp phagosomes may be enhanced and/or sustained by the failure of these organelles to recruit PTEN and SHIP. This notion is not without precedent, as exclusion of PTEN from the leading edge of motile cells prolongs accumulation of PI(3,4,5)P3 and thereby amplifies downstream signaling [60]. How PI3K signaling is terminated during phagocytosis of Hp is unknown and whether these organisms activate other 5′-phosphatases remains to be determined.
The most striking finding of this study is that PI3K activity is essential for actin polymerization beneath attached Hp. As such, our data define a new role for PI3K in phagocytosis, which is distinct from its role as a regulator of membrane trafficking events that precede closure of phagosomes containing large particles. Five lines of evidence support the notion that PI3Ks serve distinct functions in these two systems. First, Hp is considerably smaller than other particles, whose uptake requires 3′-phosphoinositides. Second, engulfment of most particles is rapid, yet type I Hp are ingested slowly [6, 36, 37, 61]. Third, Hp trigger membrane translocation of PI3Kδ, but IgG beads do not. Fourth, IgG-opsonized Hp activate PI3Ks, but their internalization is not prevented by PI3K inhibitors (ref. [36] and this study). Fifth, Hp and large particles activate distinct PI3K effectors. Amphiphysin IIm, dynamin, and myosin X control extension of pseudopodia around zymosan and IgG particles [19, 58, 62]. By contrast, Hp activates the PI3K effector PKCζ, and both enzymes are required for actin polymerization and bacterial engulfment (ref. [36] and this study). In future studies, it will be important to define how PI3K and PKCζ modulate the cytoskeleton and to determine which class IA PI3K isoforms are essential for this process.
Also of interest is the ability of Hp phagosomes to recruit and retain coronin [8], which is an actin-binding protein that is recruited to phagosomes in a PI3K-dependent manner [63]. Typically, coronin associates with forming phagosomes and is shed along with F-actin after particle internalization is complete [63, 64]. By contrast, retention of coronin on phagosomes containing Hp and Mycobacteria appears to play a role in blockade of phagosome-lysosome fusion [8, 65, 66]. Therefore, activation of PI3Ks by Hp may have effects on macrophage function that extend beyond modulation of bacterial engulfment. This notion is consistent with our finding that rapid phagocytosis of type II Hp is PI3K-independent and the fact that these less-virulent organisms neither retain coronin at the phagosome nor evade intracellular killing [6, 8].
Acknowledgments
This work was supported by funds from the Public Health Service [National Institutes of Health (NIH)/National Institute of Allergy and Infectious Diseases R01AI43617] and the Department of Veterans Affairs (Merit Review Award) to L-A. H. A. L. M. W. was supported by a NIH training grant awarded to the Division of Infectious Diseases, Department of Medicine, at the University of Iowa. We thank Dr. Marcus Thelen at the Institute for Research in Biomedicine (Bellinzona, Switzerland) for the generous gift of WTM and antibodies to p85 and PKBα.
References
- 1.Covacci A, Telford JL, Del Giudice G, Parsonnet J, Rappuoli R. Helicobacter pylori virulence and genetic geography. Science. 1999;284:1328 –1333. doi: 10.1126/science.284.5418.1328. [DOI] [PubMed] [Google Scholar]
- 2.Xiang Z, Censini S, Bayeli PF, Telford JL, Figura N, Rappuoli R, Covacci A. Analysis of expression of CagA and VacA virulence factors in 43 strains of Helicobacter pylori reveals that clinical isolates can be divided into two major types and that CagA is not necessary for expression of the vacuolating cytotoxin. Infect Immun. 1995;63:94 –98. doi: 10.1128/iai.63.1.94-98.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Allen LAH. Modulating phagocyte activation: the pros and cons of Helicobacter pylori virulence factors. J Exp Med. 2000;191:1451–1454. doi: 10.1084/jem.191.9.1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Andersen LP, Blom J, Nielsen H. Survival and ultrastructural changes of Helicobacter pylori after phagocytosis by human polymorpho-nuclear phagocytes and monocytes. APMIS. 1993;101:61–72. [PubMed] [Google Scholar]
- 5.Engstrand L, Graham D, Scheynius A, Genta RM, ElZaatari F. Is the sanctuary where Helicobacter pylori avoids antibacterial treatment intracellular? Am J Clin Pathol. 1997;108:504 –509. doi: 10.1093/ajcp/108.5.504. [DOI] [PubMed] [Google Scholar]
- 6.Allen LAH, Schlesinger LS, Kang B. Virulent strains of Helicobacter pylori demonstrate delayed phagocytosis and stimulate homotypic phagosome fusion in macrophages. J Exp Med. 2000;191:115–127. doi: 10.1084/jem.191.1.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ramarao N, Meyer TF. Helicobacter pylori resists phagocytosis by macrophages: quantitative assessment by confocal microscopy and fluorescence-activated cell sorting. Infect Immun. 2001;69:2604 –2611. doi: 10.1128/IAI.69.4.2604-2611.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zheng PY, Jones NL. Helicobacter pylori strains expressing the vacuolating cytotoxin interrupt phagosome maturation in macrophages by recruiting and retaining TACO (coronin 1) protein. Cell Microbiol. 2003;5:25– 40. doi: 10.1046/j.1462-5822.2003.00250.x. [DOI] [PubMed] [Google Scholar]
- 9.Vanhaesebroeck B, Leevers SJ, Ahmadi K, Timms J, Katso R, Driscoll PC, Woscholski R, Parker PJ, Waterfield MD. Synthesis and function of 3-phosphorylated inositol lipids. Annu Rev Biochem. 2001;70:535– 602. doi: 10.1146/annurev.biochem.70.1.535. [DOI] [PubMed] [Google Scholar]
- 10.Toker A, Cantley LC. Signaling through the lipid products of phosphoinositide-3-OH kinase. Nature. 1997;387:673– 676. doi: 10.1038/42648. [DOI] [PubMed] [Google Scholar]
- 11.Vanhaesebroeck B, Welham MJ, Kotani K, Stein R, Warne PH, Zvelebil MJ, Higashi K, Volinia S, Downward J, Waterfield MD. p110δ,a novel phosphoinositide 3-kinase in leukocytes. Proc Natl Acad Sci USA. 1997;94:4330 – 4335. doi: 10.1073/pnas.94.9.4330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fruman DA, Meyers RE, Cantley LC. Phosphoinositide kinases. Annu Rev Biochem. 1998;67:481–507. doi: 10.1146/annurev.biochem.67.1.481. [DOI] [PubMed] [Google Scholar]
- 13.Ninomiya N, Hazeki K, Fukui Y, Seya T, Okada T, Hazeki O, Ui M. Involvement of phosphatidylinositol 3-kinase in Fc γ receptor signaling. J Biol Chem. 1994;269:22732–22737. [PubMed] [Google Scholar]
- 14.Araki N, Johnson MT, Swanson JA. A role for phosphoinositide 3-kinase in the completion of macropinocytosis and phagocytosis by macrophages. J Cell Biol. 1996;135:1249 –1260. doi: 10.1083/jcb.135.5.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cox D, Tseng CC, Bjekic G, Greenberg S. A requirement for phosphatidylinositol 3-kinase in pseudopod extension. J Biol Chem. 1999;274:1240 –1247. doi: 10.1074/jbc.274.3.1240. [DOI] [PubMed] [Google Scholar]
- 16.Thelen M, Wymann MP, Langen H. Wortmannin binds specifically to 1-phosphatidylinositol 3-kinase while inhibiting guanine nucleotide-binding protein-coupled receptor signaling in neutrophil leukocytes. Proc Natl Acad Sci USA. 1994;91:4960 – 4964. doi: 10.1073/pnas.91.11.4960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wymann MP, Bulgarelli-Leva G, Zvelebil MJ, Pirola L, Vanhaese-broeck B, Waterfield MD, Panayotou G. Wortmannin inactivates phosphoinositide 3-kinase by covalent modification of Lys-802, a residue involved in the phosphate transfer reaction. Mol Cell Biol. 1996;16:1722–1733. doi: 10.1128/mcb.16.4.1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vlahos CJ, Matter WF, Hui KY, Brown RF. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002) J Biol Chem. 1994;269:5241–5248. [PubMed] [Google Scholar]
- 19.Gold ES, Underhill DM, Morrissette N, Guo J, McNiven MA, Aderem A. Dynamin 2 is required for phagocytosis in macrophages. J Exp Med. 1999;190:1849 –1856. doi: 10.1084/jem.190.12.1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leverrier Y, Ridley AJ. Requirement for Rho GTPases and PI 3-kinases during apoptotic cell phagocytosis by macrophages. Curr Biol. 2001;11:195–199. doi: 10.1016/s0960-9822(01)00047-1. [DOI] [PubMed] [Google Scholar]
- 21.Cox D, Dale BM, Kashiwada M, Helgason CD, Greenberg S. A regulatory role for src homology 2 domain-containing inositol 5′-phosphatase (SHIP) in phagocytosis mediated by Fc γ receptors and complement receptor 3 (α (M)β(2); CD11b/CD18) J Exp Med. 2001;193:61–71. doi: 10.1084/jem.193.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Celli J, Olivier M, Finlay BB. Enteropathogenic Escherichia coli mediates antiphagocytosis through the inhibition of PI 3-kinase-dependent pathways. EMBO J. 2001;20:1245–1258. doi: 10.1093/emboj/20.6.1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Forsberg M, Blomgran R, Lerm M, Sarndahl E, Sebti SM, Hamilton A, Stendahl O, Zheng LM. Differential effects of invasion by and phagocytosis of Salmonella typhimurium on apoptosis in human macrophages: potential role of Rho-GTPases and Akt. J Leukoc Biol. 2003;74:620 – 629. doi: 10.1189/jlb.1202586. [DOI] [PubMed] [Google Scholar]
- 24.Crowley MT, Costello PS, Fitzer-Attas CJ, Turner M, Meng F, Lowell C, Tybulewicz VL, DeFranco AL. A critical role for syk in signal transduction and phagocytosis mediated by Fcγ receptors on macrophages. J Exp Med. 1997;186:1027–1039. doi: 10.1084/jem.186.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Khelef N, Shuman HA, Maxfield FR. Phagocytosis of wild-type Legionella pneumophila occurs through a wortmannin-insensitive pathway. Infect Immun. 2001;69:5157–5161. doi: 10.1128/IAI.69.8.5157-5161.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim S, Watarai M, Makino S, Shirahata T. Membrane sorting during swimming internalization of Brucella is required for phagosome trafficking decisions. Microb Pathog. 2002;33:225–237. doi: 10.1006/mpat.2002.0531. [DOI] [PubMed] [Google Scholar]
- 27.Rupper AC, Rodriguez-Paris JM, Grove BD, Cardelli JA. p110-related PI 3-kinases regulate phagosome-phagosome fusion and phagosomal pH through a PKB/Akt-dependent pathway in Dictyostelium. J Cell Sci. 2001;114:1283–1295. doi: 10.1242/jcs.114.7.1283. [DOI] [PubMed] [Google Scholar]
- 28.Sly LM, Lopez M, Nauseef WM, Reiner NE. 1 α,25-Dihydroxyvitamin D-3-induced monocyte antimycobacterial activity is regulated by phosphatidylinositol 3-kinase and mediated by the NADPH-dependent phagocyte oxidase. J Biol Chem. 2001;276:35482–35493. doi: 10.1074/jbc.M102876200. [DOI] [PubMed] [Google Scholar]
- 29.Maira SM, Galetic I, Brazil DP, Kaech S, Ingley E, Thelen M, Hemmings BA. Carboxyl-terminal modulator protein (CTMP), a negative regulator of PKB/Akt and v-Akt at the plasma membrane. Science. 2001;294:374 –380. doi: 10.1126/science.1062030. [DOI] [PubMed] [Google Scholar]
- 30.Didichenko SA, Tilton B, Hemmings BA, Ballmer-Hofer K, Thelen M. Constitutive activation of protein kinase B and phosphorylation of p47phox by a membrane-targeted phosphoinositide 3-kinase. Curr Biol. 1996;6:1271–1278. doi: 10.1016/s0960-9822(02)70713-6. [DOI] [PubMed] [Google Scholar]
- 31.Kodama K, Fujioka T, Ito A, Kubota T, Murakami K, Nasu M. Expression of vacuolating cytotoxin in clinical isolates of Helicobacter pylori. J Gastroenterol. 1996;31 (Suppl 9):9 –11. [PubMed] [Google Scholar]
- 32.Ghiara P, Marchetti M, Blaser MJ, Tummuru MK, Cover TL, Segal ED, Tompkins LS, Rappuoli R. Role of the Helicobacter pylori virulence factors vacuolating cytotoxin, CagA and urease in a mouse model of disease. Infect Immun. 1995;63:4154 – 4160. doi: 10.1128/iai.63.10.4154-4160.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Langenberg W, Rauws EA, Widjojokusumo A, Tytgat GN, Zanen HC. Identification of Campylobacter pyloridis isolates by restriction endonuclease DNA analysis. J Clin Microbiol. 1986;24:414 – 417. doi: 10.1128/jcm.24.3.414-417.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McClain MS, Cao P, Iwamoto H, Vinion-Dubiel AD, Szabo G, Shao Z, Cover TL. A 12-amino-acid segment, present in type s2 but not type s1 Helicobacter pylori VacA proteins, abolishes cytotoxin activity and alters membrane channel formation. J Bacteriol. 2001;183:6499 –6508. doi: 10.1128/JB.183.22.6499-6508.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ogura K, Takahashi M, Maeda S, Ikenoue T, Kanai F, Yoshida H, Shiratori Y, Mori K, Mafune KI, Omata M. Interleukin-8 production in primary cultures of human gastric epithelial cells induced by Helicobacter pylori. Dig Dis Sci. 1998;43:2738 –2743. doi: 10.1023/a:1026671815512. [DOI] [PubMed] [Google Scholar]
- 36.Allen LAH, Allgood JA. Atypical protein kinase C-ζ is essential for delayed phagocytosis of Helicobacter pylori. Curr Biol. 2002;12:1762–1766. doi: 10.1016/s0960-9822(02)01216-2. [DOI] [PubMed] [Google Scholar]
- 37.Allen LAH, Aderem A. Molecular definition of distinct cytoskeletal structures involved in complement- and Fc receptor-mediated phagocytosis in macrophages. J Exp Med. 1996;184:627– 637. doi: 10.1084/jem.184.2.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Allen LAH, Aderem A. Protein kinase C regulates MARCKS cycling between the plasma membrane and lysosomes in fibroblasts. EMBO J. 1995;14:1109 –1121. doi: 10.1002/j.1460-2075.1995.tb07094.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hmama Z, Knutson KL, Herrera-Velit P, Nandan D, Reiner NE. Monocyte adherence induced by lipopolysaccharide involves CD14, LFA-1, and cytohesin-1—regulation by Rho and phosphatidylinositol 3-kinase. J Biol Chem. 1999;274:1050 –1057. doi: 10.1074/jbc.274.2.1050. [DOI] [PubMed] [Google Scholar]
- 40.Skorski T, Kanakaraj P, Nieborowskaskorska M, Ratajczak MZ, Wen SC, Zon G, Gewirtz AM, Perussia B, Calabretta B. Phosphatidylinositol-3 kinase activity is regulated by bcr/abl and is required for the growth of Philadelphia chromosome-positive cells. Blood. 1995;86:726 –736. [PubMed] [Google Scholar]
- 41.Corvera S, Czech MP. Direct targets of phosphoinisitide 3-kinase products in membrane traffic and signal transduction. Trends Cell Biol. 1998;8:442– 446. doi: 10.1016/s0962-8924(98)01366-x. [DOI] [PubMed] [Google Scholar]
- 42.Meier R, Thelen M, Hemmings BA. Inactivation and dephosphorylation of protein kinase B α (PKB α) promoted by hyperosmotic stress. EMBO J. 1998;17:7294 –7303. doi: 10.1093/emboj/17.24.7294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Watton SJ, Downward J. Akt/PKB localization and 3′ phosphoinositide generation at sites of epithelial cell-matrix and cell-cell interaction. Curr Biol. 1999;9:433– 436. doi: 10.1016/s0960-9822(99)80192-4. [DOI] [PubMed] [Google Scholar]
- 44.Marshall JG, Booth JW, Stambolic V, Mak T, Balla T, Schreiber AD, Meyer T, Grinstein S. Restricted accumulation of phosphatidylinositol 3-kinase products in a plasmalemmal subdomain during Fc γ receptor-mediated phagocytosis. J Cell Biol. 2001;153:1369 –1380. doi: 10.1083/jcb.153.7.1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hoppe AD, Swanson JA. Cdc42, Rac1, and Rac2 display distinct patterns of activation during phagocytosis. Mol Biol Cell. 2004;15:3509 –3519. doi: 10.1091/mbc.E03-11-0847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PRJ, Reese CB, Cohen P. Characterization of a 3-phospho-inositide-dependent protein kinase which phosphorylates and activates protein kinase b-α. Curr Biol. 1997;7:261–269. doi: 10.1016/s0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- 47.Feng J, Park J, Cron P, Hess D, Hemmings BA. Identification of a PKB/Akt hydrophobic motif Ser-473 kinase as DNA-dependent protein kinase. J Biol Chem. 2004;279:41189 – 41196. doi: 10.1074/jbc.M406731200. [DOI] [PubMed] [Google Scholar]
- 48.Virbasius JV, Guilherne A, Czech MP. Mouse p170 is a novel phosphatidylinositol 3-kinase containing a C2 domain. J Biol Chem. 1996;271:13304 –13307. doi: 10.1074/jbc.271.23.13304. [DOI] [PubMed] [Google Scholar]
- 49.Brown RA, Domin J, Arcaro A, Waterfield MD, Shepherd PR. Insulin activates the α isoform of class II phosphoinositide 3-kinase. J Biol Chem. 1999;274:14529 –14532. doi: 10.1074/jbc.274.21.14529. [DOI] [PubMed] [Google Scholar]
- 50.Cao X, Wei G, Fang HQ, Guo JP, Weinstein M, Marsh CB, Ostrowski MC, Tridandapani S. The inositol 3-phosphatase PTEN negatively regulates Fc γ receptor signaling, but supports Toll-like receptor 4 signaling in murine peritoneal macrophages. J Immunol. 2004;172:4851– 4857. doi: 10.4049/jimmunol.172.8.4851. [DOI] [PubMed] [Google Scholar]
- 51.Kim JS, Peng XD, De PK, Geahlen RL, Durden DL. PTEN controls immunoreceptor (immunoreceptor tyrosine-based activation motif) signaling and the activation of Rac. Blood. 2002;99:694 – 697. doi: 10.1182/blood.v99.2.694. [DOI] [PubMed] [Google Scholar]
- 52.Gerszten RE, Friedrich EB, Matsui T, Hung RR, Li L, Force T, Rosenzweig A. Role of phosphoinositide 3-kinase in monocyte recruitment under flow conditions. J Biol Chem. 2001;276:26846 –26851. doi: 10.1074/jbc.M011235200. [DOI] [PubMed] [Google Scholar]
- 53.Lee JS, Hmama Z, Mui A, Reiner NE. Stable gene silencing in human monocytic cell lines using lentiviral-delivered small interference RNA. Silencing of the p110α isoform of phosphoinositide 3-kinase reveals differential regulation of adherence induced by 1α,25-dihydroxy-cholecalciferol and bacterial lipopolysaccharide. J Biol Chem. 2004;279:9379 –9388. doi: 10.1074/jbc.M310638200. [DOI] [PubMed] [Google Scholar]
- 54.Leverrier Y, Okkenhaug K, Sawyer C, Bilancio A, Vanhaesebroeck B, Ridley AJ. Class I phosphoinositide 3-kinase p110 β is required for apoptotic cell and Fc γ receptor-mediated phagocytosis by macrophages. J Biol Chem. 2003;278:38437–38442. doi: 10.1074/jbc.M306649200. [DOI] [PubMed] [Google Scholar]
- 55.Vieira OV, Botelho RJ, Rameh L, Brachmann SM, Matsuo T, Davidson HW, Schreiber A, Backer JM, Cantley LC, Grinstein S. Distinct roles of class I and class III phosphatidylinositol 3-kinases in phagosome formation and maturation. J Cell Biol. 2001;155:19 –25. doi: 10.1083/jcb.200107069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Aderem A, Underhill DM. Mechanisms of phagocytosis in macrophages. Annu Rev Inmmunol. 1999;17:593– 623. doi: 10.1146/annurev.immunol.17.1.593. [DOI] [PubMed] [Google Scholar]
- 57.Underhill DM, Ozinsky A. Phagocytosis of microbes: complexity in action. Annu Rev Immunol. 2002;20:825– 852. doi: 10.1146/annurev.immunol.20.103001.114744. [DOI] [PubMed] [Google Scholar]
- 58.Gold ES, Morrissette N, Underhill DM, Guo J, Bassetti M, Aderem A. Amphiphysin IIm, a novel amphiphysin II isoform, is required for macrophage phagocytosis. Immunity. 2000;12:285–292. doi: 10.1016/s1074-7613(00)80181-8. [DOI] [PubMed] [Google Scholar]
- 59.Sue-A-Quan AK, Fialkow L, Vlahos CJ, Schelm JA, Grinstein S, Butler J, Downey GP. Inhibition of neutrophil oxidative burst and granule secretion by wortmannin: potential role of MAP kinase and renaturable kinase. J Cell Physiol. 1997;172:94 –108. doi: 10.1002/(SICI)1097-4652(199707)172:1<94::AID-JCP11>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 60.Comer FI, Parent CA. PI 3-kinases and PTEN: how opposites chemoattract. Cell. 2002;109:541–544. doi: 10.1016/s0092-8674(02)00765-1. [DOI] [PubMed] [Google Scholar]
- 61.Allen LH, Aderem A. A role for MARCKS, the α isozyme of protein kinase C and myosin I in zymosan phagocytosis by macrophages. J Exp Med. 1995;182:829 – 840. doi: 10.1084/jem.182.3.829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cox D, Berg JS, Cammer M, Chinegwundoh JO, Dale BM, Cheney RE, Greenberg S. Mysoin X is a downstream effector of PI(3)K during phagocytosis. Nat Cell Biol. 2002;4:469 – 477. doi: 10.1038/ncb805. [DOI] [PubMed] [Google Scholar]
- 63.Didichenko SA, Segal AW, Thelen M. Evidence for a pool of coronin in mammalian cells that is sensitive to PI 3-kinase. FEBS Lett. 2000;485:147–152. doi: 10.1016/s0014-5793(00)02220-1. [DOI] [PubMed] [Google Scholar]
- 64.Allen LAH, DeLeo FR, Gallois A, Toyoshima S, Suzuki K, Nauseef WM. Transient association of the nicotinamide adenine dinucleotide phosphate oxidase subunits p47phox and p67phox with phagosomes in neutrophils from patients with X-linked chronic granulomatous disease. Blood. 1999;93:3521–3530. [PubMed] [Google Scholar]
- 65.Schuller S, Neefjes J, Ottenhoff T, Thole J, Young D. Coronin is involved in uptake of Mycobacterium bovis BCG in human macrophages but not in phagosome maintenance. Cell Microbiol. 2001;3:785–793. doi: 10.1046/j.1462-5822.2001.00155.x. [DOI] [PubMed] [Google Scholar]
- 66.Ferrari G, Langen H, Naito M, Pieters J. A coat protein on phagosomes involved in the intracellular survival of mycobacteria. Cell. 1999;97:435– 447. doi: 10.1016/s0092-8674(00)80754-0. [DOI] [PubMed] [Google Scholar]