

Abstract
Mitochondria are intimately involved in the generation of and defense against reactive oxygen species (ROS). Mitochondria are themselves targets of oxidative stress and also contribute to mechanisms by which oxidative stress–related signals control cell fate. Ethanol promotes oxidative stress, both by increasing ROS formation and by decreasing cellular defense mechanisms. These effects of ethanol are prominent in the liver, the major site of ethanol metabolism in the body. The question remains to what extent this contributes to ethanol-dependent tissue damage or the susceptibility of cells to other stressors. In this review, we consider how mitochondrial actions of ethanol influence oxidative stress management of liver cells. Mitochondrial electron transport constitutes the major intracellular source of ROS, and ethanol treatment imposes conditions that promote ROS formation by mitochondria, the effects of which may be enhanced by a decrease in mitochondrial oxidative stress defenses. A significant target of ethanol-related increases in oxidative stress is mitochondrial DNA. Ethanol-induced damage to mitochondrial DNA, if not adequately repaired, impairs mitochondrial function, which further increases oxidative stress in the cell, leading to a vicious cycle of accumulating cell damage that is more apparent with advancing age. Uncontrolled mitochondrial formation of ROS promotes the inappropriate activation of the mitochondrial permeability transition, increasing the sensitivity of cells to other proapoptotic or damage signals. In combination with ethanol-induced defects in mitochondrial function, these alterations may promote both apoptotic and necrotic cell death in response to otherwise benign or beneficial challenges and contribute to the onset or progression of alcohol-induced liver diseases.
Chronic excessive alcohol consumption causes injury to the liver and other tissues, but, despite intensive study, the factors that relate ethanol intake to the onset and progression of liver disease remain controversial. There is considerable evidence from both human and animal studies that alcohol consumption enhances oxidative stress, in liver as well as in other tissues. However, opinions are divided as to why oxidative stress develops, how it affects liver function, and what its relevance is for liver pathology.1–6 Mitochondria are recognized as the major intracellular source of reactive oxygen species (ROS), which is generated as a by-product of their major metabolic activity of cellular respiration.7 Mitochondrial constituents are themselves targets of ROS, and the mitochondrial management of oxidative stress has consequences both for cellular energy metabolism and for the processes that control the onset and progression of the cell death response, whether it results in apoptosis or necrosis. Hence, an understanding of how these complementary functions of mitochondria are affected by alcohol consumption may provide important insights into the mechanisms associated with liver damage. In this review, we will focus primarily on the liver, the predominant site of ethanol metabolism, but the factors that relate mitochondrial dysfunction to cellular oxidative stress may also have relevance to ethanol’s effects on other cells and tissues.
Ethanol, Mitochondrial Energy Metabolism, and Oxidative Stress
Mitochondria synthesize most of the adenosine triphosphate (ATP) needed by mammalian cells. Oxidation of various substrates results in the formation of reduced nicotinamide adenine dinucleotide (NADH) or reduced ubiquinone, which are oxidized through the electron transport chain in the inner mitochondrial membrane. The associated extrusion of protons from the mitochondrial matrix generates a pH gradient and an electrical potential difference across the inner mitochondrial membrane, which together constitute the proton motive force, Δp (Figure 1).8 This is the driving force for ATP synthesis through the proton pumping F1F0 adenosine triphosphatase (ATP synthase), and for transport of ions, such as Ca2+, or the energy-linked pyridine nucleotide transhydrogenase, the enzyme responsible for keeping mitochondrial nicotinamide adenine dinucleotide phosphate (NADP) in a highly reduced state. Under normal conditions, the rate of oxidation of NADH through the electron transport chain is often constrained by the cellular demand for ATP. The cell employs regulatory mechanisms to link the demand for ATP to the oxidation of substrates, e.g., by regulation of the activity of Krebs cycle dehydrogenases that control the supply of NADH to the electron transport chain.9–11 However, there are conditions in which it is beneficial for the cell to relax the strict coupling of substrate oxidation and ATP synthesis.12 This may explain the widespread occurrence of uncoupling proteins, such as uncoupling protein 2 (UCP-2) and UCP-3,13,14 which dissipate Δp by permitting the controlled leakage of protons across the mitochondrial membrane and thereby allow electron transport to occur without the constraint of ATP utilization.
Figure 1.
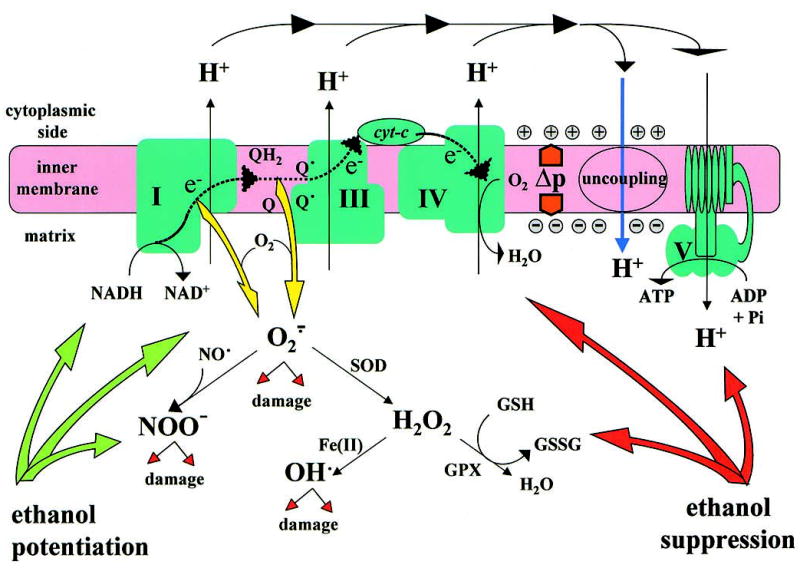
Formation of ROS as a by-product of mitochondrial oxidative phosphorylation. Electron (e−) flow through electron transport complex I, III, and IV (indicated by the dotted line) is coupled to translocation of protons to generate the proton motive force, Δp, which can drive ATP synthesis or be dissipated by enhancing inner membrane proton leak through uncoupling. Overflow of electrons occurs with 1-electron reduction of O2 at the level of Complex I or Complex III, resulting in formation of superoxide, O2., which is converted to H2O2 by superoxide dismutase (SOD) or reacts with NO• to form peroxynitrite, NOO−. H2O2 is a potential source of highly reactive hydroxyl radicals and its degradation requires glutathione peroxidase (GPX). ROS can damage mitochondrial constituents, such as phospholipids, proteins, and mitochondrial DNA (mtDNA). Ethanol increases redox pressure through NADH and promotes formation of ROS. Chronic ethanol treatment may also cause loss of mitochondrial glutathione and inactivation of GPX and respiratory complexes.
Metabolism of ethanol through alcohol dehydrogenase (ADH) generates cytosolic NADH, which is oxidized indirectly by mitochondrial electron transport depending on hydrogen shuttling mechanisms that involve metabolite carriers in the inner membrane.15 Further oxidation of the resulting acetaldehyde is mediated predominantly by the mitochondrial low Km aldehyde dehydrogenase, which also generates NADH for oxidation in the mitochondria. Thus, both steps depend on mitochondrial electron transport. Even at relatively modest circulating ethanol levels, a substantial reduction of both cytosolic and mitochondrial nicotinamide adenine dinucleotide (NAD) occurs in the liver, evidence of the strong reducing pressure exerted by its metabolism, which bypasses the metabolic control on Krebs cycle dehydrogenases. This reducing pressure poses an acute metabolic challenge for energy metabolism in the liver (and to a lesser extent in other tissues). Yet, evidence has accumulated over the past decade that much of the damaging effects of ethanol, rather than being reductive, may reflect oxidative stress on the liver and other tissues. Although ethanol oxidation through cytochrome P450 isoforms, notably cytochrome P450 2E1 (CYP2E1), contributes to the formation of ROS, mitochondrial metabolism is also likely to play a major role in this, at least in part as a direct response to the increased reduction pressure.
As illustrated in Figure 1, a fraction (1%–2%) of electrons passing through the respiratory chain is diverted to generate ROS, mostly superoxide (O2.), and this constitutes the major intracellular source of oxidative stress in many cell types.7 The major sites of ROS formation in the electron transport chain are the NADH dehydrogenase complex and the cytochrome b-c1 complex.16,17 The rate of ROS formation increases with higher Δp and, therefore, it depends on the respiratory conditions, notably on the substrate and O2 supply and the electron transport rate.12 Conditions that increase the supply of mitochondrial NAD(P)H and enhance the reducing pressure on the electron transport chain without increasing the rate of respiration promote the formation of O2. through the electron transport chain.7,12 It is not surprising, therefore, that both acute and chronic ethanol treatment enhance mitochondrial ROS formation in liver cells, where most ethanol oxidation takes place.18–20 Also not unexpectedly, the liver responds to a prolonged ethanol exposure by enhancing the rate of O2 uptake.21,22 Several mechanisms may be involved. There is some evidence of an increased respiratory activity, either by enhanced ATP utilization,21 or as a result of the induction of an uncoupling protein, such as UCP-2, which enables electron transport without a corresponding increase in ATP utilization.23,24 However, this is likely to represent only a minor contribution to the adaptation of the tissue to ethanol, and the predominant proportion of increased O2 uptake in the liver represents the activation of ethanol metabolism through alternative pathways. These include ethanol oxidation by the cytochrome P450 isoform CYP2E1, which is up-regulated by chronic ethanol intake,25 and by catalase, which can provide a significant ethanol oxidation pathway under conditions in which peroxisomal oxidation of fatty acids provides the necessary H2O2 for that reaction.26 Importantly, the increased utilization of O2 through these pathways competes with mitochondrial electron transport, and this may lead to conditions in which a localized and transient hypoxia develops in the tissue, particularly in the pericentral (downstream) region of the liver acinus.27,28 Such transient conditions of hypoxia and reoxygenation would further enhance ROS formation through the respiratory chain.
Furthermore, chronic ethanol treatment also affects mitochondrial oxidative phosphorylation in the liver by suppressing the synthesis of protein subunits that are encoded on mitochondrial DNA (mtDNA).29,30 These include subunits of the main respiratory complexes, NADH dehydrogenase (Complex I), cytochrome b-c1 (Complex III), and cytochrome oxidase (Complex IV), as well as the ATP synthase complex (Complex V). The lower capacity for oxidative phosphorylation further stresses the balance between the reducing pressure imposed by ethanol and the capacity for oxidative phosphorylation. These are precisely the conditions that would promote mitochondrial O2. formation.
Other, non-mitochondrial mechanisms contribute to an ethanol-induced increase in ROS formation. CYP2E1, the predominant cytochrome P450 isoform to oxidize ethanol, is prone to radical formation, including hydroxyethyl radicals.31 This pathway is active mostly in the endoplasmic reticulum, although a mitochondrially localized CYP2E1 isoform has recently been reported.32 Extracellular mechanisms that generate ROS may also be promoted by ethanol treatment, e.g., through an oxidative burst mediated by reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase in activated Kupffer cells or infiltrating neutrophils.33,34
ROS are potentially damaging to cellular constituents, and the cell employs a battery of defense mechanisms to keep their accumulation under control. Predominant are the superoxide dismutases (SODs) and glutathione peroxidases (GPXs). SOD converts O2. to H2O2, a substrate for peroxisomal catalase or for GPXs, which are found both in the cytosol and in the mitochondrial matrix. However, in the presence of Fe(II), H2O2 generates the highly reactive hydroxyl radical, OH•, and under conditions of increased Fe(II) in mitochondria, SOD activity may, therefore, contribute to increased free radical damage.17 Interestingly, a recent report35 showed that the delivery of the mitochondrial isoform of SOD by recombinant adenovirus infection prevented liver injury in rats receiving ethanol intragastrically. Chronic ethanol treatment is often associated with deregulation of iron metabolism in the liver,36 but whether this is required for ethanol-induced mitochondrial free radical damage remains to be established. GPXs use reduced glutathione (GSH) for reduction of H2O2 or lipid peroxides. GSH is regenerated from oxidized glutathione by the NADPH-dependent glutathione reductase. NADPH supply in mitochondria depends on the energy-linked transhydrogenase or on NADP-linked isocitrate dehydrogenase; both enzymes contribute to the protection against oxidative stress.37,38 O2. also reacts avidly with the nitric oxide radical, NO•, to form peroxynitrite, a reactive nitrogen species that can modify tyrosine residues on proteins. NO• can be generated in mitochondria and cause inhibition of cytochrome oxidase,39–41 and peroxynitrite was recently reported to affect cytochrome c activity.42
Hence, ROS and related radical species are converted in multiple ways by oxidative stress defenses both inside and outside the mitochondria, but the products of these processes are not without effect on mitochondrial metabolism and may, in fact, contribute to its regulation. Despite its potential for damage, mitochondrial ROS formation is likely to have physiologic roles, e.g., as signals by which mitochondria communicate with other cell compartments.43 Therefore, the balance between formation and conversion of ROS must be carefully controlled.
Mitochondrial defense mechanisms that dispose of ROS are also affected by chronic ethanol treatment. For instance, GPX activities reportedly are decreased after chronic ethanol intake in the liver.20 Furthermore, Fernandez-Checa and coworkers44–48 reported that chronic ethanol treatment causes a selective depletion of mitochondrial GSH levels in liver, which was progressive over long periods (up to 16 weeks) of ethanol feeding. Conditions that replenish mitochondrial glutathione or prevent its depletion were found to suppress ethanol-induced liver damage.47,48 A similar selective depletion of mitochondrial GSH was also reported in type II alveolar cells in the lung.49 However, other investigators did not observe a marked selective mitochondrial depletion of GSH in the liver with chronic ethanol feeding,20,50,51 indicating that the conditions accounting for this phenomenon still need better characterization. Also, if there is a gradual depletion of mitochondrial GSH over several months, the question remains what degree of depletion would be deleterious. Irrespective of the specific mechanism involved, however, chronic ethanol treatment appears to impair the balance of ROS formation and conversion, with implications for ethanol-induced cell damage.
Ethanol promotes ROS formation and enhances oxidative stress not only in the liver, its major site of oxidation, but also in the brain, heart, or lung, which have much less oxidative ethanol metabolism, because they lack ADH (although induction of CYP2E1 by chronic ethanol treatment has been detected in several other tissues52). Ethanol treatment affects stress-related signaling pathways even in cell lines that lack enzymes for ethanol oxidation,53,54 and these signaling systems may alter the capacity of cells to respond to other oxidative stress challenges. Thus, ethanol treatment may promote oxidative stress even in the absence of active metabolism, possibly by affecting cellular signaling systems that integrate the organism’s responses to external and internal challenges. For instance, cytokines such as tumor necrosis factor (TNF)-α, which are part of the body’s stress response, induce mitochondrial formation of ROS.55–57 There is good evidence that TNF-α release from Kupffer cells in the liver is enhanced by ethanol intake and that this plays a role in the onset of alcoholic liver disease in rats receiving ethanol by intragastric feeding.34,58
All mitochondrial constituents, proteins, lipids, and mtDNA are potential targets for ROS-mediated damage. Considerable evidence indicates that mitochondrial constituents suffer cumulative oxidative damage after chronic ethanol exposure. Peroxidation products of phospholipids, such as 4-hydroxynonenal and malondialdehyde, accumulate in the liver and other tissues after chronic ethanol exposure.59 Protein adducts form with lipid-derived aldehydes or with acetaldehyde, the primary product of ethanol oxidation.60,61 Membrane structural alterations are evident as changes in order parameter, detected by electron spin resonance or fluorescence probes62 and Fernandez-Checa and coworkers45,46 have provided evidence that this impairs mitochondrial GSH transport through an ATP-dependent GSH transport system in the inner membrane, which would account for a gradual depletion of the matrix pool of glutathione. Oxidation of mitochondrial proteins may also result from ROS exposure. For instance, oxidation of a selenocysteine group in GPX may cause its inactivation after chronic ethanol feeding.20 Through such mechanisms, a gradual impairment of oxidative defenses in mitochondria will enhance further oxidative stress. The integrity of mtDNA is also affected by ethanol treatment through mechanisms that involve ROS.63,64 To what extent ethanol-induced damage to mtDNA impairs mitochondrial function and how that affects cell and tissue injury are as yet open questions. A major consequence of these impairments may be the vulnerability of the mitochondria to processes that induce their structural demise, the mitochondrial permeability transition (MPT), with consequences for apoptosis and necrosis. In the following, we will first discuss the nature of ethanol-induced impairment of mtDNA and then consider how the different mitochondrial targets of ROS may act synergistically to promote ethanol-induced cell injury.
mtDNA as a Target for Ethanol-Induced Oxidative Stress
Oxidative Damage to mtDNA
mtDNA encodes essential subunits of electron transport complexes and ATP synthase, and its integrity is required for maintenance of oxidative phosphorylation. Damage to mtDNA may, therefore, affect mitochondrial formation of ROS. Compared with its nuclear counterpart, mtDNA is highly vulnerable to free radical attack. mtDNA is not protected by histones, and its location in the vicinity of the inner membrane, the predominant cellular source of ROS, puts high demands on the maintenance of its integrity.
Despite the sensitivity of mtDNA to ROS, oxidative defenses and mtDNA repair mechanisms normally limit accumulation of damaged mtDNA. Moreover, multiple copies of mtDNA are present in all mitochondria in a cell, and a certain level of damage to mtDNA can be tolerated, as long as intact copies are available.65 However, there is considerable literature that damage to mtDNA increases with advanced age, and the “mitochondrial theory of aging” suggests that accumulation of defective mtDNA would impair cellular energy metabolism, placing a limit on the life span of the cell (and by extension the organism).17,66 If mitochondria lack effective mechanisms to repair or dispose of excess oxidative damage, the additional production of ROS resulting from ethanol intake could exacerbate this damage, with potentially serious consequences. Alternatively, depending on the chemical nature of the changes in mtDNA, rather than accumulating, damaged mtDNA may be degraded more rapidly and the integrity of the mtDNA pool may depend on its repletion.
The most damaging form of ROS generated in mitochondria is the hydroxyl radical, OH•. All parts of the mtDNA molecule are potential targets for OH•. It attacks deoxyribose to cause release of purine and pyrimidine bases from mtDNA, accompanied by strand breaks.67 Alternatively, OH• can directly attack purine and pyrimidine bases, leading to their modification. For instance, OH•-induced modification of guanine generates 8-oxoguanine (8-oxoG), which is commonly detected as an indicator of free radical damage to DNA. However, other modifications of guanine or other bases also occur, causing their reduction or ring-opening.68 The consequences of base modifications for mtDNA integrity vary depending on the lesion. For instance, ring fragmentation of bases would block DNA replication.69 8-OxoG, on the other hand, has been shown in vitro to be misread by mtDNA polymerase-γ, resulting in it being paired with a dA residue and giving rise to G → T transversions. However, corresponding in vivo data are lacking as yet. Aging coincides with an exponential increase in 8-oxoG accumulation, but the predicted increase in G → T transversions has not been shown.
A gradual accumulation of oxidatively damaged mtDNA (detected as an increased level of 8-oxoG) and an increase in mtDNA strand breaks with increasing age have been observed in the liver, heart, and other tissues.70 It is not clear if the accumulating damage reflects an increase in ROS production with age, or a gradual defect in repair or disposal mechanisms that prevent its accumulation in younger organisms. Our group showed that accumulation of 8-oxoG and mtDNA strand breaks were significantly enhanced in the mtDNA obtained from the liver of chronically ethanol-fed rats.71 When the ethanol diet was started at a young age, accumulation of damaged mtDNA was significant only after ethanol feeding for 6 months or longer, but in older animals accumulation of oxidative damage occurred within 3 weeks of ethanol feeding (Cahill, unpublished observations). This may reflect an increase in ROS production in livers of alcoholic animals or an age-dependent decrease in repair mechanisms designed to remove oxidative lesions and strand breaks.
mtDNA Repair
Relatively little is known about the repair mechanisms that maintain mtDNA integrity. Mitochondria appear to be deficient in nucleotide excision repair, but several alternative repair processes have been identified, including base excision repair (BER), mismatch repair, recombinational repair, and DNA methyl transferases. Removal of 8-oxoG bases probably depends mostly on BER and on mismatch repair. BER is initiated by a DNA glycosylase specific for a particular modified base. A mitochondrial oxidative damage endonuclease (mtODE/OGGI) has been characterized that acts as an 8-oxoG DNA glycosylase and also possesses an associated apurinic/apyrimidinic lyase activity.72 This enzyme not only cleaves the N-glycosydic bond, but also the DNA backbone. Repair is completed by the action of apurinic/apyrimidinic endonuclease that provides a 3′ -hydroxyl group for the mtDNA polymerase-γ to extend. Any gaps are then filled in by the mtDNA ligase III. Mismatch repair is common in bacteria and is mediated by MutT, MutM, and MutY proteins. Collectively, these proteins protect bacterial DNA from the potentially mutagenic effects of 8-oxoG. In recent years, human homologs of these proteins have been discovered. The MutT homolog hMTH1 catalyzes the removal of 8-oxoguanosine triphosphate from the nucleotide pool,73 human 8-oxoG-DNA glycosylase is a MutM homolog that removes C/8-oxoG mismatches,74,75 and the MutY homolog hMYH is an adenine glycosylase repairing A/8-oxoG, A/G, and A/C mismatches.76 All 3 enzymes occur predominantly in mitochondria and likely represent the primary enzymes for repairing oxidative damage in mtDNA. It is not known if alcohol abuse impairs these repair activities, but this would have severe consequences for the stability and integrity of the mitochondrial genome and might cause metabolic defects. Interestingly, although ROS formation is presumed to increase with age, a significant decrease in rat heart mtODE/OGGI activity with advanced age was reported.77 How much this decline contributes to the accumulation of oxidative damage to mtDNA and the deterioration of metabolic capacity remains to be tested. An increased oxidative stress incurred during ethanol consumption would impose a further burden on this or other mtDNA repair mechanisms.
mtDNA Loss and Repletion
Given the multiplicity of mtDNA molecules in most cells (a single liver mitochondrion contains as many as 5–10 mtDNA molecules, and the total number of copies per cell runs into several thousands), an effective response to mtDNA damage would be the preferential degradation of defective mtDNA, i.e., mitochondrial genomes with significant damage would be removed from the gene pool rather than repaired. Recent studies suggest that this may be a significant part of the response to ethanol (see Mansouri et al.64,78 and Cahill, unpublished observations).
Mansouri et al.64 reported a marked loss of mouse hepatic mtDNA (by 50% of total mtDNA) within hours after an acute dose of ethanol (5 g/kg by gavage). This degradation was attributed to the increased oxidative stress incurred during ethanol metabolism and was prevented by inhibition of ethanol metabolism and attenuated by melatonin.64 Remarkably, mtDNA levels recovered within 24 hours of the treatment and even showed an overshoot compared with controls. Thus, the predominant response of the organism appeared to be removal of damaged mtDNA and replication of unaffected copies to maintain integrity of the overall mtDNA pool. However, the degree of damage of mtDNA was not determined in these studies, nor was the integrity of the recovered mtDNA pool tested. In a more recent publication,78 a similar transient decline in mtDNA levels was detected in other tissues, including the heart, skeletal muscle, and brain (albeit with slower kinetics in brain compared with other organs). These findings suggest that the loss of mtDNA in response to an acute ethanol challenge is not a result of ethanol metabolism through ADH, which would take place predominantly in the liver (although CYP2E1 has been detected in brain and may increase with chronic ethanol intake52).
Age may be a factor in mtDNA maintenance. Recently, our group observed mtDNA depletion in the liver of both young (2 months) and old (2 years) rats challenged with an acute dose of ethanol (Cahill, unpublished observations). However, the degree of depletion was more modest and recovery was slower than reported for mice, suggesting species differences between rats and mice in free radical defense mechanisms or mtDNA repair capacities. The animal’s age did not appear to be a factor in these effects of acute ethanol. By contrast, effects of chronic ethanol feeding on mtDNA integrity were markedly dependent on the age of the animal. Significant mtDNA degradation and depletion occurred after 3 weeks of ethanol feeding in 2-year-old animals compared with pair-fed controls, but no differences were observed in young (2-month-old) animals. An ethanol-induced net increase in ROS levels with age and/or activation of mitochondrial nucleases may account for these differences between young and old animals. Alternatively, aging may be associated with defects in maintenance of mtDNA integrity.
The mechanisms used by the cell to monitor mtDNA levels and maintain homeostasis are poorly characterized, although the mtDNA replication machinery itself likely is involved. Several proteins involved in replication have been identified that also appear to be essential for mtDNA maintenance. DNA polymerase γ and DNA ligase III (the only DNA ligase known to occur in mitochondria) are also involved in mtDNA repair.79 The mitochondrial transcription factors A80 and B81 (mtTFA and mtTFB) RNA polymerase,80 RNase MRP (mitochondrial RNA processing enzyme),82,83 DNA primase,84 DNA helicase,85 and a mitochondrial single strand binding protein86,87 are required for replication, and any alteration in their levels or activities has adverse effects on mtDNA copy number. For instance, disruption of the expression of mtTFA or DNA ligase III markedly decreases mtDNA content.88,89 Thus, mtDNA homeostasis requires an integrated functional state of the replication machinery, and changes in the activity of a variety of proteins involved in mtDNA replication or transcription may affect the steady state levels of mtDNA. Interestingly, Tang et al.90 recently suggested that regulation of mtDNA copy number may depend on the mass of mtDNA and not the number of intact genomes, implying a control mechanism that depends on mitochondrial deoxynucleoside triphosphate pools. If this is correct, imbalances in the mitochondrial nucleotide pool may also impede mtDNA replication.
If ethanol treatment would depress any of the components of the replication machinery or inhibit their activities, either directly or by ethanol-induced oxidative stress, mtDNA depletion may result. For instance, if ethanol enhances age-dependent oxidative damage to mtDNA,63,71 the defective template may affect the activity of DNA polymerase γ, impairing both mtDNA repair and replication. Quantitative polymerase chain reaction (PCR) studies have shown that damaged mtDNA templates inhibit certain polymerases. Whether mtDNA polymerase γ is inhibited by oxidatively (or otherwise) modified nucleotides has yet to be established.
In summary, the capacity of the cell to maintain mtDNA levels depends on a balance of degradation and repair or resynthesis (Figure 2). A shift in this balance with advancing age may contribute to mtDNA depletion in response to challenges, such as ethanol treatment. Because mtDNA is required for the maintenance of the cell’s bioenergetic capacity, this may ultimately impair the cellular energy supply. The mechanisms by which impaired mitochondrial function may affect the fate of a cell is considered in the following sections.
Figure 2.
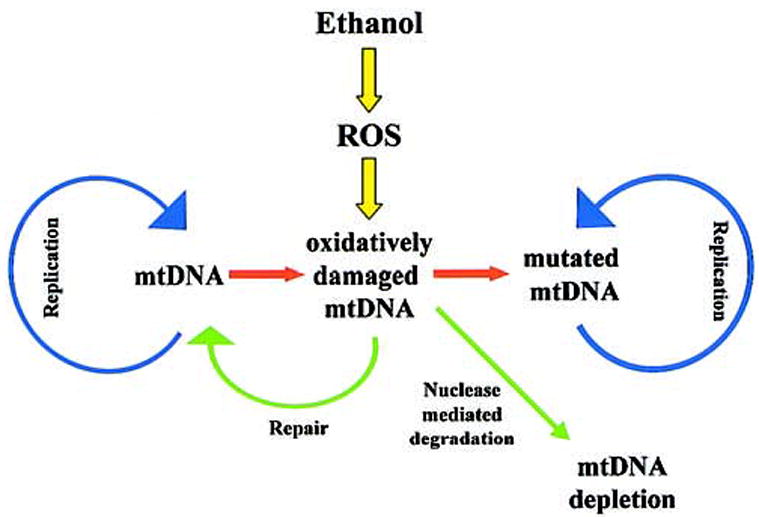
mtDNA depletion prevents accumulation of mutated mtDNA after ethanol-induced oxidative damage. Maintenance of mtDNA levels requires repair of oxidative lesions and replication of unaffected mtDNA.
Ethanol and the MPT
Mitochondria and Apoptosis
Mitochondria have major roles in cellular physiology beyond electron transport and energy conservation. The outer membrane and the intermembrane space, considered mere accessories in classical mitochondrial physiology, have turned out to be intimately involved in the control of apoptosis, a process of controlled (programmed) cell death with characteristic morphologic features that remove damaged or undesirable cells in all multicellular organisms.91 A family of proteins related to the oncogene Bcl-2 are critical regulators of the onset of apoptosis and can either protect (e.g., Bcl-2, Bcl-xL) or enhance (e.g., Bax, Bad, Bid) the process.92,93 The balance of pro- and anti-apoptotic proteins is thought to determine the fate of the cell. Much of the activity of Bcl-2 family proteins is exerted by interacting with mitochondrial membranes, through mechanisms that are still poorly characterized, resulting in the release of proteins sequestered in the mitochondrial intermembrane space, which are required for activating the degradative processes involved in apoptosis. These include cytochrome c, which acts as a cofactor for activation of the protease caspase 9, the precursor of which is also localized in the intermembrane space, apoptosis inducing factor, a flavoprotein that migrates to the nucleus after its release to activate endonucleases, and several others.91,94
Rupture or permeabilization of the outer membrane is required for the release of these proteins from mitochondria. Outer membrane rupture follows mitochondrial swelling, a characteristic of activation of the MPT, a process that involves opening of a large, nonspecific proteinaceous pore (the permeability transition pore). This pore complex requires constituents of both the outer membrane (voltage-dependent anion channel [VDAC], peripheral benzodiazepine receptor [PBR]) and the inner membrane (adenine nucleotide translocator [ANT]) and also involves the matrix protein cyclophilin D, which is characteristically inhibited by cyclosporin A (Figure 3). MPT pore opening is regulated by thiol groups,95 and formation of ROS promotes the MPT, presumably in part by oxidation of these critical thiols. Control of the MPT is closely interlinked with mitochondrial energy conservation, being enhanced by deenergization and by mitochondrial Ca2+ accumulation.96 Pore opening not only depletes the proton motive force, but also involves the loss of matrix NAD and adenine nucleotides. Cytochrome c is an essential component of the electron transport chain, and its release may impair mitochondrial electron transport. Equally, the control of apoptosis depends on the cellular energy state. Both pro- and anti-apoptotic Bcl-2 family proteins have been reported to interact with VDAC and ANT and affect Ca2+ and proton fluxes across the mitochondrial membranes.97–99 Importantly, proper execution of apoptosis depends on adequate ATP supplies in the cell targeted for destruction, and when there is a lack of ATP, the cells default to a necrotic form of cell death.100,101 Thus, although both the nature and the physiologic significance of the MPT is still hotly debated, there is little doubt that conditions that enhance MPT activation are associated with increased cell death, whether by apoptosis or necrosis.91
Figure 3.
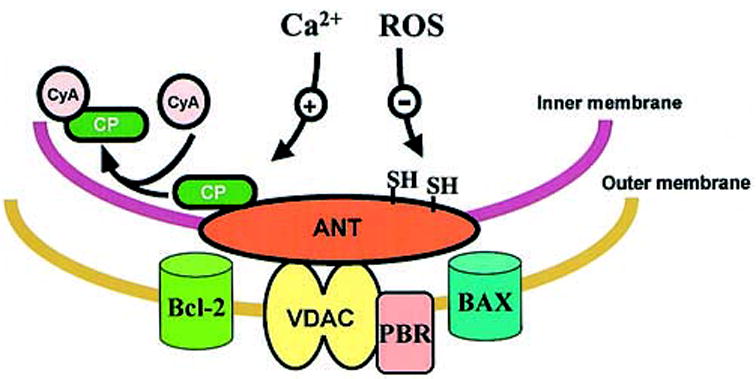
The MPT pore complex. ANT, adenine nucleotide translocator; VDAC, voltage-dependent anion channel; PBR, peripheral benzo-diazepine receptor; CyA, cyclosporin A; CP, cyclophilin D.
Ethanol and MPT Activation
There is now considerable evidence that ethanol treatment disturbs the balance of pro- and anti-apoptotic factors to enhance the susceptibility of cells and tissues to apoptotic stimuli, and that this involves facilitation of MPT activation. Oxidative stress and defects in mitochondrial energy conservation may be involved. Ishii and coworkers19 reported that acute ethanol treatment promoted apoptosis in primary hepatocyte cultures in vitro, accompanied by mitochondrial ROS formation, depolarization, and cytochrome c release, characteristic of MPT activation. HepG2 cells over-expressing CYP2E1 exhibit increased cell death when treated with ethanol and/or TNF-α,4,54,102 which is preceded by mitochondrial depolarization and prevented by Bcl-2 or cyclosporin A, suggesting involvement of the MPT. However, our group reported that ethanol treatment enhanced TNF-α induced cytotoxicity even in wild-type HepG2 cells that do not metabolize ethanol.54 An enhanced TNF-induced cytotoxicity was also observed in hepatocytes isolated from chronically ethanol-fed rats compared with pair-fed controls, even when no ethanol was present in the incubation and hepatic mitochondria isolated from these animals had an enhanced susceptibility to MPT-mediated swelling induced by a variety of stimuli.54,103 Thus, the mechanisms by which ethanol treatment facilitates the onset of the MPT may involve other factors.
The MPT potentiation after chronic ethanol intake may reflect a persistent oxidative stress. A sustained depletion of the mitochondrial pool of GSH after chronic ethanol treatment, as reported by Fernandez-Checa and coworkers,45,57 could enhance the sensitivity to TNF-α or other challenges. Lipid peroxidation products, such as malondialdehyde and 4-hydroxynonenal, which accumulate in the liver and other tissues under conditions in which liver injury occurs in association with ethanol treatment, themselves trigger MPT pore opening in isolated mitochondria and their effect would be enhanced by GSH depletion.61,104,105 Ceramide is another candidate mediator to induce mitochondrial dysfunction and onset of the MPT. Ceramide accumulates in hepatocytes in response to TNF-α and induces the onset of an MPT with consequent apoptosis by virtue of its inhibition of mitochondrial electron transport at complex III.106–108 Ceramide can be generated either from de novo ceramide synthesis in the endoplasmic reticulum, mediated by ceramide synthase, or by the action of acidic or neutral sphingomyelinases.109,110 The hydrolysis of sphingomyelin, which is activated upon engagement of death receptors such as the TNF receptor-1 (TNF R-1) or Fas, leads to equimolar formation of sphingosine-1-phosphate (SPP).111,112 In contrast to ceramide, SPP induces cell growth and protects cells from TNF-α–induced cell death.113,114 Interestingly, a recent study115 reported that astrocytes treated with 5 mmol/L ethanol for 24 hours exhibit enhanced cell killing induced by TNF-α, C2-ceramide, and sphingomyelinase. Ethanol appeared to inhibit the formation of SPP upon TNF-α treatment, but not that of ceramide, suggesting that ethanol may shift the balance of sphingolipid metabolism in TNF-α–treated astrocytes in favor of a pathway that increases ceramide levels over that of SPP, with subsequent induction of the MPT. To what extent this mechanism operates in other cell types remains to be studied.
Stress Signaling and the Control of the MPT
Ethanol treatment may enhance the oxidative stress imposed on mitochondria and thereby enhance the MPT, but it is likely that in the intact tissue, the onset of the MPT is also controlled by stress-related signaling pathways that act on proteins that form part of the MPT pore complex. Hence, the increased oxidative stress generated by ethanol treatment may synergize with other alterations brought about by ethanol to induce the MPT. Some of these signaling pathways themselves respond to oxidative stress. For instance, apoptosis signaling kinase-1 (ASK-1) is a mitogen-activated protein kinase kinase kinase (MAPK3) that can activate both the p38 –MAPK and c-Jun-N-terminal kinase (JNK) stress signaling cascades.116 Prolonged activation of either p38 or JNK has been shown to promote cell death, and the mitochondrial membrane is a potential target for these kinase cascades.117–119 The activity of ASK-1 is controlled by the cellular redox state through its effects on thioredoxin, a redox sensitive protein.120 When reduced, thioredoxin binds and inactivates ASK-1, but oxidized thioredoxin dissociates from ASK-1, thereby causing its activation and subsequent stimulation of the p38-MAPK and JNK signaling cascades. JNK localizes to mitochondria where it can phosphorylate and inactivate the anti-apoptotic proteins Bcl-2 or Bcl-xL, which relieves the suppression of the MPT.119 By contrast, p38 activity promotes the translocation of Bax, a pro-apoptotic Bcl-2 family protein, from cytosol to mitochondria, where it can trigger the MPT.121,122 However, ethanol treatment may also enhance the activity of redox sensitive proteins, such as thioredoxin. The redox state of thioredoxin is controlled by a family of thioredoxin reductases that use NADPH as the reductant.123,124 Hence, in a tissue such as the liver, where ethanol metabolism causes reduction of NAD(P), its acute effect may be to inactivate ASK-1, despite the formation of ROS. Interestingly, in a recent study on isolated hepatocytes from chronically ethanol-fed rats, we observed a marked enhancement of the activation of p38 (but not JNK) when cells were treated with TNF-α, suggesting that long-term ethanol treatment may act to enhance the sensitivity of the mitochondria to p38-mediated pathways. Whether this involves ASK-1 activity or other upstream kinases remains to be studied.
How Bax and other pro-apoptotic Bcl-2 family proteins enhance the MPT or how much of a role these processes play in the onset of apoptosis and necrosis is still a matter of considerable debate. Some current models envisage an interaction of these proteins with one or more components of the permeability transition pore, such as VDAC in the outer membrane or ANT in the inner membrane. Ethanol may enhance pore opening by altering the interactions between these proteins. Another putative component of the pore complex is the PBR in the mitochondrial outer membrane.125 This protein is thought to participate in the signaling processes that relate mitochondrial functional states to the nucleus (or vice versa). Mitochondria isolated from chronically ethanol-fed animals exhibit elevated levels of PBR and are more susceptible to undergo the MPT upon addition of PBR partial agonists like PK11195.103,126 Importantly, pro-oxidants are capable of sensitizing mitochondria to pore opening by PBR partial agonists. Hence, a shift in the balance of proteins that make up the pore complex as a consequence of chronic ethanol intake may have deleterious consequences for the control of MPT activation (or other responses related to pro- and anti-apoptotic signals). Much additional work needs to be done to gain a better picture of the permeability transition pore complex and its role in cell death by apoptosis or necrosis and in energy conservation. This work will be essential to understand the consequences of ethanol exposure.
mtDNA Defects and the MPT
Is the loss of mtDNA that occurs with chronic ethanol treatment and with aging a risk factor in ethanol-induced damage? There is considerable evidence that aging further enhances ethanol-induced cell damage through mechanisms that involve the MPT. The susceptibility of mitochondria to agents that trigger MPT onset is markedly enhanced with advanced age, and such mitochondria are less capable of handling conditions associated with ROS formation or other challenges.127,128 Various forms of mtDNA damage accumulate exponentially, and mtDNA depletion occurs with increasing age. Oxidative phosphorylation and mitochondrial membrane potential decline correspondingly.127,129,130 The mechanisms causing these age-related defects are not well characterized. The mtDNA damage may not be caused solely by an increased net ROS formation in older animals, but may reflect a decrease in mtDNA repair or replication. As mentioned above, ethanol-induced depletion of mtDNA is much more pronounced in older animals than at a young age, suggesting that the normal recovery mechanisms are defective in older animals. However, evidence is inconsistent whether damage to, or depletion of mtDNA promotes cell death by apoptosis. Several studies on cell lines that lack mtDNA (mtDNA deficient cells [ρ0 cells]) reported that apoptosis occurs normally131,132 or showed an enhanced sensitivity to agents that trigger apoptosis.133 By contrast, others reported protection against cell killing in ρ0 cells.134 It remains to be investigated to what extent these contrasting findings reflect different mechanisms by which apoptosis can be mediated, e.g., bypassing mitochondrially localized apoptotic mediators.
To what extent a sustained stress caused by chronic ethanol exposure could affect the functional threshold of mtDNA required to maintain oxidative phosphorylation capacity has not been determined. mtDNA damage or depletion is not proportionally reflected in the maintenance of oxidative phosphorylation activity (or lack thereof ). Studies on patients who suffer from mutational defects in mtDNA suggest that a threshold of functional mtDNA of <20% of normal levels may be sufficient to maintain normal tissue function.135 However, chronic ethanol treatment also suppresses respiratory capacity in the liver by impairing mitochondrial protein synthesis.29,30,136,137 This inhibition appears to be independent of ethanol-induced damage to mtDNA, because it is evident in rat liver mitochondria even after 4 – 6 weeks of ethanol feeding, well before depletion or oxidative damage to mtDNA can be detected. Indeed, it is conceivable that the decreased respiratory capacity is one of the contributing factors in the enhanced rate of formation of ROS in mitochondria from ethanol-fed animals. Thus, it is likely that any consequences of mtDNA damage in ethanol-fed animals are superimposed on the suppression of mitochondrial protein synthesis, with potentially synergistic effects on respiratory activity.
Even if cellular energy conservation capacity declines with advancing age in chronically ethanol-fed animals by accumulating mtDNA damage or depletion and in synergy with other mechanisms that suppress the oxidative phosphorylation machinery, the cellular supply of ATP may still be adequate under normal conditions, because there is considerable excess capacity in cellular respiration. For instance, the energy state of the liver of ethanol-fed rats is normal, unless the tissue is exposed to hypoxic conditions.138 However, cells may lose the ability to respond appropriately to acute stress conditions that place excess demand on the energy supply, including challenges that would normally lead to the activation of apoptosis. Under such conditions, the capacity to provide ATP for the completion of the apoptotic program may be impaired. Often, cells that abort an apoptotic process default to cell death by necrosis.100,101 A more careful scrutiny of the implications of ethanol-dependent loss of mtDNA for cellular energy supply is needed to assess to what extent this may affect the capacity of cells to induce apoptosis in response to normal stress conditions and instead shift the balance of cell death towards necrosis, with deleterious functional consequences for the tissue.
Mitochondrial Dysfunction, Liver Disease, and Aging
The cellular oxidative stress management and response systems involve a cascade of carefully maintained balancing acts that depend on mitochondrial function and that are affected by ethanol treatment at different levels (Figure 4). First, the degree of oxidative stress to which cells are exposed depends both on the formation of reactive oxygen (and nitrogen) species and on the cellular defenses that prevent their accumulation. Mitochondrial function plays a critical role in maintaining an appropriate balance between the two. Conversely, the resulting oxidative stress exerts its effects in part by directly damaging mitochondrial constituents. This may set in motion a vicious cycle, leading to more global damage. The extent to which this affects mitochondrial function is largely dependent on the capacity of the cell for repair or replacement of the affected components. This is ex-emplified by the marked effects of ethanol treatment on mtDNA, which appears to be effectively recovered or repaired in young animals, but not in older animals. The mitochondrial response is further controlled by the balance of pro- and anti-apoptotic factors that determine its release of pro-apoptotic proteins through the activation or suppression of the MPT and other mechanisms. Cellular stress response signaling systems, such as p38 and JNK MAPK cascades, presumably exert effects on mitochondrial function in part through these factors. Finally, if the stress is severe, the consequences for cell function depend on how well it can control the progression of cell death mechanisms leading to apoptosis or necrosis. Many studies have emphasized the role of mitochondria in apoptosis, but often the same conditions that initiate apoptosis can also trigger necrosis, depending on the cellular environment. Factors such as the magnitude of oxidative stress or the mitochondrial energy supply may help determine the balance between apoptotic and necrotic forms of cell death.
Figure 4.
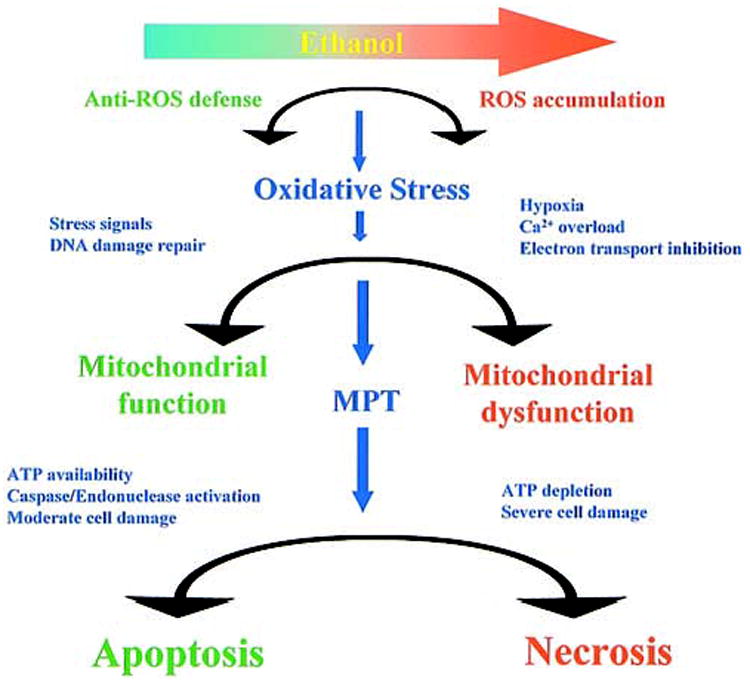
Ethanol treatment shifts the balance of cellular defense mechanisms against apoptosis and necrosis.
Furthermore, the physiological consequences of cellular dysfunction associated with oxidative stress cannot be dissociated from its interaction with its immediate environment in the liver. Cytokines, such as TNF-α and interleukin-6, are derived from Kupffer cells and other non-parenchymal cells and act on hepatocytes to generate signals that affect mitochondrial ROS formation. Conversely, the injured cells generate cytokine signals that promote neutrophil infiltration or activate stellate cells to initiate fibrogenesis.139 Inappropriate cellular responses to these signals are critical factors in the onset and progression of liver disease. Chronic ethanol consumption affects these cell-cell interactions not only by promoting the production of proinflammatory cytokines, but also by impairing the capacity of hepatocytes to respond appropriately to these cytokines. Mitochondrial dysfunction is likely to be at the core of this impairment.54 An understanding of the nature of alcohol-induced mitochondrial dysfunction and its impact on cellular stress management may, therefore, provide new insight into alcoholic liver disease that may lead to new therapeutic approaches.
A gradual deterioration in the capacity of cells to maintain their energy balance and control the level of oxidative stress appears to be associated with aging.66 This may diminish the flexibility of older organisms to compensate for the additional stress caused by ethanol exposure, thereby exacerbating the deleterious effects of ethanol. Ethanol-induced liver damage tends to have a slow onset and may require additional insults (the “second hit” hypothesis). Whether aging would itself constitute such a “second hit” remains to be firmly established, but would be compatible with observations on both the mtDNA depletion and the onset of the MPT. It is conceivable that these are not unrelated, i.e., a depletion of mtDNA as a result of a failure of repair mechanisms may decrease the capacity of oxidative phosphorylation and thereby promote the MPT, while at the same time decreasing the capacity of the cell to maintain an adequate energy supply to sustain a favorable balance of apoptotic and necrotic cell death. Much further work is needed to provide better insight into the age-related susceptibility of ethanol-induced damage to the liver and other tissues.
Abbreviations used in this paper
- ADH
alcohol dehydrogenase
- ANT
adenine nucleotide translocator
- ASK-1
apoptosis signaling kinase-1
- BER
base excision repair
- CYP2E1
cytochrome P450 2E1
- GPX
glutathione peroxidase
- GSH
reduced glutathione
- JNK
c-Jun-N-terminal kinase
- MAPK
mitogen-activated protein kinase
- MPT
mitochondrial permeability transition
- mtDNA
mitochondrial DNA
- mTFA(B)
mitochondrial transcription factor A(B)
- mtODE
mitochondrial oxidative damage endonuclease
- NAD
nicotinamide adenine dinucleotide
- NADH
reduced nicotinamide adenine dinucleotide
- NADP
nicotinamide adenine dinucleotide phosphate
- NADPH
reduced nicotinamide adenine dinucleotide phosphate
- 8-oxoG
8-oxoguanine
- Δp
mitochondrial proton motive force
- PBR
peripheral benzodiazepine receptor
- ρ0 cells
mtDNA deficient cells
- RNase MRP
ribonucleoprotein endoribo-nuclease (mitochondrial RNA processing)
- ROS
reactive oxygen species
- SOD
superoxide dismutase
- SPP
sphingosine-1-phosphate
- TNF
tumor necrosis factor
- UCP-2(3)
uncoupling protein 2(3)
- VDAC
voltage-dependent anion channel
References
- 1.Mantle D, Preedy VR. Free radicals as mediators of alcohol toxicity. Adverse Drug React Toxicol Rev. 1999;18:235–252. [PubMed] [Google Scholar]
- 2.French SW. Mechanisms of alcoholic liver injury. Can J Gastroenterol. 2000;14:327–332. doi: 10.1155/2000/801735. [DOI] [PubMed] [Google Scholar]
- 3.Cederbaum AI. Introduction-Serial review: alcohol, oxidative stress and cell injury. Free Radic Biol Med. 2001;31:1524 –1526. doi: 10.1016/s0891-5849(01)00741-9. [DOI] [PubMed] [Google Scholar]
- 4.Cederbaum AI, Wu D, Mari M, Bai J. CYP2E1-dependent toxicity and oxidative stress in HepG2 cells. Free Radic Biol Med. 2001;31:1539 –1543. doi: 10.1016/s0891-5849(01)00743-2. [DOI] [PubMed] [Google Scholar]
- 5.Tsukamoto H, Lu SC. Current concepts in the pathogenesis of alcoholic liver injury. FASEB J. 2001;15:1335–1349. doi: 10.1096/fj.00-0650rev. [DOI] [PubMed] [Google Scholar]
- 6.Bailey SM, Cunningham CC. Contribution of mitochondria to oxidative stress associated with alcoholic liver disease. Free Radic Biol Med. 2002;32:11–16. doi: 10.1016/s0891-5849(01)00769-9. [DOI] [PubMed] [Google Scholar]
- 7.Boveris A, Chance B. The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem J. 1973;134:707–716. doi: 10.1042/bj1340707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nicholls DG, Ferguson SJ. Bioenergetics 3. New York: Academic Press; 2002. (in press). [Google Scholar]
- 9.McCormack JG, Denton RM. The role of mitochondrial Ca2+ transport and matrix Ca2+ in signal transduction in mammalian tissues. Biochim Biophys Acta. 1990;1018:287–291. doi: 10.1016/0005-2728(90)90269-a. [DOI] [PubMed] [Google Scholar]
- 10.Hoek JB. Hormonal regulation of cellular energy metabolism. In: Ernster L, editor. Molecular mechanisms of bioenergetics. Vol. 23. Amsterdam, London, New York, Tokyo: Elsevier Science Publication; 1992. pp. 421–461. [Google Scholar]
- 11.Hansford RG, Zorov D. Role of mitochondrial calcium transport in the control of substrate oxidation. Mol Cell Biochem. 1998;184:359 –369. [PubMed] [Google Scholar]
- 12.Skulachev VP. Uncoupling: new approaches to an old problem of bioenergetics. Biochim Biophys Acta. 1998;1363:100 –124. doi: 10.1016/s0005-2728(97)00091-1. [DOI] [PubMed] [Google Scholar]
- 13.Porter RK. Mitochondrial proton leak: a role for uncoupling proteins 2 and 3? Biochim Biophys Acta. 2001;1504:120 –127. doi: 10.1016/s0005-2728(00)00246-2. [DOI] [PubMed] [Google Scholar]
- 14.Pecqueur C, Couplan E, Bouillaud F, Ricquier D. Genetic and physiological analysis of the role of uncoupling proteins in human energy homeostasis. J Mol Med. 2001;79:48 –56. doi: 10.1007/s001090000150. [DOI] [PubMed] [Google Scholar]
- 15.Sugano T, Handler JA, Yoshihara H, Kizaki Z, Thurman RG. Acute and chronic ethanol treatment in vivo increases malate-aspartate shuttle capacity in perfused rat liver. J Biol Chem. 1990;265:21549 –21553. [PubMed] [Google Scholar]
- 16.Bailey SM, Pietsch EC, Cunningham CC. Ethanol stimulates the production of reactive oxygen species at mitochondrial complexes I and III. Free Radic Biol Med. 1999;27:891–900. doi: 10.1016/s0891-5849(99)00138-0. [DOI] [PubMed] [Google Scholar]
- 17.Raha S, Robinson BH. Mitochondria, oxygen free radicals, disease and ageing. Trends Biochem Sci. 2000;25:502–508. doi: 10.1016/s0968-0004(00)01674-1. [DOI] [PubMed] [Google Scholar]
- 18.Kurose I, Higuchi H, Kato S, Miura S, Watanabe N, Kamegaya Y, Tomita K, Takaishi M, Horie Y, Fukuda M, Mizukami K, Ishii H. Oxidative stress on mitochondria and cell membrane of cultured rat hepatocytes and perfused liver exposed to ethanol. Gastroenterology. 1997;112:1331–1343. doi: 10.1016/s0016-5085(97)70147-1. [DOI] [PubMed] [Google Scholar]
- 19.Higuchi H, Adachi M, Miura S, Gores GJ, Ishii H. The mitochondrial permeability transition contributes to acute ethanol-induced apoptosis in rat hepatocytes. Hepatology. 2001;34:320 –328. doi: 10.1053/jhep.2001.26380. [DOI] [PubMed] [Google Scholar]
- 20.Bailey SM, Patel VB, Young TA, Asayama K, Cunningham CC. Chronic ethanol consumption alters the glutathione/glutathione peroxidase-1 system and protein oxidation status in rat liver. Alcohol Clin Exp Res. 2001;25:726 –733. [PubMed] [Google Scholar]
- 21.Israel Y, Videla L, Bernstein J. Liver hypermetabolic state after chronic ethanol consumption: hormonal interrelations and pathogenic implications. Federation Proc. 1975;34:2052–2059. [PubMed] [Google Scholar]
- 22.Rivera CA, Bradford BU, Seabra V, Thurman RG. Role of endotoxin in the hypermetabolic state after acute ethanol exposure. Am J Physiol. 1998;275:G1252–1258. doi: 10.1152/ajpgi.1998.275.6.G1252. [DOI] [PubMed] [Google Scholar]
- 23.Rashid A, Wu TC, Huang CC, Chen CH, Lin HZ, Yang SQ, Lee FY, Diehl AM. Mitochondrial proteins that regulate apoptosis and necrosis are induced in mouse fatty liver. Hepatology. 1999;29:1131–1138. doi: 10.1002/hep.510290428. [DOI] [PubMed] [Google Scholar]
- 24.Diehl AM, Hoek JB. Mitochondrial uncoupling: role of uncoupling protein anion carriers and relationship to thermogenesis and weight control: “the benefits of losing control. J Bioenerg Biomembr. 1999;31:493–506. doi: 10.1023/a:1005452624640. [DOI] [PubMed] [Google Scholar]
- 25.Ingelman-Sundberg M, Ronis MJ, Lindros KO, Eliasson E, Zhukov A. Ethanol-inducible cytochrome P4502E1: regulation, enzymology and molecular biology. Alcohol Alcohol Suppl. 1994;2:131–139. [PubMed] [Google Scholar]
- 26.Bradford BU, Enomoto N, Ikejima K, Rose ML, Bojes HK, Forman DT, Thurman RG. Peroxisomes are involved in the swift increase in alcohol metabolism. J Pharmacol Exp Ther. 1999;288:254 –259. [PubMed] [Google Scholar]
- 27.Arteel GE, Iimuro Y, Yin M, Raleigh JA, Thurman RG. Chronic enteral ethanol treatment causes hypoxia in rat liver tissue in vivo. Hepatology. 1997;25:920 –926. doi: 10.1002/hep.510250422. [DOI] [PubMed] [Google Scholar]
- 28.Cunningham CC, Ivester P. Chronic ethanol, oxygen tension and hepatocyte energy metabolism. Front Biosci. 1999;4:D551–556. doi: 10.2741/cunning. [DOI] [PubMed] [Google Scholar]
- 29.Cunningham CC, Coleman WB, Spach PI. The effects of chronic ethanol consumption on hepatic mitochondrial energy metabolism. Alcohol Alcohol. 1990;25:127–136. doi: 10.1093/oxfordjournals.alcalc.a044987. [DOI] [PubMed] [Google Scholar]
- 30.Cahill A, Cunningham CC. Effects of chronic ethanol feeding on the protein composition of mitochondrial ribosomes. Electrophoresis. 2000;21:3420 –3426. doi: 10.1002/1522-2683(20001001)21:16<3420::AID-ELPS3420>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 31.Albano E, Clot P, Morimoto M, Tomasi A, Ingelman-Sundberg M, French SW. Role of cytochrome P4502E1-dependent formation of hydroxyethyl free radical in the development of liver damage in rats intragastrically fed with ethanol. Hepatology. 1996;23:155–163. doi: 10.1002/hep.510230121. [DOI] [PubMed] [Google Scholar]
- 32.Robin MA, Anandatheerthavarada HK, Fang JK, Cudic M, Otvos L, Avadhani NG. Mitochondrial targeted cytochrome P450 2E1 (P450 MT5) contains an intact N terminus and requires mitochondrial specific electron transfer proteins for activity. J Biol Chem. 2001;276:24680 –24689. doi: 10.1074/jbc.M100363200. [DOI] [PubMed] [Google Scholar]
- 33.Kono H, Rusyn I, Yin M, Gabele E, Yamashina S, Dikalova A, Kadiiska MB, Connor HD, Mason RP, Segal BH, Bradford BU, Holland SM, Thurman RG. NADPH oxidase-derived free radicals are key oxidants in alcohol-induced liver disease. J Clin Invest. 2000;106:867–872. doi: 10.1172/JCI9020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wheeler MD, Kono H, Yin M, Nakagami M, Uesugi T, Arteel GE, Gabele E, Rusyn I, Yamashina S, Froh M, Adachi Y, Iimuro Y, Bradford BU, Smutney OM, Connor HD, Mason RP, Goyert SM, Peters JM, Gonzalez FJ, Samulski RJ, Thurman RG. The role of Kupffer cell oxidant production in early ethanol-induced liver disease. Free Radic Biol Med. 2001;31:1544 –1549. doi: 10.1016/s0891-5849(01)00748-1. [DOI] [PubMed] [Google Scholar]
- 35.Wheeler MD, Nakagami M, Bradford BU, Uesugi T, Mason RP, Connor HD, Dikalova A, Kadiiska M, Thurman RG. Overexpression of manganese superoxide dismutase prevents alcohol-induced liver injury in the rat. J Biol Chem. 2001;276:36664 –36672. doi: 10.1074/jbc.M105352200. [DOI] [PubMed] [Google Scholar]
- 36.Kurose I, Higuchi H, Kato S, Miura S, Ishii H. Ethanol-induced oxidative stress in the liver. Alcohol Clin Exp Res. 1996;20:77A–85A. doi: 10.1111/j.1530-0277.1996.tb01736.x. [DOI] [PubMed] [Google Scholar]
- 37.Hoek JB, Rydstrom J. Physiological roles of nicotinamide nucleotide transhydrogenase. Biochem J. 1988;254:1–10. doi: 10.1042/bj2540001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jo SH, Son MK, Koh HJ, Lee SM, Song IH, Kim YO, Lee YS, Jeong KS, Kim WB, Park JW, Song BJ, Huh TL, Huhe TL. Control of mitochondrial redox balance and cellular defense against oxidative damage by mitochondrial NADP+-dependent isocitrate dehydrogenase. J Biol Chem. 2001;276:16168 –16176. doi: 10.1074/jbc.M010120200. [DOI] [PubMed] [Google Scholar]
- 39.Ghafourifar P, Richter C. Nitric oxide synthase activity in mitochondria. FEBS Lett. 1997;418:291–296. doi: 10.1016/s0014-5793(97)01397-5. [DOI] [PubMed] [Google Scholar]
- 40.Kanai AJ, Pearce LL, Clemens PR, Birder LA, VanBibber MM, Choi SY, de Groat WC, Peterson J. Identification of a neuronal nitric oxide synthase in isolated cardiac mitochondria using electrochemical detection. Proc Natl Acad Sci U S A. 2001;98:14126 –14131. doi: 10.1073/pnas.241380298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brown GC. Regulation of mitochondrial respiration by nitric oxide inhibition of cytochrome c oxidase. Biochim Biophys Acta. 2001;1504:46 –57. doi: 10.1016/s0005-2728(00)00238-3. [DOI] [PubMed] [Google Scholar]
- 42.Cassina AM, Hodara R, Souza JM, Thomson L, Castro L, Ischiropoulos H, Freeman BA, Radi R. Cytochrome c nitration by peroxynitrite. J Biol Chem. 2000;275:21409 –21415. doi: 10.1074/jbc.M909978199. [DOI] [PubMed] [Google Scholar]
- 43.Thannickal VJ, Fanburg BL. Reactive oxygen species in cell signaling. Am J Physiol. 2000;279:L1005–1028. doi: 10.1152/ajplung.2000.279.6.L1005. [DOI] [PubMed] [Google Scholar]
- 44.Hirano T, Kaplowitz N, Tsukamoto H, Kamimura S, Fernandez-Checa JC. Hepatic mitochondrial glutathione depletion and progression of experimental alcoholic liver disease in rats. Hepatology. 1992;16:1423–1427. doi: 10.1002/hep.1840160619. [DOI] [PubMed] [Google Scholar]
- 45.Fernandez-Checa JC, Kaplowitz N, Garcia-Ruiz C, Colell A, Miranda M, Mari M, Ardite E, Morales A. GSH transport in mitochondria: defense against TNF-induced oxidative stress and alcohol-induced defect. Am J Physiol. 1997;273:G7–17. doi: 10.1152/ajpgi.1997.273.1.G7. [DOI] [PubMed] [Google Scholar]
- 46.Fernandez-Checa JC, Kaplowitz N, Garcia-Ruiz C, Colell A. Mitochondrial glutathione: importance and transport. Semin Liver Dis. 1998;18:389 –401. doi: 10.1055/s-2007-1007172. [DOI] [PubMed] [Google Scholar]
- 47.Garcia-Ruiz C, Morales A, Colell A, Ballesta A, Rodes J, Kaplowitz N, Fernandez-Checa JC. Feeding S-adenosyl-L-methionine attenuates both ethanol-induced depletion of mitochondrial glutathione and mitochondrial dysfunction in periportal and perivenous rat hepatocytes. Hepatology. 1995;21:207–214. doi: 10.1002/hep.1840210133. [DOI] [PubMed] [Google Scholar]
- 48.Colell A, Coll O, Garcia-Ruiz C, Paris R, Tiribelli C, Kaplowitz N, Fernandez-Checa JC. Tauroursodeoxycholic acid protects hepatocytes from ethanol-fed rats against tumor necrosis factor-induced cell death by replenishing mitochondrial glutathione. Hepatology. 2001;34:964 –971. doi: 10.1053/jhep.2001.28510. [DOI] [PubMed] [Google Scholar]
- 49.Brown LA, Harris FL, Bechara R, Guidot DM. Effect of chronic ethanol ingestion on alveolar type II cell: glutathione and inflammatory mediator-induced apoptosis. Alcohol Clin Exp Res. 2001;25:1078 –1085. [PubMed] [Google Scholar]
- 50.Deaciuc IV, Fortunato F, D’Souza NB, Hill DB, Schmidt J, Lee EY, McClain CJ. Modulation of caspase-3 activity and Fas ligand mRNA expression in rat liver cells in vivo by alcohol and lipopolysaccharide. Alcohol Clin Exp Res. 1999;23:349 –356. [PubMed] [Google Scholar]
- 51.Puzziferri I, Signorile A, Guerrieri F, Papa S, Cuomo V, Steardo L. Chronic low dose ethanol intake: biochemical characterization of liver mitochondria in rats. Life Sci. 2000;66:477–484. doi: 10.1016/s0024-3205(99)00617-7. [DOI] [PubMed] [Google Scholar]
- 52.Roberts BJ, Shoaf SE, Jeong KS, Song BJ. Induction of CYP2E1 in liver, kidney, brain and intestine during chronic ethanol administration and withdrawal: evidence that CYP2E1 possesses a rapid phase half-life of 6 hours or less. Biochem Biophys Res Commun. 1994;205:1064 –1071. doi: 10.1006/bbrc.1994.2774. [DOI] [PubMed] [Google Scholar]
- 53.Roman J, Colell A, Blasco C, Caballeria J, Pares A, Rodes J, Fernandez-Checa JC. Differential role of ethanol and acetaldehyde in the induction of oxidative stress in HEP G2 cells: effect on transcription factors AP-1 and NF-kappaB. Hepatology. 1999;30:1473–1480. doi: 10.1002/hep.510300623. [DOI] [PubMed] [Google Scholar]
- 54.Pastorino JG, Hoek JB. Ethanol potentiates tumor necrosis factor-alpha cytotoxicity in hepatoma cells and primary rat hepatocytes by promoting induction of the mitochondrial permeability transition. Hepatology. 2000;31:1141–1152. doi: 10.1053/he.2000.7013. [DOI] [PubMed] [Google Scholar]
- 55.Goossens V, Grooten J, De Vos K, Fiers W. Direct evidence for tumor necrosis factor-induced mitochondrial reactive oxygen intermediates and their involvement in cytotoxicity. Proc Natl Acad Sci U S A. 1995;92:8115–8119. doi: 10.1073/pnas.92.18.8115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pastorino JG, Simbula G, Yamamoto K, Glascott PA, Jr, Rothman RJ, Farber JL. The cytotoxicity of tumor necrosis factor depends on induction of the mitochondrial permeability transition. J Biol Chem. 1996;271:29792–29798. doi: 10.1074/jbc.271.47.29792. [DOI] [PubMed] [Google Scholar]
- 57.Colell A, Garcia-Ruiz C, Miranda M, Ardite E, Mari M, Morales A, Corrales F, Kaplowitz N, Fernandez-Checa JC. Selective glutathione depletion of mitochondria by ethanol sensitizes hepatocytes to tumor necrosis factor. Gastroenterology. 1998;115:1541–1551. doi: 10.1016/s0016-5085(98)70034-4. [DOI] [PubMed] [Google Scholar]
- 58.Yin M, Wheeler MD, Kono H, Bradford BU, Gallucci RM, Luster MI, Thurman RG. Essential role of tumor necrosis factor alpha in alcohol-induced liver injury in mice. Gastroenterology. 1999;117:942–952. doi: 10.1016/s0016-5085(99)70354-9. [DOI] [PubMed] [Google Scholar]
- 59.Chen J, Petersen DR, Schenker S, Henderson GI. Formation of malondialdehyde adducts in livers of rats exposed to ethanol: role in ethanol-mediated inhibition of cytochrome c oxidase. Alcohol Clin Exp Res. 2000;24:544 –552. [PubMed] [Google Scholar]
- 60.Tuma DJ, Thiele GM, Xu D, Klassen LW, Sorrell MF. Acetaldehyde and malondialdehyde react together to generate distinct protein adducts in the liver during long-term ethanol administration. Hepatology. 1996;23:872–880. doi: 10.1002/hep.510230431. [DOI] [PubMed] [Google Scholar]
- 61.Niemela O, Parkkila S, Pasanen M, Iimur Y, Bradford B, Thurman RG. Early alcoholic liver injury: formation of protein adducts with acetaldehyde and lipid peroxidation products, and expression of CYP2E1 and CYP3A. Alcohol Clin Exp Res. 1998;22:2118–2124. doi: 10.1111/j.1530-0277.1998.tb05925.x. [DOI] [PubMed] [Google Scholar]
- 62.Taraschi TF, Rubin E. Effects of ethanol on the chemical and structural properties of biologic membranes. Lab Invest. 1985;52:120 –131. [PubMed] [Google Scholar]
- 63.Cahill A, Wang X, Hoek JB. Increased oxidative damage to mitochondrial DNA following chronic ethanol consumption. Biochem Biophys Res Commun. 1997;235:286 –290. doi: 10.1006/bbrc.1997.6774. [DOI] [PubMed] [Google Scholar]
- 64.Mansouri A, Gaou I, De Kerguenec C, Amsellem S, Haouzi D, Berson A, Moreau A, Feldmann G, Letteron P, Pessayre D, Fromenty B. An alcoholic binge causes massive degradation of hepatic mitochondrial DNA in mice. Gastroenterology. 1999;117:181–190. doi: 10.1016/s0016-5085(99)70566-4. [DOI] [PubMed] [Google Scholar]
- 65.DiMauro S, Schon EA. Mitochondrial DNA mutations in human disease. Am J Med Genet. 2001;106:18 –26. doi: 10.1002/ajmg.1392. [DOI] [PubMed] [Google Scholar]
- 66.Hagen TM, Yowe DL, Bartholomew JC, Wehr CM, Do KL, Park JY, Ames BN. Mitochondrial decay in hepatocytes from old rats: membrane potential declines, heterogeneity and oxidants increase. Proc Natl Acad Sci U S A. 1997;94:3064 –3069. doi: 10.1073/pnas.94.7.3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Halliwell B, Aruoma OI. DNA damage by oxygen-derived species. Its mechanism and measurement in mammalian systems. FEBS Lett. 1991;281:9 –19. doi: 10.1016/0014-5793(91)80347-6. [DOI] [PubMed] [Google Scholar]
- 68.Spencer JP, Jenner A, Chimel K, Aruoma OI, Cross CE, Wu R, Halliwell B. DNA strand breakage and base modification induced by hydrogen peroxide treatment of human respiratory tract epithelial cells. FEBS Lett. 1995;374:233–236. doi: 10.1016/0014-5793(95)01117-w. [DOI] [PubMed] [Google Scholar]
- 69.Breimer LH. Ionizing radiation-induced mutagenesis. Br J Cancer. 1988;57:6 –18. doi: 10.1038/bjc.1988.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shigenaga MK, Hagen TM, Ames BN. Oxidative damage and mitochondrial decay in aging. Proc Natl Acad Sci U S A. 1994;91:10771–10778. doi: 10.1073/pnas.91.23.10771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cahill A, Stabley GJ, Wang X, Hoek JB. Chronic ethanol consumption causes alterations in the structural integrity of mitochondrial DNA in aged rats. Hepatology. 1999;30:881–888. doi: 10.1002/hep.510300434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Croteau DL, Rhys CMJ, Hudson EK, Dianov GL, Hansford RG, Bohr VA. An oxidative damage-specific endonuclease from rat liver mitochondria. J Biol Chem. 1997;272:27338 –27344. doi: 10.1074/jbc.272.43.27338. [DOI] [PubMed] [Google Scholar]
- 73.Sakumi K, Furuichi M, Tsuzuki T, Kakuma T, Kawabata S, Maki H, Sekiguchi M. Cloning and expression of cDNA for a human enzyme that hydrolyzes 8-oxo-dGTP, a mutagenic substrate for DNA synthesis. J Biol Chem. 1993;268:23524 –23530. [PubMed] [Google Scholar]
- 74.Arai K, Morishita K, Shinmura K, Kohno T, Kim SR, Nohmi T, Taniwaki M, Ohwada S, Yokota J. Cloning of a human homolog of the yeast OGG1 gene that is involved in the repair of oxidative DNA damage. Oncogene. 1997;14:2857–2861. doi: 10.1038/sj.onc.1201139. [DOI] [PubMed] [Google Scholar]
- 75.Radicella JP, Dherin C, Desmaze C, Fox MS, Boiteux S. Cloning and characterization of hOGG1, a human homolog of the OGG1 gene of Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 1997;94:8010 –8015. doi: 10.1073/pnas.94.15.8010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Parker A, Gu Y, Lu AL. Purification and characterization of a mammalian homolog of Escherichia coli MutY mismatch repair protein from calf liver mitochondria. Nucleic Acids Res. 2000;28:3206 –3215. doi: 10.1093/nar/28.17.3206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Souza-Pinto NC, Croteau DL, Hudson EK, Hansford RG, Bohr VA. Age-associated increase in 8-oxodeoxyguanosine glycosylase/AP lyase activity in rat mitochondria. Nucleic Acids Res. 1999;27:1935–1942. doi: 10.1093/nar/27.8.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mansouri A, Demeilliers C, Amsellem S, Pessayre D, Fromenty B. Acute ethanol administration oxidatively damages and depletes mitochondrial DNA in mouse liver, brain, heart, and skeletal muscles: protective effects of antioxidants. J Pharmacol Exp Ther. 2001;298:737–743. [PubMed] [Google Scholar]
- 79.Bogenhagen DF, Pinz KG, Perez-Jannotti RM. Enzymology of mitochondrial base excision repair. Prog Nucleic Acid Res Mol Biol. 2001;68:257–271. doi: 10.1016/s0079-6603(01)68105-4. [DOI] [PubMed] [Google Scholar]
- 80.Fisher RP, Clayton DA. Purification and characterization of human mitochondrial transcription factor 1. Mol Cell Biol. 1988;8:3496 –3509. doi: 10.1128/mcb.8.8.3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bogenhagen DF. Interaction of mtTFB and mtRNA polymerase at core promoters for transcription of Xenopus laevis mtDNA. J Biol Chem. 1996;271:12036 –12041. [PubMed] [Google Scholar]
- 82.Chang DD, Clayton DA. A mammalian mitochondrial RNA processing activity contains nucleus-encoded RNA. Science. 1987;235:1178 –1184. doi: 10.1126/science.2434997. [DOI] [PubMed] [Google Scholar]
- 83.Schmitt ME, Clayton DA. Yeast site-specific ribonucleoprotein endoribonuclease MRP contains an RNA component homologous to mammalian RNase MRP RNA and essential for cell viability. Genes Dev. 1992;6:1975–1985. doi: 10.1101/gad.6.10.1975. [DOI] [PubMed] [Google Scholar]
- 84.Wong TW, Clayton DA. DNA primase of human mitochondria is associated with structural RNA that is essential for enzymatic activity. Cell. 1986;45:817–825. doi: 10.1016/0092-8674(86)90556-8. [DOI] [PubMed] [Google Scholar]
- 85.Hehman GL, Hauswirth WW. DNA helicase from mammalian mitochondria. Proc Natl Acad Sci U S A. 1992;89:8562–8566. doi: 10.1073/pnas.89.18.8562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Van Dyck E, Foury F, Stillman B, Brill SJ. A single-stranded DNA binding protein required for mitochondrial DNA replication in S. cerevisiae is homologous to E. coli SSB. EMBO J. 1992;11:3421–3430. doi: 10.1002/j.1460-2075.1992.tb05421.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tiranti V, Rocchi M, DiDonato S, Zeviani M. Cloning of human and rat cDNAs encoding the mitochondrial single-stranded DNA-binding protein (SSB) Gene. 1993;126:219 –225. doi: 10.1016/0378-1119(93)90370-i. [DOI] [PubMed] [Google Scholar]
- 88.Larsson NG, Wang J, Wilhelmsson H, Oldfors A, Rustin P, Lewandoski M, Barsh GS, Clayton DA. Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nat Genet. 1998;18:231–236. doi: 10.1038/ng0398-231. [DOI] [PubMed] [Google Scholar]
- 89.Lakshmipathy U, Campbell C. Antisense-mediated decrease in DNA ligase III expression results in reduced mitochondrial DNA integrity. Nucleic Acids Res. 2001;29:668 –676. doi: 10.1093/nar/29.3.668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tang Y, Schon EA, Wilichowski E, Vazquez-Memije ME, Davidson E, King MP. Rearrangements of human mitochondrial DNA (mtDNA): new insights into the regulation of mtDNA copy number and gene expression. Mol Biol Cell. 2000;11:1471–1485. doi: 10.1091/mbc.11.4.1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kroemer G, Dallaporta B, Resche-Rigon M. The mitochondrial death/life regulator in apoptosis and necrosis. Annu Rev Physiol. 1998;60:619 –642. doi: 10.1146/annurev.physiol.60.1.619. [DOI] [PubMed] [Google Scholar]
- 92.Reed JC, Miyashita T, Takayama S, Wang HG, Sato T, Krajewski S, Aime-Sempe C, Bodrug S, Kitada S, Hanada M. BCL-2 family proteins: regulators of cell death involved in the pathogenesis of cancer and resistance to therapy. J Cell Biochem. 1996;60:23–32. doi: 10.1002/(SICI)1097-4644(19960101)60:1%3C23::AID-JCB5%3E3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 93.Chao DT, Korsmeyer SJ. BCL-2 family: regulators of cell death. Annu Rev Immunol. 1998;16:395–419. doi: 10.1146/annurev.immunol.16.1.395. [DOI] [PubMed] [Google Scholar]
- 94.Alnemri ES. Hidden powers of the mitochondria. Nat Cell Biol. 1999;1:E40 –E42. doi: 10.1038/10034. [DOI] [PubMed] [Google Scholar]
- 95.Bernardi P, Petronilli V, Di Lisa F, Forte M. A mitochondrial perspective on cell death. Trends Biochem Sci. 2001;26:112–117. doi: 10.1016/s0968-0004(00)01745-x. [DOI] [PubMed] [Google Scholar]
- 96.Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochem J. 1999;341:233–249. [PMC free article] [PubMed] [Google Scholar]
- 97.Brustovetsky N, Klingenberg M. Mitochondrial ADP/ATP carrier can be reversibly converted into a large channel by Ca2+ Biochemistry. 1996;35:8483–8488. doi: 10.1021/bi960833v. [DOI] [PubMed] [Google Scholar]
- 98.Marzo I, Brenner C, Zamzami N, Jurgensmeier JM, Susin SA, Vieira HL, Prevost MC, Xie Z, Matsuyama S, Reed JC, Kroemer G. Bax and adenine nucleotide translocator cooperate in the mitochondrial control of apoptosis. Science. 1998;281:2027–2031. doi: 10.1126/science.281.5385.2027. [DOI] [PubMed] [Google Scholar]
- 99.Shimizu S, Narita M, Tsujimoto Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature. 1999;399:483–487. doi: 10.1038/20959. [DOI] [PubMed] [Google Scholar]
- 100.Leist M, Single B, Castoldi AF, Kuhnle S, Nicotera P. Intracellular adenosine triphosphate (ATP) concentration: a switch in the decision between apoptosis and necrosis. J Exp Med. 1997;185:1481–1486. doi: 10.1084/jem.185.8.1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nicotera P, Ankarcrona M, Bonfoco E, Orrenius S, Lipton SA. Neuronal necrosis and apoptosis: two distinct events induced by exposure to glutamate or oxidative stress. Adv Neurol. 1997;72:95–101. [PubMed] [Google Scholar]
- 102.Wu D, Cederbaum AI. Ethanol-induced apoptosis to stable HepG2 cell lines expressing human cytochrome P-4502E1. Alcohol Clin Exp Res. 1999;23:67–76. [PubMed] [Google Scholar]
- 103.Pastorino JG, Marcinkeviciute A, Cahill A, Hoek JB. Potentiation by chronic ethanol treatment of the mitochondrial permeability transition. Biochem Biophys Res Commun. 1999;265:405– 409. doi: 10.1006/bbrc.1999.1696. [DOI] [PubMed] [Google Scholar]
- 104.Kristal BS, Park BK, Yu BP. 4-Hydroxyhexenal is a potent inducer of the mitochondrial permeability transition. J Biol Chem. 1996;271:6033–6038. doi: 10.1074/jbc.271.11.6033. [DOI] [PubMed] [Google Scholar]
- 105.Ramachandran V, Perez A, Chen J, Senthil D, Schenker S, Henderson GI. In utero ethanol exposure causes mitochondrial dysfunction, which can result in apoptotic cell death in fetal brain: a potential role for 4-hydroxynonenal. Alcohol Clin Exp Res. 2001;25:862–871. [PubMed] [Google Scholar]
- 106.Garcia-Ruiz C, Colell A, Mari M, Morales A, Fernandez-Checa JC. Direct effect of ceramide on the mitochondrial electron transport chain leads to generation of reactive oxygen species. Role of mitochondrial glutathione. J Biol Chem. 1997;272:11369 –11377. doi: 10.1074/jbc.272.17.11369. [DOI] [PubMed] [Google Scholar]
- 107.Arora AS, Jones BJ, Patel TC, Bronk SF, Gores GJ. Ceramide induces hepatocyte cell death through disruption of mitochondrial function in the rat. Hepatology. 1997;25:958 –963. doi: 10.1002/hep.510250428. [DOI] [PubMed] [Google Scholar]
- 108.Gudz TI, Tserng KY, Hoppel CL. Direct inhibition of mitochondrial respiratory chain complex III by cell-permeable ceramide. J Biol Chem. 1997;272:24154 –24158. doi: 10.1074/jbc.272.39.24154. [DOI] [PubMed] [Google Scholar]
- 109.Kolesnick RN, Kronke M. Regulation of ceramide production and apoptosis. Annu Rev Physiol. 1998;60:643–665. doi: 10.1146/annurev.physiol.60.1.643. [DOI] [PubMed] [Google Scholar]
- 110.Hannun YA, Luberto C, Argraves KM. Enzymes of sphingolipid metabolism: from modular to integrative signaling. Biochemistry. 2001;40:4893–4903. doi: 10.1021/bi002836k. [DOI] [PubMed] [Google Scholar]
- 111.Cuvillier O, Pirianov G, Kleuser B, Vanek PG, Coso OA, Gutkind S, Spiegel S. Suppression of ceramide-mediated programmed cell death by sphingosine-1-phosphate. Nature. 1996;381:800 –803. doi: 10.1038/381800a0. [DOI] [PubMed] [Google Scholar]
- 112.Prieschl EE, Csonga R, Novotny V, Kikuchi GE, Baumruker T. The balance between sphingosine and sphingosine-1-phosphate is decisive for mast cell activation after Fc epsilon receptor I triggering. J Exp Med. 1999;190:1–8. doi: 10.1084/jem.190.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Cuvillier O, Rosenthal DS, Smulson ME, Spiegel S. Sphingosine 1-phosphate inhibits activation of caspases that cleave poly-(ADP-ribose) polymerase and lamins during Fas- and ceramide-mediated apoptosis in Jurkat T lymphocytes. J Biol Chem. 1998;273:2910 –2916. doi: 10.1074/jbc.273.5.2910. [DOI] [PubMed] [Google Scholar]
- 114.Spiegel S. Sphingosine 1-phosphate: a prototype of a new class of second messengers. J Leukoc Biol. 1999;65:341–344. doi: 10.1002/jlb.65.3.341. [DOI] [PubMed] [Google Scholar]
- 115.De Vito WJ, Xhaja K, Stone S. Prenatal alcohol exposure increases TNFalpha-induced cytotoxicity in primary astrocytes. Alcohol. 2000;21:63–71. doi: 10.1016/s0741-8329(00)00078-1. [DOI] [PubMed] [Google Scholar]
- 116.Ichijo H, Nishida E, Irie K, ten Dijke P, Saitoh M, Moriguchi T, Takagi M, Matsumoto K, Miyazono K, Gotoh Y. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science. 1997;275:90–94. doi: 10.1126/science.275.5296.90. [DOI] [PubMed] [Google Scholar]
- 117.Chen YR, Wang X, Templeton D, Davis RJ, Tan TH. The role of c-Jun N-terminal kinase (JNK) in apoptosis induced by ultraviolet C and gamma radiation. Duration of JNK activation may determine cell death and proliferation. J Biol Chem. 1996;271:31929 –31936. doi: 10.1074/jbc.271.50.31929. [DOI] [PubMed] [Google Scholar]
- 118.Harada J, Sugimoto M. An inhibitor of p38 and JNK MAP kinases prevents activation of caspase and apoptosis of cultured cerebellar granule neurons. Jpn J Pharmacol. 1999;79:369 –378. doi: 10.1254/jjp.79.369. [DOI] [PubMed] [Google Scholar]
- 119.Kharbanda S, Saxena S, Yoshida K, Pandey P, Kaneki M, Wang Q, Cheng K, Chen YN, Campbell A, Sudha T, Yuan ZM, Narula J, Weichselbaum R, Nalin C, Kufe D. Translocation of SAPK/JNK to mitochondria and interaction with Bcl-x(L) in response to DNA damage [published erratum appears in J Biol Chem 2000 Jun 23;275(25):19433] J Biol Chem. 2000;275:322–327. doi: 10.1074/jbc.275.1.322. [DOI] [PubMed] [Google Scholar]
- 120.Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y, Kawabata M, Miyazono K, Ichijo H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998;17:2596 –2606. doi: 10.1093/emboj/17.9.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Assefa Z, Vantieghem A, Garmyn M, Declercq W, Vandenabeele P, Vandenheede JR, Bouillon R, Merlevede W, Agostinis P. p38 mitogen-activated protein kinase regulates a novel, caspase-independent pathway for the mitochondrial cytochrome c release in ultraviolet B radiation-induced apoptosis. J Biol Chem. 2000;275:21416 –21421. doi: 10.1074/jbc.M002634200. [DOI] [PubMed] [Google Scholar]
- 122.Ghatan S, Larner S, Kinoshita Y, Hetman M, Patel L, Xia Z, Youle RJ, Morrison RS. p38 MAP kinase mediates bax translocation in nitric oxide-induced apoptosis in neurons. J Cell Biol. 2000;150:335–347. doi: 10.1083/jcb.150.2.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Rigobello MP, Callegaro MT, Barzon E, Benetti M, Bindoli A. Purification of mitochondrial thioredoxin reductase and its involvement in the redox regulation of membrane permeability. Free Radic Biol Med. 1998;24:370 –376. doi: 10.1016/s0891-5849(97)00216-5. [DOI] [PubMed] [Google Scholar]
- 124.Zhang J, Li YD, Patel JM, Block ER. Thioredoxin overexpression prevents NO-induced reduction of NO synthase activity in lung endothelial cells. Am J Physiol. 1998;275:L288 –293. doi: 10.1152/ajplung.1998.275.2.L288. [DOI] [PubMed] [Google Scholar]
- 125.McEnery MW. The mitochondrial benzodiazepine receptor: evidence for association with the voltage-dependent anion channel (VDAC) J Bioenerg Biomembr. 1992;24:63–69. doi: 10.1007/BF00769532. [DOI] [PubMed] [Google Scholar]
- 126.Bidder M, Weizman R, Fares F, Grel I, Gavish M. Chronic ethanol consumption and withdrawal affects mitochondrial benzodiazepine receptors in rat brain and peripheral organs. Biochem Pharmacol. 1992;44:1335–1339. doi: 10.1016/0006-2952(92)90534-p. [DOI] [PubMed] [Google Scholar]
- 127.Kokoszka JE, Coskun P, Esposito LA, Wallace DC. Increased mitochondrial oxidative stress in the SOD2 (+/−) mouse results in the age-related decline of mitochondrial function culminating in increased apoptosis. Proc Natl Acad Sci U S A. 2001;98:2278 –2283. doi: 10.1073/pnas.051627098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Mather M, Rottenberg H. Aging enhances the activation of the permeability transition pore in mitochondria. Biochem Biophys Res Commun. 2000;273:603–608. doi: 10.1006/bbrc.2000.2994. [DOI] [PubMed] [Google Scholar]
- 129.Wallace DC. Mitochondrial genetics: a paradigm for aging and degenerative diseases? Science. 1992;256:628 –632. doi: 10.1126/science.1533953. [DOI] [PubMed] [Google Scholar]
- 130.Lenaz G. Role of mitochondria in oxidative stress and ageing. Biochim Biophys Acta. 1998;1366:53–67. doi: 10.1016/s0005-2728(98)00120-0. [DOI] [PubMed] [Google Scholar]
- 131.Jacobson MD, Burne JF, King MP, Miyashita T, Reed JC, Raff MC. Bcl-2 blocks apoptosis in cells lacking mitochondrial DNA. Nature. 1993;361:365–369. doi: 10.1038/361365a0. [DOI] [PubMed] [Google Scholar]
- 132.Jiang S, Cai J, Wallace DC, Jones DP. Cytochrome c-mediated apoptosis in cells lacking mitochondrial DNA. Signaling pathway involving release and caspase 3 activation is conserved. J Biol Chem. 1999;274:29905–29911. doi: 10.1074/jbc.274.42.29905. [DOI] [PubMed] [Google Scholar]
- 133.Wang J, Silva JP, Gustafsson CM, Rustin P, Larsson NG. Increased in vivo apoptosis in cells lacking mitochondrial DNA gene expression. Proc Natl Acad Sci U S A. 2001;98:4038 –4043. doi: 10.1073/pnas.061038798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Dey R, Moraes CT. Lack of oxidative phosphorylation and low mitochondrial membrane potential decrease susceptibility to apoptosis and do not modulate the protective effect of Bcl-x(L) in osteosarcoma cells. J Biol Chem. 2000;275:7087–7094. doi: 10.1074/jbc.275.10.7087. [DOI] [PubMed] [Google Scholar]
- 135.Wallace DC. Mitochondrial diseases in man and mouse. Science. 1999;283:1482–1488. doi: 10.1126/science.283.5407.1482. [DOI] [PubMed] [Google Scholar]
- 136.Hoek JB. Mitochondrial energy metabolism in chronic alcoholism. In: Lee CP, editor. Molecular mechanisms of mitochondrial pathology: Current topics in Bioenergetics. Vol. 17. New York: Academic Press; 1994. pp. 197–241. [Google Scholar]
- 137.Cunningham CC, Bailey SM. Ethanol consumption and liver mitochondria function. Biol Signals Recept. 2001;10:271–282. doi: 10.1159/000046892. [DOI] [PubMed] [Google Scholar]
- 138.Spach PI, Herbert JS, Cunningham CC. The interaction between chronic ethanol consumption and oxygen tension in influencing the energy state of rat liver. Biochim Biophys Acta. 1991;1056:40–46. doi: 10.1016/s0005-2728(05)80070-2. [DOI] [PubMed] [Google Scholar]
- 139.McClain CJ, Barve S, Deaciuc I, Kugelmas M, Hill D. Cytokines in alcoholic liver disease. Semin Liver Dis. 1999;19:205–219. doi: 10.1055/s-2007-1007110. [DOI] [PubMed] [Google Scholar]