

Abstract
The probability that epitope spreading occurs in multiple sclerosis (MS) and the fact that patients have been shown to respond to multiple myelin epitopes concurrently makes the use of peptide-specific tolerance therapies targeting single epitopes problematic. To attempt to overcome this limitation, we have employed cocktails of peptides in the ECDI coupled-APC tolerance system in mice to determine if T cell responses to multiple autoepitopes can be targeted simultaneously. Preventative tolerance induced with splenocytes coupled with a peptide cocktail of four distinct encephalitogenic epitopes (PLP139–151, PLP178–191, MBP84–104, and MOG92–106) inhibited initiation of active EAE induced with each individual peptide and by a mixture of the four peptides by preventing activation of autoreactive Th1 cells and subsequent infiltration of inflammatory cells into the CNS. Most relevant to treatment of clinical MS, therapeutic tolerance initiated by splenocytes coupled with the peptide cocktail administered at the peak of acute disease prevented clinical relapses due to epitope spreading and ameliorated a diverse disease induced with a mixture of the four peptides. Interestingly, therapeutic tolerance appeared to be mediated by a mechanism distinct from preventative tolerance, i.e. by significantly increasing the levels of production of the anti-inflammatory cytokines TGF-β and/or IL-10 in both the periphery and the CNS.
Keywords: Tolerance/Suppression/Anergy, Antigens/Peptides/Epitopes, Autoimmunity, T Cells, EAE/MS
Introduction
Multiple sclerosis (MS) is an immune-mediated demyelinating disease of the central nervous system (CNS) that afflicts approximately 1.1 million people worldwide[1]. The etiology of MS is unknown, but it is well-accepted that both genetic and environmental factors (likely infectious) play key roles in the susceptibility to and initiation of the disease [2–5]. While the precise pathogenic progression of MS is unknown, evidence from both MS patients [6–9] and from experimental autoimmune encephalomyelitis (EAE) [10–14], an animal model of MS, supports a major pathogenic role for autoreactive myelin-specific CD4+ T cells. It is unknown at this time how these myelin-specific T cells are initially activated, but the activated T cells produce proinflammatory cytokines, which in turn chemoattract and activate CNS-resident (microglia) and peripheral (macrophages) mononuclear cells which mediate bystander myelin destruction. Progressive disease in relapsing EAE (R-EAE) [15–20] has been shown to be primarily mediated by de novo activation of naïve T cells specific for endogenously released myelin antigens, a process known as epitope spreading. For example, in PLP139–151-induced R-EAE in the SJL mouse, there is a sequential and hierarchical order of epitope spreading in which the first relapse is associated with CD4+ Th1 cell reactivity to PLP178–191, and the second to MBP84–104. In both the SJL and (SWR × SJL)F1 R-EAE models, only tolerance to the primary spread epitope after recovery from acute clinical disease could prevent relapses and/or disease progression [17,18]. Relevant to MS therapy, a number of reports have also demonstrated epitope spreading in relapsing-remitting MS (RR-MS) patients [21–25].
Most of the currently approved therapies for the treatment of RR-MS use various antigen-nonspecific immunosuppressive and/or anti-inflammatory strategies and are only partially effective [26–28]. Currently, glatiramer acetate (GA) (copaxone®), a random polymer of L-alanine, L-glutamic acid, L-lysine, and L-tyrosine, is the only approved therapy for MS that is purported to act in a semi-antigen-specific manner [29]. GA is thought to induce T cells that produce Th2 cytokines such as IL-4, IL-5, IL-10 and IL-13 which suppress inflammatory cells [30,31]. GA treatment requires daily subcutaneous (s.c.) injections, is beneficial to only a minority of relapsing-remitting MS patients [32], and 10% of patients experience significant side effects [33]. Antigen-specific therapeutic approaches, including oral tolerance and altered peptide ligands (APLs), have shown promise in EAE, but were not particularly efficacious in subsequent patient trials [34–36]. Thus, there is clearly still a need for new antigen-specific therapies for MS and other autoimmune diseases. Ideally these would be effective with a less severe treatment schedule and have reduced side effects.
There are multiple issues which must be addressed when devising an antigen-specific therapy for MS. First, because there is clear evidence of diversity in the activated T cell repertoire in MS patients [21,24,25], any antigen-specific therapy must be capable of simultaneously targeting T cells specific for multiple myelin epitopes. Second, as the etiology of MS is unknown, disease prevention is impractical at this time, and therefore any treatment must be capable of ameliorating ongoing disease. Thirdly, because changes in the repertoire of activated T cells over time have been demonstrated [21–24], any antigen-specific therapy must be able to affect previously activated autoreactive T cells as well as naïve autoreactive T cells. We have previously employed tolerance induced by the i.v. administration of ethylene carbodiimide (ECDI)-fixed syngeneic splenic APCs (Ag-SP) coupled to individual myelin peptides or to whole myelin proteins to safely and effectively prevent the induction of active R-EAE [37], to treat active R-EAE at peak disease [38], to inhibit the expression of adoptive R-EAE [39,40], and to treat clinical relapses in SJL/J mice by tolerizing to the appropriate spread epitope after the initial acute episode [15,17,41]. However, whether multiple myelin epitopes could be simultaneously coupled to syngeneic splenocytes and presented to naïve and activated autoreactive CD4+ T cells efficiently enough to induce immune tolerance to treat established R-EAE has not been determined. The current experiments indicate that tolerance to a cocktail of peptides can be successfully induced using the ECDI-coupled cell tolerance protocol to simultaneously target T cells of multiple specificities that are actively engaged in CNS pathology. Moreover, the data indicate that tolerance in naïve vs. activated T cells is mediated by distinct mechanisms.
Materials and Methods
Mice
SJL mice, 6–7 weeks old, were purchased from Harlan Laboratories, Bethesda, MD. All mice were housed under specific pathogen-free conditions (SPF) in the Northwestern University Center for Comparative Medicine. Paralyzed animals were afforded easier access to food and water.
Reagents
Synthetic peptides PLP139–151 (HSLGKWLGHPDKF), PLP178–191 (NTWTTSQSIAFPSK), MBP84–104 (VHFFKNIVTPRTPPPSQGKGR), MOG92–106 (DEGGYTCFFRDHSYQ), PLP56–70 (DYEYLINVIHAFQYV), VP270–86 (WTSQEAFSHIRIPLP), VP1233–250 (SASVRIRYKKMKVFCPRP), and OVA323–339 (ISQAVHAAHAEINEAGR) were purchased from Genemed, San Francisco, CA.
Induction and Clinical Evaluation of EAE
Female mice 8–10 weeks old were immunized s.c. at three spots on the flank with 100 μl of an emulsion of peptide in CFA containing 200 μg Mycobacterium tuberculosis H37Ra (Difco, Detroit, MI) on day 0. In some experiments, mice also received another 100 μl peptide/CFA emulsion on day 7 and/or 200 ng Bordetella pertussis toxin (List Biological Laboratories, Campbell, CA) in 200 μl of PBS i.p. on days 0 and 2. Individual animals were observed every 1–3 days, and clinical scores were assessed on a scale of 0–4 as follows: 0 = no abnormality; 1 = limp tail or hind limb weakness; 2 = limp tail and hind limb weakness; 3 = partial hind limb paralysis; 4 = complete hind limb paralysis. Data are reported as the mean daily clinical score.
Antigen-coupled splenocyte tolerance
Peripheral tolerance induction using peptide-coupled splenocytes was performed as previously described [42]. Briefly, erythrocyte-free (Tris-NH4Cl-treated) splenocyte suspensions were coupled with peptides using ethylene carbodiimide (ECDI). Cells were washed 2X in PBS, resuspended at 3.2×106/ml in PBS with 1 mg/ml of each peptide and 30.75 mg/ml ECDI (CalBiochem, La Jolla, CA), and incubated for 1 h at 4°C with periodic shaking. Peptide-coupled cells were then washed 3X in PBS, filtered through a 70 μm cell strainer, and resuspended at 250×106 cells/ml in PBS. Each mouse received 50×106 peptide-coupled cells in 200 μl PBS i.v. Coupling efficiency has previously been determined to be approximately 30% [42].
Delayed-Type Hypersensitivity (DTH) Responses
DTH responses were quantitated using a 24 hr ear swelling assay as previously described [43]. Pre-challenge ear thickness was determined using a Mitutoyo model 7326 engineer’s micrometer (Schlesinger’s Tools, Brooklyn, New York). Immediately thereafter, DTH responses were elicited by injecting 10 μg of peptide in 10 μl of PBS into the dorsal surface of the ear using a 100 μl Hamilton syringe fitted with a 30 gauge needle. The increase in ear thickness over pre-challenge measurements was determined 24 hr after ear challenge. Results are expressed in units of 10−4 inches ± SEM.
In Vitro Antigen-Specific Recall Responses
Spleens and draining lymph nodes (axillary, brachial, and inguinal) were harvested, and single-cell suspensions were obtained by mashing through sterile 60-mesh wire screens. In 96-well microtiter plates, 5 × 105 erythrocyte-free (Tris-NH4Cl-treated) splenocytes or lymph node cells per well were incubated in supplemented culture medium with or without antigen or with anti-CD3 antibody at 37°C in 7% CO2 for 48 hr and then pulsed with 1 μCi/well [3H]Thy for the final 24 h of culture. Proliferation was determined by uptake of [3H]Thy detected using a Topcount Microplate Scintillation Counter (Packard Instruments, Meridan, CT). Results are expressed as the mean of triplicate cultures ± SEM. Alternatively, supernatants were collected at 48 hr for cytokine analysis. Cytokine measurements were performed using the Mouse Cytokine 10-Plex system and Luminex Liquidchip analyzer (Qiagen, Valencia, CA) or ELISA.
Immunohistochemistry
Mice were anesthetized and perfused with 30 ml PBS on the indicated days post-immunization. Spinal cords were removed by dissection, and 2- to 3-mm spinal cord blocks were immediately frozen in OCT (Miles Laboratories; Elkhart, IN) in liquid nitrogen. The blocks were stored at −80°C in plastic bags to prevent dehydration. Six micrometer thick cross-sections from the lumbar region (approximately L2-L3) were cut on a Reichert-Jung Cyocut CM1850 cryotome (Leica, Deerfield, IL), mounted on Superfrost Plus electrostatically charged slides (Fisher, Pittsburgh, PA), air dried, and stored at −80°C. Slides were stained using a Tyramide Signal Amplification (TSA) Direct kit (NEN, Boston, MA) according to manufacturer’s instructions. Lumbar sections from each group were thawed, air-dried, fixed in 2% paraformaldehyde at room temperature, and rehydrated in 1x PBS. Nonspecific staining was blocked using anti-CD16/CD32, (FcIII/IIR, 2.4G2; BD PharMingen), and an avidin/biotin blocking kit (Vector Laboratories) in addition to the blocking reagent provided by the TSA kit. Tissues were stained with biotin-conjugated Abs anti-mouse CD4 (H129.19) (BD Biosciences, San Jose, CA) and anti-mouse F4/80 (BM8) (Caltag, Burlingame, CA). Sections were coverslipped with Vectashield mounting medium including DAPI (Vector Laboratories, Burlingame, CA). Slides were examined and images were acquired via epifluorescence using the SPOT RT camera (Diagnostic Instruments, Sterling Heights, MI) and Metamorph imaging software (Universal Imaging, Downingtown, PA). Eight non-serial lumbar sections from each sample per group were analyzed at 100x and 200x magnification.
Statistical Analyses
Comparison of the percentage of animals showing clinical disease symptoms or of DTH responses between any two groups of mice was done by two-tailed unpaired student’s t test. p values < 0.05 were considered significant.
Results
Preventative coupled-cell tolerance to multiple myelin peptides prevents clinical EAE induced either with individual encephalitogenic myelin peptides or with multiple myelin peptides
To determine whether multiple myelin peptides could be coupled to syngeneic splenocytes and presented to naïve CD4+ T cells efficiently enough to protect against EAE induced with individual myelin peptides as well as multiple myelin peptides, SJL mice were given 50×106 syngeneic splenocytes coupled to four encephalitogenic myelin peptides (PLP139–151, PLP178–191, MBP84–104, and MOG92–106) (Tolerized) or four irrelevant peptides (PLP56–70, OVA323–339, VP1233–250, and VP270–86) (Sham) seven days before priming with each peptide individually (Figure 1), or a combination of all four encephalitogenic myelin peptides (Figure 2). Not only did the tolerization protocol completely prevent or drastically reduce clinical symptoms in every situation, but DTH responses, as an in vivo measure of myelin peptide-specific Th1/17 activity, were also significantly decreased compared to sham-tolerized controls. In addition, peripheral peptide-specific T cell recall proliferative responses were significantly decreased in tolerized vs. sham-tolerized mice (Figure 3a). This was true for mice primed with any of the peptides individually (data not shown) and for mice primed with a combination of all four peptides (Figure 3). Analysis of culture supernatants revealed a decrease in the levels of myelin peptide-induced proinflammatory cytokines IL-2 (Figure 3b) and IFN-γ (Figure 3c) produced in vitro by cells from tolerized vs. sham-tolerized mice and equivalent or slightly decreased levels of the regulatory cytokine TGF-□ (Figure 3d). IL-4, IL-5, and IL-10 responses were not seen upon peptide recall in either the tolerized or sham groups (data not shown). Together, these data indicate that preventative tolerance with Ag-SP coupled with multiple peptides leads to induction of anergy in naive myelin-specific T cells even when multiple specificities are targeted simultaneously. Furthermore, immunohistochemical evaluation of the spinal cord revealed a drastically decreased amount of infiltrating T cells and macrophages in tolerized mice compared to sham-tolerized mice (Figure 4). This is likely the result of peripheral anergy induction since only activated T cells are capable of crossing the blood-brain barrier.
Figure 1. Preventative coupled-cell tolerance to multiple myelin peptides prevents clinical EAE induced with individual encephalitogenic myelin peptides.
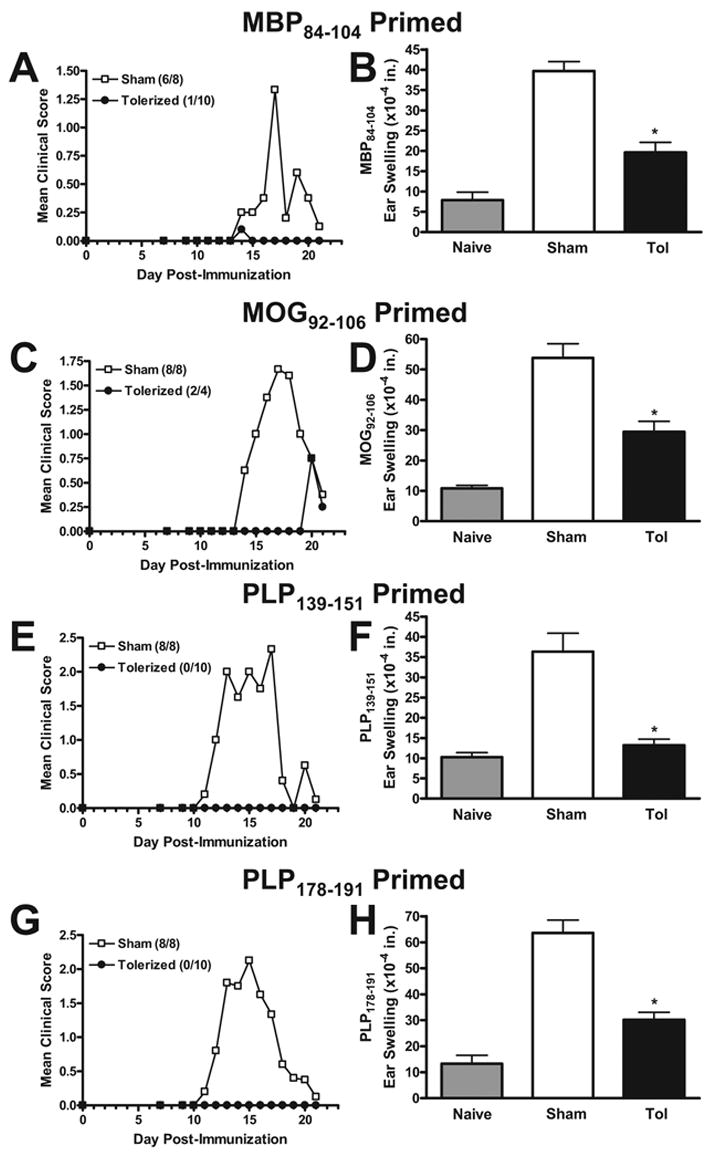
Female SJL mice were treated i.v. with 50×106 syngeneic splenocytes coupled to a myelin peptide cocktail consisting of PLP139–151, PLP178–191, MBP84–104, and MOG92–106 (Tolerized) or to a control peptide cocktail consisting of PLP56–70, OVA323–339, VP1233–250, and VP270–86 (Sham) seven days before priming. Four to ten mice per group were immunized s.c. with 200 μg MBP84–104/CFA on days 0 and 7 (A & B), 200 μg MOG92–106/CFA on days 0 and 7 (C & D), 50 μg PLP139–151/CFA on day 0 (E & F), or 100 μg PLP178–191/CFA on day 0 (G & H). Mice receiving MBP84–104 or MOG92–106 were also given 200 ng Bordatella pertussis toxin i.p. on days 0 and 2. For DTH, mice were ear challenged with 10 μg of the indicated peptides on day 20, and swelling was measured 24 h later. Data represents two separate experiments. *DTH response significantly less than sham-tolerized controls, p < 0.01.
Figure 2. Preventative coupled-cell tolerance to multiple myelin peptides prevents clinical EAE induced with a cocktail of encephalitogenic myelin peptides.
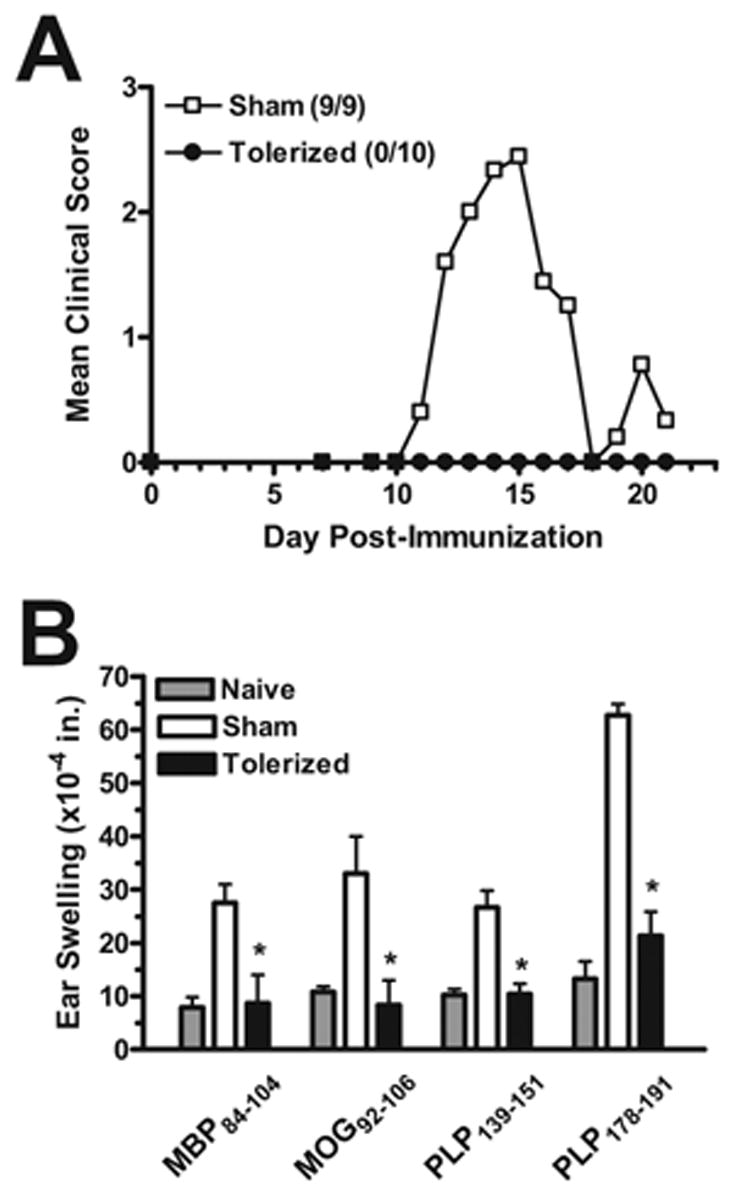
Female SJL mice were treated i.v. with 50×106 syngeneic splenocytes coupled to a myelin peptide cocktail consisting of PLP139–151, PLP178–191, MBP84–104, and MOG92–106 (Tolerized) or to a control peptide cocktail consisting of PLP56–70, OVA323–339, VP1233–250, and VP270–86 (Sham) seven days before priming. Nine to ten mice per group were immunized s.c. with a cocktail of 100 μg MBP84–104, 100 μg MOG92–106, 25 μg PLP139–151, and 50 μg PLP178–191/CFA on day 0. For DTH, mice were ear challenged with 10 μg of the indicated peptides on day 20, and swelling was measured 24 h later. *DTH response significantly less than sham-tolerized controls, p < 0.01. Data represents two separate experiments.
Figure 3. Preventative coupled-cell tolerance to multiple myelin peptides inhibits peripheral peptide-specific T cell recall responses.
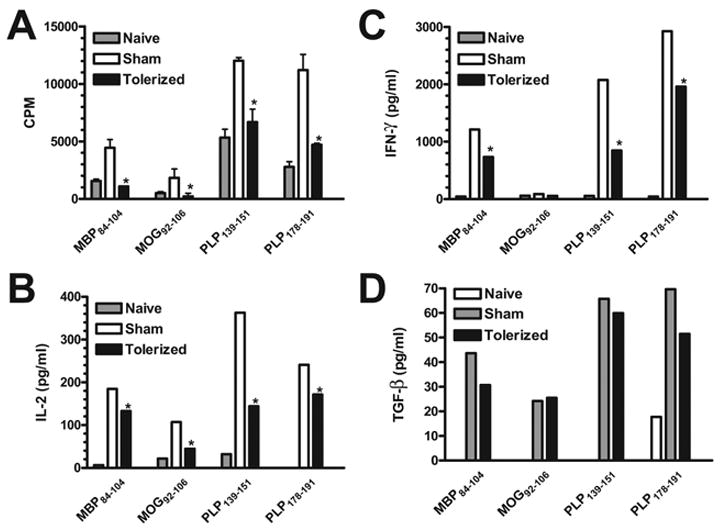
Female SJL mice were treated i.v. with 50×106 syngeneic splenocytes coupled to a myelin peptide cocktail consisting of PLP139–151, PLP178–191, MBP84–104, and MOG92–106 (Tolerized) or to a control peptide cocktail consisting of PLP56–70, OVA323–339, VP1233–250, and VP270–86 (Sham) seven days before priming. Three to five mice per group were immunized s.c. with a cocktail of 100 μg MBP84–104, 100 μg MOG92–106, 25 μg PLP139–151, and 50 μg PLP178–191/CFA on day 0. Draining lymph nodes were harvested from 3 representative mice per group on day 21. Single cell suspensions were incubated for 3 days with the indicated peptides and proliferative responses assessed by uptake of [3H] thymidine (A). Data is presented as mean CPM ± SEM. Single cell suspensions were incubated for 48 hr with the indicated antigens prior to measurement of IL-2 (B) and IFN-γ (C) levels via cytokine array analysis and TGF-β (D) levels via ELISA. Cytokine data is presented as mean pg/ml. *Responses significantly less than sham-tolerized controls, p < 0.05.
Figure 4. Preventative coupled-cell tolerance to multiple myelin peptides inhibits inflammatory spinal cord infiltrate.
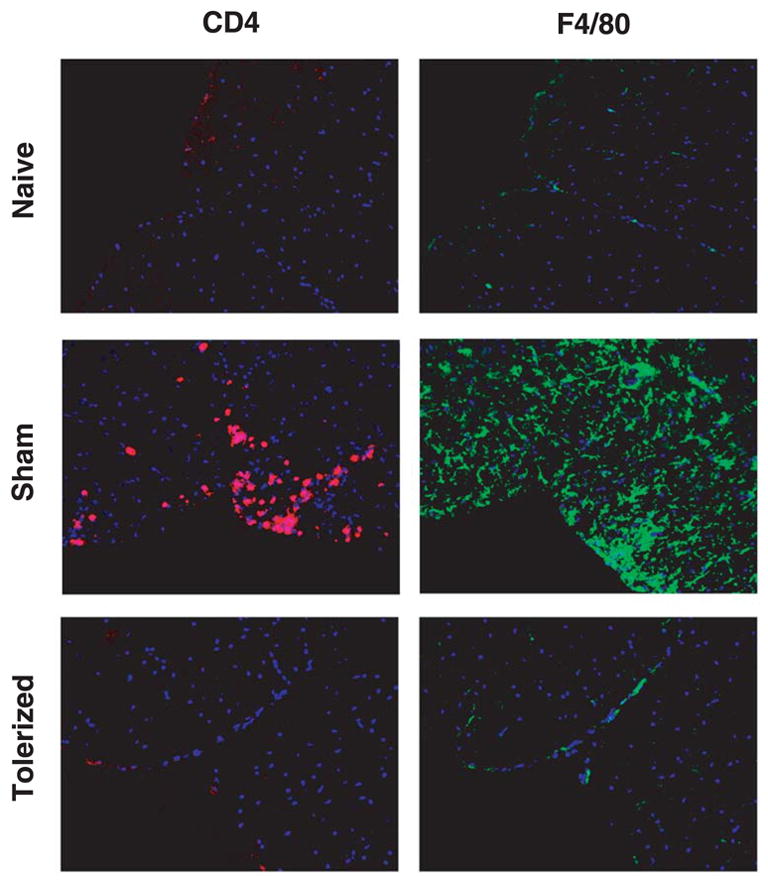
Female SJL mice were treated i.v. with 50×106 syngeneic splenocytes coupled to a myelin peptide cocktail consisting of PLP139–151, PLP178–191, MBP84–104, and MOG92–106 (Tolerized) or to a control peptide cocktail consisting of PLP56–70, OVA323–339, VP1233–250, and VP270–86 (Sham) seven days before priming. Three to five mice per group were immunized s.c. with a cocktail of 100 μg MBP84–104, 100 μg MOG92–106, 25 μg PLP139–151, and 50 μg PLP178–191/CFA on day 0. Representative mice from each group were perfused on day 21. Spinal cords were harvested and frozen in OCT. Lumbar regions were sectioned into 6 μm slices and stained with anti-CD4 (red) or anti-F4/80 (green) and counterstained with DAPI (blue). Sections shown at 200x and are representative of 3 individual mice per group.
Therapeutic coupled-cell tolerance to multiple myelin peptides in established R-EAE induced by individual myelin peptides ameliorates disease relapses
Since the etiology of MS is unknown at this time, prevention of this disease is not yet an option. That leaves treatment of ongoing disease as the only alternative for clinicians. Therefore, it is imperative to demonstrate that any experimental treatment regimen proposed as a possible therapy for MS can ameliorate ongoing disease. In addition, due to the phenomenon of epitope spreading in EAE [15–18] and its possible role in MS clinical relapses [21], it is important to demonstrate that multiple antigen coupled-cell tolerance can inhibit relapses to spread epitopes distinct from the initiating epitope. To test this, female SJL mice were primed with PLP139–151 or PLP178–191, which both result in relapsing-remitting disease in which the first relapse is due to intramolecular epitope spreading [15–17]. At the peak of acute disease, during which there is only CD4+ T cell reactivity to the initiating epitope, 50 × 106 syngeneic splenocytes coupled to four encephalitogenic myelin peptides (PLP139–151, PLP178–191, MBP84–104, and MOG92–106) (Tolerized), including both the initiating and spread epitopes, or four irrelevant peptides (PLP56–70, OVA323–339, VP1233–250, and VP270–86) (Sham) were given i.v., and mice were monitored for the development of clinical relapses. In both cases, the multi-peptide tolerization protocol significantly decreased the incidence and severity of clinical relapses to the spread epitope (Figure 5a). In addition, peripheral T cell responses were diminished to both the initiating and the spread epitopes as measured by DTH (Figure 5b). This demonstrates that both previously activated and naïve T cells can be tolerized simultaneously with splenocytes coupled to a myelin peptide cocktail.
Figure 5. Therapeutic coupled-cell tolerance to multiple myelin peptides in established R-EAE induced by individual myelin peptides ameliorates disease relapses.
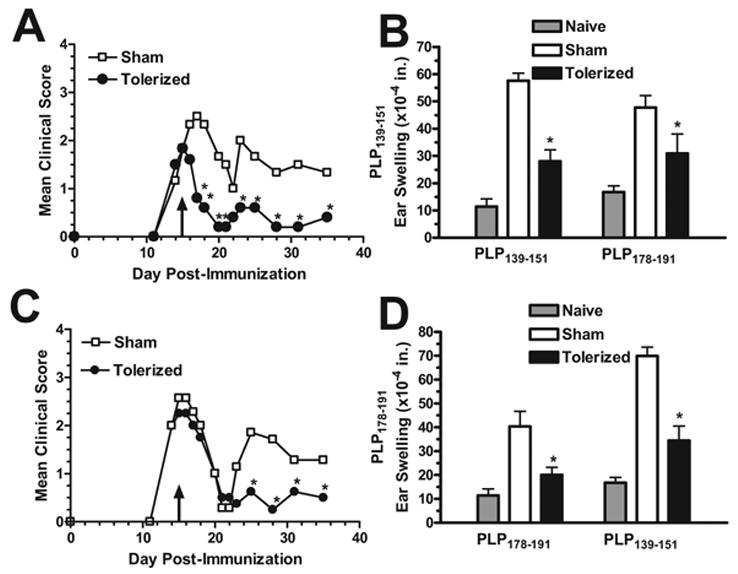
Twenty-eight female SJL mice were primed s.c. with either 50 μg PLP139–151 (A & B) or 100 μg PLP178–191 (C & D) in CFA on day 0 and monitored for clinical disease. At day 15 (indicated by arrows), during the acute phase of the disease, mice were given 50×106 syngeneic splenocytes coupled to a myelin peptide cocktail consisting of PLP139–151, PLP178–191, MBP84–104, and MOG92–106 (Tolerized) or to a control peptide cocktail consisting of PLP56–70, OVA323–339, VP1233–250, and VP270–86 (Sham) i.v. For DTH, mice were ear challenged with 10 μg peptide on day 30, and swelling was measured 24 h later. *Response significantly less than sham-tolerized controls, p < 0.01. Data is representative of two separate experiments.
Therapeutic coupled-cell tolerance to multiple myelin peptides in established R-EAE induced by immunization with a cocktail of encephalitogenic myelin peptides ameliorates clinical disease
There is clear evidence of diversity in the activated T cell repertoire in MS patients [21–25,44]. Consequently, it is necessary to establish that a specific treatment regimen can affect ongoing disease in which T cells of multiple specificities are actively playing pathologic roles. In established SJL models of relapsing-remitting EAE, the acute phase is primarily due to autoreactivity specific for only one myelin epitope. In single peptide-induced models, the acute phase is mediated by CD4+ T cells specific for the initiating epitope, and even in disease models that are initiated by priming with whole protein or mouse spinal cord homogenate (MSCH), acute disease is primarily mediated by T cells reactive to the immunodominant myelin epitope [12,15,37,43]. Therefore, in order to evaluate the ability of multiple antigen coupled-cell tolerance to ameliorate clinical symptoms caused by simultaneous reactivity to multiple self-antigens, mice were primed with a combination of PLP139–151, PLP178–191, MBP84–104, and MOG92–106/CFA. At the peak of acute disease, mice were tolerized with 50 × 106 syngeneic splenocytes coupled to a cocktail of all four encephalitogenic myelin peptides or a cocktail of four control peptides. Remarkably, clinical symptoms in tolerized mice were significantly ameliorated compared to sham-tolerized controls and remained suppressed until the termination of the experiment (Figure 6a). Additionally, DTH responses were significantly decreased to each of the four myelin antigens after tolerance induction (Figure 6b).
Figure 6. Therapeutic coupled-cell tolerance to multiple myelin peptides in established R-EAE induced by immunization with a cocktail of encephalitogenic myelin peptides ameliorates disease relapses.
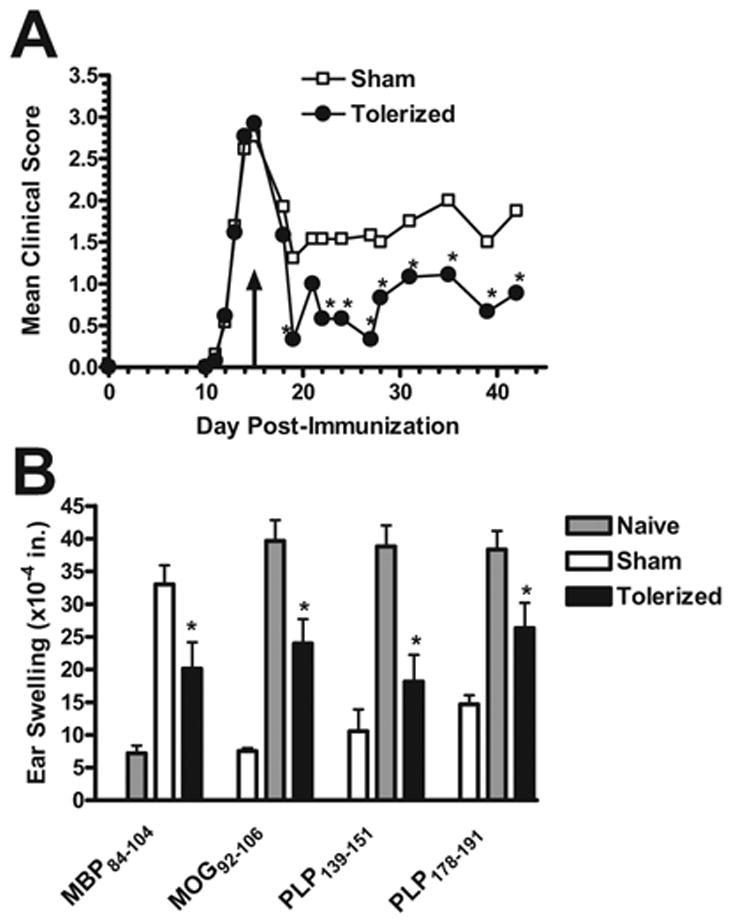
Twenty-six female SJL mice were primed s.c. with a cocktail of myelin peptides consisting of 25 μg PLP139–151, 50 μg PLP178–191, 100 μg MBP84–104, and 100 μg MOG92–106 and CFA on day 0. On day 15 (indicated by arrow), at the peak of acute disease, mice were given 50×106 syngeneic splenocytes coupled to a myelin peptide cocktail consisting of PLP139–151, PLP178–191, MBP84–104, and MOG92–106 (Tolerized) or to a control peptide cocktail consisting of PLP56–70, OVA323–339, VP1233–250, and VP270–86 (Sham) i.v. and monitored for clinical disease (A) and peptide-specific DTH responses (B). For DTH, mice were ear challenged with 10 μg peptide on day 27, and swelling was measured 24 hours later. Data is representative of three separate experiments. *Responses significantly less than sham-tolerized controls, p < 0.05.
To further examine T cell peripheral immune responses to the immunizing epitopes after therapeutic tolerization, spleens were harvested five days after coupled-cell administration and in vitro proliferation and cytokine recall responses were assayed. In response to incubation with the target antigens, the tolerized spleen cells had decreased proliferation compared to sham-tolerized spleen cells (Figure 7a). IL-2 production was decreased to MBP84–104 and PLP139–151, but not to PLP178–191 (Figure 7b). MOG92–106 did not induce recall proliferation of IL-2 production over background in cells from either tolerized or sham-tolerized mice. Interestingly, in contrast to the results after preventative tolerization (Figure 3c), IFN-γ production was either comparable, or even slightly increased in cells from tolerized mice (Figure 7c). However, there was a significant enhancement of production of transforming growth factor □ (TGF-□) by T cells from the tolerized mice in response to restimulation with all four of the myelin peptides (Figure 7d). TGF-β has been associated with the in vivo function of regulatory T cells (Tregs) [45,46]. IL-4 and IL-5 responses were not seen upon peptide recall in either the tolerized or sham groups (data not shown).
Figure 7. Effect of therapeutic coupled-cell tolerance to multiple myelin peptides on peripheral peptide-specific T cell recall responses.
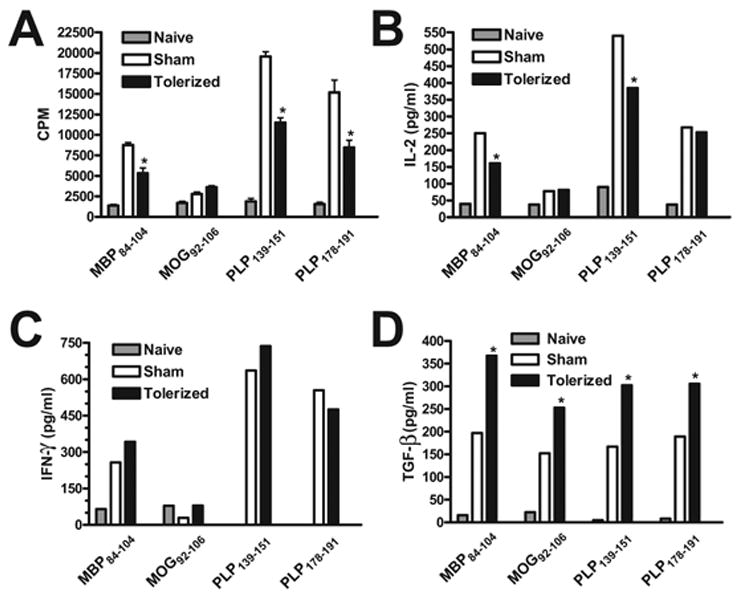
Twenty-six female SJL mice were primed s.c. with a cocktail of myelin peptides consisting of 25 μg PLP139–151, 50 μg PLP178–191, 100 μg MBP84–104, and 100 μg MOG92–106 and CFA on day 0. On day 15, at the peak of acute disease, mice were given 50×106 syngeneic splenocytes coupled to a myelin peptide cocktail consisting of PLP139–151, PLP178–191, MBP84–104, and MOG92–106 (Tolerized) or to a control peptide cocktail consisting of PLP56–70, OVA323–339, VP1233–250, and VP270–86 (Sham) i.v. Spleens were harvested from 3 representative mice from each group on day 20. Single cell suspensions were incubated for 3 days with the indicated antigens and assessed for proliferative responses by uptake of [3H] thymidine (A). Data is presented as mean CPM ± SEM. Single cell suspensions were incubated for 48 hr with the indicated antigens prior to measurement of IL-2 (B) and IFN-γ (C) levels via cytokine array analysis and TGF-β (D) levels via ELISA. Cytokine data is presented as mean pg/ml. *Responses significantly different than sham-tolerized controls, p < 0.05. Data is representative of two experiments.
To complement the analyses of peripheral immune results, we also determined the effects of therapeutic coupled-cell tolerance on T cell responses in the CNS target organ. The lumbar regions of the spinal cords of representative mice in each group were harvested and evaluated for CD4+ T cell and macrophage infiltrate via immunohistochemical staining. As opposed to preventative tolerance, mice tolerized with the cocktail of myelin peptides at the peak of acute R-EAE were found to have similar amounts of infiltrating cells as sham-tolerized controls (Figure 8). In addition, infiltrating leukocytes were isolated from whole spinal cords after tolerance induction and were restimulated in vitro with the priming peptides and supernatants collected for cytokine analysis. Surprisingly, compared to sham-tolerized controls, the levels of proinflammatory cytokines produced by cells from tolerized mice were remarkably varied, ranging from decreased levels in response to some peptides to comparable or even greater levels in response to other peptides (Figure 9). However, similar to the results from the spleen, CNS T cells isolated from therapeutically tolerized mice produced significantly greater amounts of TGF-β than those from sham-tolerized mice (Figure 9c). In addition, they also produced significantly increased levels of IL-10 (Figure 9d), which has also been associated with in vivo regulatory T cell function [47,48].
Figure 8. Effect of therapeutic coupled-cell tolerance to multiple myelin peptides on CNS inflammatory cell infiltrates in established EAE.
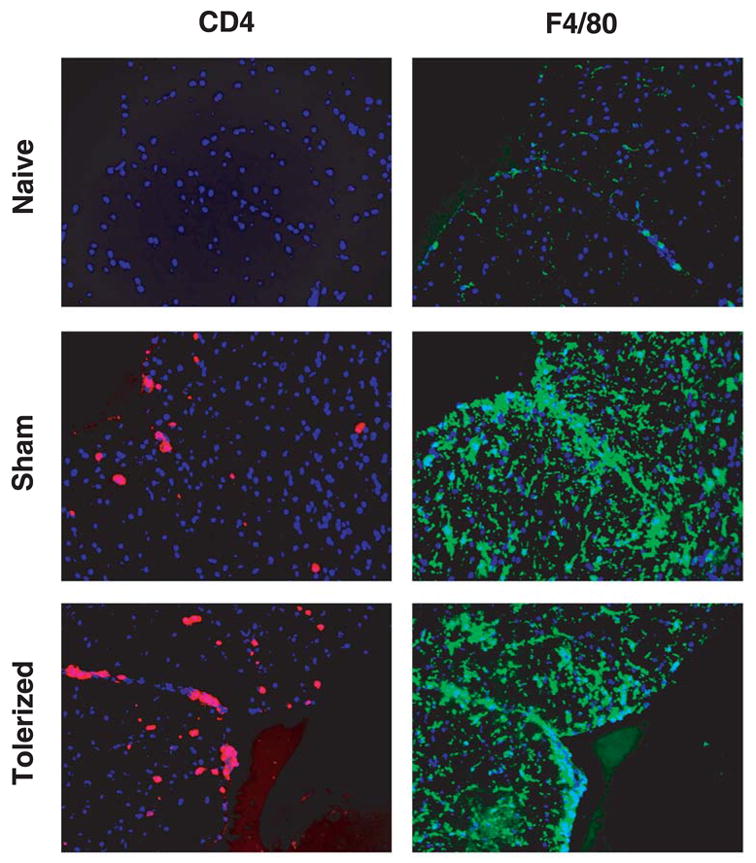
Twenty female SJL mice were primed s.c. with a cocktail of myelin peptides consisting of 25 μg PLP139–151, 50 μg PLP178–191, 100 μg MBP84–104, and 100 μg MOG92–106 and CFA on day 0. On day 15, at the peak of acute disease, mice were given 50×106 syngeneic splenocytes coupled to a myelin peptide cocktail consisting of PLP139–151, PLP178–191, MBP84–104, and MOG92–106 (Tolerized) or to a control peptide cocktail consisting of PLP56–70, OVA323–339, VP1233–250, and VP270–86 (Sham) i.v. Three mice from each group were perfused on day 31. Spinal cords were harvested, snap-frozen, sectioned into 6μm slices, stained with anti-CD4 (red) or anti-F4/80 (green) mAbs, and counterstained with DAPI (blue). Lumbar regions are shown. Sections shown at 200x and are representative of two experiments.
Figure 9. Effect of therapeutic coupled-cell tolerance to multiple myelin peptides on CNS peptide-specific T cell recall responses in established EAE.
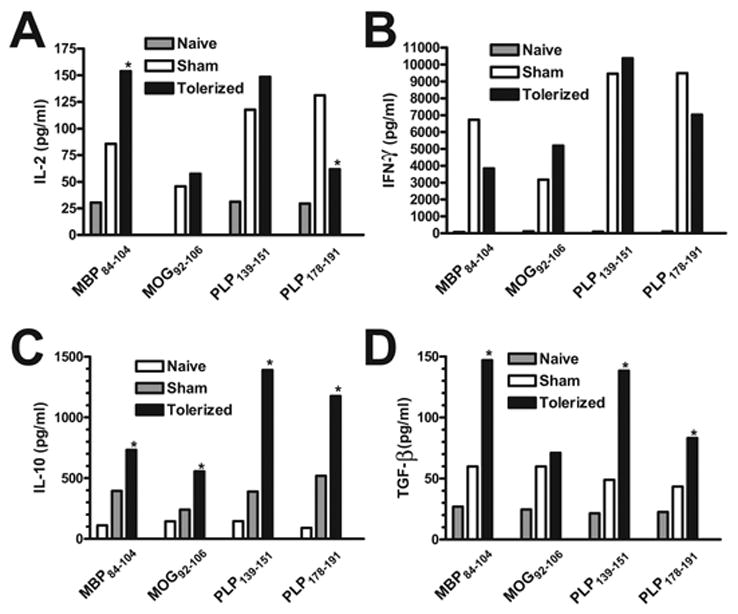
Twenty-six female SJL mice were primed s.c. with a cocktail of myelin peptides consisting of 25 μg PLP139–151, 50 μg PLP178–191, 100 μg MBP84–104, and 100 μg MOG92–106 and CFA on day 0. On day 15, at the peak of acute disease, mice were given 50×106 syngeneic splenocytes coupled to a myelin peptide cocktail consisting of PLP139–151, PLP178–191, MBP84–104, and MOG92–106 (Tolerized) or to a control peptide cocktail consisting of PLP56–70, OVA323–339, VP1233–250, and VP270–86 (Sham) i.v. On day 20, ten mice from each group were perfused, and spinal cords were harvested. Infiltrating leukocytes were isolated and single cell suspensions were incubated for 48 hr with the indicated antigens prior to measurement of IL-2 (A), IFN-γ (B) and IL-10 (C) levels via cytokine array analysis and TGF-β (D) levels via ELISA. Cytokine levels are reported as mean pg/ml and are representative of two separate experiments. *Responses significantly different than sham-tolerized controls, p < 0.05.
Discussion
In this study, multiple peptide coupled-cell tolerance was shown to be effective at preventing EAE initiated with either individual peptides or a combination of multiple peptides, at preventing relapses to spread epitopes by acting on both naïve and previously activated autoreactive T cells, and at ameliorating ongoing EAE in which T cells specific for multiple myelin antigens were simultaneously playing a pathogenic role. This provides the first demonstration that syngeneic ECDI-fixed cells coupled simultaneously with multiple peptides is an effective method of tolerance induction for both naïve and activated CD4+ T cells. This is an important demonstration relevant to the therapy of MS for several reasons. Ideally, a tolerance-inducing regimen such as this could employ whole proteins coupled to syngeneic cells [37,49] which would operate via a cross-tolerance mechanism involving re-presentation of self epitopes expressed on apoptotic ECDI-treated splenic APCs by host splenic regulatory DCs (unpublished results). By using intact proteins, all of the epitopes contained within those proteins would theoretically be available for presentation to the specific autoreactive T cells in a tolerogenic context. Unfortunately, in the case of MS, whole myelin proteins are not currently available in quantities necessary for treatment purposes. The use of intact myelin proteins isolated from human nervous tissue or produced in bacteria have safety concerns which preclude their use in the clinical setting. Therefore, it is necessary to employ synthetic myelin peptides produced under GMP conditions if coupled-cell tolerance is to be used in human patients.
The efficient use of peptide-specific tolerogenic therapies consequently requires prior knowledge of the autoepitopes involved in driving chronic disease pathogenesis. This is problematic in MS, a heterogeneous disease of unknown origin in which the specificities of both the inciting and spread epitope-specific autoreactive T cells will certainly vary from patient to patient, due to differences in HLA haplotypes. However, based on high precursor frequencies, high antigen avidities, and functional studies, Bielekova et al. have recently identified six immunodominant myelin epitopes most often discriminatory between MS patients and controls [44]. This provides a pool of candidate peptides that can be used for coupled-cell tolerance in future clinical trials in early RR-MS patients. In addition, a sensitive new technique for the functional characterization of antigen-specific T cells present in patient samples has recently been developed. This high-throughput method uses arrays of peptide-MHC complexes to capture antigen-specific T cells with antibodies against secreted cytokines, allowing for a broad-based analysis of the specificities and functional states of patient T cell repertoires [50]. This technology will no doubt facilitate the implementation of antigen-specific therapies in various autoimmune diseases.
There have been several previous studies demonstrating the ability to prevent or ameliorate EAE by targeting multiple encephalitogenic myelin antigens. These studies employed administration of either an engineered fusion protein of PLP and MBP (MP4) [51], an engineered “multiantigen/multiepitope” protein containing epitopes of PLP, MBP, and MOG (hmTAP) [52], or multiple myelin peptides [53]. However, in all of these examples of multi-epitope treatment, the soluble peptides or proteins were administered intravenously. It has recently been shown that both i.v. [38] and intraperitoneal (i.p.) [54] soluble peptide treatment as a therapy of ongoing EAE can result in fatal anaphylaxis. This puts the safety of any soluble peptide/protein treatment for human MS patients into serious question. Indeed, a recent phase II clinical trial employing subcutaneous injections of a soluble MBP APL in MS patients which was terminated due to systemic hypersensitivity reactions that developed in 13 of 142 patients [36]. We have previously demonstrated that peptide-coupled cells, as used in this study, are an effective alternative which is not complicated by the deleterious side effects of anaphylactic shock [38]. This is likely due to the fact that the cell-bound peptide fails to access IgE-coated mast cells in peripheral tissues.
Therefore, we set out to determine if multiple myelin peptides can be efficiently coupled to syngeneic cells and used to induce immune tolerance to multiple myelin antigens. Figures 1 and 2 show that multi-peptide coupled-cell tolerance is effective at preventing EAE initiated both by individual peptides and by a combination of four potent encephalitogenic peptides. In addition, because the etiology of MS is unknown, making prevention of the disease unrealistic at this time, it was essential to demonstrate that the multi-peptide coupled-cell tolerance treatment regimen can ameliorate ongoing disease. Furthermore, as there is evidence that the repertoire of encephalitogenic T cells evolves over time in MS patients, likely due to epitope spreading [21–24], it was important to verify that multi-peptide coupled-cell tolerance can specifically inhibit responses of both previously activated as well as naïve T cells. The data show that multi-peptide coupled-cell tolerance is capable of preventing relapses due to T cell reactivity to spread epitopes, tolerizing not only previously activated T cells (those specific for the priming epitopes), but also naïve T cells (those specific for the spread epitopes) (Figure 5). Finally, because there appears to be considerable diversity in the autoreactive T cell repertoire in MS patients [21,24,44], it was necessary to establish that multi-myelin peptide coupled-cell tolerance can ameliorate ongoing disease associated with multiple concurrent pathogenic reactivities. Figure 6 shows that administration of multi-peptide coupled-cells at the peak of acute EAE, in which T cells specific for four distinct encephalitogenic myelin epitopes are actively participating, can ameliorate clinical disease progression and decrease peripheral DTH responses to all four peptides. Collectively, these results are very encouraging with regard to the clinical potential of multi-peptide coupled-cell tolerance in the treatment of MS.
With regard to the mechanism(s) by which multi-peptide coupled-cell tolerance might operate, the location and activation/functional capacity of myelin-specific CD4+ T cells were evaluated after tolerance induction. Anergy induction appears to play a major functional role in protective tolerization of naïve T cells. Myelin peptide-specific recall proliferative and pro-inflammatory cytokine responses are significantly suppressed (Figure 3) in the absence of any induction of IL-4. IL-5, or IL-10 (data not shown) in T cells from peripheral immune organs of mice tolerized with multi-peptide coupled splenic APCs seven days prior to priming. In addition, there is a marked decrease in the number of inflammatory leukocytes in the spinal cords of myelin peptide-tolerized mice vs. sham-tolerized mice. This is likely a reflection of the induction of anergy in the peripheral autoreactive T cells prior to immunization, but could also be partially the result of an independent effect of the tolerization protocol on the natural pattern of T cell migration in response to pro-inflammatory stimulation. These results are similar to findings from previous studies in which either whole myelin protein or individual peptides were coupled to the syngeneic donor cells and used to induce tolerance before EAE induction [37,55–57].
In contrast, when previously activated myelin-specific T cells are tolerized, anergy appears to play a lesser role in concert with other mechanisms. This is illustrated by the finding that while myelin-specific T cells from mice tolerized during ongoing disease, in which disease progression is ameliorated (Figure 6), have a decreased ability to proliferate upon restimulation with myelin peptides, CD4+ T cells from both the spleen and spinal cord produce equivalent quantities of IFN-□ as T cells from sham-tolerized mice (Figures 7 and 9). There was also no apparent decrease of infiltrating CD4+ T cells or macrophages in the spinal cords of mice tolerized during ongoing disease. However, therapeutic tolerance led to significantly increased peptide-induced production of both TGF-β and IL-10 by spinal cord-infiltrating T cells compared to sham-tolerance (Figure 9). While IL-10 production from splenic cells of tolerized mice was inconsistent from experiment to experiment (data not shown), TGF-β production was consistently significantly upregulated in splenic T cells from tolerized mice compared to sham-tolerized mice upon myelin peptide restimulation. This pattern of regulatory cytokine production is consistent with a role for Treg cell function in therapeutic tolerance. The fact that IL-10 and TGF-β are upregulated in sham-tolerized mice compared to naive mice is likely an indication that there is a natural increase in Treg cell function during ongoing EAE in an effort to keep the inflammation resulting from the autoimmunity in check.
Although there have been several reports that in vitro CD4+CD25+ Treg cell function is contact-, but not cytokine-dependent [58–60], others have demonstrated roles for secreted IL-10 and TGF-β in in vivo Treg survival and function. For example, TGF-β1 has been shown to be necessary for CD4+CD25+ Treg homeostasis, Foxp3 expression, and regulatory function [45,46]. Furthermore, CD4+CD25+ Treg cells from wildtype, but not IL10−/−, B6 mice protect against EAE [47], and IL-10-producing CD4+CD25+ Treg cells in the CNS mediate recovery from EAE [48]. In addition, naturally occurring CD4+CD25+ Treg cells have been shown to be capable of inducing conventional T cells to become suppressive, and the suppression by the induced regulatory T cells is IL-10 and TGF-β-dependent [61–63]. Thus, it is plausible that the induction/activation/expansion of Treg cells, either in the periphery or locally at the site of inflammation, is a critical action of regulation by multi-myelin peptide coupled-cells administered at the peak of acute disease. However, it is also possible that TGF-β and IL-10 are being produced unrelated to the presence or action of Treg cells, e.g. via immune deviation. Both IL-10 and TGF-β are produced by multiple cell types and have immunoregulatory function. TGF-β suppresses the production of nitric oxide and reactive oxygen intermediates by macrophages [64–66] and has also been shown to inhibit the differentiation of T cells into Th1 and Th2 cells by blocking expression of T-bet and GATA-3 [67–69]. IL-10 has been shown to increase monocytic phagocytosis [70,71] while reducing antigen presentation and CD80/86 expression [72,73]. We are currently evaluating the requirement, or lack thereof, for natural Treg and induced Tr1 cells and/or TGF-β and IL-10 in multi-peptide coupled-cell tolerance utilizing GFP-Foxp3 knock-in mice [74].
In conclusion, multi-peptide coupled-cell tolerance is a novel approach to multiple sclerosis therapy with great clinical potential. While the mechanisms of action must be further examined, this study demonstrates the efficiency of this treatment in simultaneously tolerizing both naïve and previously activated T cells of multiple targeted specificities and to ameliorate ongoing disease associated with multiple pathogenic autoreactivities. This makes it an ideal candidate for treatment of heterogeneous autoimmune disorders such as multiple sclerosis.
Abbreviations
- Ag-SP
antigen-coupled splenocytes
- APL
altered peptide ligand
- EAE
experimental autoimmune encephalomyelitis
- ECDI
ethylene carbodiimide
- MBP
myelin basic protein
- MOG
myelin oligodendrocyte protein
- MS
multiple sclerosis
- PLP
myelin proteolipid protein
Footnotes
This work was supported in part by U.S. Public Health Service, National Institutes of Health Grants NS-026543, NS-030871-13, NS-048411-02, National Multiple Sclerosis Society Grant RG 3793-A-7, and by support from the Myelin Repair Foundation. C.E.S. was supported by NIGMS 5T32 GM08061.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hauser SL, Goodin DS. Multiple sclerosis and other demyelinating diseases. In: Kasper DL, Braunwald E, Fauci AS, Hauser SL, Longo DL, Jameson JL, Isselbacher KJ, editors. Harrison's Principles of Internal Medicine. 16. McGraw-Hill; New York: 2004. [Google Scholar]
- 2.Haines JL, Ter-Minassian M, Bazyk A, Gusella JF, Kim DJ, Terwedow H, et al. A complete genomic screen for multiple sclerosis underscores a role for the major histocompatability complex. The Multiple Sclerosis Genetics Group Nat Genet. 1996;13:469–71. doi: 10.1038/ng0896-469. [DOI] [PubMed] [Google Scholar]
- 3.Sadovnick AD, Ebers GC. Genetics of multiple sclerosis. Neurol Clin. 1995;13:99–118. [PubMed] [Google Scholar]
- 4.Barcellos LF, Oksenberg JR, Green AJ, Bucher P, Rimmler JB, Schmidt S, et al. Genetic basis for clinical expression in multiple sclerosis. Brain. 2002;125:150–8. doi: 10.1093/brain/awf009. [DOI] [PubMed] [Google Scholar]
- 5.Kurtzke JF. Multiple sclerosis in time and space--geographic clues to cause. J Neurovirol. 2000;6(Suppl 2):S134–S40. [PubMed] [Google Scholar]
- 6.Zhang J, Markovic-Plese S, Lacet B, Raus J, Weiner HL, Hafler DA. Increased frequency of interleukin 2-responsive T cells specific for myelin basic protein and proteolipid protein in peripheral blood and cerebrospinal fluid of patients with multiple sclerosis. J Exp Med. 1994;179:973–84. doi: 10.1084/jem.179.3.973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Voskuhl RR, Martin R, Bergman C, Dalal M, Ruddle NH, McFarland HF. T helper 1 (Th1) functional phenotype of human myelin-basic protein-specific T lymphocytes. Autoimmunity. 1993;15:137–43. doi: 10.3109/08916939309043888. [DOI] [PubMed] [Google Scholar]
- 8.Olsson T, Zhi WW, Hîjeberg B, Kostulas V, Jiang YP, Anderson G, et al. Autoreactive T lymphocytes in multiple sclerosis determined by antigen -induced secretion of interferon-gamma. J Clin Invest. 1990;86:981–85. doi: 10.1172/JCI114800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bielekova B, Muraro PA, Golestaneh L, Pascal J, McFarland HF, Martin R. Preferential expansion of autoreactive T lymphocytes from the memory T-cell pool by IL-7. J Neuroimmunol. 1999;100:115–23. doi: 10.1016/s0165-5728(99)00200-3. [DOI] [PubMed] [Google Scholar]
- 10.Pettinelli CB, McFarlin DE. Adoptive transfer of experimental allergic encephalomyelitis in SJL/J mice after in vitro activation of lymph node cells by myelin basic protein: requirement for Lyt 1+ 2 - T lymphocytes. J Immunol. 1981;127:1420–23. [PubMed] [Google Scholar]
- 11.Brostoff SW, Mason DW. Experimental allergic encephalomyelitis: successful treatment in vivo with a monoclonal antibody that recognizes T helper cells. J Immunol. 1984;133:1938–42. [PubMed] [Google Scholar]
- 12.Tuohy VK, Lu Z, Sobel RA, Laursen RA, Lees MB. Identification of an encephalitogenic determinant of myelin proteolipid protein for SJL mice. J Immunol. 1989;142:1523–27. [PubMed] [Google Scholar]
- 13.Goverman J, Woods A, Larson L, Weiner LP, Hood L, Zaller DM. Transgenic mice that express a myelin basic protein-specific T cell receptor develop spontaneous autoimmunity. Cell. 1993;72:551–60. doi: 10.1016/0092-8674(93)90074-z. [DOI] [PubMed] [Google Scholar]
- 14.Madsen LS, Andersson EC, Jansson L, Krogsgaard M, Andersen CB, Engberg J, et al. A humanized model for multiple sclerosis using HLA-DR2 and a human T- cell receptor. Nat Genet. 1999;23:343–47. doi: 10.1038/15525. [DOI] [PubMed] [Google Scholar]
- 15.McRae BL, Vanderlugt CL, Dal Canto MC, Miller SD. Functional evidence for epitope spreading in the relapsing pathology of experimental autoimmune encephalomyelitis. J Exp Med. 1995;182:75–85. doi: 10.1084/jem.182.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McMahon EJ, Bailey SL, Castenada CV, Waldner H, Miller SD. Epitope spreading initiates in the CNS in two mouse models of multiple sclerosis. Nat Med. 2005;11:335–39. doi: 10.1038/nm1202. [DOI] [PubMed] [Google Scholar]
- 17.Vanderlugt CL, Eagar TN, Neville KL, Nikcevich KM, Bluestone JA, Miller SD. Pathologic role and temporal appearance of newly emerging autoepitopes in relapsing experimental autoimmune encephalomyelitis. J Immunol. 2000;164:670–78. doi: 10.4049/jimmunol.164.2.670. [DOI] [PubMed] [Google Scholar]
- 18.Yu M, Johnson JM, Tuohy VK. A predictable sequential determinant spreading cascade invariably accompanies progression of experimental autoimmune encephalomyelitis: A basis for peptide-specific therapy after onset of clinical disease. J Exp Med. 1996;183:1777–88. doi: 10.1084/jem.183.4.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ellmerich S, Takacs K, Mycko M, Waldner H, Wahid F, Boyton RJ, et al. Disease-related epitope spread in a humanized T cell receptor transgenic model of multiple sclerosis. Eur J Immunol. 2004;34:1839–48. doi: 10.1002/eji.200324044. [DOI] [PubMed] [Google Scholar]
- 20.Klehmet J, Shive C, Guardia-Wolff R, Petersen I, Spack EG, Boehm BO, et al. T cell epitope spreading to myelin oligodendrocyte glycoprotein in HLA-DR4 transgenic mice during experimental autoimmune encephalomyelitis. Clin Immunol. 2004;111:53–60. doi: 10.1016/j.clim.2003.12.012. [DOI] [PubMed] [Google Scholar]
- 21.Tuohy VK, Yu M, Weinstock-Guttman B, Kinkel RP. Diversity and plasticity of self recognition during the development of multiple sclerosis. J Clin Invest. 1997;99:1682–90. doi: 10.1172/JCI119331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tuohy VK, Yu M, Yin L, Kawczak JA, Kinkel RP. Spontaneous regression of primary autoreactivity during chronic progression of experimental autoimmune encephalomyelitis and multiple sclerosis. J Exp Med. 1999;189:1033–42. doi: 10.1084/jem.189.7.1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tuohy VK, Yu M, Yin L, Kawczak JA, Kinkel PR. Regression and spreading of self-recognition during the development of autoimmune demyelinating disease. J Autoimmun. 1999;13:11–20. doi: 10.1006/jaut.1999.0293. [DOI] [PubMed] [Google Scholar]
- 24.Goebels N, Hofstetter H, Schmidt S, Brunner C, Wekerle H, Hohlfeld R. Repertoire dynamics of autoreactive T cells in multiple sclerosis patients and healthy subjects: epitope spreading versus clonal persistence. Brain. 2000;123:508–18. doi: 10.1093/brain/123.3.508. [DOI] [PubMed] [Google Scholar]
- 25.Muraro PA, Wandinger KP, Bielekova B, Gran B, Marques A, Utz U, et al. Molecular tracking of antigen-specific T cell clones in neurological immune-mediated disorders. Brain. 2003;126:20–31. doi: 10.1093/brain/awg021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hohol MJ, Olek MJ, Orav EJ, Stazzone L, Hafler DA, Khoury SJ, et al. Treatment of progressive multiple sclerosis with pulse cyclophosphamide/methylprednisolone: response to therapy is linked to the duration of progressive disease. Mult Scler. 1999;5:403–9. doi: 10.1177/135245859900500i606. [DOI] [PubMed] [Google Scholar]
- 27.Interferon beta-1b is effective in relapsing-remitting multiple sclerosis I. Clinical results of a multicenter, randomized, double-blind, placebo-controlled trial. The IFNB Multiple Sclerosis Study Group Neurology. 1993;43:655–61. doi: 10.1212/wnl.43.4.655. [DOI] [PubMed] [Google Scholar]
- 28.Ebers GC, Hommes O, Hughes RAC, Kappos L, Sandberg-Wollheim L, Palace J, et al. Randomised double-blind placebo-controlled study of interferon beta-1a in relapsing/remitting multiple sclerosis. PRISMS (Prevention of Relapses and Disability by Interferon beta-1a Subcutaneously in Multiple Sclerosis) Study Group. Lancet. 1998;352:1498–504. [PubMed] [Google Scholar]
- 29.Bornstein MB, Miller A, Slagle S, Weitzman M, Crystal H, Drexler E, et al. A Pilot Trial of Cop-1 in Exacerbating Remitting Multiple-Sclerosis. N Engl J Med. 1987;317:408–14. doi: 10.1056/NEJM198708133170703. [DOI] [PubMed] [Google Scholar]
- 30.Duda PW, Schmied MC, Cook SL, Krieger JI, Hafler DA. Glatiramer acetate (Copaxone (R)) induces degenerate, Th2-polarized immune responses in patients with multiple sclerosis. J Clin Invest. 2000;105:967–76. doi: 10.1172/JCI8970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Neuhaus O, Farina C, Yassouridis A, Wiendl H, Bergh FT, Dose T, et al. Multiple sclerosis: Comparison of copolymer-1-reactive T cell lines from treated and untreated subjects reveals cytokine shift from T helper 1 to T helper 2 cells. Proc Natl Acad Sci USA. 2000;97:7452–57. doi: 10.1073/pnas.97.13.7452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johnson KP, Brooks BR, Cohen JA, Ford CC, Goldstein J, Lisak RP, et al. Copolymer 1 reduces relapse rate and improves disability in relapsing-remitting multiple sclerosis: results of a phase III multicenter, double-blind placebo-controlled trial. The Copolymer 1 Multiple Sclerosis Study Group Neurology. 1995;45:1268–76. doi: 10.1212/wnl.45.7.1268. [DOI] [PubMed] [Google Scholar]
- 33.Korczyn AD, Nisipeanu P. Safety profile of copolymer 1: Analysis of cumulative experience in the United States and Israel. J Neurol. 1996;243:S23–S26. doi: 10.1007/BF00873698. [DOI] [PubMed] [Google Scholar]
- 34.Weiner HL, Mackin GA, Matsui M, Orav EJ, Khoury SJ, Dawson DM, et al. Double-blind pilot trial of oral tolerization with myelin antigens in multiple sclerosis. Science. 1993;259:1321–24. doi: 10.1126/science.7680493. [DOI] [PubMed] [Google Scholar]
- 35.Bielekova B, Goodwin B, Richert N, Cortese I, Kondo T, Afshar G, et al. Encephalitogenic potential of the myelin basic protein peptide (amino acids 83–99) in multiple sclerosis: results of a phase II clinical trial with an altered peptide ligand. Nat Med. 2000;6:1167–75. doi: 10.1038/80516. [DOI] [PubMed] [Google Scholar]
- 36.Kappos L, Comi G, Panitch H, Oger J, Antel J, Conlon P, et al. Induction of a non-encephalitogenic type 2 T helper-cell autoimmune response in multiple sclerosis after administration of an altered peptide ligand in a placebo-controlled, randomized phase II trial. The Altered Peptide Ligand in Relapsing MS Study Group Nat Med. 2000;6:1176–82. doi: 10.1038/80525. [DOI] [PubMed] [Google Scholar]
- 37.Kennedy MK, Tan LJ, Dal Canto MC, Tuohy VK, Lu ZJ, Trotter JL, et al. Inhibition of murine relapsing experimental autoimmune encephalomyelitis by immune tolerance to proteolipid protein and its encephalitogenic peptides. J Immunol. 1990;144:909–15. [PubMed] [Google Scholar]
- 38.Smith CE, Eagar TN, Strominger JL, Miller SD. Differential induction of IgE-mediated anaphylaxis after soluble vs. cell-bound tolerogenic peptide therapy of autoimmune encephalomyelitis. Proc Natl Acad Sci USA. 2005;102:9595–600. doi: 10.1073/pnas.0504131102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tan LJ, Kennedy MK, Miller SD. Regulation of the effector stages of experimental autoimmune encephalomyelitis via neuroantigen-specific tolerance induction. II. Fine specificity of effector T cell inhibition. J Immunol. 1992;148:2748–55. [PubMed] [Google Scholar]
- 40.Kennedy MK, Tan LJ, Dal Canto MC, Miller SD. Regulation of the effector stages of experimental autoimmune encephalomyelitis via neuroantigen-specific tolerance induction. J Immunol. 1990;145:117–26. [PubMed] [Google Scholar]
- 41.Tan LJ, Kennedy MK, Dal Canto MC, Miller SD. Successful treatment of paralytic relapses in adoptive experimental autoimmune encephalomyelitis via neuroantigen-specific tolerance. J Immunol. 1991;147:1797–802. [PubMed] [Google Scholar]
- 42.Peterson JD, Karpus WJ, Clatch RJ, Miller SD. Split tolerance of Th1 and Th2 cells in tolerance to Theiler's murine encephalomyelitis virus. Eur J Immunol. 1993;23:46–55. doi: 10.1002/eji.1830230109. [DOI] [PubMed] [Google Scholar]
- 43.McRae BL, Kennedy MK, Tan LJ, Dal Canto MC, Miller SD. Induction of active and adoptive chronic-relapsing experimental autoimmune encephalomyelitis (EAE) using an encephalitogenic epitope of proteolipid protein. J Neuroimmunol. 1992;38:229–40. doi: 10.1016/0165-5728(92)90016-e. [DOI] [PubMed] [Google Scholar]
- 44.Bielekova B, Sung MH, Kadom N, Simon R, McFarland H, Martin R. Expansion and functional relevance of high-avidity myelin-specific CD4+ T cells in multiple sclerosis. J Immunol. 2004;172:3893–904. doi: 10.4049/jimmunol.172.6.3893. [DOI] [PubMed] [Google Scholar]
- 45.Huber S, Schramm C, Lehr HA, Mann A, Schmitt S, Becker C, et al. Cutting edge: TGF-beta signaling is required for the in vivo expansion and immunosuppressive capacity of regulatory CD4+CD25+ T cells. J Immunol. 2004;173:6526–31. doi: 10.4049/jimmunol.173.11.6526. [DOI] [PubMed] [Google Scholar]
- 46.Marie JC, Letterio JJ, Gavin M, Rudensky AY. TGF-beta1 maintains suppressor function and Foxp3 expression in CD4+CD25+ regulatory T cells. J Exp Med. 2005;201:1061–7. doi: 10.1084/jem.20042276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang X, Koldzic DN, Izikson L, Reddy J, Nazareno RF, Sakaguchi S, et al. IL-10 is involved in the suppression of experimental autoimmune encephalomyelitis by CD25+CD4+ regulatory T cells. Int Immunol. 2004;16:249–56. doi: 10.1093/intimm/dxh029. [DOI] [PubMed] [Google Scholar]
- 48.McGeachy MJ, Stephens LA, Anderton SM. Natural recovery and protection from autoimmune encephalomyelitis: contribution of CD4+CD25+ regulatory cells within the central nervous system. J Immunol. 2005;175:3025–32. doi: 10.4049/jimmunol.175.5.3025. [DOI] [PubMed] [Google Scholar]
- 49.Kennedy MK, Dal Canto MC, Trotter JL, Miller SD. Specific immune regulation of chronic-relapsing experimental allergic encephalomyelitis in mice. J Immunol. 1988;141:2986–93. [PubMed] [Google Scholar]
- 50.Chen DS, Soen Y, Stuge TB, Lee PP, Weber JS, Brown PO, et al. Marked differences in human melanoma antigen-specific T cell responsiveness after vaccination using a functional microarray. PLOS Med. 2005;2:e265. doi: 10.1371/journal.pmed.0020265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Elliott EA, McFarland HI, Nye SH, Cofiell R, Wilson TM, Wilkins JA, et al. Treatment of experimental encephalomyelitis with a novel chimeric fusion protein of myelin basic protein and proteolipid protein. J Clin Invest. 1996;98:1602–12. doi: 10.1172/JCI118954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhong MC, Kerlero de RN, Ben-Nun A. Multiantigen/multiepitope-directed immune-specific suppression of "complex autoimmune encephalomyelitis" by a novel protein product of a synthetic gene. J Clin Invest. 2002;110:81–90. doi: 10.1172/JCI15692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Leadbetter EA, Bourque CR, Devaux B, Olson CD, Sunshine GH, Hirani S, et al. Experimental autoimmune encephalomyelitis induced with a combination of myelin basic protein and myelin oligodendrocyte glycoprotein is ameliorated by administration of a single myelin basic protein peptide. J Immunol. 1998;161:504–12. [PubMed] [Google Scholar]
- 54.Pedotti R, Mitchell D, Wedemeyer J, Karpuj M, Chabas D, Hattab EM, et al. An unexpected version of horror autotoxicus: anaphylactic shock to a self-peptide. Nat Immunol. 2001;2:216–22. doi: 10.1038/85266. [DOI] [PubMed] [Google Scholar]
- 55.Eagar TN, Turley DM, Padilla J, Karandikar NJ, Tan LJ, Bluestone JA, et al. CTLA-4 regulates expansion and differentiation of Th1 cells following induction of peripheral T cell tolerance. J Immunol. 2004;172:7442–50. doi: 10.4049/jimmunol.172.12.7442. [DOI] [PubMed] [Google Scholar]
- 56.Malotky MKH, Pope L, Miller SD. Epitope and functional specificity of peripheral tolerance induction in experimental autoimmune encephalomyelitis in adult Lewis rats. J Immunol. 1994;153:841–51. [PubMed] [Google Scholar]
- 57.Karpus WJ, Pope JG, Peterson JD, Dal Canto MC, Miller SD. Inhibition of Theiler's virus-mediated demyelination by peripheral immune tolerance induction. J Immunol. 1995;155:947–57. [PubMed] [Google Scholar]
- 58.Thornton AM, Shevach EM. CD4+CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J Exp Med. 1998;188:287–96. doi: 10.1084/jem.188.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Takahashi T, Kuniyasu Y, Toda M, Sakaguchi N, Itoh M, Iwata M, et al. Immunologic self-tolerance maintained by CD25+CD4+ naturally anergic and suppressive T cells: induction of autoimmune disease by breaking their anergic/suppressive state. Int Immunol. 1998;10:1969–80. doi: 10.1093/intimm/10.12.1969. [DOI] [PubMed] [Google Scholar]
- 60.Piccirillo CA, Letterio JJ, Thornton AM, McHugh RS, Mamura M, Mizuhara H, et al. CD4(+)CD25(+) regulatory T cells can mediate suppressor function in the absence of transforming growth factor beta1 production and responsiveness. J Exp Med. 2002;196:237–46. doi: 10.1084/jem.20020590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dieckmann D, Bruett CH, Ploettner, Lutz MB, Schuler G. Human CD4(+)CD25(+) regulatory, contact-dependent T cells induce interleukin 10-producing, contact-independent type 1-like regulatory T cells. J Exp Med. 2002;196:247–53. doi: 10.1084/jem.20020642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jonuleit H, Schmitt E, Kakirman H, Stassen M, Knop J, Enk AH. Infectious tolerance: human CD25(+) regulatory T cells convey suppressor activity to conventional CD4(+) T helper cells. J Exp Med. 2002;196:255–60. doi: 10.1084/jem.20020394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zheng SG, Wang JH, Gray JD, Soucier H, Horwitz DA. Natural and induced CD4+CD25+ cells educate CD4+CD25- cells to develop suppressive activity: the role of IL-2, TGF-beta, and IL-10. J Immunol. 2004;172:5213–21. doi: 10.4049/jimmunol.172.9.5213. [DOI] [PubMed] [Google Scholar]
- 64.Bogdan C, Nathan C. Modulation of macrophage function by transforming growth factor beta, interleukin-4, and interleukin-10. Ann N Y Acad Sci. 1993;685:713–39. doi: 10.1111/j.1749-6632.1993.tb35934.x. [DOI] [PubMed] [Google Scholar]
- 65.Vodovotz Y, Bogdan C. Control of nitric oxide synthase expression by transforming growth factor-beta: implications for homeostasis. Prog Growth Factor Res. 1994;5:341–51. doi: 10.1016/0955-2235(94)00004-5. [DOI] [PubMed] [Google Scholar]
- 66.Tsunawaki S, Sporn M, Nathan C. Comparison of transforming growth factor-beta and a macrophage- deactivating polypeptide from tumor cells. Differences in antigenicity and mechanism of action. J Immunol. 1989;142:3462–8. [PubMed] [Google Scholar]
- 67.Gorelik L, Constant S, Flavell RA. Mechanism of transforming growth factor beta-induced inhibition of T helper type 1 differentiation. J Exp Med. 2002;195:1499–505. doi: 10.1084/jem.20012076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gorelik L, Fields PE, Flavell RA. Cutting edge: TGF-beta inhibits Th type 2 development through inhibition of GATA-3 expression. J Immunol. 2000;165:4773–7. doi: 10.4049/jimmunol.165.9.4773. [DOI] [PubMed] [Google Scholar]
- 69.Chen CH, Seguin-Devaux C, Burke NA, Oriss TB, Watkins SC, Clipstone N, et al. Transforming growth factor beta blocks Tec kinase phosphorylation, Ca2+ influx, and NFATc translocation causing inhibition of T cell differentiation. J Exp Med. 2003;197:1689–99. doi: 10.1084/jem.20021170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Capsoni F, Minonzio F, Ongari AM, Carbonelli V, Galli A, Zanussi C. IL-10 up-regulates human monocyte phagocytosis in the presence of IL-4 and IFN-gamma. J Leukoc Biol. 1995;58:351–8. doi: 10.1002/jlb.58.3.351. [DOI] [PubMed] [Google Scholar]
- 71.Spittler A, Schiller C, Willheim M, Tempfer C, Winkler S, Boltz-Nitulescu G. IL-10 augments CD23 expression on U937 cells and down-regulates IL-4-driven CD23 expression on cultured human blood monocytes: effects of IL-10 and other cytokines on cell phenotype and phagocytosis. Immunology. 1995;85:311–7. [PMC free article] [PubMed] [Google Scholar]
- 72.Koppelman B, Neefjes JJ, de Vries JE, de Waal Malefyt R. Interleukin-10 down-regulates MHC class II alphabeta peptide complexes at the plasma membrane of monocytes by affecting arrival and recycling. Immunity. 1997;7:861–71. doi: 10.1016/s1074-7613(00)80404-5. [DOI] [PubMed] [Google Scholar]
- 73.Willems F, Marchant A, Delville JP, Gerard C, Delvaux A, Velu T, et al. Interleukin-10 inhibits B7 and intercellular adhesion molecule-1 expression on human monocytes. Eur J Immunol. 1994;24:1007–09. doi: 10.1002/eji.1830240435. [DOI] [PubMed] [Google Scholar]
- 74.Bluestone JA, Tang Q. How do CD4+CD25+ regulatory T cells control autoimmunity? Curr Opin Immunol. 2005;17:638–42. doi: 10.1016/j.coi.2005.09.002. [DOI] [PubMed] [Google Scholar]