

Abstract
Ethological assessment of murine models of Huntington’s disease (HD), an inherited neurodegenerative disorder, enables correlation between phenotype and pathophysiology. Currently, the most characterized model is the R6/2 line that develops a progressive behavioral and neurological phenotype by six weeks of age. A recently developed knock-in model with 140 CAG repeats (KI) exhibits a subtle phenotype with a longer progressive course, more typical of adult-onset HD in humans. We evaluated rotarod performance, open-field behavior, and motor activity across the diurnal cycle in KI mice during early to mid-adulthood. Although we did not observe any effects of age, relative to wild-type (WT) mice, KI mice showed significant deficits in both open-field climbing behavior and home-cage running wheel activity during the light phase of the diurnal cycle. An interesting sex difference also emerged. KI females spent more time in the open-field grooming and more time running during the diurnal dark phase than KI males and WT mice of both sexes. In striatum, the primary site of HD pathology, we measured behavior-related changes in extracellular ascorbate (AA), which is abnormally low in the R6/2 line, consistent with a loss of antioxidant protection in HD. KI males exhibited a 20–40% decrease in striatal AA from anesthesia baseline to behavioral activation that was not observed in other groups. Collectively, our results indicate behavioral deficits in KI mice that may be specific to the diurnal cycle. Furthermore, sex differences observed in behavior and striatal AA release suggest sex-dependent variation in the phenotype and neuropathology of HD.
Keywords: Huntington’s disease, sex, rotarod, open-field, diurnal cycle, ascorbate, voltammetry, oxidative stress
Introduction
Huntington’s disease (HD) is an inherited, autosomal dominant condition caused by an expanded trinucleotide (CAG) repeat resulting in degeneration of the striatum and corticostriatal pathway [10]. The HD gene encodes for huntingtin, a multi-functional protein widely expressed throughout the nervous system. Symptoms of HD include cognitive deficits and adventitious movements (e.g. chorea, tremor, dystonia). Ethological assessment of genetic murine models of HD provides correlation between phenotypic characteristics and mechanisms of HD pathophysiology. Behavioral assessments of these models also provide pre-clinical evaluations of possible therapeutic agents.
Currently, the most characterized HD model is the R6/2 transgenic line. These mice express a transgene including the huntingtin promoter region and ~150 CAG repeats [16]. R6/2 mice develop a progressive behavioral and neurological phenotype by six weeks of age, modeling juvenile-onset HD [4]. Behavioral symptoms include tremor, clasping (dystonia) and stereotyped movements similar to the chorea observed in humans with HD. R6/2 mice also exhibit neuropathological changes including intra-nuclear inclusions of huntingtin conjugated to ubiquitin, similar to that observed in neurons of HD patients [6]. Evaluation of R6/2 neurons in culture revealed changes in glutamate (GLU) receptor function, altered membrane properties, and increased striatal cell death after a single-exposure to oxidative stress [5,25]. Increased susceptibility to oxidative stress may be related to a striatal ascorbate (AA) deficit observed in these mice [26]. In fact, AA treatment and resulting increases in extracellular concentrations of AA in the striatum attenuated the behavioral phenotype [27] and normalized excessive striatal neuronal activity in R6/2 mice [28].
A recently developed mouse line with 140 CAG repeats knocked-in to the huntingtin mouse gene (KI) appears to provide a useful model of adult-onset HD. These mice not only show nuclear inclusions, but also later onset of behavioral symptoms and a longer progressive course than the R6/2 line [18]. Furthermore, unlike R6/2 mice, KI mice express the HD mutation in the appropriate genomic and protein context, similar to the manner of mutant huntingtin expression in humans [17]. Presently, the KI model has not been well characterized. In this study, we made a detailed evaluation of the behavioral phenotype during early to mid-adulthood. Rotarod performance, open-field behavior and diurnal motor activity were assessed in KI mice and wild type (WT) controls. We also used slow-scan voltammetry to monitor striatal AA release. Due to the slower rate of disease progression in this model, changes in striatal AA release at an early age may indicate a neurochemical abnormality that could be treated before overt signs of HD are present.
Materials and Methods
Animals
Homozygous male and female 140 CAG KI mice and WT controls used in this study were bred in our colony from heterozygous breeding pairs obtained from an established colony at University of California, Los Angeles [18]. The KI mice contain a chimeric mouse/human exon 1 of the huntingtin gene with ~140 CAG repeats inserted into the mouse gene via homologous targeting of W9.5 ES cells from a 129Sv background strain [18]. Mice were weaned at ~ 3 weeks, separated by sex, and housed with littermates. At ~ 8 weeks of age, tail tissue samples were obtained and mice were genotyped by standard polymerase chain reaction (PCR) protocol and placed in individual housing. All mice were housed in the departmental animal colony under standard conditions (12 hr light/dark cycle with lights on at 07:30 hr) with access to food and water ad libitum. Each group of mice assessed (n = 4–16) contained animals from three to thirteen litters. Both the housing and experimental use of the animals followed National Institutes of Health guidelines and were approved by the Institutional Animal Care and Use Committee.
Body Weight
Since a decrease in body weight is associated with disease progression in some HD models [4], KI and WT mice were weighed once a week prior to the ethological assessments.
Ethological Assessments
Rotarod performance was assessed once weekly between 09:00 and 11:00 hr in mice from 14 to 27 weeks of age. The rotarod consisted of a 6 cm diameter rubber rod ~ 22 cm long separated by metal dividers to provide running space for four mice. A metal switch plate was located under each running space. The rod was motorized and connected to a computer that recorded the time each mouse hit the switch plate. Mice were tested in random order two at a time. Mice were placed onto the rotarod facing the back of the apparatus (such that they would be running in a forward direction). Each running space was cleaned thoroughly after each animal. When both mice were situated, the rotarod was started and the speed ramped up in a linear fashion from 0 to 40 rpm over a 2-min period and then maintained 40 rpm until the mouse hit the switch plate. The maximum time on the rotarod was capped at 120 s, the end of the acceleration phase. Thus, the data presented includes only the acceleration portion of the assessment. Each weekly session consisted of three trials with at least 1 min of rest between trials.
Open-field behavior was videotaped for 10 min once every week between 12:00 to 15:00 hr in the same group of animals. At least one hour of rest was provided between assessments, and mice again were tested in random order. Mice were individually placed in an open-field arena (45 X 26 cm) with clear, Plexiglas® walls (20 cm) and standard bedding. A wire-mesh climbing box was placed in the center of the arena. Videotapes were analyzed by two independent observers who recorded several different types of behaviors using Best Analysis software (Educational Consulting Inc., Las Vegas, NV; http://www.skware.com). Observers were kept blind to the genotype of the animals. Inter-rater reliability was confirmed with Cohen’s Kappa values between ~0.8–1.0 on coding practice sessions. Each rater practiced coding behaviors until reliability was achieved before coding videotapes used in our analyses. Behaviors in the open-field were defined as follows: climbing (all four feet on the side of the climbing box), digging (burrowing into bedding with nose and/or limbs), grooming (any type of forelimb or hind limb grooming), locomotion (walking or running with all four feet on ground), rearing (hind limbs on the ground with forelimbs lifted), resting (not engaging in other behaviors), or scratching (rearing with forelimbs scratching the side of the arena).
In a separate group of animals, motor activity across the diurnal cycle was evaluated continuously from 16 to 19 weeks of age via running wheels (11.3 cm diameter) permanently placed in home cages in the animal colony. One KI female and one KI male were affected by ulcerative dermatitis early in the study and were replaced with a healthy mouse of the same age and genotype that had been habituated to the wheel. The KI male was replaced on the 3rd day of the first week (age 16 weeks) and the female was replaced on the 3rd day of the 2nd week (age 17 weeks). These two replacement mice did not exhibit significant differences in wheel revolutions per hour compared to the mice that they replaced. Location of each cage in the room was randomized by rotating cage location each week. A computer continuously recorded the number of wheel revolutions per hour for each animal.
Preparation for voltammetric recording
Voltammetric analysis of striatal ascorbate release was conducted in a separate group of mice between 8 to 30 weeks of age. Mice were anesthetized with chloropent (0.4ml/100g, i.p.) after isoflurane and were mounted in a stereotaxic frame. The skull was exposed and bilateral holes were drilled over the striatum at anterior 0.5 mm and lateral 2.0 mm from bregma. A plastic cylindrical hub (~3.1 mm diameter) sealed with a plastic cap was positioned at a 5° angle over each hole and secured to the skull with dental cement. An antibiotic cream was applied to the skin incision following surgery and animals were returned to the colony for at least 4 days of recovery.
Voltammetry
A glass-insulated carbon fiber (10 μm diameter) served as the working electrode. The active surface area extended ~ 150 μm beyond the glass. The electrode was electrochemically pretreated to separate the oxidation signal for AA from other easily oxidized compounds such as 3,4 –dihydroxyphenylacetic acid (DOPAC) [8]. Subsequent tests in citrate phosphate buffer containing 100 μm AA and 20 μm DOPAC ensured adequate sensitivity and peak separation. Generation of waveforms for staircase voltammetry and storage of sampled current was performed by a computer interfaced to a locally constructed potentiostat operating in two-electrode mode. A potential was applied in 6 mV steps from −200 to +600 mV versus reference to ensure AA and DOPAC oxidation. The scan rate was set at 20 mV/sec and scans were obtained at 60 sec intervals.
On the day of recording, both the working electrode and an Ag/AgCl reference electrode were fitted into separate microdrives, which were designed to mate with the head-mounted hubs [26]. After the microdrives were prepared, the mouse was lightly anesthetized with chloropent (0.2ml/100g, i.p.) after isoflurane and the cap over each hub was removed. If needed, fluids were cleared from the hub until the brain surface was visible. The working electrode was then manually lowered into the left or right striatum (~3.5 mm ventral to the skull surface), and the reference electrode was placed in the contralateral hub and lowered to the brain surface. A lightweight cable attached to each microdrive was connected to the voltammetry potentiostat via a swivel to allow the mouse to move freely. Voltammetry sessions were conducted between 09:00 and 15:00 hr with the majority of sessions in the morning hours. Each animal was pl aced in a circular glass arena (15 cm diameter) housed inside a sound attenuating chamber for recording.
Voltammetric scans began while mice were anesthetized, ~ 20 min after the chloropent injection and continued as they awoke (typically within 40–60 min) until ~ 150 min post-injection. Although recovery from anesthesia differed slightly among animals, there were no differences between genotype or sex in recovery from anesthesia. After the first two scans, which were obtained ~ 3–5 min apart, all subsequent scans were obtained once every 2 min. In some cases, after the recording session, the microdrives were removed and the working electrode was retested in vitro to estimate in vivo AA concentrations [3]. Most animals were returned to the colony for ~ 4–6 weeks when the recording procedure was repeated on the opposite side of the brain. In animals used for a second recording session, new electrodes were prepared and their left-right placement was reversed, allowing us to record from undisturbed striatal tissue in the same animal. The second session followed the same protocol as the first.
Histology
After the last recording session, some animals were anesthetized with isoflurane and current was passed through the working electrode to mark the recording site instead of retesting the electrode in vitro. Mice were later deeply anesthetized with chloropent (1.5 times the surgical dose), and transcardially perfused with saline followed by 10% potassium ferrocyanide in 10% formalin. Brains were removed, frozen, sectioned, stained with cresyl violet, and examined under a light microscope to confirm recording location [24].
Data Analysis
Repeated-measures analyses of variance (ANOVAs) were used to analyze body weight, mean time spent on the rotarod, and wheel revolutions per hour. These analyses compared male and female WT and KI mice across age. For the motor activity study, mean number of wheel revolutions per hour were calculated for each mouse each week for light and dark phases of the diurnal cycle. These means were used as the repeated measure. Total mean revolutions per hour for all weeks were also assessed as the dependent variable in a two-way ANOVA used to evaluate differences between genotype and light-versus-dark phase of the diurnal cycle. A regression analysis was used to evaluate slopes of mean rotarod times comparing genotype across age. Separate repeated-measures ANOVAs were run for each behavior coded during the open-field test. The analyses evaluated percent time spent engaging in each behavior, comparing male and female WT and KI mice across age. Total number of climbs and latency to climb were evaluated in the same manner. Because mice were habituated to the rotarod and open-field during the first two weeks of data collection (14–15 weeks of age), these two weeks were not included in the analyses. Statistically significant data was reported if p < 0.05. Sphericity was assumed for reporting degrees of freedom. P-values were adjusted using the Huynh-Feldt test. Contrasts on means using the Holm-Sidak adjustment were used to evaluate interaction effects.
Voltammetry scans were analyzed in 20-min segments by calculating mean AA peak height (measured from the apex). Mean brain AA concentrations were estimated by comparing in vivo scans to post calibration in vitro scans as described previously [3]. Student’s t-tests were used to evaluate: 1) estimated AA concentrations between male and female KI and WT mice; and 2) the percent change in mean AA peak height of the first 10 scans during anesthesia to the last 10 scans taken during active behavior in all KI and WT mice included in the study. A univariate ANOVA was used to evaluate the effects of age on the percent change in AA from anesthesia to behavior. Statistically significant data was reported if p < 0.05.
Results
Body Weight
Evaluation of body weight in 9 WT (4 females, 5 males) and 13 KI (5 females, 8 males) mice from 14 to 27 weeks of age showed no overall difference. A difference was observed, however, between males (mean ± SEM = 30.5 ± 0.94 g) and females (25.2 ± 1.1 g), regardless of genotype, (F1,18 = 13.35, p < 0.01). Furthermore, female KI mice weighed less (mean ± SEM = 22.7 ± 1.5 g), (F1,18 = 4.44, p < 0.05) and did not gain as much weight across time (F13,234 = 4.32, p < 0.01) compared to WT females (mean ± SEM = 27.8 ± 1.6 g) or males. There were no differences in body weight between male KI (mean ± SEM = 31.0 ± 1.2 g) and male WT (30.0 ± 1.5 g) mice. Body weight data are summarized in Fig. 1.
Fig. 1.
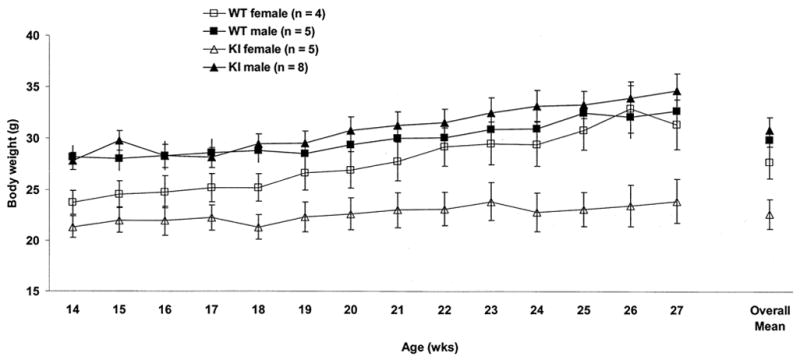
Mean ± SEM body weight (g) from 14 to 27 weeks of age. KI female mice weighed significantly less than other groups (p < 0.05) and did not gain as much weight with age (p < 0.01). There was no significant difference in body weight between male KI and male WT mice. n = number of mice.
Ethological Assessments
Rotarod performance and open-field behavior were evaluated in 9 WT (4 females, 5 males) and13 KI (5 females, 8 males) mice. Analysis of rotarod performance showed no significant differences between WT (mean time on rotarod ± SEM = 59.44 ± 7.01 s) and KI mice (mean time ± SEM = 59.94 ± 5.96 s; p = 0.7) or between males (mean time ± SEM = 59.91 ± 5.96 s) and females (mean time ± SEM = 59.46 ± 7.01 s; p = 0.4); regression analysis was also not significant (p = 0.35). In the open-field, an overall effect of age was observed for digging (F11,198 = 4.47), locomotion (F11,198 = 5.98), rearing (F11,198 = 3.50), and resting (F11,198 = 3.85; p < 0.001 in all cases). Despite fluctuations in percent time spent resting from week to week, all mice spent less time engaged in digging, locomotion, and rearing with increasing age (p < 0.05). Overall age effects were not observed for other assessed behaviors. As shown in Fig. 2, KI mice spent significantly less time climbing (F1,18= 4.61) and more time grooming (F1,18= 5.27) than WT mice (p < 0.05 in both cases). Sex differences are shown in Fig. 3 and Fig.4 (A and B). Note that the increase in grooming was restricted to KI females (F1,18= 7.84; p < 0.05) and that male WT mice climbed more than other groups (F1,18= 9.55; p < 0.01). Although there were no significant differences in latency to climb (F1,18= 2.37; p = 0.14), Fig 4. (B) shows that a similar sex effect was observed in total number of climbs (F1,18= 9.84; p < 0.05).
Fig. 2.
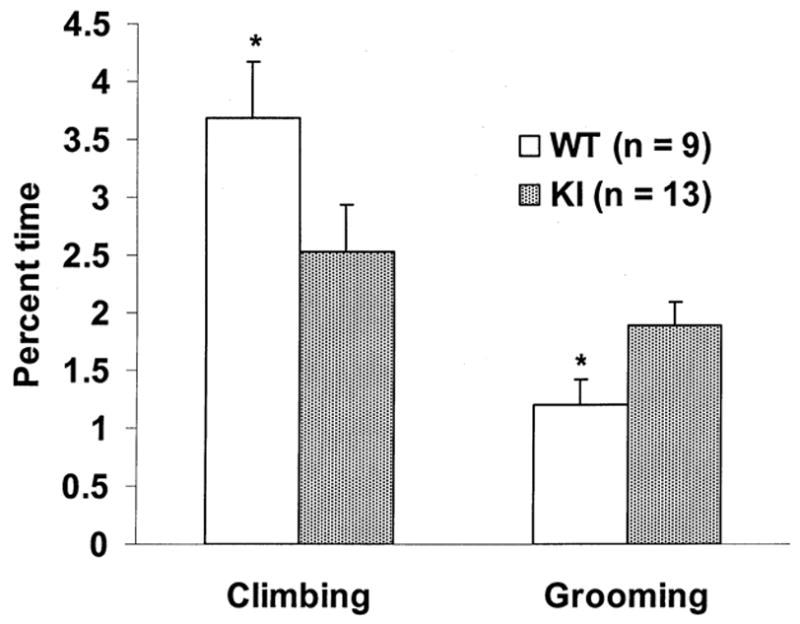
Mean ± SEM percent total activity time in the open-field spent climbing and grooming across all sessions (16 to 27 weeks of age) in WT and KI mice. ∗ = p < 0.05 compared to KI mice. n = number of mice.
Fig. 3.
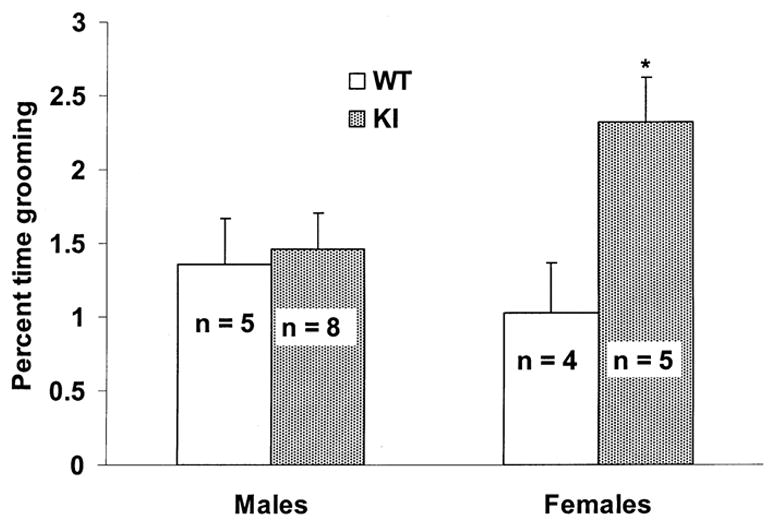
Mean ± SEM percent total activity time in the open-field spent grooming across all sessions (16 to 27 weeks of age) comparing sex and genotype. ∗ = p < 0.05 compared to males and WT females. n = number of mice.
Fig. 4.
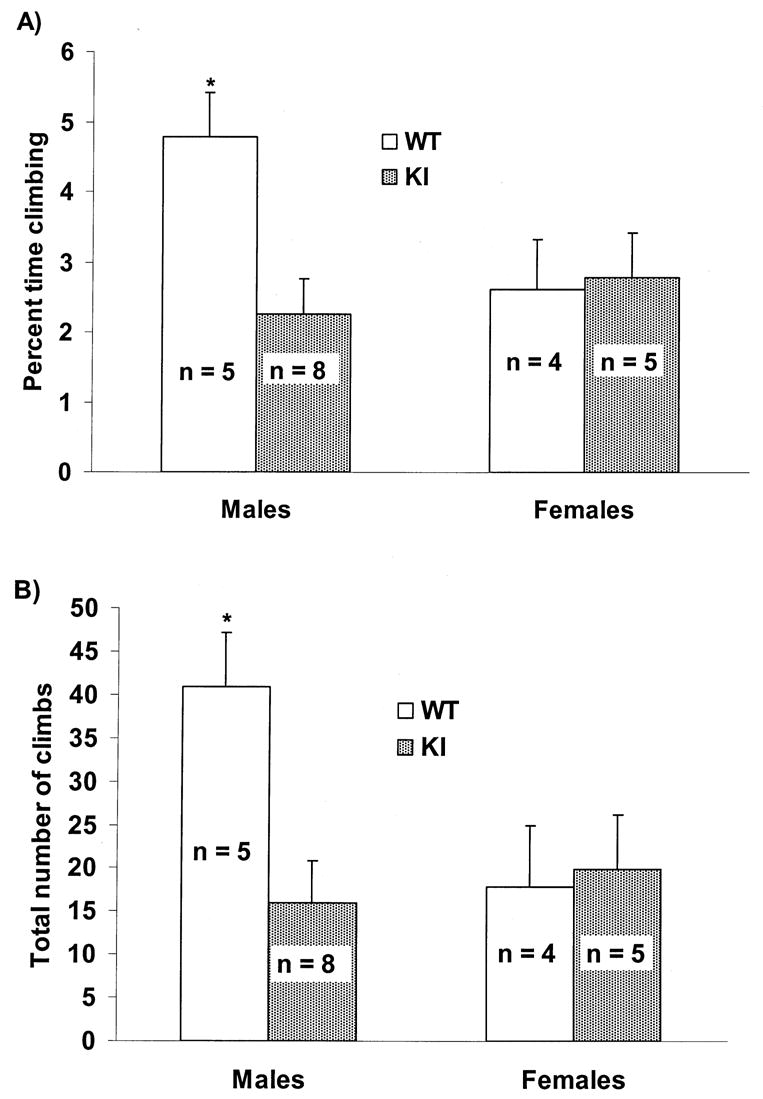
(A) Mean ± SEM percent total activity time in the open-field spent climbing across all sessions (16 to 27 weeks of age) comparing sex and genotype. ∗ = p < 0.01 compared to females and KI males. n = number of mice. (B) Mean ± SEM total number of climbs in the open-field across all sessions (16 to 27 weeks of age) comparing sex and genotype. ∗ = p < 0.05 compared to females and KI males. n = number of mice.
Overall differences between males and females were observed for locomotion (F1,18 = 6.66) and scratching (F1,18 = 7.50; p < 0.05 in each case). Males spent more time engaged in locomotion (mean percent time ± SEM = 41.33 ± 1.26) compared to females (36.30 ± 1.49). Females spent more time scratching (mean percent time ± SEM = 0.09 ± 0.019) than males (0.02 ± 0.016). Although not statistically significant, a trend toward an increase in locomotion in WT compared to KI males that was not apparent in female mice is shown in Fig. 5. No differences were observed between WT and KI mice in resting, rearing, digging, or scratching; there were no effects of genotype and sex at different ages for any of the behaviors.
Fig. 5.
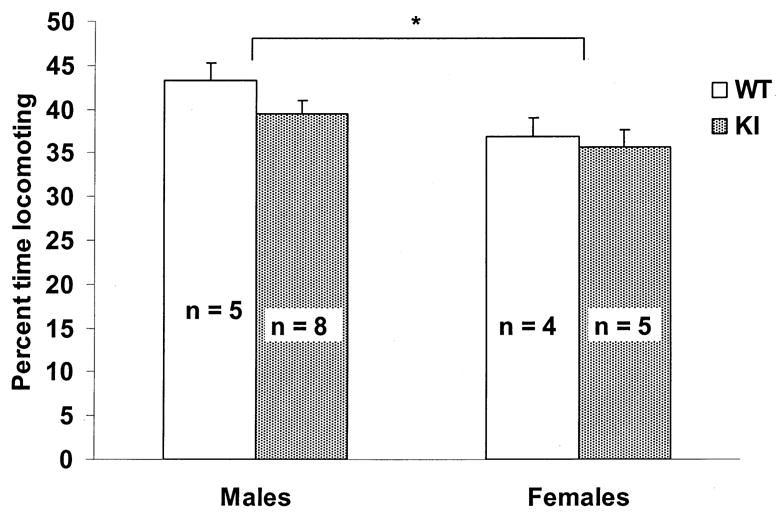
Mean ± SEM percent total activity time in the open-field spent engaging in locomotion across all sessions (16 to 27 weeks of age) comparing sex and genotype. ∗ = p < 0.05 males compared to females. Although there was no significant effect of genotype, WT males spent more time engaging in locomotion than KI males, a trend that was not observed in female mice. n = number of mice.
When provided with a running wheel in their home cage, healthy mice will spend the majority of the dark phase of the diurnal cycle running in the wheel and will rest during the light phase. Previous research has suggested that this pattern of motor activity may be disrupted in HD mice [22]. Our analysis showed no overall effect of genotype when comparing the light phase to the dark phase (F1,28 = 0.553; p = 0.46) indicating that KI mice maintained a normal diurnal activity pattern (running less during the light period and more during the dark period). Further investigation of genotype within each phase, however, revealed that KI mice ran significantly less than WT mice during the light phase (F1,12 = 5.14; p < 0.05) as shown in Fig. 6(A). During the dark phase, moreover, a sex difference emerged (F1,12 = 7.98; p < 0.05). As depicted in Fig. 6(B), KI female mice ran more than WT females and KI males (F1,12 = 10.10; p < 0.01). Interaction effects were observed between genotype and sex across age during the light phase (F3,36 = 5.76; p < 0.01). Evaluation of these effects showed that WT animals ran significantly more at 17 weeks of age (p < 0.05) (the second week of assessment) possibly indicating habituation to the wheel that was not observed in KI mice. Interaction effects also showed that WT males decreased wheel running at 18 and 19 weeks of age compared to prior weeks (p < 0.01). Interaction effects were not observed during the dark phase of the diurnal cycle.
Fig. 6.
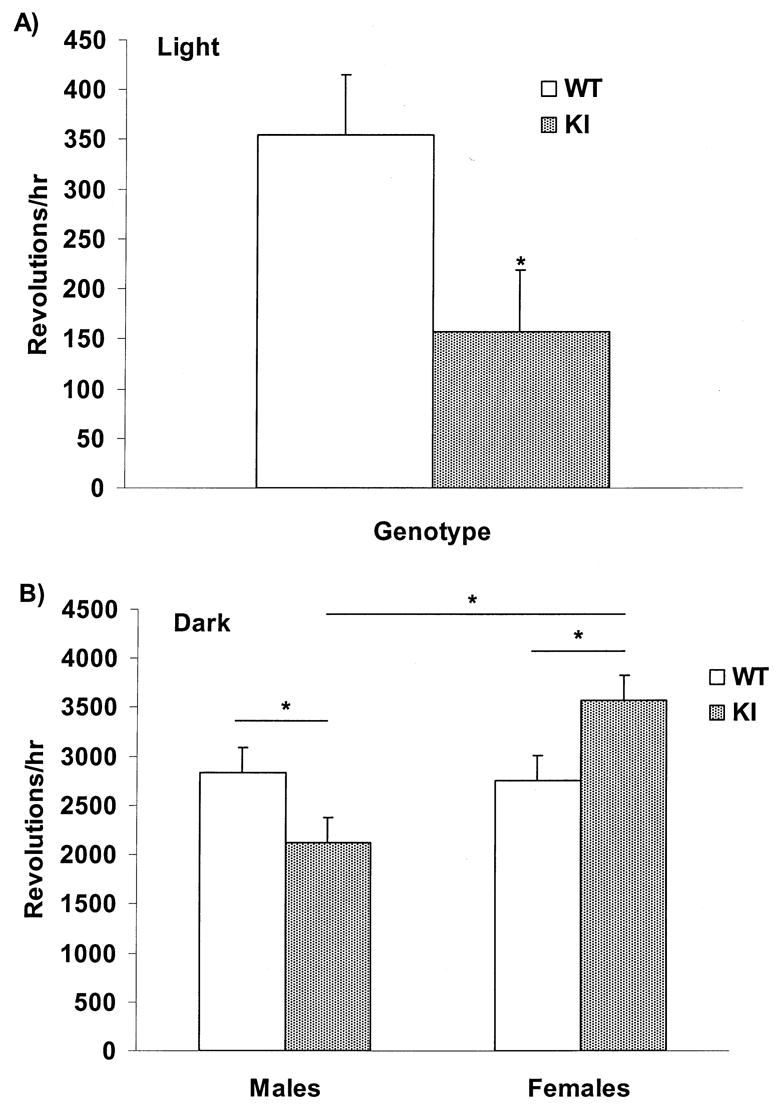
Mean ± SEM number of running wheel revolutions per hour from 16-19 weeks of age. n = 4 mice in each group (WT males, WT females, KI males, KI females) ∗ = p < 0.05. (A) Genotype is compared in mice across all weeks during the light phase of the diurnal cycle. KI mice exhibited a significantly lower number of wheel revolutions per hour compared to WT mice. (B) Genotype and sex are compared across all weeks during the dark phase. KI females ran more than WT females and KI males. KI males ran less than WT males and KI females.
Voltammetry
As shown in Table 1, evaluation of estimated AA concentrations did not reveal a difference between KI and WT mice. Like our ethological assessments, however, we found emerging evidence of a sex difference. Female KI mice exhibited a lower concentration of AA during anesthesia compared to WT females (p < 0.05). Although males did not show an AA difference during anesthesia, Fig. 7 shows a 20–40% decrease in striatal AA release in male KI mice as they became behaviorally active, while WT males increased striatal AA during the same period (p = 0.19). We also observed a KI sex difference in striatal AA release that was not apparent in WT controls. Female KI mice exhibited an increase in striatal AA release upon recovery from anesthesia compared to male KI mice (p < 0.05). No effects of age on striatal AA release were observed.
Table 1.
Mean ± SEM Baseline Ascorbate Concentrations (μM) Estimated from Re-testing In Vitro
| Group | n | AA μM |
|---|---|---|
| WT males | 12(9) | 377.41 ± 97.67 |
| KI males | 12(10) | 376.38 ± 118.6 |
| WT females | 10(6) | 283.84 ± 45.34 |
| KI females | 5(4) | 135.61 ± 26.38* |
Note.
= p < 0.05 compared to WT females.
n = number of sessions (number of mice)
Fig. 7.
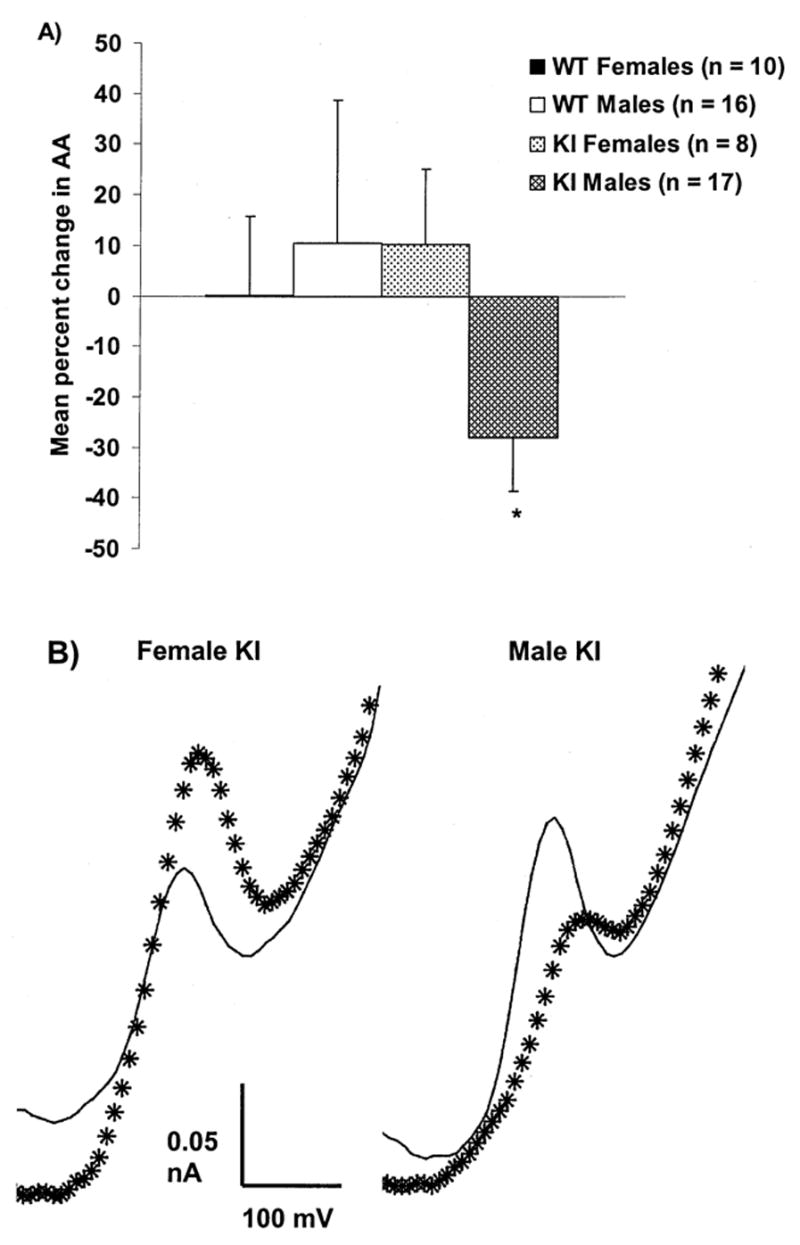
(A) Mean ± SEM percent change in AA signal from anesthesia baseline in male KI and WT mice and female KI mice. Baseline was obtained from the first 10 scans after the AA signal had stabilized during anesthesia and is compared to the last 10 scans obtained when behavioral activation was greatest. Although not significant, KI male mice exhibited a decrease in striatal AA upon recovery from anesthesia while WT males exhibited an increase (p = 0.19). The AA response in male KI mice was significantly different from the response in female KI mice (∗ = p < 0.05 compared to female KI mice). n = number of sessions in 6 WT females, 10 WT males, 5 KI females and 11 KI males. (B) Sample differential voltammograms obtained from female or male KI mice. Baseline AA peak was obtained during anesthesia (solid line) while behaving scan was obtained approximately 150 min later when mice were behaviorally active (asterisks). The ascorbate oxidation peak occurred between -100 and -200 mV versus reference. Note that the AA peak rises during behavior in the female KI mouse while it decreases in the male KI mouse.
Results of histology confirmed placement of the working voltammetry electrode in the striatum. All lesions were located approximately 0.5 mm anterior, 2 mm lateral to bregma and 3.5 mm ventral from skull surface (Fig. 8).
Fig. 8.
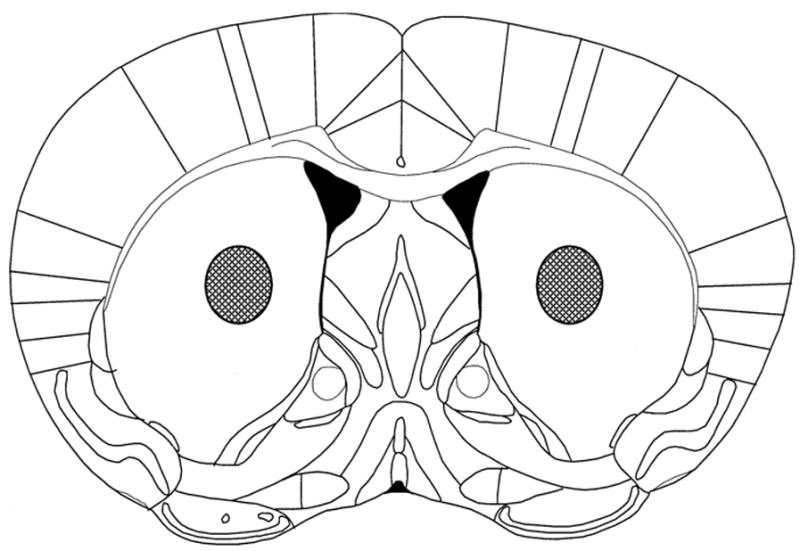
Histology confirmed placement of working voltammetry electrodes in the anterior striatum. Shaded areas indicate the location of all lesion sites, approximately 0.5 mm anterior, 2 mm lateral to bregma and 3.5 mm ventral from skull surface.
Discussion
Our results confirm and extend previous findings suggesting behavioral differences between KI and WT mice. KI mice showed deficits in both open-field climbing behavior and home-cage running wheel activity during the light phase of the diurnal cycle. Furthermore, our findings revealed sex differences between KI and WT mice in body weight, open-field behavior, running wheel activity during the dark phase and extracellular striatal AA, suggesting that sex may play a role in the phenotype and neuropathology of HD.
Similar to previous findings [18] we did not find overall differences in body weight between KI and WT mice. However, females weighed less than males, a common finding in mice [30], and an effect of HD was observed in female mice. KI females weighed less and did not gain as much weight with age. The lower body weight in KI females may be related to an increase in motor activity during the dark phase of the diurnal cycle.
Ethological assessments confirmed that KI mice exhibit behavioral deficits. Decreased motor activity during the light phase and a climbing deficit in the open-field indicates that KI mice exhibit deficits in motor control as early as 16 weeks of age. Evaluation of diurnal motor activity showed that hypoactivity in KI mice was specific to the light phase. During the dark phase, KI females exhibited hyperactivity while KI males remained less active. Hyperactivity during the dark phase and an increase in grooming in the open-field may reflect stereotypic behavior associated with HD in females. Previous research showed that the progression of HD in KI mice begins with hyperactivity followed by hypoactivity [18]. Thus, it is possible that KI females may have a later onset or slower progression than male KI mice. It is also important to note that although the normal circadian activity pattern breaks down in late stage disease in R6/2 mice and in humans with HD [22], KI mice in our study maintained a normal diurnal activity pattern from 16 to 19 weeks of age.
Previous findings have also shown differences between KI and WT mice in the open-field. Specifically, hyperactivity was observed at 1 month of age, followed by hypoactivity at 4 months of age and a decrease in rearing at 2, 4 and 6 months of age [18]. In our study, age-related differences were not apparent and we did not observe differences in rearing or locomotion in KI mice. We attribute this to our evaluation of the same group of mice for several weeks, and that animals were habituated to the open-field. The previous study evaluated separate cohorts at different ages and included younger and older groups. Furthermore, unlike our study, mice were not habituated to the open-field and were tested during the dark phase of the diurnal cycle [18]. Thus, differences in rearing and locomotion in the previous study may have reflected a response of mice to a novel environment or may only occur during the dark phase. Previous behavioral studies have not reported sex differences in HD mice. Our results suggest that the sex of the animal may contribute to the development and progression of the HD phenotype in mice.
Although expansion of the CAG repeat associated with paternal inheritance and corresponding increase in disease severity and earlier age of onset is well documented [33], the mechanisms of sex effects in HD are not well studied. In addition to expansion of the CAG repeat associated with paternal inheritance, the sex of affected progeny also affects CAG expansion and age of onset [11]. This finding was observed in a transgenic mouse model of HD (R6/1) in which siblings inherited the affected gene from the male parent [11]. Results showed that the CAG repeat was significantly expanded in the male offspring and contracted in the female offspring compared to the parent [11]. CAG repeats were the same length in both X and Y bearing sperm indicating that this change was associated with embryo development rather than changes occurring during spermatogenesis [12]. Thus, the CAG repeat is likely to be shorter in females and longer in males with successive generations. One study evaluating 1084 cases of HD found that mean age of onset was higher in females compared to males [31]. Moreover, there was no interaction effect between this sex difference and line of inheritance, suggesting that this was an overall effect of sex in HD not associated with inheritance [31].
Although the decrease in striatal AA release upon behavioral activation observed in KI males was less prominent and more variable than that previously described in male R6/2 mice, the deficit in male KI mice, ~20–40% was similar to that of R6/2 mice [26]. This deficit suggests that male HD mice may be more susceptible to oxidative stress in the striatum. The change in extracellular striatal AA associated with increasing behavioral activation in WT mice did not reach the levels previously observed in the male WT R6/2 mice [26]. Since both KI and WT mice appeared to be less active in ethological assessments (in which no anesthesia was administered) as well as during voltammetry compared to R6/2 mice, as observed in our past studies [26,27,28], a likely explanation is lower activity levels of the 129Sv strain included in the KI background compared to the C57BL/6 background strain of the R6/2 mice [16]. Behavioral differences between these strains of mice have been observed on several ethological assessments, indicating reduced activity and motivation in the 129Sv strain [2,15,19,21]. It is possible that increasing the activity level of the mice during voltammetry using a treadmill or running wheel after complete recovery from anesthesia would result in a greater increase in striatal AA release in WT males, revealing the deficit in KI males.
The difference in striatal AA release and behavior between male and female KI mice and WT controls supports evidence that striatal AA release is related to the extent of behavioral activation [29]. The increase in striatal AA release and corresponding increase in motor activity in female KI mice may reflect a hyperkinesia associated with the HD phenotype in female mice whereas the lower levels of striatal AA release and activity in KI males may be associated with hypokinesia similar to that observed previously in male R6/2 mice [26]. Furthermore, the increase in striatal AA release in female KI mice may reflect a compensatory response to oxidative stress that is not occurring in male mice. We postulate that the increase in striatal AA in female mice may be neuroprotective. These differences support the results of our ethological assessments and likely reflect sex-dependent variation in the onset and/or progression of HD.
Interestingly, sex differences in brain AA concentrations have been observed. Female rats have lower basal brain AA concentrations compared to males [1,7,13]. Sex differences are also observed in expression of AA transporters SVCT1 and SVCT2 throughout the body [14]. Although we did not see differences in basal AA concentrations in striatum, we did observe a trend toward lower levels in female mice compared to males and we observed lower levels in female KI compared to female WT mice (Table 1). Since sex differences in brain AA function are region specific [13], it is difficult to determine how past findings relate to what we have observed in the striatum. It is probable, however, that gonadal hormones such as estrogen contribute to the differences we have observed and play a role in modulating striatal AA release. Estrogen modulates brain AA levels and prevented ischemia-induced AA loss in the hippocampus [13]. Like ischemia, HD also results in oxidative stress which may account for the striatal AA loss observed in male mice. Estrogen may prevent this loss in female HD mice.
Estrogen may also exert changes in striatal neurotransmission directly contributing to the increase in AA and possibly a delay in symptoms of the disease in female mice. For example, GLU uptake in the striatum is coupled with AA release, presumably via a heteroexchange mechanism at a GLU transport site [23]. Estrogen inhibition of N-methyl-D-aspartate (NMDA) receptors and effects on GLU transmission [32,34,35] may result in increased GLU uptake and striatal AA release in female mice. Estrogen also attenuates GLU-induced Ca2+ increase [9], which may counteract the GLU excitotoxicity believed to occur in HD animals [36]. In addition, neuroprotective effects of estrogen such as upregulation of neurotrophic factors [20] may also contribute to sex differences.
The differences observed in ethological assessments and striatal AA release confirm previous findings and suggest that there are variations in the phenotype and neuropathological characteristics of HD in KI mice that are dependent on the sex of the animal. Furthermore, increased striatal AA release during behavior may be neuroprotective in female mice. Sex differences also suggest that gonadal hormones such as estrogen may modulate AA release in the striatum and probably play a significant role in the neuropathology and development of HD.
Acknowledgments
This study was supported by the National Institute of Neurological Disorders and Stroke grant #R01 NS35663 to G.V.R. and the National Science Foundation Graduate Research Fellowship to J.L.D. Michael Levine graciously provided our initial breeding pairs of KI mice. Rafael White, Michael Chapman and Anne Mattingly assisted with data collection. Faye Caylor and Paul Langley provided administrative and technical support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Allison JH, Stewart MA. Myo-inositol and ascorbic acid in developing rat brain. J Neurochem. 1973;20:1785–1788. doi: 10.1111/j.1471-4159.1973.tb00296.x. [DOI] [PubMed] [Google Scholar]
- 2.Balogh SA, McDowell CS, Stavnezer AJ, Denenberg VH. A behavioral and neuroanatomical assessment of an inbred substrain of 129 mice with behavioral comparisons to C57BL/6J mice. Brain Res. 1999;836:38–48. doi: 10.1016/s0006-8993(99)01586-3. [DOI] [PubMed] [Google Scholar]
- 3.Basse-Tomusk A, Rebec GV. Regional distribution of ascorbate and 3,4-dihydroxyphenylacetic acid (DOPAC) in rat neostriatum. Brain Res. 1991;538:29–35. doi: 10.1016/0006-8993(91)90372-3. [DOI] [PubMed] [Google Scholar]
- 4.Carter RJ, Lione LA, Humby T, Mangiarini L, Mahal A, Bates GP, et al. Characterization of progressive motor deficits in mice transgenic for the human Huntington’s disease mutation. J Neurosci. 1999;19:3248–3257. doi: 10.1523/JNEUROSCI.19-08-03248.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cepeda C, Ariano MA, Calvert CR, Flores-Hernandez, Chandler SH, Leavitt BR. NMDA receptor function in mouse models of Huntington’s disease. J Neurosci Res. 2001;66:525–539. doi: 10.1002/jnr.1244. [DOI] [PubMed] [Google Scholar]
- 6.Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, et al. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the Huntington’s disease mutation. Cell. 1997;90:537–548. doi: 10.1016/s0092-8674(00)80513-9. [DOI] [PubMed] [Google Scholar]
- 7.Ferris DC, Kume-Kick J, Russo-Menna I, Rice ME. Gender differences in cerebral ascorbate levels and ascorbate loss in ischemia. Neuroreport. 1995;6:1485–1489. doi: 10.1097/00001756-199507310-00005. [DOI] [PubMed] [Google Scholar]
- 8.Gonon F, Buda M, Cespuglio R, Jouvet M, Pujol JF. Voltammetry in the striatum of chronic freely moving rats: detection of catechols and ascorbic acid. Brain Res. 1981;223:69–80. doi: 10.1016/0006-8993(81)90807-6. [DOI] [PubMed] [Google Scholar]
- 9.Huang Y, Huang YL, Zhang S, Zhu YC, Yao T. Estradiol acutely attenuates glutamate-induced calcium overload in primarily cultured rat hippocampal neurons through a membrane receptor-dependent mechanism. Brain Res. 2004;1026:254–260. doi: 10.1016/j.brainres.2004.08.038. [DOI] [PubMed] [Google Scholar]
- 10.Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- 11.Kovtun IV, Therneau TM, McMurray CT. Gender of the embryo contributes to CAG instability in transgenic mice containing a Huntington’s disease gene. Hum Mol Genet. 2000;9:2767–2775. doi: 10.1093/hmg/9.18.2767. [DOI] [PubMed] [Google Scholar]
- 12.Kovtun IV, Welch G, Guthrie HD, Hafner KL, McMurray CT. CAG repeat lengths in X- and Y-bearing sperm indicate that gender bias during transmission of Huntington’s disease gene is determined in the embryo. J Biol Chem. 2004;279:9389–9391. doi: 10.1074/jbc.M313080200. [DOI] [PubMed] [Google Scholar]
- 13.Kume-Kick J, Rice ME. Estrogen-dependent modulation of rat brain ascorbate levels and ischemia-induced ascorbate loss. Brain Res. 1998;803:105–113. doi: 10.1016/s0006-8993(98)00628-3. [DOI] [PubMed] [Google Scholar]
- 14.Kuo SM, MacLean ME, McCormick K, Wilson JX. Gender and sodium-ascorbate transporter isoforms determine ascorbate concentrations in mice. J Nutr. 2004;134:2216–2221. doi: 10.1093/jn/134.9.2216. [DOI] [PubMed] [Google Scholar]
- 15.Logue SF, Owen EH, Rasmussen DL, Wehner JM. Assessment of locomotor activity, acoustic and tactile startle, and prepulse inhibition of startle in inbred mouse strains and F1 hybrids: implications of genetic background for single gene and quantitative trait loci analyses. Neuroscience. 1997;80:1075–1086. doi: 10.1016/s0306-4522(97)00164-4. [DOI] [PubMed] [Google Scholar]
- 16.Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- 17.Menalled LB. Knock-in mouse models of Huntington’s disease. NeuroRx. 2005;2:465–470. doi: 10.1602/neurorx.2.3.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Menalled LB, Sison JD, Dragatsis I, Zeitlin S, Chesselet MF. Time course of early motor and neuropathological anomalies in a knock-in mouse model of Huntington’s disease with 140 CAG repeats. J Comp Neurol. 2003;465:11–26. doi: 10.1002/cne.10776. [DOI] [PubMed] [Google Scholar]
- 19.Miner LL. Cocaine reward and locomotor activity in C57BL/6J and 129/SvJ inbred mice and their F1 cross. Pharmacol Biochem Behav. 1997;58:25–30. doi: 10.1016/s0091-3057(96)00465-0. [DOI] [PubMed] [Google Scholar]
- 20.Miranda RC, Sohrabji F, Toran-Allerand D. Interactions of estrogen with the neurotrophins and their receptors during neural development. Horm Behav. 1994;28:367–375. doi: 10.1006/hbeh.1994.1033. [DOI] [PubMed] [Google Scholar]
- 21.Montkowski A, Poettig M, Mederer A, Holsboer F. Behavioural performance in three substrains of mouse strain 129. Brain Res. 1997;762:12–18. doi: 10.1016/s0006-8993(97)00370-3. [DOI] [PubMed] [Google Scholar]
- 22.Morton JA, Wood NI, Hastings MH, Hurelbrink C, Barker RA, Maywood ES. Disintegration of the sleep-wake cycle and circadian timing in Huntington’s disease. J Neurosci. 2005;25:157–163. doi: 10.1523/JNEUROSCI.3842-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O’Neill RD. The measurement of brain ascorbate in vivo and its link with excitatory amino acid neurotransmission. In: Boulton A, Baker G, Adams RN, editors. Voltammetric methods in brain systems: Neuromethods. Vol. 27. Clifton New Jersey: Humana Press Inc; 1995. pp. 221–268. [Google Scholar]
- 24.Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. 2. San Diego: Academic Press; 2001. [Google Scholar]
- 25.Petersen A, Larsen KE, Behr GG, Romero N, Przedborski S, Brundin P, et al. Expanded CAG repeats in exon 1 of the Huntington’s disease gene stimulate dopamine-mediated striatal neuron autophagy and degeneration. Hum Mol Genet. 2001;10:1234–1254. doi: 10.1093/hmg/10.12.1243. [DOI] [PubMed] [Google Scholar]
- 26.Rebec GV, Barton SJ, Ennis MD. Dysregulation of ascorbate release in the striatum of behaving mice expressing the Huntington’s disease gene. J Neurosci. 2002;22:RC202. doi: 10.1523/JNEUROSCI.22-02-j0006.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rebec GV, Barton SJ, Marseilles AM, Collins K. Ascorbate treatment attenuates the Huntington behavioral phenotype in mice. Neuroreport. 2003;14:1263–1265. doi: 10.1097/00001756-200307010-00015. [DOI] [PubMed] [Google Scholar]
- 28.Rebec GV, Conroy SK, Barton SJ. Hyperactive striatal neurons in symptomatic Huntington R6/2 mice: variations with behavioral state and repeated ascorbate treatment. Neuroscience. 2006;137:327–336. doi: 10.1016/j.neuroscience.2005.08.062. [DOI] [PubMed] [Google Scholar]
- 29.Rebec GV, Wang Z. Behavioral activation in rats requires endogenous ascorbate release in striatum. J Neurosci. 2001;21:668–675. doi: 10.1523/JNEUROSCI.21-02-00668.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reinhard C, Eder G, Fuchs H, Ziesenis A, Heyder J, Schulz H. Inbred strain variation in lung function. Mamm Genome. 2002;13:429–437. doi: 10.1007/s00335-002-3005-6. [DOI] [PubMed] [Google Scholar]
- 31.Roos RA, Vegter-van der V, Hermans J, Elshove HM, Moll AC, van de Kamp JJ, et al. Age at onset in Huntington’s disease: effect of line of inheritance and patient’s sex. J Med Genet. 1991;28:515–519. doi: 10.1136/jmg.28.8.515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smith SS. Estrogen administration increases neuronal responses to excitatory amino acids as a long-term effect. Brain Res. 1989;503:354–357. doi: 10.1016/0006-8993(89)91691-0. [DOI] [PubMed] [Google Scholar]
- 33.Trottier Y, Biancalana V, Mandel JL. Instability of CAG repeats in Huntington’s disease: relation to parental transmission and age of onset. J Med Genet. 1994;31:377–382. doi: 10.1136/jmg.31.5.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weaver CEJr, Park-Chung M, Gibbs TT, Farb DH. 17beta-Estradiol protects against NMDA-induced excitotoxicity by direct inhibition of NMDA receptors. Brain Res. 1997;761:338–341. doi: 10.1016/s0006-8993(97)00449-6. [DOI] [PubMed] [Google Scholar]
- 35.Zaulyanov LL, Green PS, Simpkins JW. Glutamate receptor requirement for neuronal death from anoxia-reoxygenation: an in vitro model for assessment of the neuroprotective effects of estrogens. Cell Mol Neurobiol. 1999;19:705–718. doi: 10.1023/A:1006948921855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zeron MM, Chen N, Moshaver A, Lee ATC, Wellington CL, Hayden MR, et al. Mutant huntingtin enhances excitotoxic cell death. Mol Cell Neurosci. 2001;17:41–53. doi: 10.1006/mcne.2000.0909. [DOI] [PubMed] [Google Scholar]