

Abstract
The PARK3 locus on chromosome 2p13 has shown linkage to both the development and age of onset of Parkinson’s disease (PD). One candidate gene at this locus is sepiapterin reductase (SPR). Sepiapterin reductase catalyzes the final step in the biosynthetic pathway of tetrahydrobiopterin (BH4), an essential cofactor for aromatic amino acid hydrolases including tyrosine hydroxylase, the rate-limiting enzyme in dopamine synthesis. The expression of SPR was assayed using semiquantitative real-time RT-PCR in human post-mortem cerebellar tissue from neuropathologically confirmed PD cases and neurologically normal controls. The expression of other enzymes involved in BH4 biosynthesis, including aldose reductase (AKR1B1), carbonyl reductase (CBR1 and CBR3), GTP-cyclohydrolase I (GCH1), and 6-pyruvoyltetrahydrobiopterin (PTS), was also examined. Single-nucleotide polymorphisms around the SPR gene that have been previously reported to show association to PD affection and onset age were genotyped in these samples. Expression of SPR showed a significant 4-fold increase in PD cases relative to controls, while the expression of AKR1B1 and PTS was significantly decreased in PD cases. No difference in expression was detected for CBR1, CBR3, and GCH1. Genetic variants did not show a significant effect on SPR expression, however, this is likely due to the low frequency of rare genotypes in the sample. While the association of SPR to PD is not strong enough to support that this is the PARK3 gene, this study further implicates a role for SPR in idiopathic PD.
Keywords: Parkinson’s disease, PARK3, sepiapterin reductase, RT-PCR, human, tetrahydrobiopterin
1. Introduction
Parkinson’s disease (PD, MIM 168600) is a common adult-onset neurodegenerative disorder that affects nearly 2% of the population age 65 and older (de Rijk et al., 2000). PD is characterized clinically by resting tremor, rigidity, bradykinesia, and postural instability (Calne, 2005; Hughes et al., 2001). The pathological characteristics of this disease are a progressive loss of dopaminergic neurons, primarily in the substantia nigra pars compacta, and the presence of eosinophilic cytoplasmic inclusions, called Lewy bodies, throughout the brain (Braak et al., 2003; Gibb and Lees, 1988; Jellinger, 2001).
The PARK3 locus (MIM 602404) on chromosome 2p13 was initially reported to be associated with PD affection status in an autosomal dominant model of PD using 6 European families, each with at least 4 affected PD relatives (Gasser et al., 1998). PARK3 was later shown to be linked and associated with age of onset of PD in the GenePD Study using 103 multiplex families (DeStefano et al., 2002). Pankratz et al. replicated these results, reporting evidence of linkage and association to age of onset of PD in a region on chromosome 2p, which overlaps PARK3 (Pankratz et al., 2004).
We previously reported that a haplotype in the PARK3 region, comprised of SNPs flanking the gene SPR (MIM182125), was significantly associated with onset age of PD (Karamohamed et al., 2003). The A-T-G alleles at rs2421095-rs1876487-rs1561244, respectively, were associated with a younger onset of PD (Karamohamed et al., 2003). SNP rs1876487, located 196 base pairs upstream of SPR (according to NCBI Build 36.1), was driving the association of the aforementioned haplotype and was shown to be significantly associated to onset age of PD (Karamohamed et al., 2003). Recently, the association of rs1876487 to onset age was replicated in a European familial PD cohort and the rs2421095-rs1876487-rs1561244 haplotype was reported to be associated with PD affection status in a German sample of sporadic PD (Sharma et al., 2006). Nevertheless, while these studies implicate SPR in PD, the association is not strong enough to suggest that SPR is the PARK3 gene.
SPR is a 3 exon gene encoding the 261 amino acid protein, sepiapterin reductase (7,8-dihydrobiopterin:NADP+ oxidoreductase) (Ichinose et al., 1991; Ohye et al., 1998). SPR catalyzes the final step of the biosynthetic pathway of tetrahydrobiopterin (BH4) (Katoh and Sueoka, 1984). BH4 is an essential cofactor for nitric oxide synthase and aromatic amino acid hydrolases, including tyrosine hydroxylase (Thony et al., 2000). In addition to SPR, two other genes are essential and sufficient for BH4 biosynthesis: GTP cyclohydrolase I (GCH1) and 6-pyruvoyltetrahydropterin synthase (PTS) (Thony et al., 2000). Aldose reductase (AKR1B1) and carbonyl reductase (CBR1 and CBR3) have also been shown to participate in different stages of BH4 synthesis (Iino et al., 2003; Park et al., 1991).
Here we examine the expression of SPR and five other genes involved in BH4 biosynthesis in human PD cerebellar tissue using real-time RT-PCR. The cerebellum was a suitable brain region for our study for several reasons. First, there was an abundance of available post mortem tissue from PD cases, compared to regions more substantially affected by PD, such as the substantia nigra. Second, dopaminergic neuronal transmission occurs in the cerebellum, including dopaminergic input from the substantia nigra, while remaining largely unaffected by PD pathology and neuronal degeneration (Hurley et al., 2003; Parsons et al., 2002). We also genotyped the aforementioned SNPs using DNA extracted from the brain tissue and assessed whether any SNP had an effect on the expression of SPR.
2. Results
2.1 Real-time RT-PCR gene expression
Real-time RT-PCR was used to measure the expression of six genes in human PD cerebellar tissue: AKR1B1, CBR1, CBR3, GCH1, PTS, and SPR. Detailed information about the specific transcripts detected can be found in Table 1. Each gene was normalized to the endogenous control gene β-actin and calibrated to the mean normalized expression of the transcript in neurologically normal human cerebellar tissue. The fold difference of SPR expression was 4.46±1.07 (mean±SEM) in PD brains relative to control brains (Figure 1). The increased SPR expression in PD cases was statistically significant (p=0.01). The expression of AKR1B1 (0.76±0.06) and PTS (0.70±0.09) was significantly lower in PD cases relative to controls (p=0.03 and p=0.01, respectively). No difference in expression between PD cases and controls was detected for GCH1, CBR1, or CBR3 (Fig. 1).
Table 1.
Gene Expression Assay Information
| Gene Symbol | Gene Name | ABI Assay Number* | Chromosomal Location | NCBI RefSeq | Target Exons |
|---|---|---|---|---|---|
| ACTB | actin, beta | Hs99999903_m1 | 7p15-p12 | NM_001101 | 5’UTR/1 |
| AKR1B1 | aldo-keto reductase family 1, member B1 | Hs00739326_m1 | 7q35 | NM_001628 | 4/5 |
| CBR1 | carbonyl reductase 1 | Hs00156323_m1 | 21q22.13 | NM_001757 | 2/3 |
| CBR3 | carbonyl reductase 3 | Hs00154295_m1 | 21q22.2 | NM_001236 | 2/3 |
| GCH1 | GTP cyclohydrolase 1 | Hs00609198_m1 | 14q22.1–q22.2 | NM_000161 | 4/5 |
| PTS | 6-pyruvoyltetrahydropterin synthase | Hs00609393_m1 | 11q22.3–q23.2 | NM_000317 | 1/2 |
| SPR | sepiapterin reductase | Hs00268403_m1 | 2p14–2p12 | NM_003124 | 2/3 |
Information about the genetic transcripts detected by real-time RT-PCR. The NCBI reference sequence (RefSeq) and the target exons were provided by ABI with each TaqMan Gene Expression Assay. Gene symbols and chromosomal locations are listed according to the most recent NCBI data (http://www.ncbi.nlm.nih.gov/).
ABI TaqMan Gene Expression Assays are under proprietary license and the exact primer and probe sequences are not available.
Figure 1.
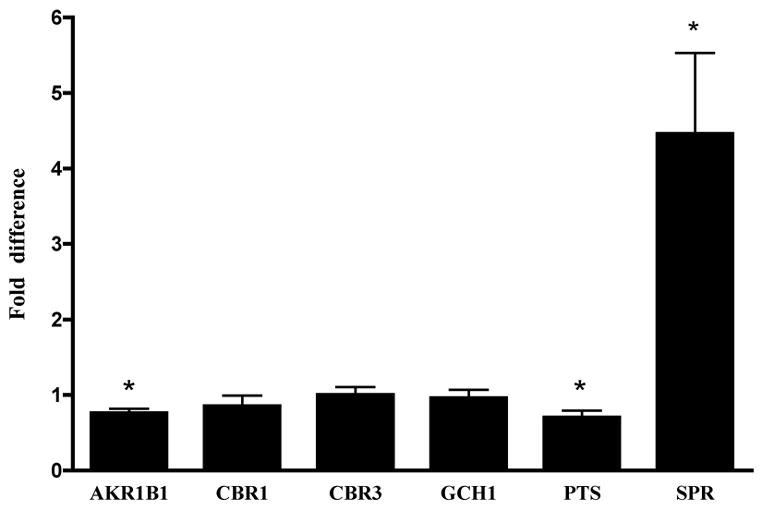
Real-time RT-PCR expression of BH4 enzymes in PD brain. Genes are listed along the x-axis. Bars represent the mean fold difference ± SEM for the PD cases compared to controls. Asterisks (*) indicate significant differences in gene expression between PD cases and controls according to a Student’s t-test with p<0.05.
2.2. SNP analysis
Three SNPs (rs2421095, rs1876487, and rs1561244) were genotyped in DNA isolated from the 60 brain samples used to study gene expression, and no difference in the distribution of genotypes between PD cases and controls was observed for any SNP (Fisher exact test, p>0.05). The genotype frequencies in cases and controls for all SNPs are listed in Table 2. There were no rare allele homozygotes detected at rs2421095 and rs1561244, therefore a dominant genetic model was used to evaluate the effect of these SNPs on SPR expression (log 2−ΔΔCt) and no significant results were found (p>0.05, data not shown).
Table 2.
SNP Genotype Frequencies in Cases and Controls
| SNP | Genotype frequency cases | Genotype frequency controls | |
|---|---|---|---|
| rs2421095* | AA | 90.6% (29) | 92.8% (26) |
| AG | 9.4% (3) | 3.6% (1) | |
| rs1876487 | GG | 59.4% (19) | 64.3% (18) |
| GT | 37.5% (12) | 32.1% (9) | |
| TT | 3.1% (1) | 3.6% (1) | |
| rs1561244 | GG | 81.3% (26) | 78.6% (22) |
| AG | 18.7% (6) | 21.4% (6) |
The genotype frequencies, displayed here as percentages, were calculated for each SNP. The number in parenthesis indicates the number of individuals with a given genotype.
The genotype for one control sample at rs2421095 could not be determined.
2.3 Sequencing of the genomic region around SPR
The sequencing of the coding region plus 5’ and 3’ regions of the SPR gene for five PD brain samples did not reveal any novel polymorphisms. Previously known polymorphisms, including rs1876487, were detected.
3. Discussion
To our knowledge, this is the first assessment of the expression of the six genes involved in biosynthesis of BH4, a cofactor in the synthesis of DA, in human PD brain. The expression of three of the genes studied, SPR, AKR1B1, and PTS, was significantly different between PD cases and controls (p<0.05, Fig.1). Interestingly, SPR was the only gene studied that showed higher expression in PD brains relative to control brains. These results suggest that it is not the entire BH4 pathway, but rather the altered expression of certain enzymes involved in the synthesis and regeneration of BH4, especially SPR, that may be contributing to the pathogenesis of certain forms of PD.
A three-SNP haplotype around the SPR gene had been previously reported to be associated with onset age of PD (Karamohamed et al., 2003) and PD affection status (Sharma et al., 2006). In unrelated individuals, haplotypes are inferred from genotype data based on statistical methods that require large samples for accurate estimates (Fallin and Schork, 2000) therefore we did not perform any haplotype analyses on the current data. We did not detect any genotype frequency differences between cases and controls. This is not surprising given the relatively small sample and the fact that the replicated association was to age at onset of PD and not PD affection status. We also sought to evaluate whether these SNPs (rs2421095, rs1876487, rs1561244) were individually associated with increased SPR expression. We did not detect a relationship between any of these SNPs and SPR expression (p>0.05, data not shown). Due to the low frequency of some of the polymorphisms in this sample, it is likely that we do not have sufficient power to detect modest effects of the genotype on expression in a dominant model.
West et al. (2001) did not detect any sequence variants in the exons and 2 kilobases upstream of SPR in two PD families, leading them to suggest that variants in SPR were not responsible for PARK3. However, genetic polymorphisms around SPR have been associated with both affection and onset age in large familial and sporadic PD cohorts (Karamohamed et al., 2003; Sharma et al., 2006). The observed significant increase in SPR mRNA in PD brain supports the hypothesis that polymorphisms affecting the expression or splicing of SPR may play a role in PD, rather than coding polymorphisms affecting the protein sequence. Additional work is needed to determine if sequence variants, either known or novel, are responsible for differences in SPR expression.
A few cases of human sepiapterin reductase deficiency, a BH4-dependent monoamine neurotransmitter deficiency without hyperphenylalaninemia, have been described in the literature (Blau et al., 2001; Bonafe et al., 2001). A SPR knockout mouse has been created to model human sepiapterin reductase deficiency (Yang et al., 2006). These mice display similar symptoms to human sepiapterin reducase deficiency patients, including growth retardation, dystonia, spasticity, and tremor (Blau et al., 2001; Yang et al., 2006). This mouse model has added to the evidence that SPR is essential for normal BH4 metabolism and the downstream function of BH4-dependent enzymes.
It has been shown that BH4 levels decrease with age in the normal brain (Furukawa and Kish, 1998) and BH4 levels in the brain and cerebrospinal fluid are lower in PD patients than controls (Lovenberg et al., 1979; Nagatsu et al., 1981). There is conflicting evidence whether BH4 is protective or toxic to dopaminergic neurons. Extracellular BH4 has been shown to increase the rate of apoptotic cell death in DA neurons (Choi et al., 2000; Choi et al., 2003; Kim et al., 2004), while intracellular BH4 appears to protect DA neurons against oxidative damage (Choi et al., 2003; Nakamura et al., 2001). Treatment of cerebellar granule neurons with sepiapterin attenuated 1-methyl-4-phenylpyridinium-induced (MPP+) toxicity (Shang et al., 2004). This protection was abolished by the presence of a SPR inhibitor, suggesting that the conversion of sepiapterin to BH4 via SPR was essential to the protective effect (Shang et al., 2004). The increase in SPR mRNA in PD brains seen here may be a compensatory reaction to protect DA neurons under oxidative stress due to higher levels of intracellular BH4.
One of the major limitations of this study is the use of post-mortem human tissue, which resulted in a small sample without detailed clinical history. The extent of levodopa therapy in the PD cases in this study is unknown. Therefore, it is possible that the changes in gene expression we observed were a result of the addition of exogenous levodopa and not PD. An additional limitation of this study is that we are not able to determine whether the mRNA levels detected by the real-time RT-PCR are localized in the dopaminergic terminals from the striatum or possibly the noradrenergic terminals from the locus coeruleus.
The results of this study suggest that further investigation of SPR and the BH4 pathway in the pathogenesis of PD is needed. Although we were able to detect a difference in SPR expression between PD cases and control brains, there may have been insufficient power to attribute these expression differences to a specific genetic variant. A larger study is needed to determine whether genetic variants around SPR are associated with expression levels. Additionally, a study of SPR activity and levels of BH4 in PD brain is recommended in order to understand the relationship, if any, between changes in SPR expression and levels of intracellular BH4 production.
4. Experimental Procedure
4.1 Human Tissue
Fresh frozen specimens of human cerebellar tissue from neuropathologically confirmed PD cases (n=30) and neurologically and neuropathologically confirmed normal controls (n=14) were obtained from the Harvard Brain Tissue Resource Center (BTRC). Additional neurologically and neuropathologically confirmed normal control specimens were obtained from the Boston University Alzheimer’s Disease Center Brain Bank (n=14). We received fresh frozen cerebellar tissue from 2 PD individuals enrolled in the GenePD Study from the Centre for Addiction and Mental Health at the University of Toronto. The age at death and post-mortem interval (PMI) measured in hours were not significantly different between the PD cases and the controls (Student’s t-test p value = 0.6 and 0.3, respectively). Table 3 is a summary of the available information on the samples used in this study.
Table 3.
Summary Sample Information
| N | Age | PMI | Male : Female | Age at Onset | |
|---|---|---|---|---|---|
| PD | 32 | 75.5±8.5 | 15.9±7.3* | 27 : 5 | 64.2±10.5‡ |
| Controls | 28 | 74.2±10.7 | 18.0±6.0† | 16 : 12 |
Information is presented as mean ± SD where N = number of samples and PMI = post-mortem interval measured in hours.
PMI available for 31 cases,
PMI available for 25 controls,
age at onset was reported for 23 cases
4.2 RNA isolation and real-time RT-PCR
RNA was isolated from brain specimens using TRIzol reagent (Invitrogen Life Technologies, Carlsbad, CA). First-strand cDNA synthesis was performed using the iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA). Real-time semiquantitative RT-PCR using Applied Biosystems’ TaqMan Gene Expression Assays was performed in triplicate for each sample on the ABI PRISM® 7900HT Sequence Detection System (Applied Biosystems, Foster City, CA). A serial dilution of pooled cDNA was used to evaluate the PCR amplification efficiency for each assay (Livak and Schmittgen, 2001).
4.3 DNA isolation, SNP genotyping, and sequencing
DNA was isolated from fresh frozen brain specimens using the PureGene DNA Purification kit (Gentra, Minneapolis, MN). Genotyping of single-nucleotide polymorphisms was performed using TaqMan SNP Genotyping Assays (Applied Biosystems) on the ABI PRISM® 7900HT. A 5,908 basepair (bp) region containing the SPR gene was sequenced using DNA from five PD samples on the Applied Biosystems 3700 DNA Analyzer. The entire 1438 bp coding region of the gene, and an additional 1002 bp 5’ of exon 1, and 166 bp 3’ of the last exon were sequenced.
4.4 Statistical Methods
The cycles to threshold (Ct), as determined by Applied Biosystems’ SDS 2.1 Software, were used to calculate the level of gene expression in PD cases relative to controls, or fold difference, using the comparative Ct method (Livak and Schmittgen, 2001). Cts of the triplicate RT-PCR reactions were averaged for each sample. In order to ensure accurate results, any sample with a range of Ct values greater than 2 across the triplicate reactions was dropped from further analysis. An assay for the endogenous control gene, β-actin, was run simultaneously with each target gene assay, and the corresponding counts to threshold were used to normalize each target gene (ΔCt = Cttarget − Ctβ-actin). Samples with a ΔCt value outside of 2 standard deviations from the mean ΔCt of cases or controls for a given gene were considered outliers and dropped from the fold change analysis of that gene. The ΔCt for each sample was calibrated to the mean ΔCt of the control brains (ΔΔCt = ΔCtsample − ΔCtmean control) and the fold difference for each sample was then calculated as 2−ΔΔCt. A Student’s t-test was used to evaluate the difference in relative expression between PD brains and control brains for each gene. The distribution of the fold difference (2−ΔΔCt ) of each gene was evaluated by a Shapiro-Wilk test and in each case was found to be significantly different than normal (p<0.05). Because this test of normality is conservative, we also visually inspected the data and examined summary statistics including mean, median and skewness to verify the non-normal distribution. Since log fold difference followed a normal distribution for all genes studied (Shapiro-Wilk p>0.05), t-tests were performed on this measure.
SNP genotypes were independently analyzed for an effect on SPR expression (log 2−ΔΔCt ) separately in PD cases and in controls using generalized linear models performed in SAS (Statistical Analysis System, Cary, North Carolina).
Acknowledgments
The authors would like to thank the Harvard Brain Tissue Resource Center, which is supported in part by PHS grant number R24 MH 068855, for providing us with tissue used in this study. We would also like to thank the Boston University Alzheimer’s Disease Center Brain Bank, supported by the National Institutes of Health, National Heart, Lung, and Blood Institute’s Framingham Heart Study, (NIH/NHLBI Contract N01-HC 25195), grants from NIA # 5R01-AG08122, NIA # 5R01-AG 16495 and NINDS # 2R01-NS17950, by the Boston University Alzheimer’s Disease Center NIAAA, P30 AG13846, and by the Department of Veteran’s Affairs; and Dr. Stephen Kish at the Centre for Addiction and Mental Health at the University of Toronto for providing us with additional brain tissue. Supported by the Bumpus Foundation, a grant from the American Parkinson Disease Association (to J. Cui), NIA grant #5-T32-AG00277-05 “Neurobiology and Neuropsychology of Aging”, and PHS grant R01-NS36711-09 “Genetic Linkage Study in Parkinson Disease”. Dr. Guttman receives support from the National Parkinson Foundation as part of the Center of Excellence Program.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Blau N, Bonafe L, Thony B. Tetrahydrobiopterin deficiencies without hyperphenylalaninemia: diagnosis and genetics of dopa-responsive dystonia and sepiapterin reductase deficiency. Mol Genet Metab. 2001;74:172–185. doi: 10.1006/mgme.2001.3213. [DOI] [PubMed] [Google Scholar]
- Bonafe L, Thony B, Penzien JM, Czarnecki B, Blau N. Mutations in the sepiapterin reductase gene cause a novel tetrahydrobiopterin-dependent monoamine-neurotransmitter deficiency without hyperphenylalaninemia. Am J Hum Genet. 2001;69:269–277. doi: 10.1086/321970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- Calne D. A definition of Parkinson’s disease. Parkinsonism Relat Disord. 2005;11(Suppl 1):S39–40. doi: 10.1016/j.parkreldis.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Choi HJ, Jang YJ, Kim HJ, Hwang O. Tetrahydrobiopterin is released from and causes preferential death of catecholaminergic cells by oxidative stress. Mol Pharmacol. 2000;58:633–640. [PubMed] [Google Scholar]
- Choi HJ, Kim SW, Lee SY, Hwang O. Dopamine-dependent cytotoxicity of tetrahydrobiopterin: a possible mechanism for selective neurodegeneration in Parkinson’s disease. J Neurochem. 2003;86:143–152. doi: 10.1046/j.1471-4159.2003.01808.x. [DOI] [PubMed] [Google Scholar]
- de Rijk MC, Launer LJ, Berger K, Breteler MM, Dartigues JF, Baldereschi M, Fratiglioni L, Lobo A, Martinez-Lage J, Trenkwalder C, Hofman A. Prevalence of Parkinson’s disease in Europe: A collaborative study of population-based cohorts. Neurologic Diseases in the Elderly Research Group. Neurology. 2000;54:S21–23. [PubMed] [Google Scholar]
- DeStefano AL, Lew MF, Golbe LI, Mark MH, Lazzarini AM, Guttman M, Montgomery E, Waters CH, Singer C, Watts RL, Currie LJ, Wooten GF, Maher NE, Wilk JB, Sullivan KM, Slater KM, Saint-Hilaire MH, Feldman RG, Suchowersky O, Lafontaine AL, Labelle N, Growdon JH, Vieregge P, Pramstaller PP, Klein C, Hubble JP, Reider CR, Stacy M, MacDonald ME, Gusella JF, Myers RH. PARK3 influences age at onset in Parkinson disease: a genome scan in the GenePD study. Am J Hum Genet. 2002;70:1089–1095. doi: 10.1086/339814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallin D, Schork NJ. Accuracy of haplotype frequency estimation for biallelic loci, via the expectation-maximization algorithm for unphased diploid genotype data. Am J Hum Genet. 2000;67:947–959. doi: 10.1086/303069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa Y, Kish SJ. Influence of development and aging on brain biopterin: implications for dopa-responsive dystonia onset. Neurology. 1998;51:632–634. doi: 10.1212/wnl.51.2.632. [DOI] [PubMed] [Google Scholar]
- Gasser T, Muller-Myhsok B, Wszolek ZK, Oehlmann R, Calne DB, Bonifati V, Bereznai B, Fabrizio E, Vieregge P, Horstmann RD. A susceptibility locus for Parkinson’s disease maps to chromosome 2p13. Nat Genet. 1998;18:262–265. doi: 10.1038/ng0398-262. [DOI] [PubMed] [Google Scholar]
- Gibb WR, Lees AJ. The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson’s disease. J Neurol Neurosurg Psychiatry. 1988;51:745–752. doi: 10.1136/jnnp.51.6.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes AJ, Daniel SE, Lees AJ. Improved accuracy of clinical diagnosis of Lewy body Parkinson’s disease. Neurology. 2001;57:1497–1499. doi: 10.1212/wnl.57.8.1497. [DOI] [PubMed] [Google Scholar]
- Hurley MJ, Mash DC, Jenner P. Markers for dopaminergic neurotransmission in the cerebellum in normal individuals and patients with Parkinson’s disease examined by RT-PCR. Eur J Neurosci. 2003;18:2668–2672. doi: 10.1046/j.1460-9568.2003.02963.x. [DOI] [PubMed] [Google Scholar]
- Ichinose H, Katoh S, Sueoka T, Titani K, Fujita K, Nagatsu T. Cloning and sequencing of cDNA encoding human sepiapterin reductase--an enzyme involved in tetrahydrobiopterin biosynthesis. Biochem Biophys Res Commun. 1991;179:183–189. doi: 10.1016/0006-291x(91)91352-d. [DOI] [PubMed] [Google Scholar]
- Iino T, Tabata M, Takikawa S, Sawada H, Shintaku H, Ishikura S, Hara A. Tetrahydrobiopterin is synthesized from 6-pyruvoyl-tetrahydropterin by the human aldo-keto reductase AKR1 family members. Arch Biochem Biophys. 2003;416:180–187. doi: 10.1016/s0003-9861(03)00295-9. [DOI] [PubMed] [Google Scholar]
- Jellinger KA. The pathology of Parkinson’s disease. Adv Neurol. 2001;86:55–72. [PubMed] [Google Scholar]
- Karamohamed S, DeStefano AL, Wilk JB, Shoemaker CM, Golbe LI, Mark MH, Lazzarini AM, Suchowersky O, Labelle N, Guttman M, Currie LJ, Wooten GF, Stacy M, Saint-Hilaire M, Feldman RG, Sullivan KM, Xu G, Watts R, Growdon J, Lew M, Waters C, Vieregge P, Pramstaller PP, Klein C, Racette BA, Perlmutter JS, Parsian A, Singer C, Montgomery E, Baker K, Gusella JF, Fink SJ, Myers RH, Herbert A. A haplotype at the PARK3 locus influences onset age for Parkinson’s disease: the GenePD study. Neurology. 2003;61:1557–1561. doi: 10.1212/01.wnl.0000095966.99430.f4. [DOI] [PubMed] [Google Scholar]
- Katoh S, Sueoka T. Sepiapterin reductase exhibits a NADPH-dependent dicarbonyl reductase activity. Biochem Biophys Res Commun. 1984;118:859–866. doi: 10.1016/0006-291x(84)91474-8. [DOI] [PubMed] [Google Scholar]
- Kim ST, Chang JW, Hong HN, Hwang O. Loss of striatal dopaminergic fibers after intraventricular injection of tetrahydrobiopterin in rat brain. Neurosci Lett. 2004;359:69–72. doi: 10.1016/j.neulet.2004.02.015. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Lovenberg W, Levine RA, Robinson DS, Ebert M, Williams AC, Calne DB. Hydroxylase cofactor activity in cerebrospinal fluid of normal subjects and patients with Parkinson’s disease. Science. 1979;204:624–626. doi: 10.1126/science.432666. [DOI] [PubMed] [Google Scholar]
- Nagatsu T, Yamaguchi T, Kato T, Sugimoto T, Matsuura S, Akino M, Nagatsu I, Iizuka R, Narabayashi H. Biopterin in human brain and urine from controls and parkinsonian patients: application of a new radioimmunoassay. Clin Chim Acta. 1981;109:305–311. doi: 10.1016/0009-8981(81)90316-8. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Bindokas VP, Kowlessur D, Elas M, Milstien S, Marks JD, Halpern HJ, Kang UJ. Tetrahydrobiopterin scavenges superoxide in dopaminergic neurons. J Biol Chem. 2001;276:34402–34407. doi: 10.1074/jbc.M103766200. [DOI] [PubMed] [Google Scholar]
- Ohye T, Hori TA, Katoh S, Nagatsu T, Ichinose H. Genomic organization and chromosomal localization of the human sepiapterin reductase gene. Biochem Biophys Res Commun. 1998;251:597–602. doi: 10.1006/bbrc.1998.9503. [DOI] [PubMed] [Google Scholar]
- Pankratz N, Uniacke SK, Halter CA, Rudolph A, Shults CW, Conneally PM, Foroud T, Nichols WC. Genes influencing Parkinson disease onset: replication of PARK3 and identification of novel loci. Neurology. 2004;62:1616–1618. doi: 10.1212/01.wnl.0000123112.51368.10. [DOI] [PubMed] [Google Scholar]
- Park YS, Heizmann CW, Wermuth B, Levine RA, Steinerstauch P, Guzman J, Blau N. Human carbonyl and aldose reductases: new catalytic functions in tetrahydrobiopterin biosynthesis. Biochem Biophys Res Commun. 1991;175:738–744. doi: 10.1016/0006-291x(91)91628-p. [DOI] [PubMed] [Google Scholar]
- Parsons RB, Smith ML, Williams AC, Waring RH, Ramsden DB. Expression of nicotinamide N-methyltransferase (E.C. 2.1.1.1) in the Parkinsonian brain. J Neuropathol Exp Neurol. 2002;61:111–124. doi: 10.1093/jnen/61.2.111. [DOI] [PubMed] [Google Scholar]
- Shang T, Kotamraju S, Kalivendi SV, Hillard CJ, Kalyanaraman B. 1-Methyl-4-phenylpyridinium-induced apoptosis in cerebellar granule neurons is mediated by transferrin receptor iron-dependent depletion of tetrahydrobiopterin and neuronal nitric-oxide synthase-derived superoxide. J Biol Chem. 2004;279:19099–19112. doi: 10.1074/jbc.M400101200. [DOI] [PubMed] [Google Scholar]
- Sharma M, Mueller JC, Zimprich A, Lichtner P, Hofer A, Leitner P, Maass S, Berg D, Durr A, Bonifati V, De Michele G, Oostra B, Brice A, Wood NW, Muller-Myhsok B, Gasser T. The sepiapterin reductase gene region reveals association in the PARK3 locus: analysis of familial and sporadic Parkinson’s disease in European populations. J Med Genet. 2006;43:557–562. doi: 10.1136/jmg.2005.039149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thony B, Auerbach G, Blau N. Tetrahydrobiopterin biosynthesis, regeneration and functions. Biochem J. 2000;347(Pt 1):1–16. [PMC free article] [PubMed] [Google Scholar]
- West AB, Zimprich A, Lockhart PJ, Farrer M, Singleton A, Holtom B, Lincoln S, Hofer A, Hill L, Muller-Myhsok B, Wszolek ZK, Hardy J, Gasser T. Refinement of the PARK3 locus on chromosome 2p13 and the analysis of 14 candidate genes. Eur J Hum Genet. 2001;9:659–666. doi: 10.1038/sj.ejhg.5200698. [DOI] [PubMed] [Google Scholar]
- Yang S, Lee YJ, Kim JM, Park S, Peris J, Laipis P, Park YS, Chung JH, Oh SP. A murine model for human sepiapterin-reductase deficiency. Am J Hum Genet. 2006;78:575–587. doi: 10.1086/501372. [DOI] [PMC free article] [PubMed] [Google Scholar]