

Abstract
A rodent model of diencephalic amnesia, pyrithiamine-induced thiamine deficiency (PTD), was used to investigate diencephalic-limbic interactions. In-vivo acetylcholine (ACh) efflux, a marker of memory-related activation, was measured in the hippocampus and the amygdala of PTD-treated and pair-fed (PF) control rats while they were tested on a spontaneous alternation task. During behavioral testing, all animals displayed increases in ACh efflux in both the hippocampus and amygdala. However, during spontaneous alternation testing ACh efflux in the hippocampus and the alternation scores were higher in PF rats relative to PTD-treated rats. In contrast, ACh efflux in the amygdala was not suppressed in PTD treated rats, relative to PF rats, prior to or during behavioral testing. In addition, unbiased stereological estimates of the number of choline acetyltransferase (ChAT) immunopositive neurons in the medial septal/diagonal band (MS/DB) and nucleus basalis of Meynert (NBM) also reveal a selective cholinergic dysfunction: In PTD-treated rats a significant loss of ChAT-immunopositive cells was found only in the MS/DB, but not in the NBM. Significantly, these results demonstrate that thiamine deficiency causes selective cholinergic dysfunction in the septohippocampal pathway.
Keywords: diencephalon, hippocampus, amygdala, microdialysis, acetylcholine, stereology
Selective septohippocampal- but not forebrain amygdalarcholinergic dysfunction in diencephalic amnesia
Diencephalic lesions are seen in a range of disorders: malnourishment, ischemic infarcts, viral infections, tumors and traumatic damage. Damage to certain nuclei and fiber systems within the diencephalon interrupts the flow of information between key memory structures and thus causes severe and long-lasting amnesia [25, 29, 45]. Although there is evidence that lesions to particular diencephalic nuclei result in memory impairment in their own right, there is also evidence that damage to diencephalic nuclei can disrupt memory circuits leading to dysfunction in other regions of the brain [4, 29, 45, 49, 53].
In cases of diencephalic amnesia associated with thiamine deficiency (i.e., Wernicke-Korsakoff Syndrome [WKS]) there is neuronal loss and lesions in multiple limbic thalamic nuclei, mammillary bodies [25, 26, 28, 29], as well as degeneration in key fiber tracts connecting limbic structures [fornix and mammillothalamic tract; 24, 27]. The damage to both key diencephalic nuclei and fiber tracts likely results in a “disconnection syndrome” within the limbic system [see 2, 30, 59].
At one point, WKS patients were labeled as “temporal lobe-related amnesiacs” based on their memory performance [10]. This clinical population was critical for the development of the memory system classifications (i.e., declarative vs. nondeclarative memory [51]). However, it is clear from neuroimaging studies that the most consistent neuropathology of WKS is damage to the cortex and the diencephalon—particularly the thalamus and mammillary bodies [19, 23]. One of the best predictors of memory loss in WKS is pathology of the anterior thalamus [16]. In contrast, the hippocampus is damaged in less than 10% of WKS cases. Although the two types of amnesia differ in structural correlates, the functional distinction between temporal lobe amnesia and diencephalic amnesia has been questioned, given the behavioral similarities and the neural connections between the diencephalon and the hippocampus [2, 51].
Using a rodent model of WKS, pyrithiamine-induced thiamine deficiency (PTD), we have demonstrated that the hippocampus is functionally impaired: When PTD-rats are performing a spontaneous alternation task [49] or learning a nonmatching-to-position task [46] the rise in hippocampal acetylcholine (ACh) efflux is blunted relative to control rats. This impairment in hippocampal ACh efflux is only evident during behavioral testing as baseline levels of hippocampal ACh are normal in the PTD model. Given the known importance of ascending cholinergic systems in a range of behaviors and neural physiology within the limbic system, understanding the role of cholinergic dysfunction in diencephalic amnesia will both further our understanding of the complex role of ACh in learning and memory and aid in the development of pharmacotherapies.
The impairment of functional ACh hippocampal release during behavioral testing in the PTD model could be driven by a number of neuroanatomical changes associated with thiamine deficiency and diencephalic amnesia. We have previously documented a loss of choline acetyltransferase (ChAT) immunopositive neurons in the medial septal/diagonal band (MS/DB) in the PTD model using a profile counting method [43, 44]. Furthermore, Nakagawasai and colleagues [36, 37, 38] have demonstrated a decrease in the intensity of ChAT positive fibers in the hippocampus, cortex and thalamus after thiamine deficiency. However, this decrease in ChAT immunoflorescence is not observed in the amygdala. Pires and colleagues have also demonstrated that after mild thiamine deficiency [41] or PTD treatment [42] stimulated ACh release is decreased in the cortex. Furthermore, these authors found that acetylcholinesterase (AChE) activity was decreased in both the hippocampus and cortex after PTD treatment [42]. In addition, compounds that enhance ACh levels decrease mortality and sickness behaviors (attacking, antinociception, altered startle response) as well as improve learning in thiamine-deficient mice [36] and spontaneous alternation in PTD-treated rats [unpublished data].
Reviews of the pathology produced by the PTD treatment have consistently demonstrated that although mild to moderate cell loss occurs outside the diencephalon, the anterior and midline thalamic damage are critical and responsible for the majority of the loss of learning and memory function that occurs after thiamine deficiency [26, 29]. These changes, along with the white matter loss that occurs in key fiber tracts from the diencephalon after PTD treatment [27], suggest that thiamine deficiency likely causes system level dysfunction. There is neurobiological evidence that even discrete diencephalic damage alters the activation of other limbic regions – in particular the hippocampus [see 20, 45, 49]. Thus, damage to the diencephalon produced by thiamine deficiency could produce dysfunction in other limbic regions.
In the current experiment we examined whether thiamine deficiency, which causes diencephalic pathology, alters both hippocampal and amygdalar ACh efflux when rats are actively exploring a maze. Measurement of ACh efflux in the brains of rats during learning appears to be a useful marker of activation of a given neural system—particularly the hippocampus and amygdala [8, 14, 31, 32, 33]. We chose a task, spontaneous alternation, which is sensitive to diencephalic damage [25] and to changes in release of ACh in both the hippocampus and amygdala [8, 31, 34]. In addition, to understand the neuroanatomical correlates of this dysfunction we used unbiased stereological techniques to estimate ChAT cell populations in the MS/DB (that projects to the hippocampus) as wells as the nucleus basalis of Meynert (NBM; that projects to the amygdala). Given our behavioral data [47], that suggests intact amygdala functioning in the PTD model, we expect that during behavioral testing there will be a blunted rise in hippocampal—but not amygdalar—ACh efflux in the PTD treated rats. Furthermore, we expect PTD-treated rats to display a loss of ChAT immunopositive cells in the MS/DB [43, 44]. However, no one has examined cell loss in the NBM of this model. Our goal was to determine the selectivity of cholinergic dysfunction by measuring both ACh efflux during behavioral testing in two structures and ChAT immunopositive cell loss in the regions that project to these structures. The data from this study which combines behavioral, neurochemical and histological measures will give a more complete picture of the role of cholinergic abnormalities to the learning and memory problems associated with the thiamine deficiency form of diencephalic amnesia.
Results
Statistical Analyses
The mean percent alternation and total number of arm entries during spontaneous alternation, as well as ChAT immunopositive cell counts (MS/DB, NBM) were analyzed with a 1-factor (Group= PF vs. PTD) ANOVA. The neurochemical microdialysis/HPLC data was analyzed with a one-between (Group), two-within factors repeated measures (phase, sample) AVOVA. Simple regression was used to correlate ACh efflux in the hippocampus and amygdala with alternation scores.
Histology
Figure 1-AB displays the PTD neuropathology compared to a PF brain. Figure 1-CD displays representative cannula placement in the hippocampus and amygdala. Two PTD and one PF subjects had misplaced amygdalar cannulae and data from those subjects was discarded.
Figure 1.
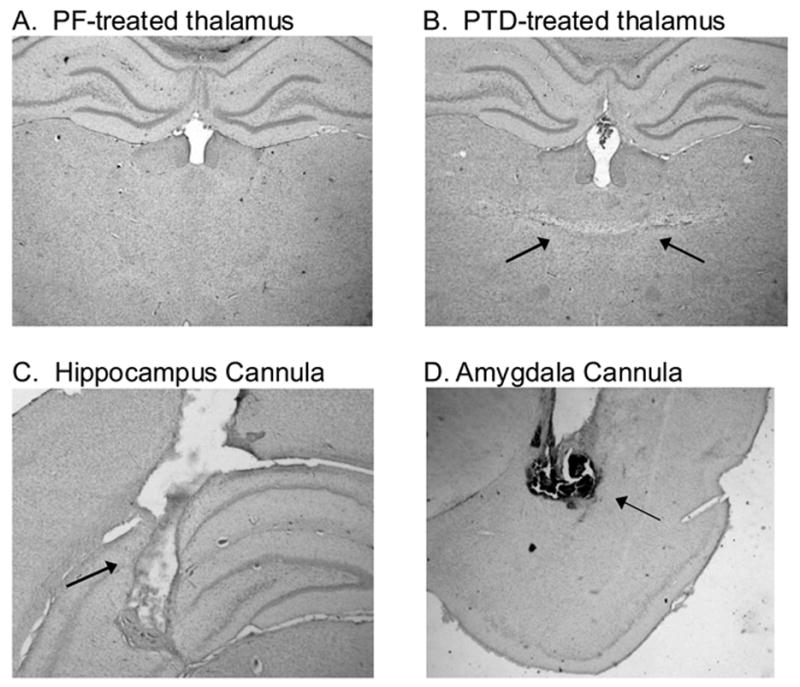
Photomicrographs illustrating a midline thalamic lesion produced by PTD treatment (2–B), relative to a PF control (2–A). The second row of photomicrographs are examples of acceptable probe placement in the hippocampus (C) and amygdala (D).
Stereological estimates of ChAT positive cells
In the MS/DB region, the PTD rats (n=7) had significantly fewer ChAT positive cells than the PF rats (n=7; F[1,12]=9.29, p<.01). The PTD group displayed about a 20% loss of ChAT positive cells in the MS/DB region (see Table 1). However, in the NBM region, the loss of ChAT positive cells (−5%) in the PTD model was nonsignificant (F[1,12]=1.42, p>.2). Figure 2 displays the regions sampled and the localization of the ChAT immunopositive staining.
Table 1.
Estimated total numbers of ChAT immunopositive cells (Mean±SEM) in the medial-septum-diagonal band (MS/DB) and the nucleus basalis (NB) of PF and PTD treated male rats.
| Group | MS/DB | NB | Total |
|---|---|---|---|
| PF (n=7) | 10187.14±488.44 (s=4.71) | 6848.57±340.75 (s=5.00) | 17035.71±510.08 |
| PTD (n=7) | 8173.57±445.01* (s=4.71) | 6334.86±262.64 (s=5.14) | 14508.43±444.85* |
n= number of subjects in the Group; s=mean number of brain sections from the region from each subject;
= significant difference (p<.01)
Figure 2.
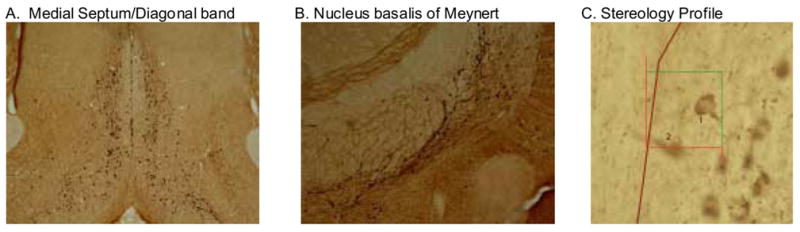
Sections of the medial septum/diagonal band (A) and nucleus basalis of Meynert (B) stained for choline acetyltransferase (ChAT) immunoreactivity as a marker for cholinergic neurons. The third panel is a high magnification (40X) demonstrating the disector counting rule (cell 1 counted, cell 2 was not) within the counting frame (50 μm × 50 μm).
Spontaneous alternation
As shown in Figure 3–A, PTD-treated rats (n=8) had a significant decrease in spontaneous alternation rates relative to PF (n=8) control rats (F[1, 14]=8.56, p<.01). However, the PTD rats were more active on the maze than the PF rats (see Figure 3–B; F[1, 14]=5.10, p<.05). Thus, we ran a second analysis including alteration rates for only the first 22 trials. Equating number of arms entered across PF and PTD-treated rats on the maze still resulted in impaired alternation rates in the PTD-treated rats relative to the PF rats (F[1, 14]=8.27, p<.02).
Figure 3.
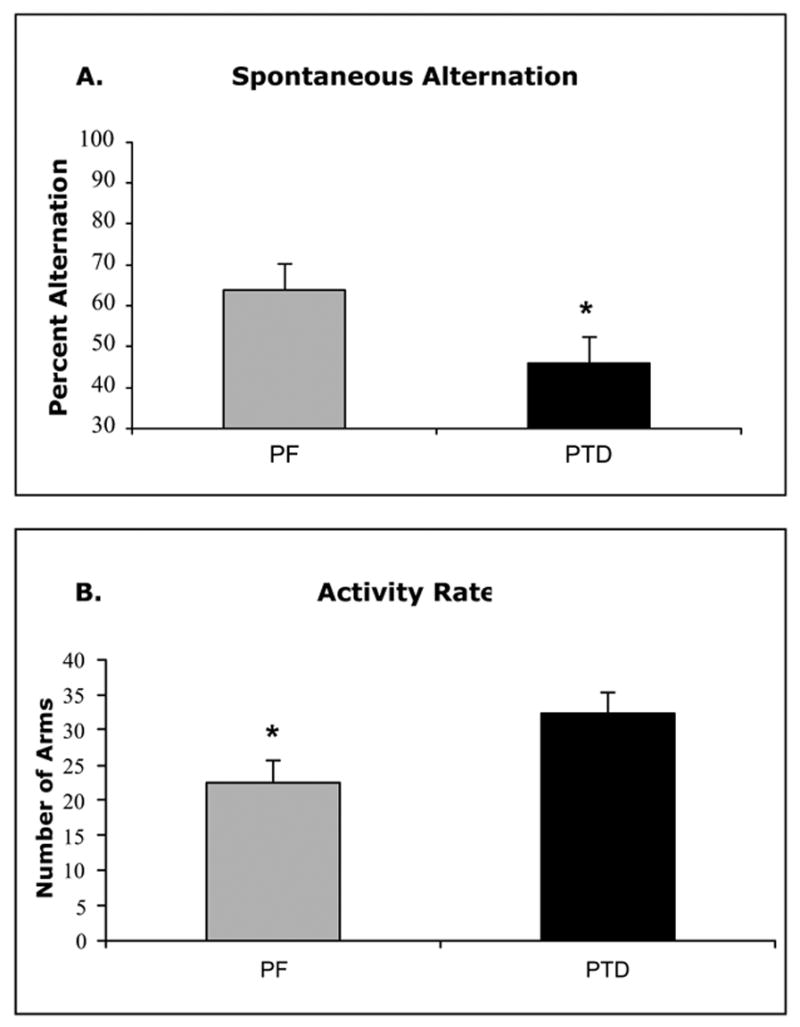
Behavioral data (Mean ± SEM) from a single session of spontaneous alternation testing for PTD and PF rats. Panel A shows the significant difference between PTD and PF rats in alternation behavior. Panel B shows that the PTD rats transversed more arms during the session.
Acetylcholine efflux
Hippocampal and amygdalar ACh levels increased during behavioral testing relative to baseline levels in the holding cage (both F’s [1, 14] >164.00, p’s<.0001; see Figure 4–AB). Basal amounts of ACh in the hippocampus (58.73±15.5 fM) and amygdala (34.43±10.2 fM) were not different as a function of Group (both F’s [1, 14] <1.5). However, there was a significant Group (PF vs. PTD) X Phase (Baseline, Maze) interaction (F[1, 14]=13.78, p<.01) for the hippocampal ACh samples. There was no significant main effect of Sample time (p>.10); however, the Group X Phase X Sample interaction approached significance (F[3, 42]=2.46, p<.08). Figure 4–A shows that PTD-treated rats had a significantly lower level of hippocampal ACh release (expressed as percent increase above baseline values) during behavioral testing, relative to PF rats (F[1,14]=13.18, p<0.01). Furthermore, there was a significant correlation between hippocampal ACh efflux and alternation rate (r=.50, p<.05).
Figure 4.
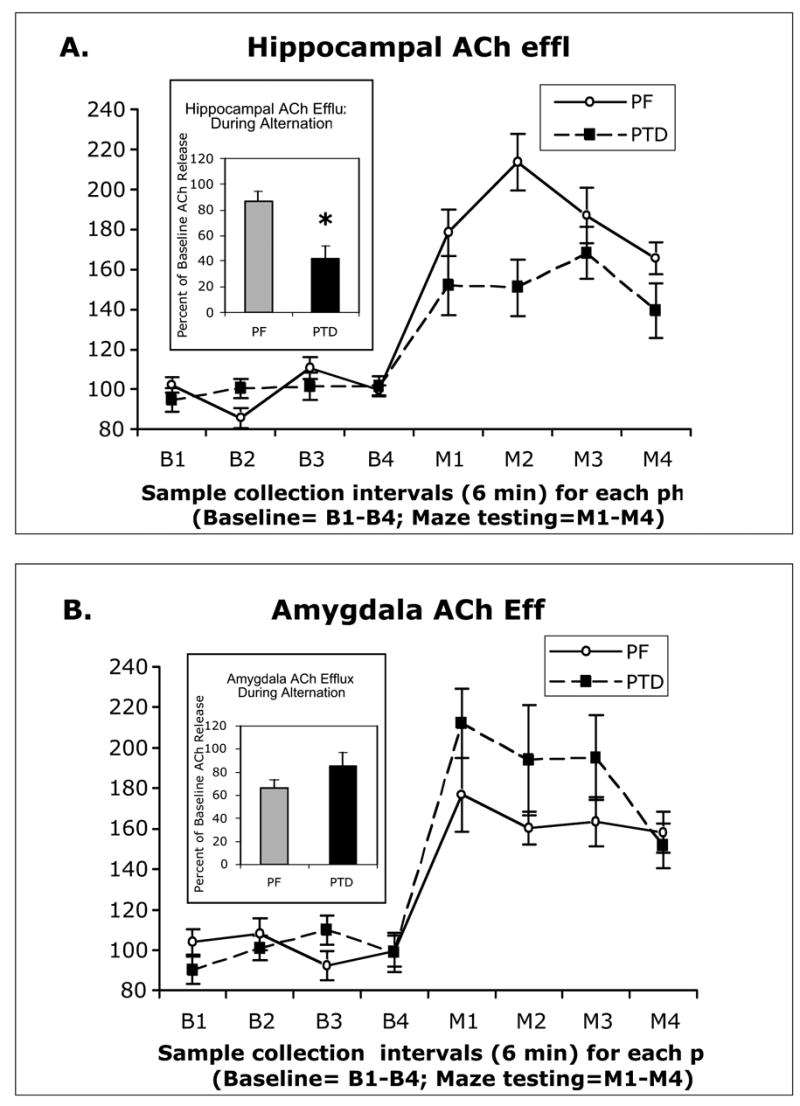
Profiles of ACh efflux (Mean ± SEM) from the hippocampus (A) and the amygdala (B) during 6-minute sample bins prior to and during spontaneous alternation. The PF rats’ hippocampal ACh efflux increased to 80% above baseline values, whereas the PTD rats displayed a significantly blunted ACh efflux of only 40% above baseline values. In the amydala, the PF rats had about 65% increase in ACh efflux above baseline and the PTD rats displayed a greater 84% ACh efflux, but this change was not significant. Insert graphs are the mean increase of ACh efflux across samples obtained from the Group X Phase interactions.
In contrast, the Group X Phase interaction did not reach significance for the amygdala ACh samples (F[1,14]=3.17, p=.09). The rise in amygdalar ACh efflux during behavioral testing in the PTD group was slightly higher than the amygdalar ACh rise in the PF groups. The variable of Sample time was not significant (p=.08) and it did not interact with any variables (all p’s >.2). Thus, in contrast to hippocampal functioning, amygdalar ACh functioning was not suppressed in the PTD model. In addition, there was no relationship between amygdalar ACh efflux and alternation score (r=−.31, p>.2).
Discussion
This study provides evidence that the cholinergic dysfunction in the PTD model is selective to the septohippocampal axis. The data replicate that PTD rats have impaired hippocampal ACh release that is evident only under conditions of spatial exploration or place learning [46, 49]. In addition, the unbiased stereological estimate of cell counts also replicates the profile counting method of assessment of loss of ChAT immunopositive cells in the MS/DB of PTD treated rats [43, 44]. The novel findings from this study are (1) Amygdalar functioning is intact in the PTD-model: The behavioral relevant increase in amygdalar ACh efflux was not suppressed. (2) The number of ChAT positive neurons in the NBM region, which projects to the amygdala, was not significantly altered in PTD rats. These results together suggest that the forebrain cholinergic innervation of limbic structures likely modulate the activation of these structures during environmental-behavioral demand and the degree of activation precipitates behavioral adaptation.
Although there have been previous data that suggests normal amygdala functioning in the PTD model, until the microdialysis data from the present study there has been no direct evidence for that claim. In a previous study we modified the reward contingencies on a matching-to-position task such that reward expectancies, rather than spatial location, could be used to solve the task. This slight manipulation completely eliminated the learning impairment in PTD treated rats [47]. Modifying a task such that reward expectancies can be used to solve the task requires the amygdala [5, 48]. Second, the intensity of ChAT positive fibers in the amygdala remains unchanged after thiamine deficiency in mice [36]. Our ChAT immunocytochemistry data provide evidence that the loss (hippocampus) or sparing (amygdala) of ChAT-positive fibers reported in the Nakagawasai paper is due to a decrease of cells staining positive for ChAT in the MS/DB, but not the NBM. We do not know if the reduction in the number of neurons staining positive for ChAT in the PTD model represents specific or general cell death in the MS/DB region. Analysis of total (cresyl violet) or other cell population (such as GABA) would shed light on this question.
It was interesting that there was a trend (p=.09) for the ACh efflux in the amygdala to be higher in the PTD-treated rats than that observed in the PF controls. This suggests that there is some effort towards a compensatory response in PTD-treated rats. As stated above, PTD rats do show a compensatory response on a matching-to-position task if they can use reward information (amygdala-dependent process) rather than spatial location (hippocampal-dependent process) to solve the task [47]. ACh efflux in the hippocampus, amygdala and dorsal striatum has provided a neurochemical marker of differential activation during maze exploration and learning [7, 15, 32]. Although a previous study found a correlation between alternation performance and amygdala ACh efflux in normal rats [32], we did not observe such an effect in our study that included the behaviorally impaired PTD rats. Increases in amygdalar ACh levels are unique in that they can be achieved under either complementary or compensatory reactions [32, 33]. However, our data suggest that when the hippocampus is dysfunctional, activation of the amygdala does not lead to behavioral recovery on a spatial exploration task without reward in the PTD model. Thus, given the demands of the task, level of training and integrity of the brain, amygdalar ACh response can vary to correlate with behavior or not.
This example exemplifies that the role of ACh in learning and memory is not a simple story [15, 17, 34, 39]. A current theory is that the ACh level sets the appropriate physiological tone in the hippocampus (and potentially other structures) for learning and later memory as well as related neural adaptation [13, 15, 17]. Thus, ACh tone across structures modulates adaptation of behavioral strategies [14, 15].
The ascending cholinergic pathways of the rat brain are important for providing tonic increases in excitability in cortical and subcortical structures—primarily those that are part of the limbic system [21, 22, 50]. Specifically, the basal forebrain complex (MS/DB, NBM, substantia innominata) provides cholinergic input to a number of limbic structures. As noted in the introduction, the MS/DB provides the major cholinergic input to the hippocampus and the NBM provides the major cholinergic input into the amygdala as well as the cortex [57]. Interestingly, these sub-populations of cholinergic neurons respond differently to some neural toxic events and appear to have differential vulnerability to oxidative stress [34]. The cholinergic neurons in the NBM that project to the amygdala are not damaged by the immunotoxin IgG-saporin, presumably because they do not express the p75 receptor [6, 18]. Our results suggest that these neurons are also less sensitive to the neural insults caused by thiamine deficiency. Our population estimates of ChAT immunopositive cells in the NBM, and the MS/DB, are similar to other published results using stereological estimation techniques [35, 56]. It should be pointed out that significantly lower number of ChAT immunopositive cells does not necessarily indicate cell death, although it is commonly used as a measure of cholinergic cell loss [34, 56]. The exact mechanisms of cholinergic neural degeneration remain controversial [34].
Multiple mechanisms of cell death, as a result thiamine deficiency, have been proposed for the diencephalic damage. The data suggest that after thiamine deficiency there is mitochondrial dysfunction that leads to impairments in cerebral energy metabolism, lactic acidosis, glutamate receptor-mediated excitotoxicity and oxidative stress [52]. Thus, the two main theories to explain neuronal death in the PTD model are the excitotoxicity hypothesis and the oxidative stress hypothesis, but these mechanisms are not mutually exclusive [12]. Currently, we do not know the mechanisms of ChAT-immunopositive cell loss in the PTD model. However, degeneration of basal forebrain cholinergic neurons also involves mitochondrial abnormalities and oxidative stress [34].
Another question that remains to be answered is: Why is the cholinergic activation of the hippocampus selectively impaired during behavioral testing in the PTD model? However, to answer that question we must also understand the molecular mechanisms responsible for the cholinergic modulation of hippocampal activity that regulates behavior. Our data suggests that baseline cholinergic tone in the hippocampus and amygdala is normal in the PTD model. However, when environmental or behavioral demands are placed on the hippocampal system there is not enough of an increase in ACh efflux to produce successful behavioral change on spatial tasks. This lack of adaptation in the hippocampal cholinergic response may be due to the loss of MS/DB cells in the PTD model. There is evidence that both cholinergic and GABAergic cells in the MS/DB modulate neural activation of the hippocampus [1, 56, 60]. Cholinergic activation in the hippocampus triggers rhythmic activity that can persist for hours and such rhythmic activity is related to both learning and neural plasticity [11]. Furthermore, our recent data demonstrated that when the hippocampal ACh levels are increased in the hippocampus by direct application of physostigmine, the behavioral impairment in spontaneous alternation in PTD rats is eliminated (unpublished data). Limited clinical trails have also revealed that anticholinesterase inhibitors improve cognitive functioning in patients with WKS [3, 9]. Thus, although cholinergic dysfunction may be an over looked feature in thiamine deficiency-induced amnesia, increasing ACh levels does lead to significant behavioral recovery in the human disorder of WKS and the PTD model.
In the PTD model there is impairment on “hippocampal dependent” tasks- with the most common behavioral measures being assessment of spatial abilities [25, 28, 47]. There are two disease features of thiamine deficiency-induced amnesia that could result in this selective hippocampal dysfunction: (1) As the present study suggests, the loss of ChAT positive neurons in the MS/DB produces an inadequate activation of the hippocampus during behavioral testing; (2) Lesions of discrete diencephalic regions (anterior thalamus, mammillary bodies) can lead to memory impairment as well as changes in hippocampal physiological activity [20, 45, 54, 55, 58]. Additional research is needed comparing the two factors before we can identify the primary mechanisms for ACh dysfunction in diencephalic amnesia.
In summary, a picture is emerging that suggests there are significant cholinergic abnormalities in thiamine-deficiency related amnesia. However, these abnormalities appear selective to the septohippoccampal axis. The present findings further demonstrate the complexity of the role of ACh in learning and memory—particularly across different memory-related structures. Brain integrity interacts with task demands to determine whether compensatory mechanisms or strategies will be successful in behavioral adaptation.
Methods
Subjects
Twenty-two young male Sprague-Dawley rats (3–4 months; 250–300 g) were used as subjects in this study. They were housed one per cage with unlimited access to water and Purina rodent chow in a colony room with a 12- hour/12- hour light-dark cycle (onset at 7:00 am). All rats were fasted the night before behavioral testing.
PTD treatment
Animals were first randomly assigned to one of the following treatments: (i) pair-fed control (PF, n = 11), or (ii) pyrithiamine-induced thiamine deficiency (PTD, n = 11) groups. Subjects in the PTD group were free-fed a thiamine-deficient chow (Teklad Diets, Madison, WI) and given daily injections (0.25 mg/kg, i.p.) of pyrithiamine hydrobromide (Sigma, St. Louis, MO). On days 14–16 of treatment, animals display signs of local tonoclonic movement of the front and hind limbs, and generalized convulsions (seizures). Within 4 hours after observing the onset of seizure, PTD-treated animals were given an injection of thiamine (100 mg/kg, i.p.) every 8 hours until the seizure activity disappeared and the rats regained upright posture. The PF animals were fed an amount of thiamine-deficient chow equivalent to the average amount consumed by the PTD groups on the previous day of treatment, and were given daily injections of thiamine (0.4 mg/kg i.p.). After treatment all subjects were placed on regular chow and allowed to regain the weight lost during treatment.
Surgery
Sixteen days after recovering from PTD or PF treatments stereotaxic surgery (David Kopf Instruments, USA) was performed on animals anesthetized with an i.p. injection (.1ml/kg) of a Ketamine (83 mg/kg)/Xylazine (17 mg/kg) mixture. Each subject was implanted with two plastic guide cannulae (CMA/12 mm, Carnegie Medicine Associates, Chelmsford, MA). One cannula was lowered into the left hippocampus (5.0 mm posterior to bregma, 5.0 mm lateral to the midline, and 4.2 mm DV) and another guide cannula (CMA/12) was lowered into the lateral amygdala (3.0 mm AP, 4.8 mm ML, 6.8 mm DV) according to the atlas of Paxinos & Watson [40]. Cannula placement and cannulae size were based on a previous study assessing ACh efflux in the amygdala and hippocampus [32]. Two days after surgery animals were handled daily (5 min/day) for 5 days prior to behavioral testing.
Microdialysis/HPLC
One week following surgery two microdialysis probes (CMA/12) were inserted into the guide cannulae (hippocampus-3 mm; amygdala-2 mm). The probes were connected to plastic tubing and driven by a microinfusion system (CMA/100 pump). The dialysis probes were perfused continuously at a rate of 2.0 μl/min with artificial CSF (in mM: 128 NaCl, 2.5 KCL, 1.3 CaCl2, 2.1 MgCl2, NaH2PO4, 1.3 NaHPO4, and 1.0 glucose, brought to a pH of 7.4), which contained the acetylcholinesterase inhibitor neostigmine (500 nM). Prior to maze testing, the microdialysis probes were inserted into the hippocampal and amygdalar cannulae and the animal was placed into the holding cage (41 cm × 30 cm × 35 cm) located in the testing room. After 60 min of stabilization, dialysis samples (sample volume 12 μl) were collected every 6 minutes for a period of 24 minutes in the holding cage to determine basal levels of ACh in awake rats. During this initial baseline phase the animal was free to move about the holding cage. After initial exploration, most animals sat in a corner and occasionally groomed. After 4 baseline samples were collected, the rat was gently picked up and placed on the center of the maze. The plus-maze used for behavioral testing was made of wood with clear Plexiglas sidewalls (12 cm high) and a painted black floor with the four arms of equal distance (55 cm) was used for training. It was elevated 80 cm from the floor. The rat was allowed to transverse the maze freely for 24 minutes. The number and sequence of arms entered were recorded to determine alternation scores [see 8, 49].
An alternation was defined as the choice of an arm to the left or right of the rat when it reached the center area, not the arm straight ahead or a re-enter in to the same arm. The percent alteration score is equal to the ratio of: (actual alternations/possible alterations) × 100. The maze testing room contained various extramaze cues (posters, doors, tables, etc). Upon completion of 24 minutes of maze testing, rats were transferred back to the holding cage for an additional 24 min. One PTD and 2 PF rats had microdialysis collectection problems during behavioral testing and their data was discarded.
Dialysate samples were assayed for ACh using HPLC with electrochemical detection (Bio Analytic Systems, West Lafayette, IN). The system included an ion-exchange microbore analytical column, a microbore ACh/Ch immobilized enzyme reactor containing acetylcholinesterase and choline oxidase, and a peroxidase wired working electrode. The detection limit of this system is 10 fmol. ACh peaks were quantified by comparison to peak heights of standard solutions and corrected for in-vitro recovery of the probe.
Tissue fixation and histological analysis
At the conclusion of behavioral testing, animals were anesthetized with 0.5 mg/kg, i.p. injection of Sleep-away (26% sodium pentobarbital in 7.8% isopropyl alcohol and 20.7% propylene glycol solution; Fort Dodge, Iowa) and perfused transcardially using 0.9% saline solution and 4% phosphate-buffered paraformaldehyde. Their brains were removed, post fixed in a 10% formalin solution for at least 72 h and then transferred to a 30% sucrose solution. Coronal sections from the brains were cut (60 μm thick) on a vibratome from the level of the anterior commissure to the level of the posterior pontine tegmentum, sections were collected into wells in sequential order and every fifth section was stained with cresyl violet for assessment of probe location.
Immunocytochemical (ICC) analysis
Every 6th section (with a random start point within the first six sections) through out the extent of the MS/DB and the NBM were systematically collected and were used for immunocytochemical analysis of choline acetyltransferase (ChAT) to assess the integrity of the basal forebrain cholinergic system. For this assay, 0.1 M phosphate buffer (PB) of pH 7.3 was used for all washes, and all steps until the chromogenic reaction were carried out at 4°C. Sections were first placed in PB overnight; the next day, sections were washed in PB three times for 10 min each. A quench of 30% H2O2 in 60 ml of PB was then undertaken for 20 min. Three 5-min PB washes were performed, after which the sections were incubated in the primary antibody (goat anti-ChAT, 1:100; Chemicon Inc, CA) for approximately 24 h. Next, sections were washed in PB three times for 5 min each, then incubated in the secondary antibody (biotinylated rabbit anti-goat, 1:100; Vector Laboratories, CA) for 1 h, followed by three more 5-min PB washes. Sections were then incubated in avidin-biotin complex (Vector) at 4°C for 1 h followed again by three 5-min PB washes. The sections were placed in the chromogenic (DAB fast tab; Sigma-Aldrich, IL) reaction for approximately 1 min. The reaction was stopped with three 5-min PB washes, after which the sections were mounted on gelatinized slides, air-dried overnight, dehydrated through ascending alcohols, and cover-slipped. To prevent variability in staining due to ICC procedures, the brains of 6–8 animals, equated across experimental groups, were processed at the same time using the same reagents and temperature conditions. In each ICC run, control for specificity was conducted using 4 sections (2 MS/DB, 2 NMB) from a randomly selected PF and PTD rat, using the above procedures with the exception that the primary antibody was replaced by normal goat serum. Under those conditions no immunostaining was observed.
One of the PF subject’s tissue was in poor condition and would not withstand immunocytochemistry procedure. Thus, the brains from seven subjects each from the PF and PTD groups were used to estimate ChAT cell populations.
Stereology
Unbiased stereology was used to estimate ChAT immunopositive cell counts in the MS/DB and NBM. Parameters for sampling were based on previously published studies assessing the number of ChAT positive neuron in the basal forebrain [35, 56]. A low power image was captured with a digital camera (DVC-1310; DVC Company, Austin Texas) with a 5X lens on a Zeiss microscope (Axioscope 2- plus) with an attached 3-axis motorized stage. A contour was drawn around the region of interest using StereoInvestigator software (MicroBrightField Inc; Williston, VT). These cholinergic nuclei are well defined and have clear boundaries particularly when stained with a ChAT antibody (see Figure 1–AB). Anatomical boundaries used to define the basal forebrain were the corpus callosum on the dorsal aspect and the walls of the lateral ventricles. Sections began with the appearance of the rostral genu of the corpus callosum crossing hemispheres and ended caudally with the last section containing the full anterior commissure (approximately IA 10.6–8.74 mm [Paxinos & Watson, 1986]). For the NBM the anatomical landmarks were the appearance of the fornix and the anterior thalamus. The boundaries were set by the internal capsule, optic tract and globus pallidus (approximately IA 7.7–5.86 mm [Paxinos & Watson, 1986]). ChAT positive neurons must have an immunosignal in the soma to be counted. The neurons were counted with a 40X objective using optical fractionator approach (see Figure 1–C). The fractionator sampling consisted of a section sampling fraction (ssf=1/6), an area sampling fraction representing ratio between counting frame size and grid size (asf= 50μm × 50μm/100μm × 100μm) and a height sampling fraction (hsf= 20μm/60μm). The equation for determining cell estimates was: N= σQ− × 1/ssf × 1/asf × 1/hsf, where σQ is the number of neurons actually counted in the specimens, whereas N represents the total cell estimation.
Figure 5.
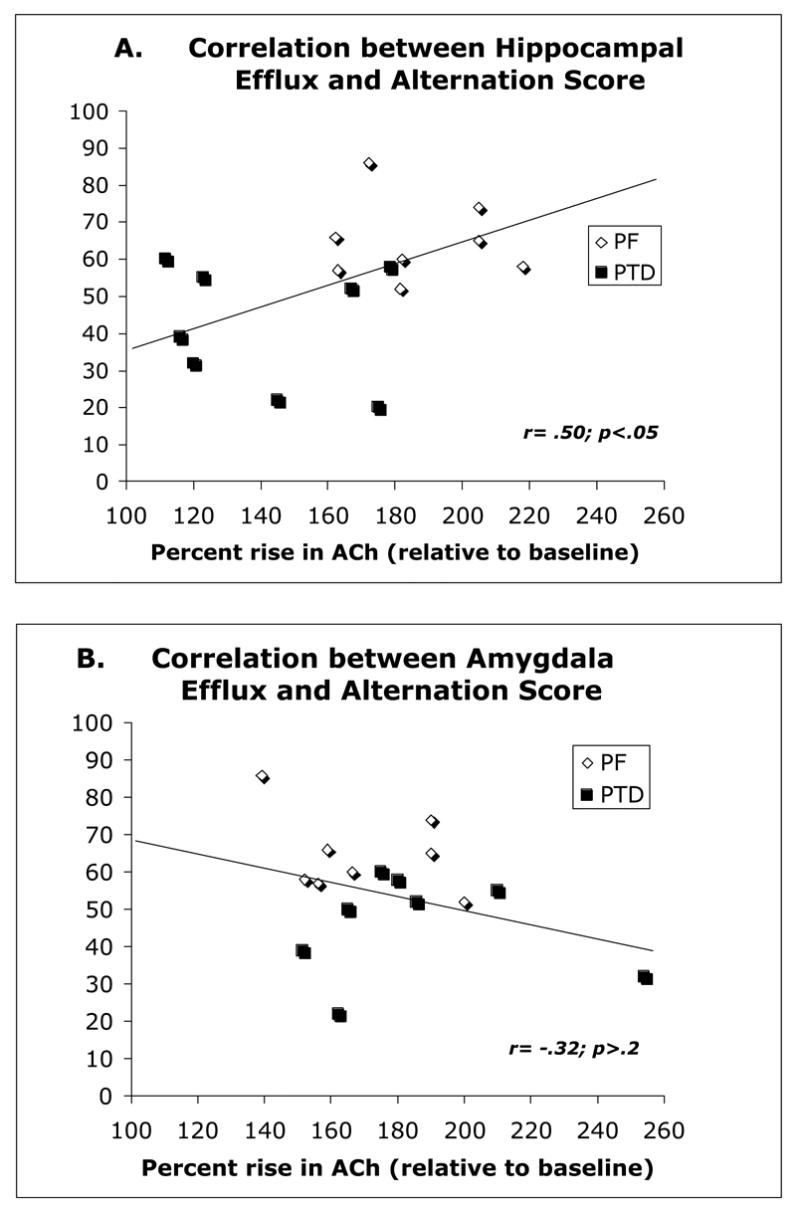
Correlative relationship between spontaneous alternation rate and ACh efflux in the hippocampus (top) and amygdala (bottom). Trend lines on each graph represents the overall combined regression analysis for the PTD and PF groups.
Acknowledgments
This work was supported by research grant NINDS 054272 to LMS. We would like to thank Samual Cruz for his role in brain sectioning as well as Jess Blackwolf and Osman Halarou for their assistance in behavioral testing.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Alreja M, Wu M, Liu W, Atkins JB, Leranth C, Shanabrough M. Muscarinic tone sustains impulse flow in the septohippocampal GABA but not cholinergic pathway: implications for learning and memory. The Journal of Neuroscience. 2000;20:8103–8110. doi: 10.1523/JNEUROSCI.20-21-08103.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aggleton JP, Brown MW. Episodic memory, amnesia, and the hippocampal-anterior thalamic axis. Behavioral Brain Science. 1999;22:425–489. [PubMed] [Google Scholar]
- 3.Angunawela I, Barker A. Anticholinesterase drugs for alcoholic Korsakoff syndrome. International journal of geriatric psychiatry. 2001;16:338–339. doi: 10.1002/gps.338. [DOI] [PubMed] [Google Scholar]
- 4.Bentivoglio M, Aggleton JP, Mishkin M. In: The thalamus and memory formation. Thalamus M, Steriade EJ, Jones DA, McCormick, editors. Elsevier; Amsterdam: 1997. pp. 689–720. [Google Scholar]
- 5.Blundell P, Hall G, Killcross S. Lesions of the basolateral amygdala disrupt selective aspects of reinforcer representation in rats. The Journal of Neuroscience. 2001;21:9018–9026. doi: 10.1523/JNEUROSCI.21-22-09018.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berger-Sweeney J, Heckers S, Mesulam MM, Wiley RG, Lappi DA, Sharma M. Differential effects on spatial navigation of immunotoxin- induced cholinergic lesions of the medial septal area and nucleus basalis magnocellularis. The Journal of Neuroscience. 1994;14:4507–4519. doi: 10.1523/JNEUROSCI.14-07-04507.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chang Q, Gold PE. Switching memory systems during learning: changes in patterns of brain acetylcholine release in the hippocampus and striatum in rats. 2003 doi: 10.1523/JNEUROSCI.23-07-03001.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chang Q& Gold PE. Impaired and spared cholinergic functions in the hippocampus after lesions of the medial septum/vertical limb of the diagonal band with 192 IgG-saporin. Hippocampus. 2004;14:170–179. doi: 10.1002/hipo.10160. [DOI] [PubMed] [Google Scholar]
- 9.Cochrane M, Cochrane A, Jauhar P, Ashton E. Acetylcholinesterase inhibitors for the treatment of Wernicke-Korsakoff Syndrome-Three further cases show response to donepezil. Alcohol & Alcoholism. 2005;40:151–154. doi: 10.1093/alcalc/agh127. [DOI] [PubMed] [Google Scholar]
- 10.Cohen J, Squire LR. Preserved learning and retention of pattern-analyzing skill in amnesia: Dissociation of knowing how and knowing that. Science. 1980;210:207–210. doi: 10.1126/science.7414331. [DOI] [PubMed] [Google Scholar]
- 11.Colgin LL, Kubota D, Lynch G. Cholinergic plasticity in the hippocampus. Proceedings of the National Academy of Sciences. 2003;100:2872–2877. doi: 10.1073/pnas.0530289100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Desjardins P, Butterworth RF. Role of mitochondrial dysfunction and oxidative stress in the pathogenesis of selective neuronal loss in Wernicke’s encephalopathy. Molecular neurobiology. 2005;31:17–25. doi: 10.1385/MN:31:1-3:017. [DOI] [PubMed] [Google Scholar]
- 13.Elvander E Schött PA, Sandin J, Bjelke B, Kehr J, Yoshitake T, Ogren SO. Intraseptal muscarinic ligands and galanin: influence on hippocampal acetylcholine and cognition. Neuroscience. 2004;126:541–57. doi: 10.1016/j.neuroscience.2004.03.058. [DOI] [PubMed] [Google Scholar]
- 14.Gold PE. Acetylcholine modulation of neural systems involved in learning and memory. Neurobiology of Learning and Memory. 2003;80:194–210. doi: 10.1016/j.nlm.2003.07.003. [DOI] [PubMed] [Google Scholar]
- 15.Gold PE. Coordination of multiple memory systems. Neurobiology of Learning and Memory. 2004;82:230–242. doi: 10.1016/j.nlm.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 16.Harding A, Halliday G, Caine D, Kril J. Degeneration of anterior thalamic nuclei differentiates alcoholics with amnesia. Brain. 2000;123:141–54. doi: 10.1093/brain/123.1.141. [DOI] [PubMed] [Google Scholar]
- 17.Hasselmo ME. The role of acetylcholine in learning and memory. Current Opinions in Neurobiology. 2006;16:1–6. doi: 10.1016/j.conb.2006.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heckers S, Ohtake T, Wiley RG, Lappi DA, Geula C, Mesulam MM. Complete and selective cholinergic denervation of rat neocortex and hippocampus but not amygdala by an immunotoxin against the p75 NGF receptor. The Journal of Neuroscience. 1994;14:1271–1289. doi: 10.1523/JNEUROSCI.14-03-01271.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jacobson RR, Lishman WA. Selective memory loss and global intellectual deficits in alcoholic Korsakoff’s syndrome. Psychological Medicine. 1997;17:649–655. doi: 10.1017/s0033291700025885. [DOI] [PubMed] [Google Scholar]
- 20.Jenkins TA, Dias RAmin E, Brown MW& Aggleton JP. Fos imaging reveals that lesions of the anterior thalamic nuclei produce widespread limbic hypoactivity in rats. Neuroscience. 2002;22:5230–5238. doi: 10.1523/JNEUROSCI.22-12-05230.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kirk IJ& Mackay JC. The role of theta-range oscillations in synchronizing and integrating activity in distributed mnemonic networks. Cortex. 2003;39:993–1008. doi: 10.1016/s0010-9452(08)70874-8. [DOI] [PubMed] [Google Scholar]
- 22.Kolmac C, Mitrofanis J. Organization of the basal forebrain projection to the thalamus in rats. Neuroscience Letters. 1999;272:151–154. doi: 10.1016/s0304-3940(99)00614-x. [DOI] [PubMed] [Google Scholar]
- 23.Kopelman MD. The Korsakoff syndrome. British Journal of Psychiatry. 1995;166:154–173. doi: 10.1192/bjp.166.2.154. [DOI] [PubMed] [Google Scholar]
- 24.Langlais PJ, Zhang SX. Extracellular glutamate is increased in the thalamus during thiamine deficiency-induced lesions and is blocked by MK-801. Journal of Neurochemistry. 1993;61:2175–2182. doi: 10.1111/j.1471-4159.1993.tb07457.x. [DOI] [PubMed] [Google Scholar]
- 25.Langlais PJ, Savage LM. Thiamine deficiency in rats produces cognitive and memory deficits on spatial tasks which correlate with tissue loss in diencephalon, cortex and white matter. Behavioral Brain Research. 1995;68:75–89. doi: 10.1016/0166-4328(94)00162-9. [DOI] [PubMed] [Google Scholar]
- 26.Langlais PJ, Zhang SX, Savage LM. Neuropathology of thiamine deficiency: An update on the comparative analysis of human disorders and experimental models. Metabolic Brain Disease. 1996;11:19–37. doi: 10.1007/BF02080929. [DOI] [PubMed] [Google Scholar]
- 27.Langlais PJ, Zhang SX. Cortical and subcortical white matter damage without Wernicke’s encephalopathy after recovery from thiamine deficiency in the rat. Alcoholism: Clinical and Experimental Research. 1997;68:75–89. doi: 10.1111/j.1530-0277.1997.tb03788.x. [DOI] [PubMed] [Google Scholar]
- 28.Mair RG, Anderson CD, Langlais PJ, McEntee WJ. Behavioral impairments, brain lesions, and monoaminergic activity in the rat following recovery from a bout of thiamine deficiency. Behavioural Brain Research. 1988;27:223–39. doi: 10.1016/0166-4328(88)90119-2. [DOI] [PubMed] [Google Scholar]
- 29.Mair RG. On the role of thalamic pathology in diencephalic amnesia. Reviews in Neuroscience. 1994;5:105–40. doi: 10.1515/revneuro.1994.5.2.105. [DOI] [PubMed] [Google Scholar]
- 30.Markowitsch HJ, Pritzel M. The neuropathology of amnesia. Progress in Neurobiology. 1985;25:189–287. doi: 10.1016/0301-0082(85)90016-4. [DOI] [PubMed] [Google Scholar]
- 31.McIntyre CK, Pal SN, Marriott LK, Gold PE. Competition between memory systems: acetylcholine release in the hippocampus correlates negatively with good performance on an amygdala-dependent task. Journal of Neuroscience. 2002;22:1171–1176. doi: 10.1523/JNEUROSCI.22-03-01171.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McIntyre CK, Marriott LK, Gold PE. Cooperation between memory systems: acetylcholine release in the amygdala correlates positively with performance on a hippocampus-dependent task. Behavioral Neuroscience. 2003a;117:320–326. doi: 10.1037/0735-7044.117.2.320. [DOI] [PubMed] [Google Scholar]
- 33.McIntyre CK, Marriott LK, Gold PE. Patterns of brain acetylcholine release predict individual differences in preferred learning strategies in rats. Neurobiology of Learning and Memory. 2003b;79:177–183. doi: 10.1016/s1074-7427(02)00014-x. [DOI] [PubMed] [Google Scholar]
- 34.McKinney M, Jacksonville MC. Brain cholinergic vulnerability: relevance to behavior and disease. Biochemical pharmacology. 2005;70:1115–1124. doi: 10.1016/j.bcp.2005.05.019. [DOI] [PubMed] [Google Scholar]
- 35.Miettinen RA, Kalesnykas G, Koivisto EH. Estimations of the total number of cholinergic neurons containing estrogen receptors-α in the rat basal forebrain. The Journal of Histochemistry & Cytochemistry. 2002;50:891–902. doi: 10.1177/002215540205000703. [DOI] [PubMed] [Google Scholar]
- 36.Nakagawasai O, Tadano T, Hozumi S, Tan-No K, Niijima F, Kisara K. Immunohistochemical estimation of brain choline acetyltransferase and somatostatin related to the impairment of avoidance learning induced by thiamine deficiency. Brain Research Bulletin. 2000;52:189–196. doi: 10.1016/s0361-9230(00)00248-3. [DOI] [PubMed] [Google Scholar]
- 37.Nakagawasai O, Yamadera FIwasaki K, Arai H, Taniguchi R, Tan-No K, Sasaki H, Tadano T. Effect of Kamiuntan-to on impairment of learning and memory induced by thiamine-deficient feeding in mice. Neuroscience. 2004;125:233–241. doi: 10.1016/j.neuroscience.2003.10.051. [DOI] [PubMed] [Google Scholar]
- 38.Nakagawasai O. Behavioral and Neurochemical alterations following thiamine deficiency in rodents: relationship to functions of cholinergic neurons. The Pharmaceutical Society of Japan. 2005;125:549–554. doi: 10.1248/yakushi.125.549. [DOI] [PubMed] [Google Scholar]
- 39.Parent MB, Baxter MG. Septohippocampal acetylcholine: involved in but not necessary for learning and memory? Learning & Memory. 2004;11:9–20. doi: 10.1101/lm.69104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. Academic Press; San Diego, CA: 1986. [Google Scholar]
- 41.Pires RG, Pereira SRC, Pittella EJH, Franco GC, Ferreira CLM, Fernandes PA, Ribeiro AM. The contribution of mild thiamine deficiency and ethanol consumption to central cholinergic parameter dysfunction and rats’ open-field performance impairment. Pharmacology, Biochemistry, and Behavior. 2001;70:227–235. doi: 10.1016/s0091-3057(01)00593-7. [DOI] [PubMed] [Google Scholar]
- 42.Pires RG, Pereira SRC Oliveira-Silva IF, Franco GC, Ribeiro AM. Cholinergic parameters and the retrieval of learned and re-learned spatial information: a study using a model of Wernicke-Korsakoff Syndrome. Behavioural Brain Research. 2005;162:11–21. doi: 10.1016/j.bbr.2005.02.032. [DOI] [PubMed] [Google Scholar]
- 43.Pitkin SR, Savage LM. Aging potentiates acute neurological symptoms and brain pathology in pyrithiamine-induced thiamine deficient rats. Behavioural Brain Research. 2001;119:167–177. doi: 10.1016/s0166-4328(00)00350-8. [DOI] [PubMed] [Google Scholar]
- 44.Pitkin SR, Savage LM. Age-related vulnerability to diencephalic amnesia produced by thiamine deficiency: the role of time of insult. Behavioural Brain Research. 2004;148:93–105. doi: 10.1016/s0166-4328(03)00208-0. [DOI] [PubMed] [Google Scholar]
- 45.Reed LJ, Lasserson D, Marsden P, Stanhope N, Stevens T, Bello F, Kingsley D, Colchester A, Kopelman MD. FDG-PET findings in the WernickeKorsakoff Syndrome. Cortex. 2003;39:1027–1045. doi: 10.1016/s0010-9452(08)70876-1. [DOI] [PubMed] [Google Scholar]
- 46.Roland JJ, Savage LM. Hippocampal and striatal acetylcholine efflux during learning in diencephalic-lesioned rats. Neurobiology of Learning and Memory. 2007;87:123–132. doi: 10.1016/j.nlm.2006.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Savage LM, Langlais PJ. Differential outcomes attenuates spatial memory impairments seen in pyrithiamine-induced thiamine deficiency in rats. Psychobiology. 1995;23:153–160. [Google Scholar]
- 48.Savage L. In search of the neurobiological correlates of the Differential Outcomes Effect. Integrative Physiological and Behavioral Science. 2001;36:182–195. doi: 10.1007/BF02734092. [DOI] [PubMed] [Google Scholar]
- 49.Savage LM, Chang Q, Gold PE. Diencephalic-lesions impair spontaneous alternation behavior and reduce behaviorally-activated acetylcholine release in the hippocampus. Learning & Memory. 2003;10:242–246. doi: 10.1101/lm.60003. [DOI] [PubMed] [Google Scholar]
- 50.Sotty F, Danik M, Manseau F, Laplante F, Quirion R, Williams S. Distinct electrophysiological properties of glutamatergic, cholinergic and GABAergic rat septohippocampal neurons: novel implications for hippocampal rhythmicity. Journal of Physiology. 2003;551:927–943. doi: 10.1113/jphysiol.2003.046847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Squire LR. The neuropsychology of human memory. Annual Review of Neuroscience. 1982;5:241–273. doi: 10.1146/annurev.ne.05.030182.001325. [DOI] [PubMed] [Google Scholar]
- 52.Todd K, Butterworth RF. Mechanisms of selective neuronal cell death due to thiamine deficiency. Annals of the New York Academy of Sciences. 1999;893:404–411. doi: 10.1111/j.1749-6632.1999.tb07866.x. [DOI] [PubMed] [Google Scholar]
- 53.Vann SD, Aggleton JP. Evidence of a spatial encoding deficit in rats with lesions of the mammillary bodies or mammillothalamic tract. Journal of Neuroscience. 2003;23:3506–3514. doi: 10.1523/JNEUROSCI.23-08-03506.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vann SD, Aggleton JP. The mammillary bodies: two memory systems in one? Nature Reviews. 2004;5:35–44. doi: 10.1038/nrn1299. [DOI] [PubMed] [Google Scholar]
- 55.Vertes RP, Hoover WB, Di Prisco GV. Theta rhythm of the hippocampaus: subcortical control and functional significance. Behavioral and Cognitive Neuroscience Reviews. 2004;3:173–200. doi: 10.1177/1534582304273594. [DOI] [PubMed] [Google Scholar]
- 56.Yoder RM, Pang CH. Involvement of GABAergic and cholinergic Medial septal neurons in hippocampal theta rhythm. Hipppocampus. 2005;15:381–392. doi: 10.1002/hipo.20062. [DOI] [PubMed] [Google Scholar]
- 57.Wainer BH, Mesulam MM. Asending cholinergic pathways in the rat brain. In: Steriade D, Biesold, editors. Brain Cholinergic Systems. M. Oxford Science Publications; Oxford: 1990. pp. 65–119. [Google Scholar]
- 58.Warburton EC, Baird A, Morgan A, Muir JL, Aggleton JP. The conjoint importance of the hippocampus and anterior thalamic nuclei for allocentric spatial learning: evidence from a disconnection study in the rat. Journal of Neuroscience. 2001;21:7323–7330. doi: 10.1523/JNEUROSCI.21-18-07323.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Warrington EK, Weiskrantz L. Amnesia: A disconnection syndrome? Neuropsychologia. 1982;20:233–248. doi: 10.1016/0028-3932(82)90099-9. [DOI] [PubMed] [Google Scholar]
- 60.Wu M, Newton SS, Atkins JB, Xu C, Duman RS, Alreja M. Acetylcholinesterase inhibitors activate septohippocampal GABAergic neurons via muscarinic but not nicotinic receptors. The Journal of Pharmacology and Experimental Therapeutics. 2003;307:535–543. doi: 10.1124/jpet.103.052514. [DOI] [PubMed] [Google Scholar]