

Abstract
Presence of neuritic plaques and neurofibrillary tangles in the brain are two neuropathological hallmarks of Alzheimer’s disease (AD), although the molecular basis of their coexistence remains elusive. The neurofibrillary tangles are composed of microtubule binding protein Tau, whereas neuritic plaques consist of amyloid-β peptides derived from amyloid precursor protein (APP). Recently, the peptidyl-prolyl cis/trans isomerase Pin1 has been identified to regulate the function of certain proteins after phosphorylation and to play an important role in cell cycle regulation and cancer development. New data indicate that Pin1 also regulates the function and processing of Tau and APP, respectively, and is important for protecting against age-dependent neurodegeneration. Furthermore, Pin1 is the only gene known so far that, when deleted in mice, can cause both tau and Aβ-related pathologies in an age-dependent manner, resembling many aspects of human Alzheimer’s disease. Moreover, in the human AD brain Pin1 is downregulated or inhibited by oxidative modifications and/or genetic changes. These results suggest that Pin1 deregulation may provide a link between formation of tangles and plaques in AD.
Pin1 in cell cycle regulation and cancer
Transition through the cell cycle in eukaryotic cells is regulated by highly orchestrated and intertwined processes of protein synthesis, degradation and post-translational modification. For its rapid activating/inhibiting effect, phosphorylation of regulatory molecules by cell cycle kinases plays a key role among the post-translational processes. Several families of the cell cycle kinases can be distinguished, the most prominent being cyclin dependent kinase (cdk), Polo, aurora and never in mitosis A (NIMA) families (for review see [1]). Activation of the protein kinases during the cell cycle triggers phosphorylation cascades that drive transition from one phase of the cell cycle to another. For example, activation of the cyclin-dependant kinase Cdc2 during the G2/M transition leads to phosphorylation of a large number of proteins on Ser/Thr-Pro motifs, which has been shown in some cases to regulate mitotic events [2–4].
With the discovery of Pin1, another level of cell cycle regulation has been uncovered [5]. Pin1 has been originally identified as a binding partner and suppressor of the mitotic kinase NIMA [5]. It contains two functional domains, an N-terminal WW domain and a C-terminal peptidyl-prolyl cis/trans isomerase (PPIase) domain [5–7]. The WW domain is a phosphorylation-specific protein interaction module that directs Pin1 to its substrates – proteins phosphorylated at a certain serine or threonine residue followed by proline (pSer/Thr-Pro motif) [7–9]. Upon this binding, the PPIase domain catalyzes conformational change of the Pin1 substrates by isomerizing specific pSer/Thr-Pro bonds [6, 10]. The specific binding to and isomerization of pSer/Thr-Pro motifs distinguishes Pin1 from the other known PPIase families such as cyclophilins and FK506-binding proteins. To date, Pin1-type PPIases are the only known pSer/Thr-Pro-specific isomerases [5, 7, 8]. The isomerization of pSer/Thr-Pro motifs represents an important regulatory mechanism since several protein kinases (e.g. CDK2, MAPK) and phosphatases (e.g. PP2A) are conformation specific, recognizing only trans Ser/Thr-Pro isomers [10–12]. Furthermore, phosphorylation slows the already protracted isomerization reaction of Ser/Thr-Pro bonds [8, 13], and renders the phosphopeptide bond resistant to the catalytic action of cyclophilin, FKBP or parvulin [8, 14]. Thus, conformation of a Ser/Thr-Pro motif can have a profound effect on phosphorylation signaling.
Due to a large number of Pin1 substrates, Pin1 is involved in multiple cellular processes. The discovery of Pin1’s regulatory function in the cell cycle and signaling has been followed by its important function in DNA damage responses, transcription, splicing, and germ cell development [5, 6, 9, 10, 15–32]. The involvement of Pin1 in the regulation of the cell cycle, cell signaling and responses to DNA damage suggests that its deregulation might contribute to some medical conditions in humans. Indeed, Pin1 is overexpressed in many tumors and its overexpression correlates with poor clinical outcome [20, 33–35]. Furthermore, Pin1 is an E2F target gene that is critical for activation of multiple upstream oncogenic pathways [20, 21, 27, 33, 36, 37] and also for coordination of some downstream cell cycle events such as centrosome duplication [38]. Moreover, Pin1 overexpression results in centrosome amplification and tumorigenesis in vitro and in vivo [38]. In contrast, Pin1 knockout in mice prevents certain oncogenes from inducing tumors [39] and Pin1 knockout in cancer cells suppresses cell growth in vitro and tumor growth in vivo [40]. These and other results indicate that Pin1 plays a major role in cancer development and is an attractive anticancer target [41, 42].
Pin1 in Alzheimer’s Disease
Many phospho-proteins recognized by Pin1 are recognized also by a phospho-specific monoclonal antibody mitotic phospho-protein monoclonal-2 (MPM-2), which strongly reacts with mitotic protein extracts [43] and with neurofibrillary tangles (NFTs), neuritic processes, and neurons in the brains of Alzheimer disease (AD) patients [44–46]. Reappearance of the MPM-2 epitopes in the AD brains is concomitant with aberrant expression of some kinases, e.g. Cdc2 - a mitotic kinase prosphorylating Ser/Thr-Pro motif during the G2/M phase of the cell cycle but absent in the healthy brain [45]. Consequently, while in the healthy brains Pin1 is expressed mainly in the neuronal soluble fraction [5, 20, 32, 47, 48], in the brains of AD patients it co-localizes and co-purifies with NFTs resulting in depletion of soluble Pin1 [47–50]. Moreover, Pin1−/− mice develop progressive age-dependent neuropathy characterized by Tau hyper-phosphorylation, Tau filament formation, amyloid precursor protein (APP) amyloidogenesis, intracellular Aβ42 accumulation and neuronal degeneration [51, 52]. Pin1 inhibition in the brain, therefore, may be an important factor in development of neurodegenerative disorders and AD, in particular. In the following section we will focus on the role of Pin1 in Tau- and APP-related pathologies.
Pin1 and tauopathies
Tauopathies are a heterogeneous group of diseases characterized by the presence of NFTs, a pathological structure composed of hyper-phosphorylated microtubule-associated protein Tau organized in dense arrays of paired helical filaments (PHFs). NFTs have been discovered in AD, frontotemporal dementia parkinsonism linked to chromosome 17 (FTDP-17), progressive supranuclear palsy (PSP) Pick disease and cortico-basal degeneration (for review see [53]). Even though the presence of hyper-phosphorylated Tau is clearly pathological, it has been a matter of debate whether it causes the diseases or whether it is a consequence of a common pathologic process. The fact that Tau mutations have been found in patients suffering from FTDP-17 indicates that at least in some cases mutant Tau can trigger a disease [54–56]. Several mouse models have been created mimicking pathologic features of human tauopathies. Overexpression of human wild-type Tau or especially Tau FDTP-17 mutants causes progressive and age-dependent formation of NFTs in mice [57–59]. Transgenic mice overexpressing a different version of human Tau (namely the smallest isoform, four repeats of microtubule domain isoform, Pro301Leu mutant, Arg406Trp mutant, or Val337Met mutant) all exhibit hyper-phosphorylation of Tau [57–63], although different mutations or splice variants of Tau have demonstrated different levels and patterns of neuronal loss or axonal degeneration in brain and spinal cords [57–63].
Tangle formation in AD appears to be preceded by increased phosphorylation of Tau and other proteins on serine or threonine residues followed by proline (pSer/Thr-Pro). Even though the exact role of hyper-phosphorylation for development of tauopathies has not been clearly defined, analysis of transgenic mouse models overexpressing p25 activator of CDK5 kinase has shown that an increased phosphorylation can induce tauopathy in mice [64]. Importantly, hyper-phosphorylated Tau can activate mitotic signaling pathways [65], as demonstrated in Drosophila. Since re-entry into the cell cycle is toxic for neurons, and activation of CDKs and other mitotic proteins has been correlated with neurodegeneration in AD proteins (reviewed by [66]), hyper-phosphorylated Tau activating mitotic signaling could in some cases lead to neuronal cell death independent of NFT formation. Hyper-phosphorylation of Tau may trigger multiple cellular responses. These include cell cycle re-entry and deposition of NFTs since there are many phosphorylation sites on Tau protein which may interact with various signaling molecules contributing separately or in combination to neuronal toxicity. Notably, among the phosphorylation sites in Tau there are 15 pSer/Thr-Pro motifs [67], i.e. putative Pin1 binding sites.
Pin1 binds to Tau in a phosphorylation-dependent manner specifically to its pThr231 residue [48, 68]. Interestingly, the levels of Tau-P-T231 have been shown to correlate with the progression of the AD [69, 70]. Upon its binding to pThr231 Pin1 catalyzes cis/trans isomerization of pSer/Thr–Pro, thereby inducing conformational changes in Tau. Such conformational changes can directly restore the ability of phosphorylated Tau to bind microtubules and promote microtubule assembly [71] and/or facilitate Tau dephosphorylation by its phosphatase PP2A, as PP2A activity is conformation-specific [10]. Hamdane et al. recently showed that Pin1 level was strongly increased during neuronal differentiation and tightly correlated with Tau dephosphorylation at Thr231 [72]. In their cellular model, Pin1 facilitated Tau dephosphorylation of Thr231 specifically, whereas other phosphorylation sites were not affected by Pin1 [72]. To investigate whether Pin1 could function similarly in Tau overexpressing animal models, would be of a great interest.
When Pin1 protein was analyzed in human brain using immunohistochemical staining, its expression in normal brain was relatively higher in hippocampal CA3, CA2 and CA4 regions and in presubiculum and lower in hippocampal CA1 region and subiculum [51]. In the parietal cortex, expression of Pin1 is relatively higher in layer IIIb-c neurons and lower in layer V neurons [51]. In the AD brain, Pin1 expression in the hippocampus and parietal cortex is relatively high in tangle-sparing subregions, but low in the tangle-rich subregions. [51]. Furthermore, even within the tangle-prone CA1 region and subiculum of the hippocampus, Pin1 expression in most tangle-bearing neurons is still relatively lower than that in tangle-fee neurons [51]. Thus, Pin1 expression level is inversely correlated with the neuronal vulnerability to degeneration in normal brain and with actual neurofibrillary degeneration in AD brain.
The significance of the differential Pin1 expression is further demonstrated by analyzing neuronal phenotypes of Pin1 knockout mice. Pin1 knockout mice develop normally but they suffer from progressive retinal degeneration with the onset at around 4–6 months [21]. Aged Pin1−/− mice, but not their wild-type littermates, show progressive age-dependent motor and behavioral deficits, which includes abnormal limb-clasping reflexes, hunched postures, and reduced mobility [51]. These deficits have also been reported in Tau transgenic mice studies [58, 73]. The phenotype seems to be caused by neuronal loss as the number of neurons is significantly decreased in the parietal cortex and spinal cords of old (over 1 year), but not young Pin1−/− mice. Correspondingly, Tau hyperphosphorylation has been observed in aged Pin1−/− mice [51]. The various phosphorylated forms of Tau in Pin1−/− mice were also detected by a range of phospho-specific or Alzheimer-conformation-specific antibodies, such as AT180 and TG3 [51]. In aged Pin1−/− mice, immunohistochemical staining of the hippocampus, cortex and spinal cord with specific pTau antibodies showed pathological localization of Tau in soma and dendrites of neurons [51]. The hyper-phosphorylation of Tau eventually leads to Tau aggregation and Tau filament formation in Pin1−/− mice [51]. Additionally, NFT-like Tau filaments decorated by AT180 gold label can be isolated from sarkosyl insoluble fractions of Pin1−/− mice [51]. The entorhinal cortex and hippocampus, two brain regions which show prominent degeneration in AD, were strongly immunopositive for stains that label NFTs including Gallyas and thioflavin-S in Pin1−/− mice [51].
Together with the in vitro data, analysis of Pin1−/− mice demonstrates that Pin1 regulates the function of Tau both in vivo and in vitro likely through catalysis of its conformational change. Importantly, Tau hyperphosphorylation and NFT formation can be induced by Aβ challenge and/or PS1 mutation [74–78]. Thus, Pin1 functioning in APP processing could also contribute to the development of Tau phenotypes in Pin1−/− mice.
Pin1 and APP processing and Aβ production
APP is a transmembrane protein consisting of a large extracellular and short transmembrane and intracellular domains. It can be processed by two alternative pathways: non-amyloidogenic, which involves cleavage by α-secretase, or amyloidogenic, which involves cleavage by β- and γ-secretases and leads to production of plaque forming β-amyloid peptides (Aβ) (for review see [79]). Several factors can influence APP processing and shift it towards amyloid/non-amyloid pathway. Recently, phosphorylation of APP has been found to be one of those factors [52, 80, 81].
To date only four phosphorylation sites have been confirmed at the intracellular domain of APP: S655, T654, T668, and Y682 [82–86]. Even though APP has not been identified as an MPM-2 epitope, it does contain a pThr-Pro motif (T668) which has been shown to be phosphorylated during mitosis by CDC2 [83, 87], in neurons by CDK5 [88] and in vitro by GSK-3β [89] and which is located at the consensus Pin1 binding sequence [8, 9]. In addition, APP has been found to undergo a conformational change following phosphorylation of T668 [90] making it a promising candidate for a Pin1 substrate. Indeed, we have shown that Pin1 interacts with APP isolated from mitotic cells and that the interaction is phosphorylation dependent [52]. Furthermore, direct NMR measurement demonstrated that Pin1 catalyzes the cis/trans isomerization of pThr668-Pro [52], since it accelerates both kcis to transcat and ktrans to ciscat by over 1000 fold over the typical uncatalyzed isomerization rates for pThr-Pro peptides [13]. The catalyzed cis to trans rate is 10-fold faster than the catalyzed trans to cis rate [52]. Change of the conformation of pThr668-Pro may represent an important regulatory mechanism since it may influence the interaction between APP and its binding partners such as Fe65. The binding of Fe65 to APP has been shown to be prosphorylation-dependent through pT668 residue of APP and influence production of Aβ [91]. Thus, Pin1 may have a direct impact on this regulatory process.
Pin1 subcellular localization is driven by the presence of its substrates [6, 31, 92, 93]. Processing of APP, an integral membrane protein, is influenced by APP subcellular localization and occurs through non-amyloidogenic α-secretases mainly at the plasma membrane and amyloidogenic β-secretases at endosomes and other subsequent structures [94–98]. Recently, it has been shown that Pin1 co-localizes with APP primarily in vesicles localized at the plasma membrane and in AP-2 coated clathrin-coated vesicles, but not at endosomes [52]. This suggests that Pin1 may influence APP intracellular localization, affecting its processing and Aβ production. This hypothesis has been corroborated by additional data. In the cell culture experiments, overexpression of Pin1 has been shown to reduce Aβ secretion, and its effect was particularly pronounced in the mitotic cells where Thr668 phosphorylation was increased [52].
Sequential proteolysis of APP by β- and γ-secretases generates mainly 40- and 42-residue Aβ peptides (Aβ40 and Aβ42). While Aβ40 is a major secreted product, Aβ42 is more toxic and is the major contributor to the plaque formation in AD brains [96–98]. Familial AD-linked (FAD) mutations in the APP or presenilin genes selectively increase Aβ42 levels in humans and mice [99–102]. Pin1 ablation seems not to have a significant effect on soluble Aβ40 or Aβ42 levels, but we found a significant increase of insoluble Aβ42 in Pin1−/− brains over Pin1+/+ littermates [52]. Additionally, Pin1 ablation causes prominent localization of Aβ42 to multivesicular bodies (MVB) [52], as in human AD and APP-Tg2576 mice before β-amyloid plaque pathology [103]. Thus, although it is not clear yet whether and how Aβ42 at MVB contributes to plaque formation, the data suggest that Pin1 may be involved in Aβ-related pathology.
Strong support for a direct role of Pin1 in APP processing came from analysis of a classic mouse AD model: APP-Tg2576 mice overexpressing the human APP KM670/671NL (Swedish) mutant [76]. APP processing is initiated by either non-amyloidogenic α- or amyloidogenic β-secretases, which cleave the extracellular/lumenal domain, generating soluble NH2-terminal fragments, αAPPs or βAPPs, and a membrane-anchored 83-residue or 99/89-residue COOH-terminal fragments (αCTFs or βCTFs), respectively [94–98]. In APP-Tg2576 mouse brains, Pin1 deletion leads to a significant increase of soluble total APPs and βAPPs, but to a decrease of αAPPs in an age-dependent manner [52]. More importantly, these age-dependent effects are also accompanied by the age-dependent increase in insoluble Aβ42 [52]. Thus, Pin1 depletion seems to favor the amyloidogenic versus non-amyloidogenic processing of APP.
The view of Pin1 as an important factor contributing to development of AD has been supported by several studies. Recently, a new genetic locus associated with late-onset AD has been identified on chromosome 19p13.2 where the Pin1 gene is located [104]. Furthermore, the Pin1 promoter polymorphisms at -842 bp and -667 bp have been found to be associated with reduced Pin1 levels and increased risk for late-onset AD in Italian cohorts [105], although apparently not in French cohorts [106]. Moreover, proteomic approaches have confirmed downregulation of Pin1 in AD neurons, and also uncovered that Pin1 is inhibited by oxidation in AD hippocampus even in patients with mild cognitive impairment [107, 108]. Finally, our findings of the opposite effects of Pin1 on the pathogenesis of cancer versus on AD suggest an interesting inverse relationship between these two major age-dependent diseases, which is also supported by an epidemiological study [109]. Roe et al. have found that the risk of developing cancer decreased among participants with Alzheimer type dementia versus non-demented participants and that the risk of developing Alzheimer type dementia may be lower for participants with a history of cancer [109]. Together these results indicate that Pin1 inhibition may be an important contributing factor to the development of AD.
A model of the role of Pin1 in the healthy and Alzheimer’s neurons
The above results suggest a model of the function of Pin1 in the healthy and AD neurons (Fig. 1): In the healthy neurons (Fig. 1A), Pin1 binds to Tau and APP after their phosphorylation at Thr231 or Thr668, respectively (which drastically slows down their cis to trans isomerization) and greatly accelerates their cis to trans isomerization rates, thereby inducing conformational changes. This might facilitate dephosphorylation of Tau, and promote the non-amyloidogenic pathway and reduce Aβ production (presumably through affecting APP intracellular localization and its interactions with specific binding partners). However, under pathological conditions (Fig. 1B), Pin1 function may be absent as in Pin1−/− mice or inhibited as seen in AD (due to down-regulation, oxidation of and/or genetic changes in Pin1, or relatively due to excess APP phosphorylation caused by upstream regulators including over-activation of JNKs, Cdks and/or GSKs). In these cases, isomerization rate of pThr231-Pro of Tau and pT668-Pro of APP may be reduced, which may lead to accumulation of phosphorylated Tau and promote the amyloidogenic pathway of APP. Increased levels of phosphorylated Tau may lead to tangle formation as well as trigger pathological re-entry into the cell cycle and cell death of the affected neurons, as demonstrated in the Drosophila model [65], while increased Aβ production might enhance the plaque pathology.
Fig. 1.
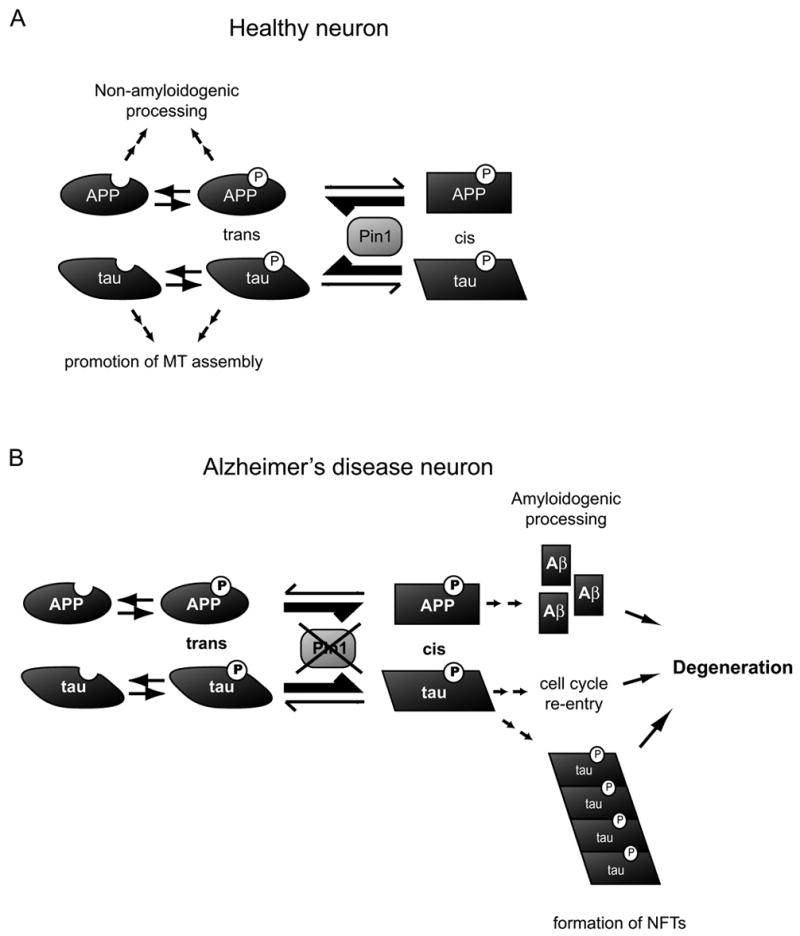
Pin1 catalyzes cis to trans conformational change of APP phosphorylated at T668-P and Tau phosphorylated at T231-P. In the presence of functional Pin1 (A) isomerization of pT668-P and pT231-P is shifted towards the trans conformation, which promotes non-amyloidogenic cleavage of APP and dephosphorylation of Tau. In Alzheimer’s disease (B), downregulation, oxidation or mutation of Pin1 reduces the isomerization rate that may, in the case of APP, promote the amyloidogenic pathway and increase Aβ production. In the case of Tau, reduced isomerization rate may promote aggregation of hyperphosphorylated Tau, inducing formation of NFTs and pathological re-entry into cell cycle.
Conclusions
Neurofibrillary tangles and senile plaques are two neuron-pathological hallmarks of AD, but the molecular basis of their coexistence remains elusive. Recent results indicate that the peptidyl-prolyl cis/trans isomerase Pin1 acts on both Tau and APP to regulate their dephosphorylation, processing and biological function [51, 52]. Furthermore, loss of Pin1 function in mice can cause both tau and Aβ-related pathologies in an age-dependent manner, resembling many aspects of human Alzheimer’s disease [51, 52]. As a support for the deleterious effect of Pin1 depletion, Pin1 has been found to be oxidized and inhibited in AD brains even with mild cognitive impairment [107, 108] and AD neurons have been shown to be depleted of the soluble form of Pin1 [48, 49]. Moreover, the Pin1 promoter polymorphisms have been found to be associated with reduced Pin1 levels and an increased risk for late-onset AD in a certain population [105]. Thus, Pin1 plays a pivotal role in protecting against age-dependent neurodegeneration, and Pin1 downregulation or inhibition may provide a link between tangle and plaque formation in AD. While it is possible that depletion of Pin1 modulates additional AD related molecular pathways, its impact on the function and metabolism of the two major AD related molecules Tau and APP is likely an important factor in AD development. Furthermore, since the ablation of the Pin1 gene alone or in combination with mutant APP overexpression leads to the age-dependent accumulation of insoluble Aβ42 in multivesicular bodies of neurons, an early sign of plaque pathology in human AD, it can be speculated that Pin1 plays a role in the initial steps of β-amyloid pathology and plaque formation. Moreover, Pin1 prevents tau hyperphosphorylation, which again precedes tangle formation and neurodegeneration. These findings suggest that Pin1 may be an attractive new target to be modulated for the treatment of Alzheimer’s disease at early stages. Further analysis of the regulation of Pin1 expression in neurons will be necessary to achieve this goal.
Acknowledgments
We are grateful to members of K.P. Lu’s and G. Wolf’s laboratories for stimulating discussions and particularly to Prudence Lam for editing of the manuscript. J. L. is a Human Frontier Research Program Fellow. Work in the authors’ laboratory has been supported by NIH grants GM56230, GM58556, AG17870 and AG22082 and a gift from Merck Research Laboratories-Boston to K.P.L.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Nigg EA. Mitotic kinases as regulators of cell division and its checkpoints. Nat Rev Mol Cell Biol. 2001;2:21–32. doi: 10.1038/35048096. [DOI] [PubMed] [Google Scholar]
- 2.Blangy A, Lane HA, d’Herin P, Harper M, Kress M, Nigg EA. Phosphorylation by p34cdc2 regulates spindle association of human Eg5, a kinesin-related motor essential for bipolar spindle formation in vivo. Cell. 1995;83:1159–1169. doi: 10.1016/0092-8674(95)90142-6. [DOI] [PubMed] [Google Scholar]
- 3.Qiao M, Shapiro P, Fosbrink M, Rus H, Kumar R, Passaniti A. Cell cycle-dependent phosphorylation of the RUNX2 transcription factor by cdc2 regulates endothelial cell proliferation. J Biol Chem. 2006;281:7118–7128. doi: 10.1074/jbc.M508162200. [DOI] [PubMed] [Google Scholar]
- 4.Lowe M, Rabouille C, Nakamura N, Watson R, Jackman M, Jamsa E, Rahman D, Pappin DJ, Warren G. Cdc2 kinase directly phosphorylates the cis-Golgi matrix protein GM130 and is required for Golgi fragmentation in mitosis. Cell. 1998;94:783–793. doi: 10.1016/s0092-8674(00)81737-7. [DOI] [PubMed] [Google Scholar]
- 5.Lu KP, Hanes SD, Hunter T. A human peptidyl-prolyl isomerase essential for regulation of mitosis. Nature. 1996;380:544–547. doi: 10.1038/380544a0. [DOI] [PubMed] [Google Scholar]
- 6.Lu KP, Liou YC, Zhou XZ. Pinning down proline-directed phosphorylation signaling. Trends Cell Biol. 2002;12:164–172. doi: 10.1016/s0962-8924(02)02253-5. [DOI] [PubMed] [Google Scholar]
- 7.Ranganathan R, Lu KP, Hunter T, Noel JP. Structural and functional analysis of the mitotic rotamase Pin1 suggests substrate recognition is phosphorylation dependent. Cell. 1997;89:875–886. doi: 10.1016/s0092-8674(00)80273-1. [DOI] [PubMed] [Google Scholar]
- 8.Yaffe MB, Schutkowski M, Shen M, Zhou XZ, Stukenberg PT, Rahfeld JU, Xu J, Kuang J, Kirschner MW, Fischer G, Cantley LC, Lu KP. Sequence-specific and phosphorylation-dependent proline isomerization: a potential mitotic regulatory mechanism. Science. 1997;278:1957–1960. doi: 10.1126/science.278.5345.1957. [DOI] [PubMed] [Google Scholar]
- 9.Lu PJ, Zhou XZ, Shen M, Lu KP. Function of WW domains as phosphoserine- or phosphothreonine-binding modules. Science. 1999;283:1325–1328. doi: 10.1126/science.283.5406.1325. [DOI] [PubMed] [Google Scholar]
- 10.Zhou XZ, Kops O, Werner A, Lu PJ, Shen M, Stoller G, Kullertz G, Stark M, Fischer G, Lu KP. Pin1-dependent prolyl isomerization regulates dephosphorylation of Cdc25C and tau proteins. Mol Cell. 2000;6:873–883. doi: 10.1016/s1097-2765(05)00083-3. [DOI] [PubMed] [Google Scholar]
- 11.Weiwad M, Kullertz G, Schutkowski M, Fischer G. Evidence that the substrate backbone conformation is critical to phosphorylation by p42 MAP kinase. FEBS Lett. 2000;478:39–42. doi: 10.1016/s0014-5793(00)01794-4. [DOI] [PubMed] [Google Scholar]
- 12.Brown NR, Noble ME, Endicott JA, Johnson LN. The structural basis for specificity of substrate and recruitment peptides for cyclin-dependent kinases. Nat Cell Biol. 1999;1:438–443. doi: 10.1038/15674. [DOI] [PubMed] [Google Scholar]
- 13.Schutkowski M, Bernhardt A, Zhou XZ, Shen M, Reimer U, Rahfeld JU, Lu KP, Fischer G. Role of phosphorylation in determining the backbone dynamics of the serine/threonine-proline motif and Pin1 substrate recognition. Biochemistry. 1998;37:5566–5575. doi: 10.1021/bi973060z. [DOI] [PubMed] [Google Scholar]
- 14.Uchida T, Fujimori F, Tradler T, Fischer G, Rahfeld JU. Identification and characterization of a 14 kDa human protein as a novel parvulin-like peptidyl prolyl cis/trans isomerase. FEBS Lett. 1999;446:278–282. doi: 10.1016/s0014-5793(99)00239-2. [DOI] [PubMed] [Google Scholar]
- 15.Ayala G, Wang D, Wulf G, Frolov A, Li R, Sowadski J, Wheeler TM, Lu KP, Bao L. The prolyl isomerase Pin1 is a novel prognostic marker in human prostate cancer. Cancer Res. 2003;63:6244–6251. [PubMed] [Google Scholar]
- 16.Shen M, Stukenberg PT, Kirschner MW, Lu KP. The essential mitotic peptidyl-prolyl isomerase Pin1 binds and regulates mitosis-specific phosphoproteins. Genes Dev. 1998;12:706–720. doi: 10.1101/gad.12.5.706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Crenshaw DG, Yang J, Means AR, Kornbluth S. The mitotic peptidyl-prolyl isomerase, Pin1, interacts with Cdc25 and Plx1. Embo J. 1998;17:1315–1327. doi: 10.1093/emboj/17.5.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fujimori F, Takahashi K, Uchida C, Uchida T. Mice lacking Pin1 develop normally, but are defective in entering cell cycle from G(0) arrest. Biochem Biophys Res Commun. 1999;265:658–663. doi: 10.1006/bbrc.1999.1736. [DOI] [PubMed] [Google Scholar]
- 19.Winkler KE, Swenson KI, Kornbluth S, Means AR. Requirement of the prolyl isomerase Pin1 for the replication checkpoint. Science. 2000;287:1644–1647. doi: 10.1126/science.287.5458.1644. [DOI] [PubMed] [Google Scholar]
- 20.Ryo A, Nakamura N, Wulf G, Liou YC, Lu KP. Pin1 regulates turnover and subcellular localization of beta-catenin by inhibiting its interaction with APC. Nature Cell Biol. 2001;3:793–801. doi: 10.1038/ncb0901-793. [DOI] [PubMed] [Google Scholar]
- 21.Liou YC, Ryo R, Huang HK, Lu PJ, Bronson R, Fujimori F, Uchidafl U, Hunter T, Lu KP. Loss of Pin1 function in the mouse causes phenotypes resembling cyclin D1-null phenotypes. Proc Natl Acad Sci USA. 2002;99:1335–1340. doi: 10.1073/pnas.032404099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stukenberg PT, Kirschner MW. Pin1 acts catalytically to promote a conformational change in Cdc25. Mol Cell. 2001;7:1071–1083. doi: 10.1016/s1097-2765(01)00245-3. [DOI] [PubMed] [Google Scholar]
- 23.Hsu T, McRackan D, Vincent TS, Gert De Couet H. Drosophila Pin1 prolyl isomerase Dodo is a MAP kinase signal responder during oogenesis. Nat Cell Biol. 2001;3:538–543. doi: 10.1038/35078508. [DOI] [PubMed] [Google Scholar]
- 24.Wulf GM, Liou YC, Ryo A, Lee SW, Lu KP. Role of Pin1 in the regulation of p53 stability and p21 transactivation, and cell cycle checkpoints in response to DNA damage. J Biol Chem. 2002;277:47976–47979. doi: 10.1074/jbc.C200538200. [DOI] [PubMed] [Google Scholar]
- 25.Zheng H, You H, Zhou XZ, Murray SA, Uchida T, Wulf G, Gu L, Tang X, Lu KP, Xiao ZXJ. The prolyl isomerase Pin1 is a regulator of p53 in genotoxic response. Nature. 2002;419:849–853. doi: 10.1038/nature01116. [DOI] [PubMed] [Google Scholar]
- 26.Zacchi P, Gostissa M, Uchida T, Salvagno C, Avolio A, Voliniak S, Ronai Z, Blandino G, Schneider C, Del Sal G. The prolyl isomerase Pin1 reveals a mechanism to control p53 functions after genotoxic insults. Nature. 2002;419:853–857. doi: 10.1038/nature01120. [DOI] [PubMed] [Google Scholar]
- 27.Ryo A, Suizu F, Yoshida Y, Perrem K, Liou YC, Wulf G, Rottapel R, Yamaoka S, Lu KP. Regulation of NF-kappaB signaling by Pin1-dependent prolyl isomerization and ubiquitin-mediated proteolysis of p65/RelA. Mol Cell. 2003;12:1413–1426. doi: 10.1016/s1097-2765(03)00490-8. [DOI] [PubMed] [Google Scholar]
- 28.Atchison FW, Capel B, Means AR. Pin1 regulates the timing of mammalian primordial germ cell proliferation. Development. 2003;130:3579–3586. doi: 10.1242/dev.00584. [DOI] [PubMed] [Google Scholar]
- 29.Atchison FW, Means AR, Capel B. Spermatogonial depletion in adult Pin1-deficient mice: Pin1 regulates the timing of mammalian primordial germ cell proliferation. Biol Reprod. 2003;20:1989–1997. doi: 10.1095/biolreprod.103.020859. [DOI] [PubMed] [Google Scholar]
- 30.Xu YX, Hirose Y, Zhou XZ, Lu KP, Manley JL. Pin1 modulates the structure and function of human RNA polymerase II. Genes Dev. 2003;17:2765–2776. doi: 10.1101/gad.1135503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lu KP. Pinning down cell signaling, cancer and Alzheimer’s disease. Trends Biochem Sci. 2004;29:200–209. doi: 10.1016/j.tibs.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 32.Wulf GM, Ryo A, Wulf GG, Lee SW, Niu T, Petkova V, Lu KP. Pin1 is overexpressed in breast cancer and cooperates with Ras signaling in increasing the transcriptional activity of c-Jun towards cyclin D1. Embo J. 2001;20:3459–3472. doi: 10.1093/emboj/20.13.3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wulf GM, Ryo A, Wulf GG, Lee SW, Niu T, Lu KP. Pin1 is overexpressed in breast cancer and potentiates the transcriptional activity of phosphorylated c-Jun towards the cyclin D1 gene. EMBO J. 2001;20:3459–3472. doi: 10.1093/emboj/20.13.3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ayala G, Wang D, Wulf G, Frolov A, Le R, Wheeler T, Sowadski JM, Lu KP, Bao L. Pin1 is a novel prognostic marker in prostate cancer. Cancer Research. 2003;63:6244–6251. [PubMed] [Google Scholar]
- 35.Bao L, Sauter G, Sowadski J, Lu KP, Wang D. Prevalent overexpression of prolyl isomerase Pin1 in human cancers. Am J Pathol. 2004;164:1727–1737. doi: 10.1016/S0002-9440(10)63731-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ryo A, Liou YC, Wulf G, Nakamura N, Lee SW, Lu KP. Pin1 is an E2F target gene essential for the Neu/Ras-induced transformation of mammary epithelial cells. Mol Cell Biol. 2002;22:5281–5295. doi: 10.1128/MCB.22.15.5281-5295.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dougherty MK, Müller J, Ritt DA, Zhou XZ, Zhou M, Copeland TD, Conrads TP, Veenstra T, Lu KP, Morrison DK. Regulation of Raf-1 by direct feedback phosphorylation. Mol Cell. 2005;17:215–224. doi: 10.1016/j.molcel.2004.11.055. [DOI] [PubMed] [Google Scholar]
- 38.Suizu F, Ryo A, Wulf G, Lim J, Lu KP. Pin1 regulates centrosome duplication and its overexpression induces centrosome amplification, chromosome instability and oncogenesis. Mol Cell Biol. 2006;26:1463–1479. doi: 10.1128/MCB.26.4.1463-1479.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wulf G, Garg P, Liou YC, Iglehart D, Lu KP. Modeling breast cancer in vivo and ex vivo reveals an essential role of Pin1 in tumorigenesis. EMBO J. 2004;23:3397–3407. doi: 10.1038/sj.emboj.7600323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ryo A, Uemura H, Ishiguro H, Saitoh T, Yamaguchi A, Perrem P, Kubota Y, Lu KP, Aoki I. Stable suppression of tumorigenicity by Pin1-targeted RNA interference in prostate cancer. Clin Cancer Res. 2005;11:7523–7531. doi: 10.1158/1078-0432.CCR-05-0457. [DOI] [PubMed] [Google Scholar]
- 41.Lu KP. Prolyl isomerase Pin1 as a molecular target for cancer diagnostics and therapeutics. Cancer Cell. 2003;4:175–180. doi: 10.1016/s1535-6108(03)00218-6. [DOI] [PubMed] [Google Scholar]
- 42.Lu KP, Suizu F, Zhou XZ, Finn G, Lam P, Wulf G. Targeting carcinogenesis: A role for the prolyl isomerase Pin1? Mol Carcinog. 2006;45:397–402. doi: 10.1002/mc.20216. [DOI] [PubMed] [Google Scholar]
- 43.Davis FM, Tsao TY, Fowler SK, Rao PN. Monoclonal antibodies to mitotic cells. Proc Natl Acad Sci U S A. 1983;80:2926–2930. doi: 10.1073/pnas.80.10.2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vincent I, Rosado M, Davies P. Mitotic mechanisms in Alzheimer’s disease? J Cell Biol. 1996;132:413–425. doi: 10.1083/jcb.132.3.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vincent I, Jicha G, Rosado M, Dickson DW. Aberrant expression of mitotic cdc2/cyclin B1 kinase in degenerating neurons of Alzheimer’s disease brain. J Neurosci. 1997;17:3588–3598. doi: 10.1523/JNEUROSCI.17-10-03588.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lu KP, Liou YC, Vincent I. Proline-directed phosphorylation and isomerization in mitotic regulation and in Alzheimer’s Disease. Bioessays. 2003;25:174–181. doi: 10.1002/bies.10223. [DOI] [PubMed] [Google Scholar]
- 47.Thorpe JR, Mosaheb S, Hashemzadeh-Bonehi L, Cairns NJ, Kay JE, Morley SJ, Rulten SL. Shortfalls in the peptidyl-prolyl cis-trans isomerase protein Pin1 in neurons are associated with frontotemporal dementias. Neurobiol Dis. 2004;17:237–249. doi: 10.1016/j.nbd.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 48.Lu PJ, Wulf G, Zhou XZ, Davies P, Lu KP. The prolyl isomerase Pin1 restores the function of Alzheimer-associated phosphorylated tau protein. Nature. 1999;399:784–788. doi: 10.1038/21650. [DOI] [PubMed] [Google Scholar]
- 49.Thorpe JR, Morley SJ, Rulten SL. Utilizing the peptidyl-prolyl cis-trans isomerase pin1 as a probe of its phosphorylated target proteins. Examples of binding to nuclear proteins in a human kidney cell line and to tau in Alzheimer’s diseased brain. J Histochem Cytochem. 2001;49:97–108. doi: 10.1177/002215540104900110. [DOI] [PubMed] [Google Scholar]
- 50.Ramakrishnan P, Dickson DW, Davies P. Pin1 colocalization with phosphorylated tau in Alzheimer’s disease and other tauopathies. Neurobiol Dis. 2003;14:251–264. doi: 10.1016/s0969-9961(03)00109-8. [DOI] [PubMed] [Google Scholar]
- 51.Liou YC, Sun A, Ryo A, Zhou XZ, Yu ZX, Huang HK, Uchida T, Bronson R, Bing G, Li X, Hunter T, Lu KP. Role of the prolyl isomerase Pin1 in protecting against age-dependent neurodegeneration. Nature. 2003;424:556–561. doi: 10.1038/nature01832. [DOI] [PubMed] [Google Scholar]
- 52.Pastorino L, Sun A, Lu PJ, Zhou XZ, Balastik M, Finn G, Wulf G, Lim J, Li SH, Li X, Xia W, Nicholson L, Lu KP. The prolyl isomerase Pin1 regulates amyloid precursor protein processing and amyloid-beta production. Nature. 2006;440:528–534. doi: 10.1038/nature04543. [DOI] [PubMed] [Google Scholar]
- 53.Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci. 2001;24:1121–1159. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- 54.Rizzu P, Van Swieten JC, Joosse M, Hasegawa M, Stevens M, Tibben A, Niermeijer MF, Hillebrand M, Ravid R, Oostra BA, Goedert M, van Duijn CM, Heutink P. High prevalence of mutations in the microtubule-associated protein tau in a population study of frontotemporal dementia in the Netherlands. Am J Hum Genet. 1999;64:414–421. doi: 10.1086/302256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Petersen RC, Stevens M, de Graaff E, Wauters E, van Baren J, Hillebrand M, Joosse M, Kwon JM, Nowotny P, Che LK, Norton J, Morris JC, Reed LA, Trojanowski J, Basun H, Lannfelt L, Neystat M, Fahn S, Dark F, Tannenberg T, Dodd PR, Hayward N, Kwok JB, Schofield PR, Andreadis A, Snowden J, Craufurd D, Neary D, Owen F, Oostra BA, Hardy J, Goate A, van Swieten J, Mann D, Lynch T, Heutink P. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- 56.Hong M, Zhukareva V, Vogelsberg-Ragaglia V, Wszolek Z, Reed L, Miller BI, Geschwind DH, Bird TD, McKeel D, Goate A, Morris JC, Wilhelmsen KC, Schellenberg GD, Trojanowski JQ, Lee VM. Mutation-specific functional impairments in distinct tau isoforms of hereditary FTDP-17. Science. 1998;282:1914–1917. doi: 10.1126/science.282.5395.1914. [DOI] [PubMed] [Google Scholar]
- 57.Spittaels K, Van den Haute C, Van Dorpe J, Bruynseels K, Vandezande K, Laenen I, Geerts H, Mercken M, Sciot R, Van Lommel A, Loos R, Van Leuven F. Prominent axonopathy in the brain and spinal cord of transgenic mice overexpressing four-repeat human tau protein. Am J Pathol. 1999;155:2153–2165. doi: 10.1016/S0002-9440(10)65533-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lewis J, McGowan E, Rockwood J, Melrose H, Nacharaju P, Van Slegtenhorst M, Gwinn-Hardy K, Paul Murphy M, Baker M, Yu X, Duff K, Hardy J, Corral A, Lin WL, Yen SH, Dickson DW, Davies P, Hutton M. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet. 2000;25:402–405. doi: 10.1038/78078. [DOI] [PubMed] [Google Scholar]
- 59.Ishihara T, Hong M, Zhang B, Nakagawa Y, Lee MK, Trojanowski JQ, Lee VM. Age-dependent emergence and progression of a tauopathy in transgenic mice overexpressing the shortest human tau isoform. Neuron. 1999;24:751–762. doi: 10.1016/s0896-6273(00)81127-7. [DOI] [PubMed] [Google Scholar]
- 60.Brion JP, Tremp G, Octave JN. Transgenic expression of the shortest human tau affects its compartmentalization and its phosphorylation as in the pretangle stage of Alzheimer’s disease. Am J Pathol. 1999;154:255–270. doi: 10.1016/S0002-9440(10)65272-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gotz J, Chen F, Barmettler R, Nitsch RM. Tau filament formation in transgenic mice expressing P301L tau. J Biol Chem. 2001;276:529–534. doi: 10.1074/jbc.M006531200. [DOI] [PubMed] [Google Scholar]
- 62.Gotz J, Probst A, Spillantini MG, Schafer T, Jakes R, Burki K, Goedert M. Somatodendritic localization and hyperphosphorylation of tau protein in transgenic mice expressing the longest human brain tau isoform. Embo J. 1995;14:1304–1313. doi: 10.1002/j.1460-2075.1995.tb07116.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tanemura K, Murayama M, Akagi T, Hashikawa T, Tominaga T, Ichikawa M, Yamaguchi H, Takashima A. Neurodegeneration with tau accumulation in a transgenic mouse expressing V337M human tau. J Neurosci. 2002;22:133–141. doi: 10.1523/JNEUROSCI.22-01-00133.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cruz JC, Tseng HC, Goldman JA, Shih H, Tsai LH. Aberrant Cdk5 activation by p25 triggers pathological events leading to neurodegeneration and neurofibrillary tangles. Neuron. 2003;40:471–483. doi: 10.1016/s0896-6273(03)00627-5. [DOI] [PubMed] [Google Scholar]
- 65.Khurana V, Lu Y, Steinhilb ML, Oldham S, Shulman JM, Feany MB. TOR-mediated cell-cycle activation causes neurodegeneration in a Drosophila tauopathy model. Curr Biol. 2006;16:230–241. doi: 10.1016/j.cub.2005.12.042. [DOI] [PubMed] [Google Scholar]
- 66.Raina AK, Zhu X, Rottkamp CA, Monteiro M, Takeda A, Smith MA. Cyclin’ toward dementia: cell cycle abnormalities and abortive oncogenesis in Alzheimer disease. J Neurosci Res. 2000;61:128–133. doi: 10.1002/1097-4547(20000715)61:2<128::AID-JNR2>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 67.Reynolds CH, Betts JC, Blackstock WP, Nebreda AR, Anderton BH. Phosphorylation sites on tau identified by nanoelectrospray mass spectrometry: differences in vitro between the mitogen-activated protein kinases ERK2, c-Jun N-terminal kinase and P38, and glycogen synthase kinase-3beta. J Neurochem. 2000;74:1587–1595. doi: 10.1046/j.1471-4159.2000.0741587.x. [DOI] [PubMed] [Google Scholar]
- 68.Lim J, Lu KP. Pinning down phosphorylated tau and tauopathies. Biochim Biophys Acta. 2005;1739:311–322. doi: 10.1016/j.bbadis.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 69.Hampel H, Buerger K, Kohnken R, Teipel SJ, Zinkowski R, Moeller HJ, Rapoport SI, Davies P. Tracking of Alzheimer’s disease progression with cerebrospinal fluid tau protein phosphorylated at threonine 231. Ann Neurol. 2001;49:545–546. [PubMed] [Google Scholar]
- 70.Kohnken R, Buerger K, Zinkowski R, Miller C, Kerkman D, DeBernardis J, Shen J, Moller HJ, Davies P, Hampel H. Detection of tau phosphorylated at threonine 231 in cerebrospinal fluid of Alzheimer’s disease patients. Neurosci Lett. 2000;287:187–190. doi: 10.1016/s0304-3940(00)01178-2. [DOI] [PubMed] [Google Scholar]
- 71.Lu PJ, Wulf G, Zhou XZ, Davies P, Lu KP. The prolyl isomerase Pin1 restores the function of Alzheimer-associated phosphorylated tau protein. Nature. 1999;399:784–788. doi: 10.1038/21650. [DOI] [PubMed] [Google Scholar]
- 72.Hamdane M, Dourlen P, Bretteville A, Sambo AV, Ferreira S, Ando K, Kerdraon O, Begard S, Geay L, Lippens G, Sergeant N, Delacourte A, Maurage CA, Galas MC, Buee L. Pin1 allows for differential Tau dephosphorylation in neuronal cells. Mol Cell Neurosci. 2006;32:155–160. doi: 10.1016/j.mcn.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 73.Allen B, Ingram E, Takao M, Smith MJ, Jakes R, Virdee K, Yoshida H, Holzer M, Craxton M, Emson PC, Atzori C, Migheli A, Crowther RA, Ghetti B, Spillantini MG, Goedert M. Abundant tau filaments and nonapoptotic neurodegeneration in transgenic mice expressing human P301S tau protein. J Neurosci. 2002;22:9340–9351. doi: 10.1523/JNEUROSCI.22-21-09340.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F, et al. Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature. 1995;373:523–527. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- 75.Gotz J, Chen F, van Dorpe J, Nitsch RM. Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science. 2001;293:1491–1495. doi: 10.1126/science.1062097. [DOI] [PubMed] [Google Scholar]
- 76.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 77.Lewis J, Dickson DW, Lin WL, Chisholm L, Corral A, Jones G, Yen SH, Sahara N, Skipper L, Yager D, Eckman C, Hardy J, Hutton M, McGowan E. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science. 2001;293:1487–1491. doi: 10.1126/science.1058189. [DOI] [PubMed] [Google Scholar]
- 78.Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 79.Nunan J, Small DH. Proteolytic processing of the amyloid-beta protein precursor of Alzheimer’s disease. Essays Biochem. 2002;38:37–49. doi: 10.1042/bse0380037. [DOI] [PubMed] [Google Scholar]
- 80.da Cruz e Silva EF, da Cruz e Silva OA. Protein phosphorylation and APP metabolism. Neurochem Res. 2003;28:1553–1561. doi: 10.1023/a:1025630627319. [DOI] [PubMed] [Google Scholar]
- 81.Pastorino L, Lu KP. Phosphorylation of the Amyloid Precursos Protein (APP): is this a mechanism in favor or agains Alzheimer’s disease? Neuroscience Research Communications. 2004;35:213–231. [Google Scholar]
- 82.Gandy S, Czernik AJ, Greengard P. Phosphorylation of Alzheimer disease amyloid precursor peptide by protein kinase C and Ca2+/calmodulin-dependent protein kinase II. Proc Natl Acad Sci U S A. 1988;85:6218–6221. doi: 10.1073/pnas.85.16.6218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Suzuki T, Oishi M, Marshak DR, Czernik AJ, Nairn AC, Greengard P. Cell cycle-dependent regulation of the phosphorylation and metabolism of the Alzheimer amyloid precursor protein. Embo J. 1994;13:1114–1122. doi: 10.1002/j.1460-2075.1994.tb06360.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tarr PE, Contursi C, Roncarati R, Noviello C, Ghersi E, Scheinfeld MH, Zambrano N, Russo T, D’Adamio L. Evidence for a role of the nerve growth factor receptor TrkA in tyrosine phosphorylation and processing of beta-APP. Biochem Biophys Res Commun. 2002;295:324–329. doi: 10.1016/s0006-291x(02)00678-2. [DOI] [PubMed] [Google Scholar]
- 85.Zambrano N, Bruni P, Minopoli G, Mosca R, Molino D, Russo C, Schettini G, Sudol M, Russo T. The beta-amyloid precursor protein APP is tyrosine-phosphorylated in cells expressing a constitutively active form of the Abl protoncogene. J Biol Chem. 2001;276:19787–19792. doi: 10.1074/jbc.M100792200. [DOI] [PubMed] [Google Scholar]
- 86.Liu F, Su Y, Li B, Zhou Y, Ryder J, Gonzalez-DeWhitt P, May PC, Ni B. Regulation of amyloid precursor protein (APP) phosphorylation and processing by p35/Cdk5 and p25/Cdk5. FEBS Lett. 2003;547:193–196. doi: 10.1016/s0014-5793(03)00714-2. [DOI] [PubMed] [Google Scholar]
- 87.Oishi M, Nairn AC, Czernik AJ, Lim GS, Isohara T, Gandy SE, Greengard P, Suzuki T. The cytoplasmic domain of Alzheimer’s amyloid precursor protein is phosphorylated at Thr654, Ser655, and Thr668 in adult rat brain and cultured cells. Mol Med. 1997;3:111–123. [PMC free article] [PubMed] [Google Scholar]
- 88.Iijima K, Ando K, Takeda S, Satoh Y, Seki T, Itohara S, Greengard P, Kirino Y, Nairn AC, Suzuki T. Neuron-specific phosphorylation of Alzheimer’s beta-amyloid precursor protein by cyclin-dependent kinase 5. J Neurochem. 2000;75:1085–1091. doi: 10.1046/j.1471-4159.2000.0751085.x. [DOI] [PubMed] [Google Scholar]
- 89.Aplin AE, Gibb GM, Jacobsen JS, Gallo JM, Anderton BH. In vitro phosphorylation of the cytoplasmic domain of the amyloid precursor protein by glycogen synthase kinase-3beta. J Neurochem. 1996;67:699–707. doi: 10.1046/j.1471-4159.1996.67020699.x. [DOI] [PubMed] [Google Scholar]
- 90.Ramelot TA, Nicholson LK. Phosphorylation-induced structural changes in the amyloid precursor protein cytoplasmic tail detected by NMR. J Mol Biol. 2001;307:871–884. doi: 10.1006/jmbi.2001.4535. [DOI] [PubMed] [Google Scholar]
- 91.Ando K, Iijima KI, Elliott JI, Kirino Y, Suzuki T. Phosphorylation-dependent regulation of the interaction of amyloid precursor protein with Fe65 affects the production of beta-amyloid. J Biol Chem. 2001;276:40353–40361. doi: 10.1074/jbc.M104059200. [DOI] [PubMed] [Google Scholar]
- 92.Rippmann JF, Hobbie S, Daiber C, Guilliard B, Bauer M, Birk J, Nar H, Garin-Chesa P, Rettig WJ, Schnapp A. Phosphorylation-dependent proline isomerization catalyzed by Pin1 is essential for tumor cell survival and entry into mitosis. Cell Growth Differ. 2000;11:409–416. [PubMed] [Google Scholar]
- 93.Lu PJ, Zhou XZ, Liou YC, Noel JP, Lu KP. Critical role of WW domain phosphorylation in regulating its phosphoserine-binding activity and the Pin1 function. J Biol Chem. 2002;277:2381–2384. doi: 10.1074/jbc.C100228200. [DOI] [PubMed] [Google Scholar]
- 94.Selkoe DJ, Yamazaki T, Citron M, Podlisny MB, Koo EH, Teplow DB, Haass C. The role of APP processing and trafficking pathways in the formation of amyloid beta-protein. Ann N Y Acad Sci. 1996;777:57–64. doi: 10.1111/j.1749-6632.1996.tb34401.x. [DOI] [PubMed] [Google Scholar]
- 95.De Strooper B, Annaert W. Proteolytic processing and cell biological functions of the amyloid precursor protein. J Cell Sci. 2000;113:1857–1870. doi: 10.1242/jcs.113.11.1857. [DOI] [PubMed] [Google Scholar]
- 96.Esler WP, Wolfe MS. A portrait of Alzheimer secretases--new features and familiar faces. Science. 2001;293:1449–1454. doi: 10.1126/science.1064638. [DOI] [PubMed] [Google Scholar]
- 97.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 98.Mattson MP. Pathways towards and away from Alzheimer’s disease. Nature. 2004;430:631–639. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, Bird TD, Hardy J, Hutton M, Kukull W, Larson E, Levy-Lahad E, Viitanen M, Peskind E, Poorkaj P, Schellenberg G, Tanzi R, Wasco W, Lannfelt L, Selkoe D, Younkin S. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat Med. 1996;2:864–870. doi: 10.1038/nm0896-864. [DOI] [PubMed] [Google Scholar]
- 100.Duff K, Eckman C, Zehr C, Yu X, Prada CM, Perez-tur J, Hutton M, Buee L, Harigaya Y, Yager D, Morgan D, Gordon MN, Holcomb L, Refolo L, Zenk B, Hardy J, Younkin S. Increased amyloid-beta42(43) in brains of mice expressing mutant presenilin 1. Nature. 1996;383:710–713. doi: 10.1038/383710a0. [DOI] [PubMed] [Google Scholar]
- 101.Borchelt DR, Thinakaran G, Eckman CB, Lee MK, Davenport F, Ratovitsky T, Prada CM, Kim G, Seekins S, Yager D, Slunt HH, Wang R, Seeger M, Levey AI, Gandy SE, Copeland NG, Jenkins NA, Price DL, Younkin SG, Sisodia SS. Familial Alzheimer’s disease-linked presenilin 1 variants elevate Abeta1-42/1–40 ratio in vitro and in vivo. Neuron. 1996;17:1005–1013. doi: 10.1016/s0896-6273(00)80230-5. [DOI] [PubMed] [Google Scholar]
- 102.Citron M, Westaway D, Xia W, Carlson G, Diehl T, Levesque G, Johnson-Wood K, Lee M, Seubert P, Davis A, Kholodenko D, Motter R, Sherrington R, Perry B, Yao H, Strome R, Lieberburg I, Rommens J, Kim S, Schenk D, Fraser P, St George Hyslop P, Selkoe DJ. Mutant presenilins of Alzheimer’s disease increase production of 42-residue amyloid beta-protein in both transfected cells and transgenic mice. Nat Med. 1997;3:67–72. doi: 10.1038/nm0197-67. [DOI] [PubMed] [Google Scholar]
- 103.Takahashi RH, Milner TA, Li F, Nam EE, Edgar MA, Yamaguchi H, Beal MF, Xu H, Greengard P, Gouras GK. Intraneuronal Alzheimer abeta42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am J Pathol. 2002;161:1869–1879. doi: 10.1016/s0002-9440(10)64463-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wijsman EM, Daw EW, Yu CE, Payami H, Steinbart EJ, Nochlin D, Conlon EM, Bird TD, Schellenberg GD. Evidence for a novel late-onset Alzheimer disease locus on chromosome 19p13.2. Am J Hum Genet. 2004;75:398–409. doi: 10.1086/423393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Segat L, Pontillo A, Annoni G, Trabattoni D, Vergani C, Clerici M, Arosio B, Crovella S. Pin1 promoter polymorphisms are associated with Alzheimer’s disease. Neurobiol Aging. 2005 Dec 26; doi: 10.1016/j.neurobiolaging.2005.11.009. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 106.Lambert JC, Bensemain F, Chapuis J, Cottel D, Amouyel P. Association study of the PIN1 gene with Alzheimer’s disease. Neurosci Lett. 2006;402:259–261. doi: 10.1016/j.neulet.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 107.Sultana R, Boyd-Kimball D, Poon HF, Cai J, Pierce WM, Klein JB, Markesbery WR, Zhou XZ, Lu KP, Butterfield DA. Oxidative modification and down-regulation of Pin1 in Alzheimer’s disease hippocampus: A redox proteomics analysis. Neurobiol Aging. 2006;27:918–925. doi: 10.1016/j.neurobiolaging.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 108.Butterfield DA, Poon HF, St Clair D, Keller JN, Pierce WM, Klein JB, Markesbery WR. Redox proteomics identification of oxidatively modified hippocampal proteins in mild cognitive impairment: insights into the development of Alzheimer’s disease. Neurobiol Dis. 2006;22:223–232. doi: 10.1016/j.nbd.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 109.Roe CM, Behrens MI, Xiong C, Miller JP, Morris JC. Alzheimer disease and cancer. Neurology. 2005;64:895–898. doi: 10.1212/01.WNL.0000152889.94785.51. [DOI] [PubMed] [Google Scholar]