

Summary
TNF receptor 1 (TNFR1) can trigger opposing responses within the same cell: one leads to a pro-survival response whereas the other leads to cell death [1, 2]. The pro-survival response is mediated primarily by activation of the NF-κB signaling pathway, which leads to the expression of pro-survival genes such as c-FLIP, c-IAPs, TRAFs and BCL-2 family members [3–9]. On the other hand, TNF can also trigger activation of CASPASE-8 and 3 leading eventually to apoptosis [1]. In most cell types, stimulation with TNF does not lead to apoptosis unless NF-κB signaling is blocked [5, 7–9]. Hence, NF-κB-mediated transcription of pro-survival genes acts as a cell death switch during TNF signaling. This current study demonstrates the existence of another cell death switch elsewhere in the pathway that is independent of NF-κB. Our results show that lysine 63-linked ubiquitination of RIP1 on lysine 377 inhibits TNF-induced apoptosis first through an NF-κB-independent mechanism, and subsequently, through an NF-κB-dependent mechanism. In contrast, in the absence of ubiquitination, RIP1 serves as a pro-apoptotic signaling molecule by engaging CASPASE-8. Therefore, RIP1 is a dual-function molecule that can be either pro-survival or pro-death depending on its ubiquitination state and this serves as an NF-κB-independent cell death switch early in TNF signaling. These results provide an explanation for the conflicting reports on the role of RIP1 in cell death, which was previously implicated to be both pro-survival and pro-death [10–12]. Since TRAF2 is the E3 ligase for RIP1 [13], these observations also provide an explanation for the NF-κB-independent anti-apoptotic function of TRAF2 previously described [14–16].
Results and discussion
TNFR1 ligation can induce the pro-survival NF-κB pathway or the cell death pathway. The molecular mechanism that determines which of these two opposing signaling pathways is triggered is unclear. Lysine 377 of RIP1 was recently shown to be an acceptor site for K63-linked polyubiquitination and this mediates efficient NF-κB activation by TNF ([17, 18] and Supplementary Figure 1). Whether ubiquitination of lysine 377 regulates the opposing cell death pathway has not been fully examined. To do so, TNF-induced apoptosis was quantified in RIP1-null Jurkat cells or RIP1-null cells reconstituted with RIP1-WT or RIP1-K377R. After 5 hours of TNF treatment, there was no significant difference in the amount of apoptosis of RIP1-null and RIP1-WT cells (Figure 1A & Supplementary Figure 1D). This comparison did not reveal a role for RIP1 in apoptosis. However, a comparison of the RIP1-K377R cells to the RIP1-WT showed a clear enhancement in sensitivity to TNF-mediated killing in RIP1-K377R cells suggesting that ubiquitination of RIP1 serves to inhibit apoptosis. The surprising observation was that RIP1-K377R cells are more sensitive to apoptosis even though they activate more NF-κB than RIP1-null cells (Supplementary Figure 1A & B). This suggests that ubiquitination of lysine 377 has an anti-apoptotic effect at this early time point that may be NF-κB-independent since there is no correlation between induction of NF-κB signaling and sensitivity to apoptosis. After 24 hours of TNF stimulation, the RIP1-K377R cells remained more susceptible to TNF-mediated killing when compared to RIP1-null cells. However, the RIP1-WT cells are now less susceptible than RIP1-null cells suggesting that upon prolonged stimulation, RIP1-WT provides an additional anti-apoptotic signal (Figure 1A). A time-course experiment showed that RIP1-K377R cells have a discernable enhancement in sensitivity to apoptosis compared to either RIP1-null or RIP1-WT cells by 4 hours, which persisted for the duration of the experiment (Figure 1B). The resistance of the RIP1-WT cells to apoptosis relative to RIP1-null cells was not apparent during the first 12 hours but is discernable after 24 hours. Thus, ubiquitination of RIP1 on lysine 377 appears to provide at least two distinct survival signals: one that is apparent after a short period of receptor ligation, and a second one that is evident only after prolonged stimulation.
Figure 1. Lysine 377 of RIP1 regulates cell death.
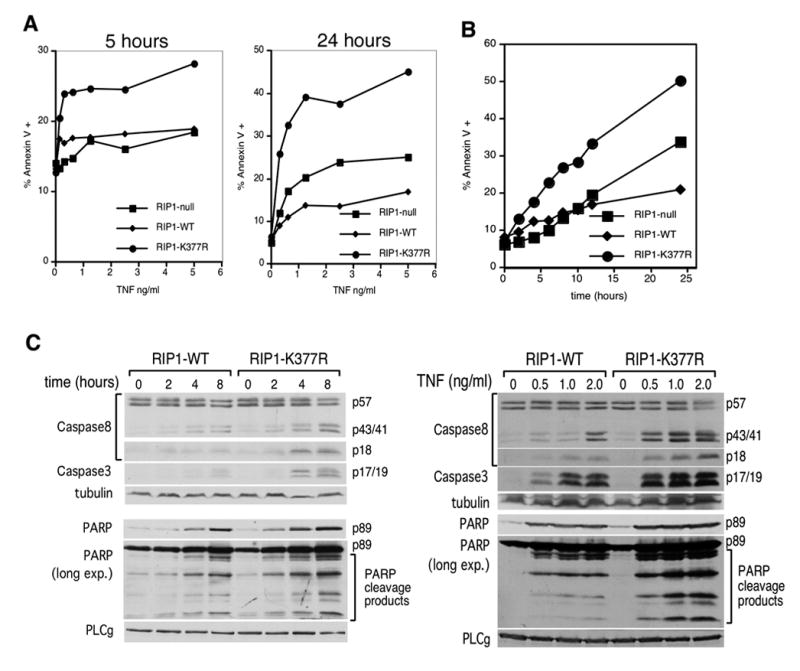
(A) RIP1-null cells and RIP1-null cells reconstituted with RIP1-WT or RIP1-K377R were stimulated with the indicated doses of TNF for 5 and 24 hours. Cells were stained with annexin V-PE and analyzed by flow cytometry. The percentage of total cells that were annexin V-reactive is shown. (B) The three cell lines in (A) were stimulated with 10 ng/ml TNF for the indicated times and annexin-V levels were analyzed. (C) RIP1-WT and RIP1-K377R cells were stimulated with 10 ng/ml TNF for different periods of time (left panel) or the indicated doses of TNF for 24 hours (right panel). Cell lysates were blotted with antibodies specific for CASPASE-8, cleaved CASPASE-3 or cleaved PARP. The anti-PARP blots were also overexposed to visualize additional PARP cleavage products. Re-blotting with anti-tubulin and PLCγ was also performed to demonstrate equivalent loading. These western blots are representative of at least three similar experiments.
TNF-mediated apoptosis requires CASPASE-8, which initiates cleavage and activation of the executioner caspase, CASPASE-3 to cleave substrates including poly (ADP Ribose) polymerase (PARP). RIP1-K377R cells exhibit faster CASPASE-8 processing to the active p18 fragment and enhanced CASPASE-3 and PARP cleavage compared to RIP1-WT cells (Figure 1C, left panel). Consistent with the time-course experiment, more CASPASE-8, -3 and PARP cleavage were observed in response to increasing concentrations of TNF in the RIP1-K377R cells (Figure 1C, right panel). Therefore, the enhanced apoptosis observed in RIP1-K377R cells compared to RIP1-WT cells (Figure 1A) is due to increased activation of caspases upon TNFR1 ligation.
We next hypothesized that NF-κB activation is not required for the early pro-survival signal mediated by RIP1’s ubiquitination but that the second pro-survival signal is dependent on NF-κB transcription factors. We tested these hypotheses by blocking NF-κB activation in RIP1-null, RIP1-WT and RIP1-K377R cells by stable transfection of the IκBαSR (Supplementary Figure 2). NF-κB blockade had no effect on the sensitivity to apoptosis of the three different cell lines after a short TNF stimulation of 5 hours; the RIP1-K377R cells remained more sensitive to apoptosis than the RIP1-null and RIP1-WT cells in the presence of the IκBαSR (Figure 2A, panels I & II & Supplementary Figure 2C). Therefore, we conclude that the enhanced sensitivity in the RIP1-K377R cells is not due to its reduced capacity to activate the NF-κB signaling pathway. Rather, this reflects the loss of an NF-κB-independent anti-apoptotic signal in the RIP1-K377R cells consistent with the hypothesis that ubiquitination of RIP1 provides an NF-κB-independent anti-apoptotic signal early in TNFR1 signaling. Next, we analyzed the effect of NF-κB blockade after a prolonged 24-hour stimulation with TNF. In the presence of the IκBαSR, both the RIP1-null and RIP1-WT cells were equally sensitive to TNF-mediated apoptosis (Figure 2A, panel IV & Supplementary Figure 2C) confirming that the second, delayed anti-apoptotic effect of RIP1-WT is dependent on NF-κB signaling. Moreover, the continued heightened sensitivity of the RIP1-K337R cells to apoptosis after extended TNF stimulation indicates the NF-κB independence of the early anti-apoptotic effect of RIP1 ubiquitination.
Figure 2. Lysine 377 of RIP1 provides an early NF-κB-independent and a late NF-κB-dependent anti-apoptotic signal.
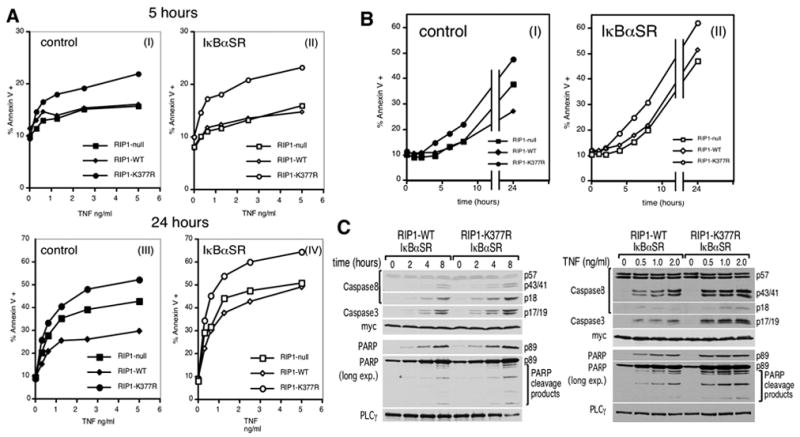
(A) RIP1-null, RIP1-WT or RIP1-K377R cells were transduced with retroviruses encoding either GST as a negative control (left panels) or IκBαSR (right panels). These cells were stimulated with the indicated doses of TNF for 5 hours (panels I & II) and 24 hours (panels III & IV). Apoptosis was analyzed by annexin V staining and flow cytometry as in Figure 1C. (B) The cell lines in (A) were stimulated with 10 ng/ml TNF and apoptosis analyzed at the indicated times. (C) RIP1-WT and RIP1-K377R cells stably transfected with the myc-tagged IκBαSR were stimulated with the indicated doses of TNF for 24 hours (left panel) or 10 ng/ml TNF for different periods of time (right panel). Cell lysates were blotted as in Figure 1C. The anti-myc blot serves as both a loading control and to show equivalent expression of the IκBαSR in the two cell lines. These western blots are representative of at least three similar experiments.
A time-course experiment was performed to determine the kinetic relationship of the two anti-apoptosis signals (Figure 2B). In both the control and the IκBαSR-transfected cells, we consistently observed enhanced apoptosis in the RIP1-K377R cells starting at 4 hours post-stimulation, indicating that the initial NF-κB-independent anti-apoptotic signal is having an effect by that time. Consistent with the time-course experiment in Figure 1B, the sensitivity of the RIP1-WT and RIP1-null cells only showed a difference after 24 hours (Figure 2B, panel I). This pro-survival effect of RIP1-WT was abrogated in the presence of IκBαSR (Figure 2B, panel II). Thus, the NF-κB-dependent anti-apoptotic signal generated by RIP1 comes into effect only after prolonged stimulation. This kinetic analysis confirms that ubiquitination of RIP1 on lysine 377 provides first an anti-death signal that is NF-κB-independent, which is then followed by one that is NF-κB-dependent. To gain insight into whether the NF-κB-independent anti-apoptotic signal inhibits caspases, we compared CASPASE-8, -3 and PARP cleavage in the RIP1-WT and RIP1-K377R cells transfected with the IκBαSR. The RIP1-K377R/IκBαSR cells showed enhanced cleavage in a time and dose-dependent manner (Figure 2C). Therefore, the NF-κB-independent, anti-apoptosis signal provided by ubiquitination of lysine 377 of RIP1 inhibits the activation of CASPASE-8.
Loss-of-function of TRAF2 (the E3 ligase for RIP1[13]), or its upstream E2 ubiquitin conjugating enzyme UBC13 [19] should result in the same phenotype as loss of the K377 ubiquitin acceptor site i.e., enhanced apoptosis in an NF-κB-independent manner. The IKKγ-deficient Jurkat T cell line 8321 which lacks NF-κB signaling [20], was transduced with a TRAF2 dominant negative (DN) or the UBC13DN. As predicted, the TRAF2DN and UBC13DN expressing cells are both more sensitive to apoptosis than control cells after either 5 or 24 hours of TNF treatment (Figure 3A). In agreement with this, more activation of the caspase cascade was observed in the TRAF2DN and UBC13DN cells compared to the negative control in a dose-dependent (Supplementary Figure 3A) and time-dependent manner (Figure 3B). Consistent with published studies [14–16], Traf2−/− MEFs also exhibited a similar NF-κB-independent enhancement in apoptosis compared to Traf2+/+ cells (Supplementary Figure 4). We conclude that K63-linked ubiquitination of RIP1 by UBC13 and TRAF2 provides an NF-κB-independent anti-apoptotic signal. Conversely, the loss of K63-linked ubiquitination, either through blocking the E2 and E3 ligase, or through the loss of the ubiquitin acceptor site on RIP1, generates a pro-death signal.
Figure 3. UBC13 and TRAF2 have a NF-κB-independent anti-apoptotic function.
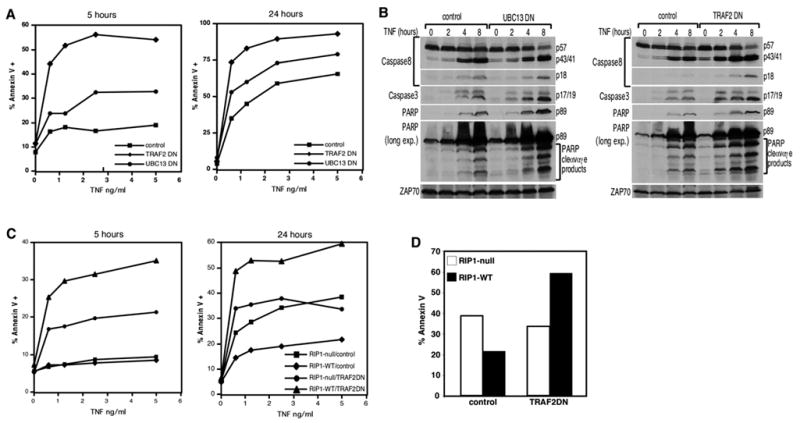
(A) IKKγ/NEMO-deficient Jurkat T cells stably expressing the control protein GST, UBC13DN or TRAF2DN were treated with indicated concentrations of TNF for 5 and 24 hours. Apoptosis was analyzed by annexin V staining. (B) The cell lines in (A) were treated with 2 ng/ml of TNF for the indicated times. 150 ug of cell extracts were analyzed by western blot analysis as in Figure 1C. Anti-ZAP70 blotting was performed to confirm equivalent loading. (C) RIP1-null or RIP1-WT cells were transduced with control GST or TRAF2DN-encoding retroviruses. Stably selected cells were treated with indicated concentrations of TNF for 5 or 24 hours and stained with annexin V-PE. (D) The data from (C) where the cells are stimulated with 5 ng/ml TNF for 24 hours are plotted in a bar graph to highlight the dual functional role of RIP1.
The observations up to this point suggest that TRAF2 loss-of-function enhances apoptosis because RIP1 becomes a death-signaling molecule in the absence of ubiquitination. If so, TRAF2 loss-of-function should have less of an effect in RIP1-null cells relative to RIP1-sufficient cells. We expressed the TRAF2DN in both the RIP1-null and RIP1-WT cell lines. After either 5 or 24 hours of stimulation, the RIP1-null/TRAF2DN cells clearly displayed lower sensitivity to apoptosis than the RIP1-WT/TRAF2DN cells (Figure 3C). Therefore, TRAF2 loss-of-function potentiates cell death in a RIP1-dependent manner. The 24-hour experiment highlights further the dual functionality of RIP1. In control-transfected cells, the presence of RIP1 confers protection against apoptosis whereas in TRAF2DN-transfected cells, the presence of RIP1 confers sensitivity to apoptosis (Figure 3D).
Lastly, we hypothesized that ubiquitination of RIP1 or the lack thereof may regulate its interaction with CASPASE-8 based on a report showing a direct interaction between the PRO domain of CASPASE-8 with RIP1 [21]. Furthermore, CASPASE-8 can cleave RIP1 which is also suggestive of a direct interaction [22–24]. Co-immunoprecipitation studies were carried out to test this hypothesis. In RIP1-WT cells, a low level of RIP1-PROCASPASE-8 interaction was observed and this was not altered upon TNF stimulation. In contrast, there is a stimulus-dependent association of PROCASPASE-8, as well as processed CASPASE-8, with RIP1 in the RIP1-K377R cells (Figure 4A). Therefore, ubiquitination of RIP1 prevents RIP1 from forming a complex with CASPASE-8. Conversely, in the absence of ubiquitination, RIP1 is able to form a complex with CASPASE-8 to initiate apoptosis.
Figure 4. Ubiquitination of RIP1 regulates association with CASPASE-8.
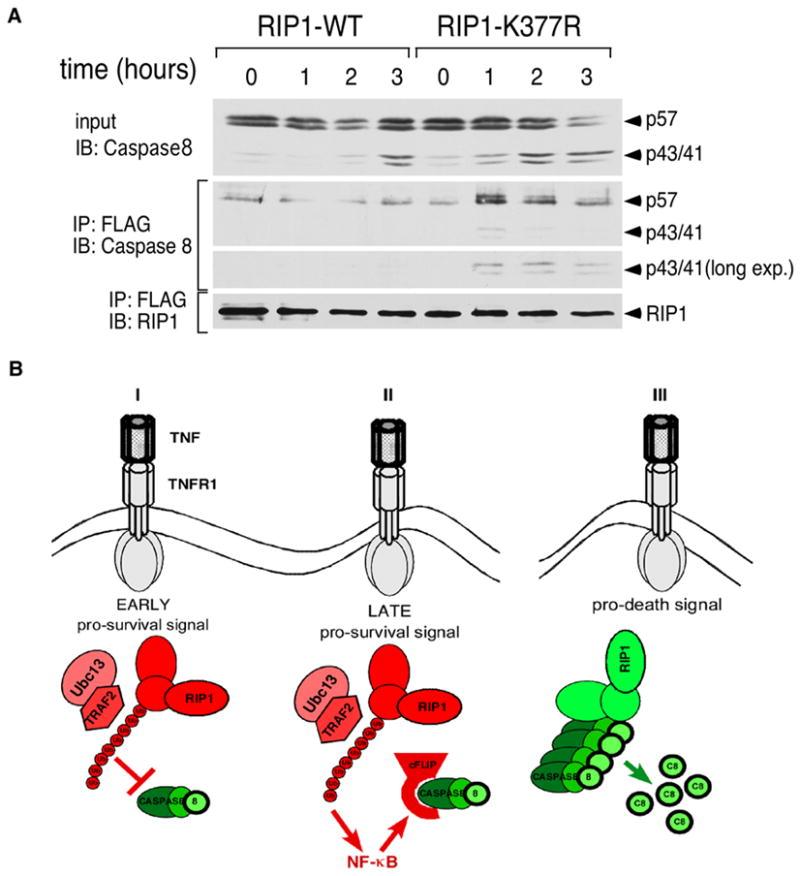
(A) RIP1-WT and RIP1-K377R cells were stimulated with 10 ng/ml TNF for the indicated times. FLAG-tagged RIP1 and associated proteins were immunoprecipitated with anti-FLAG beads and eluted with the FLAG peptide. The immune complexes were blotted sequentially with anti-CASPASE-8 and anti-RIP1 as a loading control. The anti-CASPASE-8 blot was also overexposed to visualize the p43/p41 cleaved form. (B) A model for the role of RIP1 in TNF-mediated apoptosis. (I) In the early phase of TNFR1 signaling, TRAF2-mediated ubiquitination of lysine 377 functions as a brake to prevent RIP1 from associating with CASPASE-8. This does not require NF-κB-mediated gene transcription. (II) In the late phase of TNFR1 signaling, a second pro-survival signal provided by the ubiquitination of lysine 377 becomes effective via the up-regulation of NF-κB-dependent anti-apoptotic genes such as c-FLIP. (III) In the absence of K63-linked poly-ubiquitination, RIP1 becomes a death-signaling molecule by forming a complex with CASPASE-8 to initiate apoptosis. Red indicates anti-apoptotic molecules and green indicates pro-apoptotic molecules.
The observations presented here indicate that RIP1 is a dual-functional signaling molecule that is capable of either pro-survival or pro-death signaling depending on its ubiquitination state. We propose the model that RIP1, ubiquitinated by TRAF2 on lysine 377, provides two distinct anti-apoptotic signals (Figure 4B). The first anti-apoptotic signal occurs early after receptor ligation to prevent RIP1 from complexing with CASPASE-8 and is not dependent on the NF-κB pathway. The second anti-apoptotic signal emerges at a later time point and it is dependent on NF-κB-mediated up-regulation of anti-apoptotic genes such as c-FLIP. In the absence of ubiquitination, RIP1 converts to a death-signaling molecule that actively engages CASPASE-8 independently of its effect on NF-κB signaling. Since ubiquitination of RIP1 occurs prior to the induction of NF-κB-mediated gene transcription and can regulate cell death in an NF-κB-independent manner, we propose that the ubiquitination status of lysine 377 is a cell death decision switch early in TNF signaling.
The role of RIP1 in TNF-mediated apoptosis has been controversial with reports of either pro-survival or pro-death effects [10–12]. Our observation that RIP1 can generate either pro-survival or pro-death signals depending on the ubiquitination state of lysine 377 provides an explanation for these conflicting reports. It is also well appreciated that TRAF2 has an NF-κB-independent anti-apoptotic function [14–16, 25] but the underlying mechanism has been unclear. Our results now suggest that the anti-apoptotic function of TRAF2 is due in part to its ubiquitination of RIP1, which acts as a brake on the pro-death effect of RIP1. The results described here raise the possibility that altering the ubiquitination state of RIP1 will influence cell survival, which can be achieved physiologically by altering the level of TRAF2 in the cell. In this regard, stimulation of TNFR2 is known to lead to TRAF2 degradation [12, 26, 27] and to potentiate TNFR1-mediated cell death in a RIP1-dependent manner [12, 28]. It is likely in cells that express both TNFR1 and TNFR2, TRAF2 degradation triggered by TNFR2 creates a situation akin to the RIP1-K377R mutation, which then enables TNFR1 to engage the apoptotic cascade more efficiently via non-ubiquitinated RIP1.
The observations in this study raise additional questions regarding the regulation of the RIP1-dependent apoptotic pathway. For instance, since only a fraction of the total pool of RIP1 in a wildtype cell is ubiquitinated in response to TNF, why isn’t the non-ubiquitinated pool of RIP1 interacting with CASPASE-8. Since these death signaling molecules are constitutively expressed in cells, it is in the interest of the cell to prevent these interactions from occurring in unstimulated cells. These preventive mechanisms are poorly understood and could involve differential subcellular localization of the molecules, the requirement for post-translational modifications or conformational changes for the interactions to occur, or the presence of inhibitors bound to RIP1 and CASPASE-8. In the case of RIP1, it is likely that only a fraction of the total RIP1 is released from inhibition to become active whereas the rest of the pool remains in an inactive state and does not interact with CASPASE-8. It has been shown that RIP1 is recruited to TNFR1 in both Traf2+/+ and Traf2−/− cells [29] suggesting that ubiquitination is not required for this process. The receptor-associated RIP1 may therefore constitute the active pool of RIP1. The observation that RIP1-WT fails to form a complex with CASPASE-8 whereas RIP1-K377R is able to do so also raises the question as to how the ubiquitination status of RIP1 determines its association with CASPASE-8. One possibility is that RIP1 molecules recruited to the receptor are retained at the plasma membrane if they undergo ubiquitination via ubiquitin-dependent binding to molecules such as TAB2/3 and IKKγ [17, 18, 30]. In the absence of ubiquitination, RIP1 molecules recruited to the receptor are not retained at the plasma membrane and may now translocate to the cytosolic compartment where CASPASE-8 is located. Alternatively, ubiquitination of RIP1 may block its interaction with CASPASE-8 via steric hindrance, either by the poly-ubiquitin chains themselves or because binding of IKKγ and TAB2/3 to the poly-ubiquitin chains occludes CASPASE-8 from binding. These possibilities are currently being investigated.
While the initial anti-apoptotic signal provided by ubiquitination of RIP1 does not require NF-κB signaling, it is likely that NF-κB-mediated gene transcription can modulate this ubiquitination event. For instance, several ubiquitin-modifying proteins are induced by NF-κB, including TRAF2, c-IAPs, A20 and CYLD [3, 31, 32]. Some of these molecules are known to interact with RIP1 and therefore, the net amount of ubiquitination on lysine 377 is likely to be dependent on the levels and activity of these molecules. As we have shown in this report, the level of ubiquitination on this critical lysine residue in turn determines whether the net outcome is survival or death.
Supplementary Material
Acknowledgments
We are very grateful to Drs. Zhijian Chen (UT Southwestern), Paul Leibson (Mayo Clinic) and Brian Seed (Massachusetts General Hospital) for providing critical reagents. We would also like to thank Dr. Michelle Kelliher and Yibin Yang (UMASS Worchester) for sharing data, and Drs. Ben Chen, Jay Unkeless and Ramnik Xavier for critical reading of the manuscript. This work was supported by National Institute of Health grants R21-57997 and R01-AI52417 (A.T.T.), and was aided by a grant from the New York Chapter of the Arthritis Foundation (A.T.T.). M.A.O’D. is a recipient of a Research Fellowship Award from the Crohn’s and Colitis Foundation of America. W.C.Y. is an employee of Amgen Inc.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003;114:181–190. doi: 10.1016/s0092-8674(03)00521-x. [DOI] [PubMed] [Google Scholar]
- 2.Aggarwal BB. Signalling pathways of the TNF superfamily: a double-edged sword. Nat Rev Immunol. 2003;3:745–756. doi: 10.1038/nri1184. [DOI] [PubMed] [Google Scholar]
- 3.Wang CY, Mayo MW, Korneluk RG, Goeddel DV, Baldwin AS., Jr NF-kappaB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science. 1998;281:1680–1683. doi: 10.1126/science.281.5383.1680. [DOI] [PubMed] [Google Scholar]
- 4.Wang CY, Mayo MW, Baldwin AS., Jr TNF- and cancer therapy-induced apoptosis: potentiation by inhibition of NF-kappaB. Science. 1996;274:784–787. doi: 10.1126/science.274.5288.784. [DOI] [PubMed] [Google Scholar]
- 5.Van Antwerp DJ, Martin SJ, Kafri T, Green DR, Verma IM. Suppression of TNF-alpha-induced apoptosis by NF-kappaB. Science. 1996;274:787–789. doi: 10.1126/science.274.5288.787. [DOI] [PubMed] [Google Scholar]
- 6.Micheau O, Lens S, Gaide O, Alevizopoulos K, Tschopp J. NF-kappaB signals induce the expression of c-FLIP. Mol Cell Biol. 2001;21:5299–5305. doi: 10.1128/MCB.21.16.5299-5305.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kreuz S, Siegmund D, Scheurich P, Wajant H. NF-kappaB inducers upregulate cFLIP, a cycloheximide-sensitive inhibitor of death receptor signaling. Mol Cell Biol. 2001;21:3964–3973. doi: 10.1128/MCB.21.12.3964-3973.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Doi TS, Marino MW, Takahashi T, Yoshida T, Sakakura T, Old LJ, Obata Y. Absence of tumor necrosis factor rescues RelA-deficient mice from embryonic lethality. Proc Natl Acad Sci U S A. 1999;96:2994–2999. doi: 10.1073/pnas.96.6.2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beg AA, Baltimore D. An essential role for NF-kappaB in preventing TNF-alpha-induced cell death. Science. 1996;274:782–784. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- 10.Kelliher MA, Grimm S, Ishida Y, Kuo F, Stanger BZ, Leder P. The death domain kinase RIP mediates the TNF-induced NF-kappaB signal. Immunity. 1998;8:297–303. doi: 10.1016/s1074-7613(00)80535-x. [DOI] [PubMed] [Google Scholar]
- 11.Stanger BZ, Leder P, Lee TH, Kim E, Seed B. RIP: a novel protein containing a death domain that interacts with Fas/APO-1 (CD95) in yeast and causes cell death. Cell. 1995;81:513–523. doi: 10.1016/0092-8674(95)90072-1. [DOI] [PubMed] [Google Scholar]
- 12.Pimentel-Muinos FX, Seed B. Regulated commitment of TNF receptor signaling: a molecular switch for death or activation. Immunity. 1999;11:783–793. doi: 10.1016/s1074-7613(00)80152-1. [DOI] [PubMed] [Google Scholar]
- 13.Lee TH, Shank J, Cusson N, Kelliher MA. The kinase activity of Rip1 is not required for tumor necrosis factor-alpha-induced IkappaB kinase or p38 MAP kinase activation or for the ubiquitination of Rip1 by Traf2. J Biol Chem. 2004;279:33185–33191. doi: 10.1074/jbc.M404206200. [DOI] [PubMed] [Google Scholar]
- 14.Yeh WC, Shahinian A, Speiser D, Kraunus J, Billia F, Wakeham A, de la Pompa JL, Ferrick D, Hum B, Iscove N, Ohashi P, Rothe M, Goeddel DV, Mak TW. Early lethality, functional NF-kappaB activation, and increased sensitivity to TNF-induced cell death in TRAF2-deficient mice. Immunity. 1997;7:715–725. doi: 10.1016/s1074-7613(00)80391-x. [DOI] [PubMed] [Google Scholar]
- 15.Natoli G, Costanzo A, Guido F, Moretti F, Bernardo A, Burgio VL, Agresti C, Levrero M. Nuclear factor kB-independent cytoprotective pathways originating at tumor necrosis factor receptor-associated factor 2. J Biol Chem. 1998;273:31262–31272. doi: 10.1074/jbc.273.47.31262. [DOI] [PubMed] [Google Scholar]
- 16.Lee SY, Kaufman DR, Mora AL, Santana A, Boothby M, Choi Y. Stimulus-dependent synergism of the antiapoptotic tumor necrosis factor receptor-associated factor 2 (TRAF2) and nuclear factor kappaB pathways. J Exp Med. 1998;188:1381–1384. doi: 10.1084/jem.188.7.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ea CK, Deng L, Xia ZP, Pineda G, Chen ZJ. Activation of IKK by TNFalpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol Cell. 2006;22:245–257. doi: 10.1016/j.molcel.2006.03.026. [DOI] [PubMed] [Google Scholar]
- 18.Li H, Kobayashi M, Blonska M, You Y, Lin X. Ubiquitination of RIP Is Required for Tumor Necrosis Factor {alpha}-induced NF-{kappa}B Activation. J Biol Chem. 2006;281:13636–13643. doi: 10.1074/jbc.M600620200. [DOI] [PubMed] [Google Scholar]
- 19.Deng L, Wang C, Spencer E, Yang L, Braun A, You J, Slaughter C, Pickart C, Chen ZJ. Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell. 2000;103:351–361. doi: 10.1016/s0092-8674(00)00126-4. [DOI] [PubMed] [Google Scholar]
- 20.He KL, Ting AT. Essential role for IKKgamma/NEMO in TCR-induced IL-2 expression in Jurkat T cells. Eur J Immunol. 2003;33:1917–1924. doi: 10.1002/eji.200323650. [DOI] [PubMed] [Google Scholar]
- 21.Shikama Y, Yamada M, Miyashita T. Caspase-8 and caspase-10 activate NF-kappaB through RIP, NIK and IKKalpha kinases. Eur J Immunol. 2003;33:1998–2006. doi: 10.1002/eji.200324013. [DOI] [PubMed] [Google Scholar]
- 22.Lin Y, Devin A, Rodriguez Y, Liu ZG. Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes Dev. 1999;13:2514–2526. doi: 10.1101/gad.13.19.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martinon F, Holler N, Richard C, Tschopp J. Activation of a pro-apoptotic amplification loop through inhibition of NF-kappaB-dependent survival signals by caspase-mediated inactivation of RIP. FEBS Lett. 2000;468:134–136. doi: 10.1016/s0014-5793(00)01212-6. [DOI] [PubMed] [Google Scholar]
- 24.Chaudhary PM, Eby MT, Jasmin A, Kumar A, Liu L, Hood L. Activation of the NF-kappaB pathway by caspase 8 and its homologs. Oncogene. 2000;19:4451–4460. doi: 10.1038/sj.onc.1203812. [DOI] [PubMed] [Google Scholar]
- 25.Tada K, Okazaki T, Sakon S, Kobarai T, Kurosawa K, Yamaoka S, Hashimoto H, Mak TW, Yagita H, Okumura K, Yeh WC, Nakano H. Critical roles of TRAF2 and TRAF5 in tumor necrosis factor-induced NF-kappa B activation and protection from cell death. J Biol Chem. 2001;276:36530–36534. doi: 10.1074/jbc.M104837200. [DOI] [PubMed] [Google Scholar]
- 26.Fotin-Mleczek M, Henkler F, Samel D, Reichwein M, Hausser A, Parmryd I, Scheurich P, Schmid JA, Wajant H. Apoptotic crosstalk of TNF receptors: TNF-R2-induces depletion of TRAF2 and IAP proteins and accelerates TNF-R1-dependent activation of caspase-8. J Cell Sci. 2002;115:2757–2770. doi: 10.1242/jcs.115.13.2757. [DOI] [PubMed] [Google Scholar]
- 27.Li X, Yang Y, Ashwell JD. TNF-RII and c-IAP1 mediate ubiquitination and degradation of TRAF2. Nature. 2002;416:345–347. doi: 10.1038/416345a. [DOI] [PubMed] [Google Scholar]
- 28.Chan FK, Shisler J, Bixby JG, Felices M, Zheng L, Appel M, Orenstein J, Moss B, Lenardo MJ. A role for tumor necrosis factor receptor-2 and receptor-interacting protein in programmed necrosis and antiviral responses. J Biol Chem. 2003;278:51613–51621. doi: 10.1074/jbc.M305633200. [DOI] [PubMed] [Google Scholar]
- 29.Devin A, Cook A, Lin Y, Rodriguez Y, Kelliher M, Liu Z. The distinct roles of TRAF2 and RIP in IKK activation by TNF-R1: TRAF2 recruits IKK to TNF-R1 while RIP mediates IKK activation. Immunity. 2000;12:419–429. doi: 10.1016/s1074-7613(00)80194-6. [DOI] [PubMed] [Google Scholar]
- 30.Wu CJ, Conze DB, Li T, Srinivasula SM, Ashwell JD. Sensing of Lys 63-linked polyubiquitination by NEMO is a key event in NF-kappaB activation [corrected] Nat Cell Biol. 2006;8:398–406. doi: 10.1038/ncb1384. [DOI] [PubMed] [Google Scholar]
- 31.Jono H, Lim JH, Chen LF, Xu H, Trompouki E, Pan ZK, Mosialos G, Li JD. NF-kappaB is essential for induction of CYLD, the negative regulator of NF-kappaB: evidence for a novel inducible autoregulatory feedback pathway. J Biol Chem. 2004;279:36171–36174. doi: 10.1074/jbc.M406638200. [DOI] [PubMed] [Google Scholar]
- 32.Krikos A, Laherty CD, Dixit VM. Transcriptional activation of the tumor necrosis factor alpha-inducible zinc finger protein, A20, is mediated by kappa B elements. J Biol Chem. 1992;267:17971–17976. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.