

Abstract
The basis for mutations at A:T base pairs in immunoglobulin hypermutation and defining how AID interacts with the DNA of the immunoglobulin locus are major aspects of the immunoglobulin mutator mechanism where questions remain unanswered. Here, we examined the pattern of mutations generated in mice deficient in various DNA repair proteins implicated in A:T mutation and found a previously unappreciated bias at G:C base pairs in spectra from mice simultaneously deficient in DNA mismatch repair and uracil DNA glycosylase. This suggests a strand-biased DNA transaction for AID delivery which is then masked by the mechanism that introduces A:T mutations. Additionally, we asked if any of the known components of the A:T mutation machinery underscore the basis for the paucity of A:T mutations in the Burkitt lymphoma cell lines, Ramos and BL2. Ramos and BL2 cells were proficient in MSH2/MSH6-mediated mismatch repair, and express high levels of wild-type, full-length DNA polymerase η. In addition, Ramos cells have high levels of uracil DNA glycosylase protein and are proficient in base excision repair. These results suggest that Burkitt lymphoma cell lines may be deficient in an unidentified factor that recruits the machinery necessary for A:T mutation or that AID-mediated cytosine deamination in these cells may be processed by conventional base excision repair truncating somatic hypermutation at the G:C phase. Either scenario suggests that cytosine deamination by AID is not enough to trigger A:T mutation, and that additional unidentified factors are required for full spectrum hypermutation in vivo.
Keywords: somatic hypermutation, AID, immunoglobulin, class switch recombination, mismatch repair, B cells, Burkitt lymphoma, translesion synthesis
Introduction
Somatic hypermutation (SHM) of immunoglobulin (Ig) genes is triggered by the Activation Induced Deaminase (AID) -mediated deamination of cytosines in the DNA encoding the variable (V) regions of Ig genes ( Petersen-Mahrt et al., 2002, Rada et al., 2002, Muramatsu et al., 2000, Revy et al., 2000). The resulting G:U mismatch is an intermediate which can be replicated to yield C →T and G → A transitions, or whose U is removed by uracil DNA glycosylase (UNG) to generate a mutagenic abasic site (Neuberger et al., 2003, Storb and Stavnezer 2002). In a SHM-specific manner, it appears that the G:U pair may be recognized by the MSH2-MSH6 heterodimer (Wilson et al., 2005, Bardwell et al., 2004, Martin et al., 2003, Wiesendanger et al., 2000, Phung et al., 1999, Jacobs et al., 1998, Rada et al., 1998, Shen et al., 2006), initiating an error-prone mismatch repair (MMR) reaction that involves re-synthesis by translesion synthesis (TLS) DNA polymerases such as DNA polymerase η, ζ, θ, and ι (Diaz and Lawrence 2005, Zan et al., 2005, Delbos et al., 2005, Mayorov et al., 2005, Faili et al., 2002a, Diaz et al., 2001, Rogozin et al., 2001, Zan et al., 2001, Zeng et al., 2001), however, this model remains speculative. Therefore, while it is likely that AID-mediated deamination of cytosines in Ig DNA is the trigger to SHM, other aspects of the reaction that remain poorly understood include: 1) How is AID specifically targeted to the Ig locus?, 2) In what context does AID interact with the DNA of Ig regions (transcription, repair, etc.)? 3) How does cytosine deamination lead to mutations at A:T base pairs, especially when uracil in the DNA is not typically mutagenic to adjacent bases? (Diaz and Lawrence 2005).
AID deaminates single-stranded DNA but not double-stranded DNA in vitro ( Yu et al., 2004, Chaudhuri et al., 2004, Chaudhuri et al., 2003, Dickerson et al., 2003, Pham et al., 2003, Bransteitter et al., 2003, Sohail et al., 2003). Recent work by Alt and colleagues revealed that AID interacts with replication protein A (RPA) and that this interaction enhances the deamination reaction (Chaudhuri et al., 2004). Work by Goodman and colleagues demonstrated that AID can act processively in vitro (Bransteitter et al., 2004), a finding that may be consistent with a process involving elongation, such as transcription. The pattern of mutations generated by SHM revealed hotspots of hypermutation (DGYW) that are AID-mediated, strand bias in mutations at A:T base pairs but not at G:C base pairs, and a predominance of base substitutions (Larijani et al., 2005, Beale et al., 2004, Rogozin and Diaz 2004, Yu et al., 2004, Bransteitter et al., 2003, Foster et al., 1999, Diaz and Flajnik 1998, Dorner et al 1998, Rogozin and Kolchanov 1992). While these characteristics are clues to the interaction between AID and the TLS polymerases with IgV regions, there is significant controversy regarding the mechanism that delivers AID to the Ig V regions. Considerable evidence suggests that AID may interact directly with the transcriptional machinery (Delpy et al., 2004, Shen and Storb 2004, Nambu et al., 2003, Ramiro et al., 2003, Yoshikawa et al., 2002, Bachl et al., 2001, Fukita et al., 1998, Rada et al., 1997, Tumas-Brundage and Manser 1997, Peters and Storb 1996). However, a transcription-based model has been difficult to reconcile with the paucity of strand bias at G:C base pairs in SHM. Here, we examined strand bias at G:C base pairs in a variety of contexts such as the Ig locus of DNA repair-deficient mice. We reasoned that it is possible that AID is delivered to the DNA of the Ig V regions in a strand-biased manner, such as through a transcription-mediated process, but that this may be masked by the actions of subsequent proteins. We demonstrate that the Ig V regions from mice simultaneously deficient in both MSH2 and UNG (Rada et al., 2004) display significant strand bias in mutations at G:C base pairs.
Hypermutation in mammalian Ig and in the nurse shark antigen receptor, NAR, is proportionally distributed among G:C and A:T base pairs (Diaz et al., 1999, Rogozin and Diaz 2004). However, in some species such as the frog and in bona-fide IgM in the shark, there is a strong bias towards mutations at G:C base pairs ( Wilson et al., 1995, Hinds-Frey et al., 1993). This G:C bias is also seen in Burkitt lymphoma cell lines such as Ramos that undergo SHM constitutively (Harris et al., 2001) or BL2 that can be induced to hypermutate (Denepoux et al., 1997, Poltoratsky et al., 2001) . Indeed, there is seldom an excess of mutations at A:T base pairs; if there is a bias it favors G:C base pairs, probably a result of the requirement for AID-mediated deamination of cytosine to initiate SHM. We examined the possibility that the paucity of mutations at A:T base pairs in cell lines may be due to defects in known components of the A:T mutational machinery such as the MMR proteins, base excision repair (BER) or DNA polymerase η. We found that Ramos is proficient at BER and MSH2-MSH6-mediated MMR, and that DNA polymerase η sequence and expression in Ramos cells is intact. In addition, MMR and DNA polymerase η expression and sequence are intact in BL2 cells. These results suggest the existence of unknown factors that contribute to A:T hypermutation of Ig genes.
Material and Methods
Cell lines
The human Burkitt lymphoma cell line, Ramos, and a human MMR-proficient lymphoblastic cell line, TK6 (Wei et al., 2003) were obtained from ATCC, while BL2 cells were obtained from Matthew Scharff and Claude-Agnes Reynaud. A cell line representing a late stage of B cell development, OCYLy8c3 (Stiernholm and Berinstein, 1994) was a gift of Laurent Verkoczy. Cells were grown in RPMI 1640 (GiBCO) medium supplemented with 10% fetal calf serum (FCS), penicillin and streptomycin (1:100 dilution) (RPMI 1640 feeding medium) at 37°C. The mouse embryonic stem (MES) cell line that is deficient in MMR was grown on gelatinized plates in DMEM containing 10% fetal calf serum, penicillin, and streptomycin, β-mercaptoethanol, 1000 units/ml leukemia inhibitory factor (ESGRO from Chemicon), and supplemented with nucleosides and nonessential amino acids. BL2 cells were induced to undergo hypermutation as described previously (Faili et al., 2002b). Briefly, 2 million cells were incubated in 500 μl of RPMI medium containing 4 μl of biotinylated anti-human IgM (Caltag laboratories, CA), 40 μl of fluorescein isothiocyanate-anti-CD19 (Immunotech, France) and 40 μl of of phycoerythrin-anti-CD21 (BD Pharmingen, CA) for 20 minutes at 4°C, excess antibodies were removed by washing once, and the cells were resuspended in RPMI containing streptavidin-conjugated magnetic beads (Dynabeads M280, Dynal, Norway) and incubated at 4°C for 20 min. Complete RPMI medium (10 ml) was added to the activated B cells, followed by incubation at 37°C for 90 min.
RT-PCR, cloning and sequencing of the Ramos and BL2 Vh and of DNA polymerase η coding regions
RNA was isolated by using TRIZOL reagent (Invitrogen) and first-strand cDNA was synthesized using SuperScript TM First-Strand Synthesis system (Invitrogen) with the oligo (dT) primer following the manufacturer’s protocol. The rearranged Ig Vh from Ramos and BL2 was amplified by PCR from first strand cDNA using Pfu Turbo DNA polymerase (Stratagene) as follows: 94°C for 2min, then 35 cycles of: 94°C for 30sec, 52°C for 30sec, and 72°C for 45 sec. A final extension at 72°C was done for 7 min. Primers for Ramos used were: Vh1F:5′-GGGCGCAGGACTGTTGAAGCC-3′ andVh1R 5′-GTGGTCCCTTGGCCCCAGACG-3′ based on those designed by Papavasiliou and Schatz (Papavasiliou and Schatz, 2000). The primers for BL2 were: Vh4-n: 5′-GAGGCTGCCTCTGATCCCAG-3′, and Jh5-n: 5′-CCTGGCAAGCTGAGTCTCCC-3′. Full length DNA Polymerase η cDNA was amplified by using Accuprimer™ Pfx DNA polymerase (Invitrogen) with the primers: Etaf- 5′-ATGGCTACTGGACAGGATCGAGTGGTTGCTC-3′, and EtaR- 5′-CTAATGTGTTAATGGCTTAAAAAATGATTCC-3′. All PCR products were cloned into Topo PCR cloning vector (Invitrogen), and sequenced with T7 primer (Invitrogen) as well as primers designed for sequencing the entire coding region of DNA Polymerase η as follows: S5 5′-AGCCAGTGTTGAAGTGATGG-3′,S8 5′-TTCACACAATAAGGTCCTGGC-3′, S13 5′-AGCTGGTTGTGAGCATTCG-3′, S17 5′-CCATGAGCAATTCACCATCC-3′, S21 5′-GGATATGCCAGAACACATGG-3′. The amino acid sequence of DNA polymerase η deduced from mRNA extracted from Ramos and BL2 cells was aligned with the known human DNA polymerase η sequence using the CLUSTALW web interface at the European Bioinformatics Institute website (http://www.ebi.ac.uk/clustalw/#).
Analysis of DNA polymerase η splice variants
Primers were specifically designed spanning exon II of polymerase η to identify alternative splicing site as follows: SPLF- 5′-GCCAGGTGTTTGTTACCTTGA-3′ SPLR- 5′-GCACGTTCAATCACAGCAAAAC-3′. PCR was conducted with REDTaq TM ReadyMix PCR reaction with MgCl2 (Sigma).
Preparation of cell extracts
Extracts for base excision repair assays were prepared as previously described (Prasad et al., 2000). Briefly, Ramos cells (5 X 106) were suspended in 100 μl Buffer I (10 mM Tris–HCl, pH 7.8, 200 mM KCl), and an equal volume of Buffer II (10 mM Tris–HCl, pH 7.8, 200 mM KCl, 2 mM EDTA, 40% glycerol, 0.2% NP-40, 2 mM dithiothreitol (DTT), 0.5 mM phenylmethylsulfonyl fluoride, 10 mg/ml apoprotinin, 5 mg/ml leupeptin, 1 mg/ml pepstatin A) was added. Cell suspensions were then rotated at 4°C for 1h and centrifuged at 14,000rpm for 10min. The supernatant fraction was stored at −80°C.
Cytosolic extracts for mismatch repair assays were prepared from Ramos, BL2R, TK6 and MES msh2-/- cell lines (Thomas et al., 1991). Cells (1 × 108) were washed in ice-cold isotonic buffer (20 mM HEPES, pH 7.9, containing 5 mM KCl, 1.5 mM MgCl2, 1 mM dithiothreitol, 250 mM sucrose ) followed by ice-cold hypotonic buffer (as isotonic buffer above but minus sucrose, plus 0.5 mM phenylmethylsulfonyl fluoride). Cells were re-suspended in hypotonic buffer at concentration of ~ 0.7-1 X 108 cells/ml and then were lysed using a glass homogenizer (Bellco). After 85% of the cells were lysed, the lysates were incubated on ice for 30 min. The nuclei were pelleted at 2500 x g for 10 min. and the supernatant was cleared by centrifugation at 12 000 x g for 10 min. Aliquots of the lysate were frozen in liquid nitrogen and stored at −80°C prior to analysis. Protein concentration was measured using the Bradford protein assay (Bio-Rad).
Preparation of DNA substrates by 5′-end labeling and primer-template annealing
Preparation of DNA substrates for the BER assay and 5′-end labeling were carried out as described previously (Sambrook 2001). The sequence of the deoxy-uracil (U)-containing DNA was as follows: 5′-CTGCAGCTGATGCGCUGTACGGATCCCCGGGTAC-3′. Oligodeoxynucleotide was 5′-32P-phosphorylated with [γ-32P]ATP (7000 Ci/mmol) and T4 polynucleotide kinase. After incubation at 37°C for 45 min. the reaction was terminated by heating the reaction mixture in boiling water for 3 min. Complementary oligodeoxynucleotide was mixed in equimolar concentration and annealed in 10 mM Tris–HCl, pH 7.4, and 1 mM EDTA by heating the solution to 90° C for 3 min, followed by slow cooling to room temperature. Unreacted [γ-32P]ATP was removed by a MicroSpin™ G-25 column (GE Healthcare) using the manufacturer’s protocol. The DNA was stored at −30°C.
BER assay
The uracil DNA glycosylase assay was performed as described previously (Prasad et al., 2000). Briefly, a reaction mixture (10μl) was assembled on ice that contained 50 mM Hepes, pH 7.5, 20 mM KCl, 2 mM DTT, 50 nM 32P-labeled 34-base-pair DNA with uracil at position 16 (sequence of oligo in Fig. 2B1). The reaction was initiated by adding either 10 nM UNG or Ramos extracts, as indicated, and incubated at 37°C for 20 min. 1 μl NaOH (1M) was then added to the mixture and heated for 5 min at 95°C, and an equal volume of gel loading buffer (40 mM EDTA, 80 % formamide, 0.02 % bromophenol blue and 0.02 % xylene Cyanol) was added and heated again for 2 min at 95°C. The reaction products were separated by electrophoresis in a 15% polyacrylamide gel containing 8M urea in 89 mM Tris-HCl, 89 mM boric acid and 2 mM EDTA, pH 8.8. To quantify the reaction products, the gels were scanned on a Phosphorimager (Molecular Dynamics, Model 450) and the data were analyzed using Image Quant software.
Figure 2.
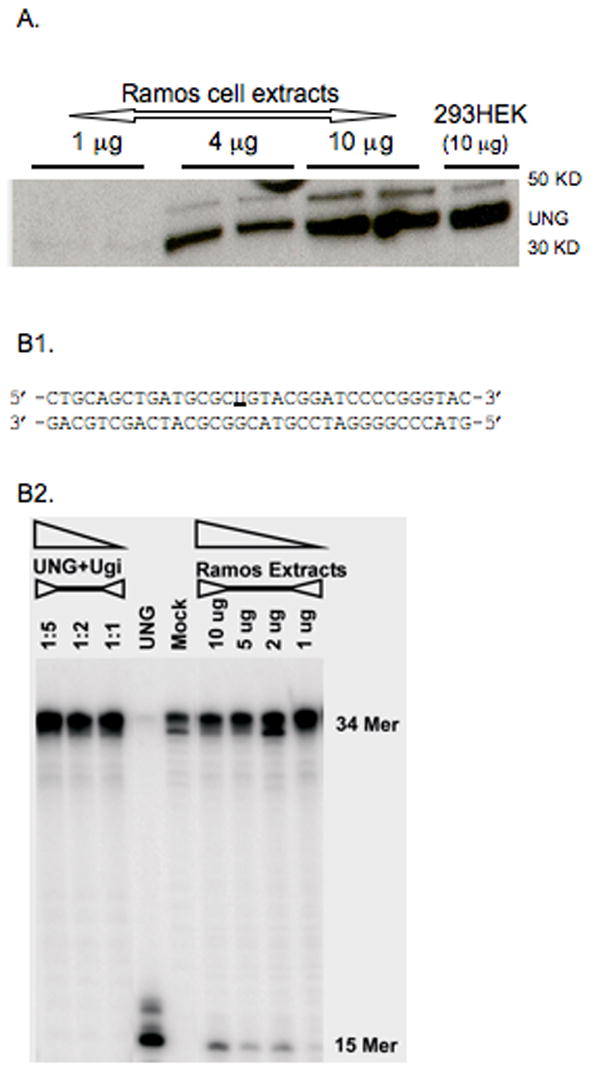
BER activity in Ramos cells. (A) Western blot of UNG found in extracts from Ramos cells. (B1) The sequence of 5′-end-32P-labeling DNA substrates containing a single uracil (underlined) used in the assays. (B2) Ramos cells are proficient in BER. Exogenous UNG + UGI (UNG inhibitor) (first 3 lanes, at molar ratio 1:1, 1:2, 1:5) and UNG alone was added to oligo as controls. “Mock” represents oligo alone without the extract or exogeneous UNG.
MMR assay
The ability of cytosolic extracts to repair DNA mismatches was assessed as described previously (Thomas et al., 1991). Briefly, wild-type and mutant M13mp2 phage derivatives were used to prepare heteroduplex substrates that contain a nick in the (–)-strand and mismatched or unpaired bases in the lacZ α-complementation gene (β-galactosidase gene). Five nanograms (1 fmol) of an M13mp2 phage heteroduplex substrate, containing a G:G- mispair, was incubated with 50 μg cytosolic extract in a 25 μl reaction for 15 min. The purified product was electroporated into MMR-deficient E.coli NR 9162. Transformed bacteria were plated with the α-complementation E.coli strain CSH50 onto minimal agar plates supplemented with isopropyl-β-D-thiogalactopyranoside and X-Gal (Sigma, Dorset, UK). Repair efficiency was calculated as 1 − (% mixed plaques in test sample ÷ % mixed plaques in a mock no-extract control). Assays were performed using cytosolic extracts from the mismatch repair-proficient human TK6 cell line as a positive control and mouse (MES) Msh2-deficient cells as a negative control.
Western blot analysis for UNG expression
After SDS-PAGE electrophoresis, proteins were transferred to polyvinylidene difluoride membranes (Millopore) using a semi-dry transfer apparatus (Pharmacia). The membranes were blocked in 1x TBST (10 mM Tris-HCl [pH 7.5], 150 mM NaCl, 0.05% Tween 20, 5% nonfat dry milk) for 1 h and incubated at room temperature in 5 ml anti-UNG (IMGENEX)(1:500 dilution) primary polyclonal antibody for 1 hour. The membranes were then washed four times (15 min each) in 1x TBST and incubated with the goat anti- rabbit horseradish peroxidase conjugate (1:5000 dilution) (Amersham) in 5 ml 1x TBST. The membranes were washed as described above, developed with the Enhanced Chemiluminescence Kit (Amersham) and exposed to Kodak film.
Computational analysis of mutations and strand bias
Spectra of somatic mutations in various prokaryotic and eukaryotic genes were analyzed, spectra are listed in the Supplemental Table. The data are available upon request from Igor Rogozin (rogozin@ncbi.nlm.nih.gov). All mutations are shown from the non-transcribed strand. Frequencies of substitutions were corrected to represent a sequence with equal amounts of the four bases. The Fisher exact test was used to compare frequencies of substitutions in A, T, G, and C sites. Calculations were done using the COLLAPSE program (Khromov-Borisov et al., 1999).
Results
Strand-bias at G:C base pairs in cells lacking A:T mutation
It has been difficult to reconcile the apparent lack of strand bias at G:C mutations with a polarized process such as transcription for the delivery of AID into the Ig V regions. Therefore, we considered the possibility that AID may be delivered in a strand-biased fashion, but that this effect is masked by the proteins that follow the initial deamination, i.e. those involved in the A:T mutation phase. We examined strand bias at G:C and A:T base pairs from a variety of spectra, including Ig V regions from mice incapable of generating A:T mutations, such as UNG and MSH2 doubly-deficient mice (Table 1). While most of the spectra revealed strand bias at A:T base pairs, we found that strand bias at G:C base pairs is undetectable in most spectra, including UNG and MSH2 singly- deficient mice (Table 1). However, mice simultaneously deficient in UNG and MSH2, the Burkitt lymphoma cell line BL2, and the GFP gene from fibroblasts expressing AID, displayed significant strand-bias at G:C base pairs (Table 1). Previous work by Reynaud and colleagues had demonstrated that in BL2, AID deamination is strand-biased (Faili et al., 2002b). These three spectra have the lowest mutation frequency at A:T base pairs suggesting that the A:T mutational machinery masks strand bias during AID-mediated deamination, and that G:C base pairs are also mutated during the “A:T” phase of hypermutation.
Table 1.
Analysis of strand bias in Somatic Hypermutation in various genes
| Spectrum | G→N | C→N | PGC1 | A→N | T→N | PAT2 |
|---|---|---|---|---|---|---|
| In vivo mammalian SHM: | ||||||
| VkOx1 | 0.31 | 0.23 | 0.11 | 0.32 | 0.14 | 0.0001 |
| neo | 0.21 | 0.17 | 0.52 | 0.40 | 0.22 | 0.02 |
| gpt | 0.23 | 0.30 | 0.65 | 0.25 | 0.22 | 0.32 |
| globin | 0.21 | 0.29 | 0.61 | 0.27 | 0.23 | 0.18 |
| lambda | 0.26 | 0.22 | 0.26 | 0.37 | 0.18 | 0.000005 |
| MMjh4 intron | 0.18 | 0.21 | 0.55 | 0.38 | 0.23 | 0.003 |
| HSjh4 intron | 0.27 | 0.23 | 0.54 | 0.33 | 0.17 | 0.02 |
| msh2-/- | 0.50 | 0.33 | 0.41 | 0.11 | 0.06 | 0.47 |
| iota-/- | 0.25 | 0.20 | 0.35 | 0.36 | 0.19 | 0.001 |
| ung-/- | 0.26 | 0.25 | 0.86 | 0.33 | 0.16 | 0.00004 |
| ung-/-msh2-/- | 0.39 | 0.60 | 0.002 | <0.01 | <0.01 | 1.00 |
| In vitro mammalian SHM: | ||||||
| Ramos | 0.41 | 0.37 | 0.26 | 0.12 | 0.10 | 0.88 |
| AID-tg-hP1-5 | 0.37 | 0.45 | 0.52 | 0.11 | 0.07 | 0.43 |
| BL2 | 0.71 | 0.21 | 0.0005 | 0.08 | 0.00 | 0.10 |
| GFP | 0.55 | 0.44 | 0.05 | 0.01 | <0.01 | 0.59 |
| In vivo non-mammalian SHM: | ||||||
| Chicken | 0.30 | 0.16 | 0.14 | 0.40 | 0.14 | 0.05 |
| Xenopus | 0.38 | 0.54 | 0.31 | 0.03 | 0.05 | 0.73 |
| Shark NAR | 0.23 | 0.25 | 0.71 | 0.29 | 0.23 | 0.43 |
PGC represents the probability of absence of strand-bias at G,C bases.
PAT represents the probability of absence of strand-bias at A,T bases.
Underlined frequencies depict significant bias.
The Burkitt lymphoma cell lines, Ramos and BL2, are proficient in MMR and express unmutated, full-length DNA polymerase η
Burkitt lymphoma cell lines that undergo SHM display a strong bias in favor of mutations at G:C over A:T (Denepoux et al., 1997, Harris et al., 2001, Poltoratsky et al., 2001). The reasons why some B cells display only the initial phase of hypermutation, i.e. the deamination step, are unknown but can be multiple. For example, both the MSH2-MSH6 heterodimer and DNA polymerase η are required for A:T mutation in vivo and could be deficient in cell lines. Given that A:T mutation resulting from cytosine deamination is unique to B cells undergoing SHM, it is likely that some components of the A:T mutational machinery in SHM may be novel and unique to hypermutating B cells (Diaz and Lawrence, 2005). Thus, defects in MMR, DNA polymerase η, or novel components of A:T mutation could cause the paucity of mutation at A:T base pairs in the cell lines. Therefore, we examined mutation in Ramos and in induced BL2 and found it to have similarly low levels of A:T mutation as described previously (Table 2). We then examined MMR in both cell lines. We used a G•G mismatch substrate that is efficiently repaired by the MSH2/MSH6-dependent pathway of DNA mismatch repair. Extracts prepared from Ramos and BL2 cells were proficient in MSH2/MSH6-mediated MMR (Fig. 1). In addition, Neuberger and colleagues demonstrated previously that BER may provide a secondary pathway for A:T mutation (Rada et al., 2004), and recent data suggest that that BER contribution may vary depending on DNA sequence (Shen et al., 2006). This pathway contributes only a small fraction of A:T mutations, but may account for the low yet significantly higher A:T mutation frequency in Ramos over most Burkitt lymphoma cell lines. However, Ramos cells express high levels of UNG (Fig. 2a) and are proficient in BER (Fig. 2b). We also sequenced and examined the expression of mRNA transcripts encoding DNA polymerase η in Ramos and BL2 cells. DNA polymerase η expression (Fig. 3) and coding sequence (supplemental Figure 1) are intact in these cells. A differentially spliced form of DNA polymerase η, predicted to encode a non-functional protein, has been reported and appears to be a major source of regulation of this protein (Thakur et al., 2001). However, polymerase η mRNA in Ramos and in BL2 is not subject to alternative splicing because the smaller fragments were not detectable by PCR (Fig. 3). Thus, there is no obvious evidence of polymerase η dysfunction in Ramos or BL2 cells.
Table 2.
Distribution of mutation at A:T and G:C base pairs in Ramos and activated BL2 cells*
| G:C mutations | A:T mutations | |
|---|---|---|
| RAMOS (48 clones) | 80 (85%) | 14 (15%) |
| BL2 (30 clones) | 5 (100%) | 0 (0%) |
Spectra similar as reported previously and is available upon request. These results are representative of at least three experiments.
Figure 1.
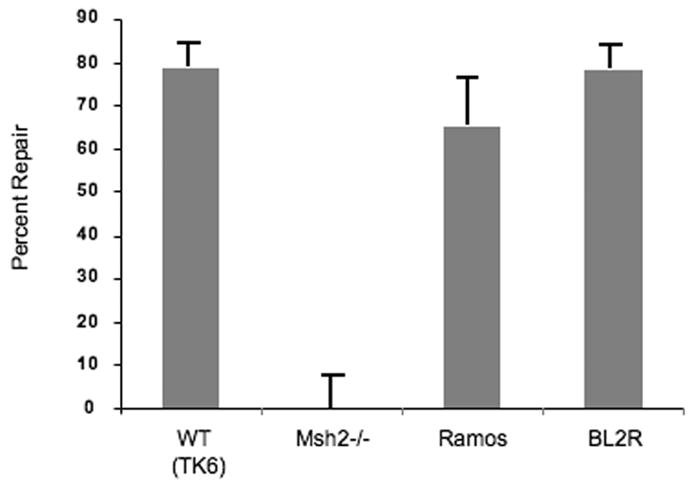
Ramos and BL2 cell extracts are proficient in MSH2/MSH6-mediated mismatch repair. The M13 heteroduplex substrate used in mismatch repair reactions (Thomas et al., 1991) contained a G•G mismatch. A 5′ nick in the (–) strand at the Bsu36I restriction site serves as the strand discrimination signal for the repair. The results are averages based on counting more than 500 M13 plaques per variable in three independent experiments.
Figure 3.

The truncated inactive form of DNA polymerase η is not expressed in mature B cell lines. A schematic model for the cDNA of Polymerase η, indicating exon II deletion is depicted. Primers were synthesized for exon I and exon IV to generate a 358-bp product in the full-length configuration. Loss of the 140-bp exon II would lead to a 218-bp fragment. The lower bright band in the size marker (TrackIt 100 bp DNA Ladder, Invitrogen) is 600 base pairs.
Discussion
To assess whether AID-mediated deamination is a strand-biased process we examined mutations at G:C base pairs in Ig V regions from a variety of databases. We found that when phase 2 (the A:T phase) of hypermutation is almost completely absent, such as in mice simultaneously deficient in UNG and in MMR and in the BL2 cell line, significant strand bias at G:C bases was revealed. This implicates a strand-biased process in the delivery of AID to Ig V regions. One candidate process is transcription. Storb and colleagues and others have reported that e-box-like sequences or the E2A proteins themselves enhance hypermutation (Michael et al., 2003, Schoetz et al., 2006) and that supercoiled DNA may help direct AID to Ig V regions (Shen and Storb, 2004). Alternatively, other DNA transactions such as DNA repair patches (Brar et al., 2004, Bross and Jacobs, 2003, Zan et al., 2003, Kong and Maizels, 2001, Bross et al., 2000, Papavasiliou and Schatz, 2000) or the formation of AID substrates by a novel mechanism (similar to the R-loops of switch regions) could account for a strand-biased delivery of AID. Recent evidence, however, suggests that AID might directly associate with the transcriptional machinery (Besmer et al., 2006, Duquette et al., 2005, Nambu et al., 2003).
An obvious conclusion from these results is that the A:T mutational machinery alters the strand bias generated during AID-mediated deamination. Indeed, the three spectra with significant G:C strand bias were those displaying the lowest level of A:T mutation: MSH2-UNG double knockouts, the BL2 cell line, and the GFP gene from fibroblasts expressing AID (Table 1). Since the single UNG or MSH2 knockouts did not display G:C strand bias, the explanation cannot be due to a role by these molecules individually but rather as a part of a complex involved in A:T mutation. An attractive explanation may lie in the participation of various error-prone DNA polymerases in addition to DNA polymerase η in the A:T phase, some which may contribute to mutation at G:C base pairs in a strand-unbiased manner (Diaz and Lawrence 2005).
The reason why Burkitt lymphoma cell lines display mutations predominantly at G:C bases is an unresolved issue. The most straightforward interpretation is that these cell lines lack components of the A:T mutational machinery. Here we demonstrate that Ramos cells are proficient in MSH2/MSH6-mediated MMR and express wild-type, full-length DNA polymerase η. In addition, a lack of contribution to A:T mutation by UNG and BER, a secondary pathway that plays a smaller role in A:T mutation, is an unlikely explanation for the lower A:T mutation in Ramos, since these cells are proficient in BER and express high levels of UNG. BL2 cells, where the mutations are near 100% at G:C base pairs, are proficient in MSH2/MSH6-mediated MMR, and express full-length, wild-type DNA polymerase η. A BER defect in BL2 would not explain the severe paucity of mutations at A:T base pairs in BL2, since BER contributes only a fraction of A:T mutations (Rada et.al. 2004, Shen et al., 2006). The combined results suggest that all the known components of the A:T mutational machinery are in place in at least some of the Burkitt lymphoma cell lines displaying a strong G:C bias in Ig hypermutation, implicating deficiency of unknown components for the paucity of mutations at A:T base pairs in their Ig loci. An A:T hypermutation recruiting factor, and additional TLS DNA polymerases are potential candidates, specially considering mounting evidence for the involvement of multiple DNA polymerases in Ig hypermutation, and not just DNA polymerase η (Diaz et al., 2001, Zan et al., 2005, Masuda et al., 2005). However, it is also possible that AID-mediated cytosine deamination is processed through a conventional BER pathway in Burkitt lymphoma cell lines, leading to mutations predominantly at G:C base pairs. For example, if removal of the uracil occurs prior to the recruitment of the MMR proteins, mutations might be limited to or predominantly found at G:C base pairs. Indeed, expression of AID in non-lymphoid tissues results in mutations predominantly at G:C base pairs (Okazaki et al., 2003, Yoshikawa et al., 2002), and Ig hypermutation in cell lines may resemble this conventional outcome of cytosine deamination. This suggests SHM-specific factors that cause hypermutation to occur at bases adjacent to deaminated cytosines in the DNA (Diaz and Lawrence, 2005). Perhaps, if SHM occurs at a different stage of the cell cycle in vitro than in vivo, the DNA repair pathway recruited to deal with the uracil might be different, leading to the frequent early removal of the uracil in cell lines and the ensuing conventional BER reaction prior to recruitment of the A:T machinery. Such competition for the uracil between a SHM-specific mechanism and conventional BER could attenuate A:T mutation in at least some of the Burkitt lymphoma cell lines. Interestingly, these scenarios implicate a unique B cell mechanism in vivo: one which promotes high frequency mutation at bases adjacent to deaminated cytosines.
Supplementary Material
Supplemental Figure 1. The deduced amino acid sequence of Ramos and Bl2-derived DNA polymerase η. The sequence was obtained from at least 2 clones derived from products of RT-PCR reaction and from direct sequencing of PCR product.
Acknowledgments
We are grateful to Jan Drake and Vladimir Poltoratsky for critical reading of the manuscript. We also thank Laurent Verkoczy, Mathew Scharff, and Claude-Agnes Reynaud for providing various B cell lines, Cristina Rada and Mathew Scharff for kindly providing sequence data and Thomas Kunkel for discussions. We are indebted to Rajendra Prasad and Samuel Wilson for providing expertise and technical assistance with the BER assays. This research was supported by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bachl J, Carlson C, Gray-Schopfer V, Dessing M, Olsson C. Increased transcription levels induce higher mutation rates in a hypermutating cell line. J Immunol. 2001;166:5051–5057. doi: 10.4049/jimmunol.166.8.5051. [DOI] [PubMed] [Google Scholar]
- Bardwell PD, Woo CJ, Wei K, Li Z, Martin A, Sack SZ, Parris T, Edelmann W, Scharff MD. Altered somatic hypermutation and reduced class-switch recombination in exonuclease 1-mutant mice. Nat Immunol. 2004;5:224–229. doi: 10.1038/ni1031. [DOI] [PubMed] [Google Scholar]
- Beale RC, Petersen-Mahrt SK, Watt IN, Harris RS, Rada C, Neuberger MS. Comparison of the differential context-dependence of DNA deamination by APOBEC enzymes: correlation with mutation spectra in vivo. J Mol Biol. 2004;337:585–596. doi: 10.1016/j.jmb.2004.01.046. [DOI] [PubMed] [Google Scholar]
- Besmer E, Market E, Papavasiliou FN. The transcription elongation complex directs activation-induced cytidine deaminase-mediated DNA deamination. Mol Cell Biol. 2006;26:4378–4385. doi: 10.1128/MCB.02375-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bransteitter R, Pham P, Calabrese P, Goodman MF. Biochemical analysis of hypermutational targeting by wild type and mutant activation-induced cytidine deaminase. J Biol Chem. 2004;279:51612–51621. doi: 10.1074/jbc.M408135200. [DOI] [PubMed] [Google Scholar]
- Bransteitter R, Pham P, Scharff MD, Goodman MF. Activation-induced cytidine deaminase deaminates deoxycytidine on single-stranded DNA but requires the action of RNase. Proc Natl Acad Sci U S A. 2003;100:4102–4107. doi: 10.1073/pnas.0730835100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brar SS, Watson M, Diaz M. Activation-induced cytosine deaminase (AID) is actively exported out of the nucleus but retained by the induction of DNA breaks. J Biol Chem. 2004;279:26395–26401. doi: 10.1074/jbc.M403503200. [DOI] [PubMed] [Google Scholar]
- Bross L, Fukita Y, McBlane F, Demolliere C, Rajewsky K, Jacobs H. DNA double-strand breaks in immunoglobulin genes undergoing somatic hypermutation. Immunity. 2000;13:589–597. doi: 10.1016/s1074-7613(00)00059-5. [DOI] [PubMed] [Google Scholar]
- Bross L, Jacobs H. DNA double strand breaks occur independent of AID in hypermutating Ig genes. Clin Dev Immunol. 2003;10:83–89. doi: 10.1080/10446670310001626571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri J, Khuong C, Alt FW. Replication protein A interacts with AID to promote deamination of somatic hypermutation targets. Nature. 2004;430:992–998. doi: 10.1038/nature02821. [DOI] [PubMed] [Google Scholar]
- Chaudhuri J, Tian M, Khuong C, Chua K, Pinaud E, Alt FW. Transcription-targeted DNA deamination by the AID antibody diversification enzyme. Nature. 2003;422:726–730. doi: 10.1038/nature01574. [DOI] [PubMed] [Google Scholar]
- Delbos F, De Smet A, Faili A, Aoufouchi S, Weill JC, Reynaud CA. Contribution of DNA polymerase eta to immunoglobulin gene hypermutation in the mouse. J Exp Med. 2005;201:1191–1196. doi: 10.1084/jem.20050292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delpy L, Sirac C, Le Morvan C, Cogne M. Transcription-dependent somatic hypermutation occurs at similar levels on functional and nonfunctional rearranged IgH alleles. J Immunol. 2004;173:1842–1848. doi: 10.4049/jimmunol.173.3.1842. [DOI] [PubMed] [Google Scholar]
- Denepoux S, Razanajaona D, Blanchard D, Meffre G, Capra JD, Banchereau J, Lebecque S. Induction of somatic mutation in a human B cell line in vitro. Immunity. 1997;6:35–46. doi: 10.1016/s1074-7613(00)80240-x. [DOI] [PubMed] [Google Scholar]
- Diaz M, Flajnik MF. Evolution of somatic hypermutation and gene conversion in adaptive immunity. Immunol Rev. 1998;162:13–24. doi: 10.1111/j.1600-065x.1998.tb01425.x. [DOI] [PubMed] [Google Scholar]
- Diaz M, Lawrence C. An update on the role of translesion synthesis DNA polymerases in Ig hypermutation. Trends Immunol. 2005;26:215–220. doi: 10.1016/j.it.2005.02.008. [DOI] [PubMed] [Google Scholar]
- Diaz M, Velez J, Singh M, Cerny J, Flajnik MF. Mutational pattern of the nurse shark antigen receptor gene (NAR) is similar to that of mammalian Ig genes and to spontaneous mutations in evolution: the translesion synthesis model of somatic hypermutation. Int Immunol. 1999;11:825–833. doi: 10.1093/intimm/11.5.825. [DOI] [PubMed] [Google Scholar]
- Diaz M, Verkoczy LK, Flajnik MF, Klinman NR. Decreased frequency of somatic hypermutation and impaired affinity maturation but intact germinal center formation in mice expressing antisense RNA to DNA polymerase zeta. J Immunol. 2001;167:327–335. doi: 10.4049/jimmunol.167.1.327. [DOI] [PubMed] [Google Scholar]
- Dickerson SK, Market E, Besmer E, Papavasiliou FN. AID mediates hypermutation by deaminating single stranded DNA. J Exp Med. 2003;197:1291–1296. doi: 10.1084/jem.20030481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorner T, Foster SJ, Farner NL, Lipsky PE. Somatic hypermutation of human immunoglobulin heavy chain genes: targeting of RGYW motifs on both DNA strands. Eur J Immunol. 1998;28:3384–96. doi: 10.1002/(SICI)1521-4141(199810)28:10<3384::AID-IMMU3384>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Duquette ML, Pham P, Goodman MF, Maizels N. AID binds to transcription-induced structures in c-MYC that map to regions associated with translocation and hypermutation. Oncogene. 2005;24:5791–5798. doi: 10.1038/sj.onc.1208746. [DOI] [PubMed] [Google Scholar]
- Faili A, Aoufouchi S, Flatter E, Gueranger Q, Reynaud CA, Weill JC. Induction of somatic hypermutation in immunoglobulin genes is dependent on DNA polymerase iota. Nature. 2002a;419:944–947. doi: 10.1038/nature01117. [DOI] [PubMed] [Google Scholar]
- Faili A, Aoufouchi S, Gueranger Q, Zober C, Leon A, Bertocci B, Weill JC, Reynaud CA. AID-dependent somatic hypermutation occurs as a DNA single-strand event in the BL2 cell line. Nat Immunol. 2002b;3:815–821. doi: 10.1038/ni826. [DOI] [PubMed] [Google Scholar]
- Foster SJ, Dorner T, Lipsky PE. Somatic hypermutation of VkappaJkappa rearrangements: targeting of RGYW motifs on both DNA strands and preferential selection of mutated codons within RGYW motifs. Eur J Immunol. 1999;29:4011–4021. doi: 10.1002/(SICI)1521-4141(199912)29:12<4011::AID-IMMU4011>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Fukita Y, Jacobs H, Rajewsky K. Somatic hypermutation in the heavy chain locus correlates with transcription. Immunity. 1998;9:105–114. doi: 10.1016/s1074-7613(00)80592-0. [DOI] [PubMed] [Google Scholar]
- Harris RS, Croom-Carter DS, Rickinson AB, Neuberger MS. Epstein-Barr virus and the somatic hypermutation of immunoglobulin genes in Burkitt’s lymphoma cells. J Virol. 2001;75:10488–10492. doi: 10.1128/JVI.75.21.10488-10492.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinds-Frey KR, Nishikata H, Litman RT, Litman GW. Somatic variation precedes extensive diversification of germline sequences and combinatorial joining in the evolution of immunoglobulin heavy chain diversity. J Exp Med. 1993;178:815–824. doi: 10.1084/jem.178.3.815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs H, Fukita Y, van der Horst GT, de Boer J, Weeda G, Essers J, de Wind N, Engelward BP, Samson L, Verbeek S, de Murcia JM, de Murcia G, te Riele H, Rajewsky K. Hypermutation of immunoglobulin genes in memory B cells of DNA repair-deficient mice. J Exp Med. 1998;187:1735–1743. doi: 10.1084/jem.187.11.1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khromov-Borisov NN, Rogozin IB, Pegas Henriques JA, de Serres FJ. Similarity pattern analysis in mutational distributions. Mutat Res. 1999;430:55–74. doi: 10.1016/s0027-5107(99)00148-7. [DOI] [PubMed] [Google Scholar]
- Kong Q, Maizels N. DNA breaks in hypermutating immunoglobulin genes: evidence for a break-and-repair pathway of somatic hypermutation. Genetics. 2001;158:369–378. doi: 10.1093/genetics/158.1.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larijani M, Frieder D, Basit W, Martin A. The mutation spectrum of purified AID is similar to the mutability index in Ramos cells and in ung(-/-)msh2(-/-) mice. Immunogenetics. 2005;56:840–845. doi: 10.1007/s00251-004-0748-0. [DOI] [PubMed] [Google Scholar]
- Martin A, Li Z, Lin DP, Bardwell PD, Iglesias-Ussel MD, Edelmann W, Scharff MD. Msh2 ATPase activity is essential for somatic hypermutation at a-T basepairs and for efficient class switch recombination. J Exp Med. 2003;198:1171–1178. doi: 10.1084/jem.20030880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda K, Ouchida R, Takeuchi A, Saito T, Koseki H, Kawamura K, Tagawa M, Tokuhisa T, Azuma T, O-Wang J. DNA polymerase theta contributes to the generation of C/G mutations during somatic hypermutation of Ig genes. Proc Natl Acad Sci U S A. 2005;102:13986–13991. doi: 10.1073/pnas.0505636102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayorov VI, Rogozin IB, Adkison LR, Gearhart PJ. DNA polymerase eta contributes to strand bias of mutations of A versus T in immunoglobulin genes. J Immunol. 2005;174:7781–7786. doi: 10.4049/jimmunol.174.12.7781. [DOI] [PubMed] [Google Scholar]
- Michael N, Shen HM, Longerich S, Kim N, Longacre A, Storb U. The E box motif CAGGTG enhances somatic hypermutation without enhancing transcription. Immunity. 2003;19:235–242. doi: 10.1016/s1074-7613(03)00204-8. [DOI] [PubMed] [Google Scholar]
- Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102:553–563. doi: 10.1016/s0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- Nambu Y, Sugai M, Gonda H, Lee CG, Katakai T, Agata Y, Yokota Y, Shimizu A. Transcription-coupled events associating with immunoglobulin switch region chromatin. Science. 2003;302:2137–2140. doi: 10.1126/science.1092481. [DOI] [PubMed] [Google Scholar]
- Neuberger MS, Harris RS, Di Noia J, Petersen-Mahrt SK. Immunity through DNA deamination. Trends Biochem Sci. 2003;28:305–312. doi: 10.1016/S0968-0004(03)00111-7. [DOI] [PubMed] [Google Scholar]
- Okazaki IM, Hiai H, Kakazu N, Yamada S, Muramatsu M, Kinoshita K, Honjo T. Constitutive expression of AID leads to tumorigenesis. J Exp Med. 2003;197:1173–1181. doi: 10.1084/jem.20030275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papavasiliou FN, Schatz DG. Cell-cycle-regulated DNA double-stranded breaks in somatic hypermutation of immunoglobulin genes. Nature. 2000;408:216–221. doi: 10.1038/35041599. [DOI] [PubMed] [Google Scholar]
- Peters A, Storb U. Somatic hypermutation of immunoglobulin genes is linked to transcription initiation. Immunity. 1996;4:57–65. doi: 10.1016/s1074-7613(00)80298-8. [DOI] [PubMed] [Google Scholar]
- Petersen-Mahrt SK, Harris RS, Neuberger MS. AID mutates E. coli suggesting a DNA deamination mechanism for antibody diversification. Nature. 2002;418:99–103. doi: 10.1038/nature00862. [DOI] [PubMed] [Google Scholar]
- Pham P, Bransteitter R, Petruska J, Goodman MF. Processive AID-catalysed cytosine deamination on single-stranded DNA simulates somatic hypermutation. Nature. 2003;424:103–107. doi: 10.1038/nature01760. [DOI] [PubMed] [Google Scholar]
- Phung QH, Winter DB, Alrefai R, Gearhart PJ. Hypermutation in Ig V genes from mice deficient in the MLH1 mismatch repair protein. J Immunol. 1999;162:3121–3124. [PubMed] [Google Scholar]
- Poltoratsky V, Woo CJ, Tippin B, Martin A, Goodman MF, Scharff MD. Expression of error-prone polymerases in BL2 cells activated for Ig somatic hypermutation. Proc Natl Acad Sci U S A. 2001;98:7976–7981. doi: 10.1073/pnas.141222198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad R, Dianov GL, Bohr VA, Wilson SH. FEN1 stimulation of DNA polymerase beta mediates an excision step in mammalian long patch base excision repair. J Biol Chem. 2000;275:4460–4466. doi: 10.1074/jbc.275.6.4460. [DOI] [PubMed] [Google Scholar]
- Rada C, Di Noia JM, Neuberger MS. Mismatch recognition and uracil excision provide complementary paths to both Ig switching and the A/T-focused phase of somatic mutation. Mol Cell. 2004;16:163–171. doi: 10.1016/j.molcel.2004.10.011. [DOI] [PubMed] [Google Scholar]
- Rada C, Ehrenstein MR, Neuberger MS, Milstein C. Hot spot focusing of somatic hypermutation in MSH2-deficient mice suggests two stages of mutational targeting. Immunity. 1998;9:135–141. doi: 10.1016/s1074-7613(00)80595-6. [DOI] [PubMed] [Google Scholar]
- Rada C, Williams GT, Nilsen H, Barnes DE, Lindahl T, Neuberger MS. Immunoglobulin isotype switching is inhibited and somatic hypermutation perturbed in UNG-deficient mice. Curr Biol. 2002;12:1748–1755. doi: 10.1016/s0960-9822(02)01215-0. [DOI] [PubMed] [Google Scholar]
- Rada C, Yelamos J, Dean W, Milstein C. The 5′ hypermutation boundary of kappa chains is independent of local and neighbouring sequences and related to the distance from the initiation of transcription. Eur J Immunol. 1997;27:3115–3120. doi: 10.1002/eji.1830271206. [DOI] [PubMed] [Google Scholar]
- Ramiro AR, Stavropoulos P, Jankovic M, Nussenzweig MC. Transcription enhances AID-mediated cytidine deamination by exposing single-stranded DNA on the nontemplate strand. Nat Immunol. 2003;4:452–456. doi: 10.1038/ni920. [DOI] [PubMed] [Google Scholar]
- Revy P, Muto T, Levy Y, Geissmann F, Plebani A, Sanal O, Catalan N, Forveille M, Dufourcq-Labelouse R, Gennery A, Tezcan I, Ersoy F, Kayserili H, Ugazio AG, Brousse N, Muramatsu M, Notarangelo LD, Kinoshita K, Honjo T, Fischer A, Durandy A. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the Hyper-IgM syndrome (HIGM2) Cell. 2000;102:565–575. doi: 10.1016/s0092-8674(00)00079-9. [DOI] [PubMed] [Google Scholar]
- Rogozin IB, Diaz M. Cutting edge: DGYW/WRCH is a better predictor of mutability at G:C bases in Ig hypermutation than the widely accepted RGYW/WRCY motif and probably reflects a two-step activation-induced cytidine deaminase-triggered process. J Immunol. 2004;172:3382–3384. doi: 10.4049/jimmunol.172.6.3382. [DOI] [PubMed] [Google Scholar]
- Rogozin IB, Kolchanov NA. Somatic hypermutagenesis in immunoglobulin genes. II Influence of neighbouring base sequences on mutagenesis. Biochim Biophys Acta. 1992;1171:11–18. doi: 10.1016/0167-4781(92)90134-l. [DOI] [PubMed] [Google Scholar]
- Rogozin IB, Pavlov YI, Bebenek K, Matsuda T, Kunkel TA. Somatic mutation hotspots correlate with DNA polymerase eta error spectrum. Nat Immunol. 2001;2:530–536. doi: 10.1038/88732. [DOI] [PubMed] [Google Scholar]
- Sambrook DWR. Molecular Cloning, a Laboratory Manual. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY, Cold Spring Harbor, NY: 2001. [Google Scholar]
- Shen HM, Storb U. Activation-induced cytidine deaminase (AID) can target both DNA strands when the DNA is supercoiled. Proc Natl Acad Sci U S A. 2004;101:12997–13002. doi: 10.1073/pnas.0404974101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen HM, Tanaka A, Bozek G, Nicolae D, Storb U. Somatic hypermutation and class switch recombination in msh6-/-ung-/- double-knockout mice. J Immunol. 2006;177:5386–5392. doi: 10.4049/jimmunol.177.8.5386. [DOI] [PubMed] [Google Scholar]
- Schoetz U, Cervelli M, Wang YD, Fiedler P, Buerstedde JM. E2A expression stimulates Ig hypermutation. J Immunol. 2006;177:395–400. doi: 10.4049/jimmunol.177.1.395. [DOI] [PubMed] [Google Scholar]
- Sohail A, Klapacz J, Samaranayake M, Ullah A, Bhagwat AS. Human activation-induced cytidine deaminase causes transcription-dependent, strand-biased C to U deaminations. Nucleic Acids Res. 2003;31:2990–2994. doi: 10.1093/nar/gkg464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiernholm NB, Berinstein NL. Immunoglobulin somatic variation; studies of receptor editing in a human B cell lymphoma. Leuk Lymphoma. 1994;12:333–341. doi: 10.3109/10428199409073774. [DOI] [PubMed] [Google Scholar]
- Storb U, Stavnezer J. Immunoglobulin genes: generating diversity with AID and UNG. Curr Biol. 2002;12:R725–R727. doi: 10.1016/s0960-9822(02)01247-2. [DOI] [PubMed] [Google Scholar]
- Thakur M, Wernick M, Collins C, Limoli CL, Crowley E, Cleaver JE. DNA polymerase eta undergoes alternative splicing, protects against UV sensitivity and apoptosis, and suppresses Mre11-dependent recombination. Genes Chromosomes Cancer. 2001;32:222–235. doi: 10.1002/gcc.1186. [DOI] [PubMed] [Google Scholar]
- Thomas DC, Roberts JD, Kunkel TA. Heteroduplex repair in extracts of human HeLa cells. J Biol Chem. 1991;266:3744–3751. [PubMed] [Google Scholar]
- Tumas-Brundage K, Manser T. The transcriptional promoter regulates hypermutation of the antibody heavy chain locus. J Exp Med. 1997;185:239–250. doi: 10.1084/jem.185.2.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei K, Clark AB, Wong E, Kane MF, Mazur DJ, Parris T, Kolas NK, Russell R, Hou H, Jr, Kneitz B, Yang G, Kunkel TA, Kolodner RD, Cohen PE, Edelmann W. Inactivation of Exonuclease 1 in mice results in DNA mismatch repair defects, increased cancer susceptibility, and male and female sterility. Genes Dev. 2003;17:603–614. doi: 10.1101/gad.1060603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiesendanger M, Kneitz B, Edelmann W, Scharff MD. Somatic hypermutation in MutS homologue (MSH)3-, MSH6-, and MSH3/MSH6-deficient mice reveals a role for the MSH2-MSH6 heterodimer in modulating the base substitution pattern. J Exp Med. 2000;191:579–584. doi: 10.1084/jem.191.3.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson M, Marcuz A, du Pasquier L. Somatic mutations during an immune response in Xenopus tadpoles. Dev Immunol. 1995;4:227–234. doi: 10.1155/1995/38639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson TM, Vaisman A, Martomo SA, Sullivan P, Lan L, Hanaoka F, Yasui A, Woodgate R, Gearhart PJ. MSH2-MSH6 stimulates DNA polymerase eta, suggesting a role for A:T mutations in antibody genes. J Exp Med. 2005;201:637–645. doi: 10.1084/jem.20042066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikawa K, Okazaki IM, Eto T, Kinoshita K, Muramatsu M, Nagaoka H, Honjo T. AID enzyme-induced hypermutation in an actively transcribed gene in fibroblasts. Science. 2002;296:2033–2036. doi: 10.1126/science.1071556. [DOI] [PubMed] [Google Scholar]
- Yu K, Huang FT, Lieber MR. DNA substrate length and surrounding sequence affect the activation-induced deaminase activity at cytidine. J Biol Chem. 2004;279:6496–6500. doi: 10.1074/jbc.M311616200. [DOI] [PubMed] [Google Scholar]
- Zan H, Komori A, Li Z, Cerutti A, Schaffer A, Flajnik MF, Diaz M, Casali P. The translesion DNA polymerase zeta plays a major role in Ig and bcl-6 somatic hypermutation. Immunity. 2001;14:643–653. doi: 10.1016/s1074-7613(01)00142-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zan H, Wu X, Komori A, Holloman WK, Casali P. AID-dependent generation of resected double-strand DNA breaks and recruitment of Rad52/Rad51 in somatic hypermutation. Immunity. 2003;18:727–738. doi: 10.1016/s1074-7613(03)00151-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zan H, Shima N, Xu Z, Al-Qahtani A, Evinger AJ, Zhong Y, Schimenti JC, Casali P. The translesion DNA polymerase theta plays a dominant role in immunoglobulin gene somatic hypermutation. EMBO J. 2005;24:3757–3769. doi: 10.1038/sj.emboj.7600833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng X, Winter DB, Kasmer C, Kraemer KH, Lehmann AR, Gearhart PJ. DNA polymerase eta is an A-T mutator in somatic hypermutation of immunoglobulin variable genes. Nat Immunol. 2001;2:537–541. doi: 10.1038/88740. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. The deduced amino acid sequence of Ramos and Bl2-derived DNA polymerase η. The sequence was obtained from at least 2 clones derived from products of RT-PCR reaction and from direct sequencing of PCR product.