

Abstract
Sympathetic-induced vasoconstriction is mediated by various adrenergic receptor (AR) subtypes located on membranes of vascular smooth muscle cells (VSMC) located on the arterial wall, but is mostly attributed to activation of the α1D-AR. In order to study interaction and crosstalk among AR genes, we induced post-transcriptional silencing of the α1D-AR gene in cultured VSMC using the RNAi technique. A pSEC neo expression plasmid vector containing a small interfering RNA (siRNA) sequence selected to bind to the targeted mRNA of the α1D-AR gene was transfected into cultured VSMC from rat aorta. The RNA expression of all AR-subtype genes was assessed by Q-RT-PCR and the α1D and α2A-AR proteins quantified by Western blot. In siRNA transfected cells, the α1D-AR protein levels decreased by 55%, 69% and 75% at 24h, 48h and 72h, respectively (p < 0.03–0.01) with progressive increases in its gene expression by 50%–61% and concurrent increase in α2A-AR protein peaking at 48 h. Decreases were noted in expression of the α1A, α2A, and β3 AR genes. We conclude that post-transcriptional silencing of the α1D-AR gene leads to significant decrease in receptor protein despite reactive increase in gene expression. However, suppression of one AR leads to reactive changes in other subtypes, indicating that cross-talk among related genes, whose products have overlapping functions, may partly offset anticipated effects in vivo.
1. Introduction
The sympathoadrenal system exerts its effects via three major types of adrenergic receptors (AR): α1, α2 and β; for each one of these, three subtypes have been identified and cloned: α1A, α1B, α1D, α2A, α2B, α2C and β1 β2, β3. All are members of the guanidine nucleotide (G) regulatory protein-coupled receptor superfamily (Bylund, 1992; Lefkowitz and Caron, 1988). All are activated to variable extent by catecholamines or other sympathomimetic agents, which elicit type-specific and tissue specific responses. In the past we have been particularly interested in the role of various α2-AR subtypes in hypertension and heart failure (Gavras et al., 2001). In those studies we employed either gene knockout mice with deleted gene for a specific α2-AR subtype (Makaritsis et al., 2000) or gene treatment in rats using antisense technology to inhibit a specific subtype’s gene (Kintsurashvili et al., 2001; Kintsurashvili et al., 2003).
Activation of postjunctional α1-ARs contributes to a number of sympathoadrenal actions relevant to hypertension (Gavras et al., 1995) including renal sodium handling (Jeffries et al., 1988), cardiac rhythm (Kurz et al., 1991), contractility (Endoh et al., 1991) and hypertrophy (Ikeda et al., 1991), as well as vascular smooth muscle cell (VSMC) contraction, resulting in systemic vasoconstriction (Ruffolo et al., 1991; Young et al., 1988). Indeed, stimulation of α1-ARs accounts for about 70% of the vasopressor response to catecholamines, whereas the post-synaptic α2-ARs account for the remainder 30% (Duka et al., 2000). Ample evidence suggests that the α1-AR subtype mostly responsible for vascular contractile responses in big arteries (aorta, femoral and iliac) is the α1D-AR (Gisbert et al., 2000; Hrometz et al., 1999; Piascik et al., 1995; Villalobos-Molina and Ibarra, 1999).
The current experiments were designed to further explore the function of the α1D-AR located in VSMC and its interaction with other ARs. Our hypothesis was that silencing of this AR’s gene would elicit compensatory changes in expression of related AR subtypes. To this aim, we employed the RNAi technology (McManus and Sharp, 2002) that involves post-transcriptional gene silencing by use of short RNA nucleotide sequences complementary to selected 19–21 base regions of the target mRNA. The small interfering RNA (siRNA) sequence binds to the targeted mRNA, thus inducing its cleavage, degradation and ultimately loss of the encoded protein (Elbashir et al., 2001; Gitlin et al., 2002). By adapting this technology to the α1D-AR gene, we achieved the post-transcriptional inhibition of this gene in cultured VSMC and assessed α1D-AR protein production, as well as the effects on the possible cross-talk with other related AR subtype genes.
2. Material and Methods
2.1. siRNA Constructs
To design α1D-AR-specific siRNA duplex, we selected 4 siRNA target sequences at different positions through the α1D-AR gene according to the Guidelines for siRNA target selection using Ambion’s online target finder tool. Three of these are located in the ORF part and one in the 3'UTR. The sequences are 5'-GCTCAAGTACCCAGCCATTAT-3', 5-GCCAAAGGATATCCCGGAACA-3', 5-GCTGTCATCTGCCAGGCTTAT-3' and 5'-AAACTACTTAGTCAACTCCTA-3'. The selected sequences were submitted to a BLAST search against the rat genome to ensure specificity of targeted sequences. Sense and antisense templates were synthesized (Invitrogen, Carlsbad, CA), and then generated the precursor short hairpin RNA with mouse U6 RNA polymerase using the Silencer Express Cassette (Ambion, Austin, TX), which was cloned into pSEC neo expression vector to produce siRNA by in vitro transcription. A GAPDH siRNA expression plasmid was also constructed by pSEC neo vector as a positive control. Block-it U6 RNAi entry vector, which also expresses short hairpin RNA, was used to construct the α1D-AR/siRNA plasmids containing these 4 different target sequences and to compare the knock-down efficiency.
2.2. Cells and Transfection
VSMC were isolated from the rat thoracic aorta of 8-week-old male Wistar rats by an explant method (Ross R, 1971) and were stained with smooth muscle-specific alpha-actin, indicating a smooth muscle phenotype. Cells were cultured in Dulbecco's modified Eagle's medium (DMEM Glucose 4.5mg/ml) supplemented with 10% fetal bovine serum, penicillin (100U/ml), and streptomycin (100μg/ml) in humidified atmosphere at 37°C with 5% carbon dioxide. The cells were trypsinized every 5 to 7 days. All experiments were performed on cells between passage 6 to 10.
One day before transfection, cells were trypsinized and resuspended in a new medium without antibiotics, and transferred onto a 100mm dish at a density of 5×106 cells per dish. The transfection was performed using Lipofectamine 2000, and the whole procedure and conditions were according to the manufacturer’s protocol. (Invitrogen). Each tranfection was performed on three groups under the same conditions; an α1D-AR/pSEC transfection group, a GAPDH/pSEC transfection group and a control non-transfection group. Twenty-four, 48 and 72 hours after transfection, all cells from the three groups were harvested for RT-PCR and Western blot analysis to investigate the level of RNA and endogenous protein.
2.3. Total RNA Isolation
Total RNA was prepared from siRNA-transfected rat VSMC cells or non-transfected cells using the TRIzol Reagents (GIBCO-BRL; Gaithersburg, MD) according to the manufacturer’s instructions. This was followed by DNase treatment and removal of contaminating DNA from the RNA preparation with DNA-free (Ambion, Austin, TX).
2.4. Analysis of adrenergic receptor mRNA levels by RT-PCR
The RNA expression of α1D-AR gene as well as α1A, α1β and α2A, α2B, α2C, β1, β2, β3-AR genes in rat VSMC was examined by quantitative reverse transcriptase-polymerase chain reaction (RT-PCR) techniques. From each sample, 1μg of total RNA was converted to cDNA using a RNA PCR kit (Perkins-Elmer; Branchburg, NJ), and PCR was performed with oligonucleotide primers complementary to each rat AR subtype cDNA. The primers of each AR subtype and the length of PCR product fragments are shown in Table 1. The reverse transcription was performed by first incubating at room temperature for 10 minutes and then at 42°C for 15 minutes followed by heating at 95°C for 5 minutes. To establish conditions that allow comparison of the amounts of cDNA produced by RT-PCR, we varied the number of cycles from 24 to 40; a cycle number of 30 was chosen to compare the different levels of expression of various mRNAs. Three-step cycling was performed as follows: denaturation for 45s at 95°C, annealing for 45s at 60°C and extension for 1 minute at 72°C. The 3-step cycle was repeated for 35 cycles and followed by a final extension at 72°C for 5 minutes. Each of the PCR products was separated on 3 % agarose gels and visualized by ethidium bromide staining. The resulting gels were scanned with a Pdi scanner (model 420oe, Pdi; Huntington Station, NY) and the densitometry analyzed with the ImageJ 1.32 program. We also chose the housekeeping gene 18S rRNA as an endogenous standard. The RT-PCR product from 18S rRNA-specific primers (Ambion) produced a 488-base pair fragment.
Table 1.
The forward and reverse primers and the length of PCR product in each AR-subtype for RT-PCR
| AR-Subtype | Sequence | Fragment Length |
|---|---|---|
| α1A | F 5'-GAATGTCCTGCGAATCCAGT-3'
R 5'-GATTGGTCCTTTGGCACTGT-3' |
237 |
| α1B | F 5’-AACCTTGGGCATTGTAGTCG-3’
R 5’ACCCAAGGATACGCATGAAG-3’ |
212 |
| α1D | F 5’-AGCCTCTGCACCATCTCTGT-3’
R 5’-AAGGAGCACACGGAAGAGAA-3’ |
233 |
| α2A | F 5’-GGTAAGGTGTGGTGCGAGAT-3’
R 5’-CAGCGCCCTTCTTCTCTATG-3’ |
229 |
| α2C | F 5’-TACTGTGCTGGTTCCCCTTC-3’
R 5’-CAGAGGCCCAGTTGTCTCTC-3’ |
245 |
| β1 | F 5’-CATCACGCTGCCCTTTCGC-3’
R 5’-CGGTTGGTGACGAAATCGC-3’ |
256 |
| β2 | F 5’-AAGTTCGAGCGACTACAAACCGT-3’
R 5’-TGAAGAAGTCACAGCAAGTCTCC-3’ |
279 |
| β3 | F 5’-ACAGACCATAACCAACGTGTTCG-3’
R 5’-GAACACTCGAGCATAGACGAAGA-3’ |
284 |
2.5. Membrane Protein Preparation and Western Blot Analysis
Both transfected and non-transfected VSMCs were washed twice with ice-cold phosphate-buffered saline, then dislodged and homogenized in ice-cold Tris-HCl buffer (50mmol/L Tris, 5mmol/L EDTA, 150mmol/L NaCl) and centrifuged at 51500g for 30 minutes at 4°C. The pellet was resuspended in the same buffer and homogenized again. The protein concentration was determined by the Bio-Rad DC protein assay. Samples (in equal protein concentration) were mixed with 3×loading buffer (0.2M Tris-Cl PH 6.8, 6% SDS, 30% glycerol, 2% DTT, 0.03% bromophenol blue) and heated at 95°C for 5 minutes. Gel electrophoresis was performed at 5% stacking, 10% separating SDS-polyacrylamide gels. Proteins were subsequently transferred to a pure nitrocellulose membrane (Bio-Rad, Hercules, CA) for 1 hr at 90V. Following this, the blot membrane was incubated for 1 hr in blocking buffer (PBS containing 5% dry non-fat milk and 0.1% Tween-20). After washing the membrane was incubated overnight at 4°C with polyclonal anti- α1D-AR (Santa Cruz Biotechnology, Santa Cruz, California) or anti- α2A-AR (Affinity Bioreagents, Golden, CO), or β-actin (Sigma-Aldrich, Milwaukee, WI) antibody. The antibody solution was removed, and the membranes were washed 3 times with PBS that contained 0.1% Tween-20. Incubation with horseradish peroxidase-conjugated donkey anti-goat immunoglobulin G (IgG) secondary antibody (Santa Cruz Biotechnology) at 1:5,000 dilution was carried out for 45 min at room temperature. Chemiluminescence was detected immediately using the ECL Western Blotting Detection Kit (Amersham Bioscience, Buckinghamshire, UK). The quantification of α1D-AR, α2A-AR and β-actin expression was determined by densitometry scanning followed by computer analysis using the ImageJ 1.32 program.
2.6. Statistical Analysis
All data are expressed as means ± SD. The statistical analyses were carried out with SAS 8.1 (SAS Institute, Inc, Cary, NC). We used the General Linear Models (GLM) procedure in SAS to compare mean differences across groups, with Duncan adjustment. The control group was set as reference. A P < 0.05 level of significance was chosen in all of our analyses.
3. Results
3.1. Effects of α1D-AR siRNA transfection on AR protein levels
After preliminary transfections with α1D-AR/Block-it plasmid containing the 4 different target sequences, we chose the one located in 3'UTR (5'-AAACTACTTAGTCAACTCCTA-3') because it produced the maximal inhibition in protein level. Therefore, we used this sequence to make the α1D-AR/pSEC expression plasmid for all subsequent experiments. In non-transfection groups, at either mRNA or protein level, gene expressions remained the same at 24 h, 48 h and 72 h. Therefore, we present one value of non-transfected control for comparison with the transfected groups.
The successful post-transcriptional silencing of the α1D-AR gene by the selected siRNA nucleotide sequence was demonstrated by the highly significant decrease in the level of α1D-AR protein generation. Figure 1 shows the time-dependent decrease of α1D-AR protein levels in VSMCs evaluated by Western blot analysis at 24 h, 48 h and 72 h after transfection. Compared to the non-transfected VSMCs, the α1D-AR protein levels in siRNA-transfected cells at these time points were decreased by 55% (p < 0.07), 69% (p < 0.03) and 75% (p < 0.01), respectively. In parallel, in the siRNA transfected cells there was a significant increase in α2A-AR protein level that peaked at 48 h and tended to return to baseline thereafter (Figure 2).
Figure 1.
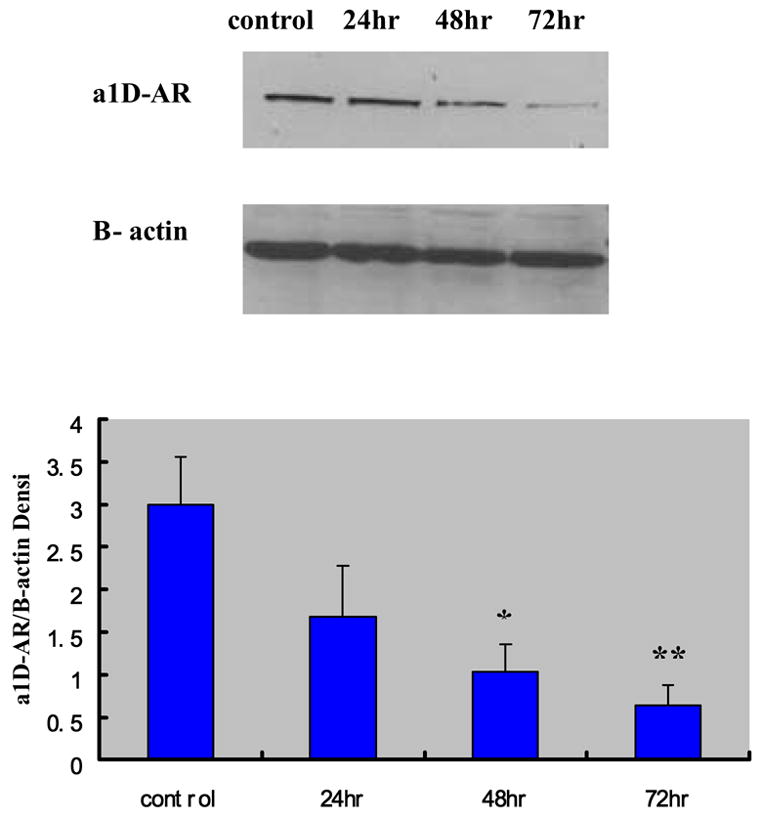
Effects of siRNA on the protein level of α1D-AR at 24 hrs, 48 hrs and 72 hrs after transfection (mean ± standard deviation from six independent experiments) The densitometry of different groups was adjusted according to that of β-actin. *P < 0.05; **P < 0.01 compared to the non transfection control group.
Figure 2.
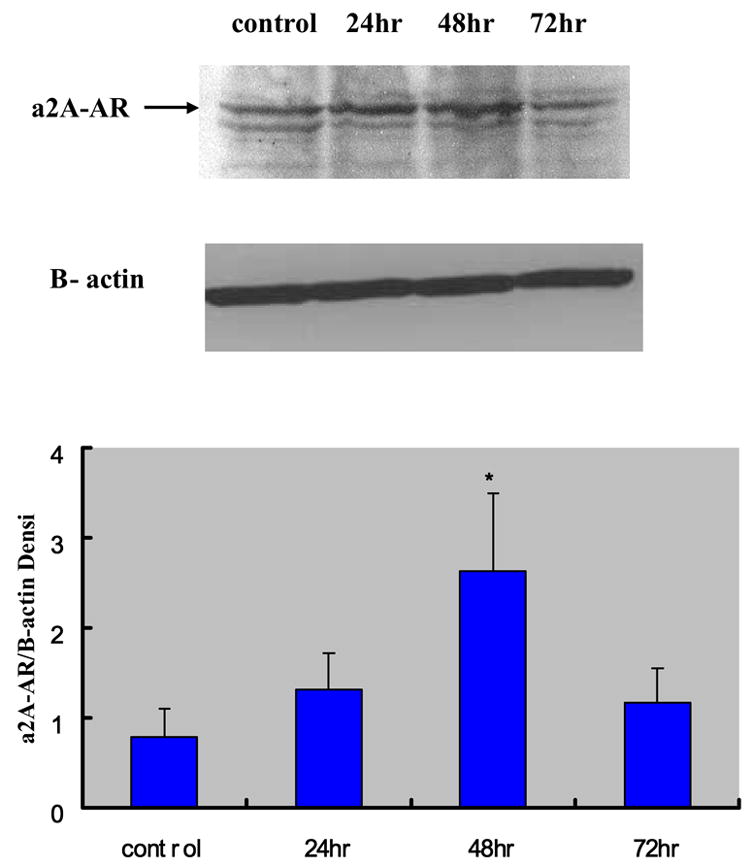
Effects of α1D-AR/siRNA on the protein expression level of α2A-AR at 24 hrs, 48 hrs and 72 hrs after transfection (mean ± standard deviation from six independent experiments). The densitometry of different groups was adjusted according to that of β-actin. *P < 0.05 compared to the control group.
3.2. Effects of α1D-AR siRNA transfection on AR gene expression
Using quantitative RT-PCR we evaluated the changes in AR-gene expression at 24 h, 48 h and 72h after transfection of VSMCs by the α1D-AR specific siRNA. Figure 3 shows the progressive increase in α1D-AR gene expression, which tended to rise at 48 h and increased significantly by 61% at 72 h (p < 0.05), in parallel to the progressive decline in generation of its protein. At the same time, there were changes in gene expression of other related ARs. Table 2 shows that the post-transcriptional silencing of the α1D-AR gene was associated with a steep decrease in expression of the α1A-AR gene (p < 0.05), the α2A-AR gene (p < 0.01) and the α2C-AR gene (although the last did not attain significance), between 24 h and 48 h, with no further change at 72 h; whereas the α1B-AR gene expression remained unchanged (The α2B-AR gene was undetectable in aortic VSMC in accordance with our previous finding (Handy et al., 1998)). The expression of the β3-AR gene was also significantly decreased at 48h (p < 0.05) and 72h (p < 0.01), whereas the β1-AR gene tended also to decrease its expression and the β2-AR gene remained unchanged.
Figure 3.
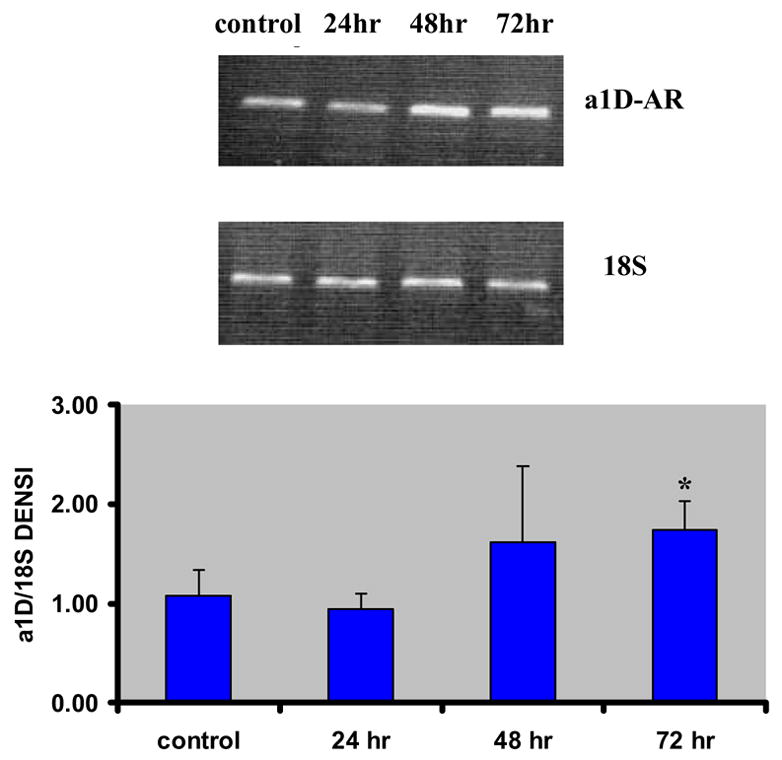
Effects of siRNA on the level of mRNA expression of α1D-AR gene at 24 hrs, 48 hrs and 72 hrs after transfection (mean ± standard deviation from six independent experiments). The densitometry of different groups was adjusted according to that of 18S. *P < 0.05 compared to the control group.
Table 2.
Effect of α1D-AR/siRNA on the level of mRNA expression of other AR genes at 24 hrs, 48 hrs and 72 hrs after transfection assessed by densitometry
| Control | 24h | 48h | 72h | |
|---|---|---|---|---|
| α1A | 1.72±0.64 | 1.83±0.16 | 0.88±0.14 | 0.79±0.18* |
| α1B | 1.69±0.26 | 1.32±0.20 | 1.30±0.21 | 1.39±0.24 |
| α2A | 1.99±0.25 | 1.90±0.05 | 1.06±0.10** | 0.79±0.12** |
| α2C | 1.24±0.71 | 1.38±0.56 | 0.62±0.27 | 0.59±0.47 |
| β1 | 0.71±0.55 | 1.27±0.93 | 0.95±0.36 | 0.44±0.14 |
| β2 | 1.48±1.06 | 1.56±0.99 | 1.43±0.82 | 0.82±0.22 |
| β3 | 0.95±0.18 | 0.97±0.48 | 0.30±0.04* | 0.16±0.94** |
Each value is the mean ± SD of six independent experiments. The densitometry of different groups was adjusted according to that of 18S.
P < 0.05,
P<0.01 as compared to the control group.
4. Discussion
Gene therapy has been recently proposed as a new approach for the treatment of hypertension (Phillips, 2001), aiming at altering the levels or function of various vasoactive systems. Approaches have included the delivery of extra copies of candidate genes, whose products contribute to vasodilation, and the inhibition of expression of genes, whose products contribute to vasoconstriction. The latter approach has consisted mostly of antisense oligodeoxynucleotides targeting a specific gene sequence, in order to suppress the gene’s expression. The antisense sequence is delivered either naked or attached to a plasmid vector or an adeno-associated viral vector, in order to prolong its action. In recent years, we have explored all three methods in order to reverse salt-induced hypertension by targeting the α2B-AR gene (Kintsurashvili et al., 2001; Kintsurashvili et al., 2003; Shenouda et al., 2006). The current experiments were designed to explore in vitro an alternative method of inhibiting gene expression of an α-AR subtype, namely, that of post-transcriptional silencing by RNAi, and we chose as target the VSMC α1D-AR. Although different α1-AR subtypes have been reported to predominate in different arteries of various animal species, the α1D-AR is predominant in the aortic wall and has been shown to be mostly responsible for catecholamine-induced systemic vasoconstriction. We selected the post-transcriptional silencing of the gene by siRNA, which requires only a few molecules of double-stranded RNA (dsRNA) to almost completely abolish the encoded product of a gene homologous to the dsRNA. We demonstrated that transfection of VSMCs with the selected α1D/siRNA nucleotide sequence, progressively decreased the production of α1D-AR protein (assessed by Western blot) up to 75% over the next three days. In parallel, there was a gradual increase of up to 61% in the degree of expression of the α1D-AR gene (as assessed by RT-PCR), probably reflecting an unsuccessful effort to counteract the loss of receptor protein resulting from post-transcriptional silencing. Of particular interest, however, was the concurrent increase in α2A-AR protein level within these VSMCs, with a subsequent decrease in the expression of that gene. This appears to be a compensatory reaction aiming at preserving to a large extent the contractile capacity of the VSMCs in response to adrenergic stimuli, since the postsynaptic α2A-AR is also a major contributor to catecholamine-induced vasoconstriction (Duka et al., 2000).
Several other AR genes were also found to exhibit diminished expression over the next 72 h, most pronounced in the α1A, α2A and β3-AR genes. All AR subtypes belong to the same G protein-coupled receptor superfamily of membrane proteins (Bylund, 1992; Lefkowitz and Caron, 1988) with overlapping functions in response to various ligands. Furthermore, they are encoded by genes with similar features, suggesting that all may have evolved from a single ancestral gene, which would explain conservation of particular sequences and spacing between key functional amino acids. Therefore, it should not be surprising that cross-talk exists among these genes (Deng et al., 1998), so that suppressing the activity of one would lead to compensatory reactions from others, whose activation elicits similar responses (Daly et al., 2002; Devic et al., 2001; Hutchinson et al., 2001; Trendelenburg et al., 2001). In selecting the target sequence for α1D-AR RNAi, we ensured that there was no homologous sequence with other off-target genes. Accordingly, any changes in related other AR genes should be attributable to cross-talk rather than to inadvertent interference with those genes’ RNA. The specificity of siRNA-mediated RNAi for the α1D-AR was further ascertained by the use of a GAPDH siRNA vector as a positive control; by demonstrating that the 21-nucleotide sequence selected uniquely against the GAPDH mRNA had no influence on the α1D-AR gene expression or protein production, we proved that changes in expression of various AR genes following transfection of α1D-AR siRNA were not unexpected non-specific reactions, but rather the results of cross-talk among genes of receptors with overlapping structural and functional characteristics (Ruffolo et al., 1991).
Compensatory alterations have been described before in related receptors when one receptor is blocked or its gene is deleted: examples are upregulation of α-AR or β-AR subtypes in gene knockout mice with deletion of other α or β-AR subtypes (Daly et al., 2002; Devic et al., 2001; Hutchinson et al., 2001; Rohrer et al., 1999; Trendelenburg et al., 2001). In fact, when a normal AR-receptor population is genetically suppressed, another AR-subtype can replace it with a similar cellular distribution. Thus, in α1B-AR knockout mice, hepatic function of α1B-ARs is assumed by α1A-AR (Deighan et al., 2004); in α1D-AR knockout mice, the expression of α1A- and α1B-AR tended to increase in brain tissues compared to wildtype mice (Tanove et al., 2002), and so did the mesenteric artery expression of α1A-AR in double α1D- and α1B-AR knockouts (Hosoda et al., 2005). This is by no means unique to ARs: in a series of experiments investigating the properties of bradykinin receptors, we also demonstrated that pharmacologic blockade or genetic engineering to eliminate one type of bradykinin receptor leads to upregulation of the other, which compensates for many of the affected receptor’s functions (Duka et al., 2001; Duka et al., 2003).
5. Conclusions
In summary, in these experiments we employed the method of RNAi via siRNA for post-transcriptional silencing of the α1D-AR gene in rat VSMCs. We demonstrated that the approach can profoundly suppress the production of the targeted protein despite subsequent significant reactive upregulation of the target gene, and that such interference leads to cross-talk among related AR-genes with significant concurrent alterations in their expression patterns. Further experiments in vivo are needed to explore whether this approach to gene treatment would yield the hemodynamic results expected from blockade of the selected gene, or whether compensatory changes from genes of receptors with overlapping structure/function characteristics might minimize the anticipated effects. The potential therapeutic implications of this treatment are of great interest, since pharmacologic blockade of α1-ARs is widely used as adjunct treatment for hypertension, as well as urinary bladder and sphincter disorders.
Acknowledgments
This study was partially supported by NIH Grant R01 HL 65311.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bylund DB. Subtypes of α1- and α 2-adrenergic receptors. Faseb J. 1992;6:832–839. doi: 10.1096/fasebj.6.3.1346768. [DOI] [PubMed] [Google Scholar]
- Daly CJ, Deighan C, McGee A, Mennie D, Ali Z, McBride M, McGrath JC. A knockout approach indicates a minor vasoconstrictor role for vascular α1B-adrenoceptors in mouse. Physiol Genomics. 2002;9:85–91. doi: 10.1152/physiolgenomics.00065.2001. [DOI] [PubMed] [Google Scholar]
- Deighan C, Woollhead AM, Colston JF, McGrath JC. Hepatocytes from α1B-adrenoceptor knockout mice reveal compensatory adrenoceptor subtype substitution. Br J Pharmacol. 2004;142:1031–1037. doi: 10.1038/sj.bjp.0705872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng XF, Sculptoreanu A, Mulay S, Peri KG, Li JF, Zheng WH, Chemtob S, Varma DR. Crosstalk between α-1A and α-1B adrenoceptors in neonatal rat myocardium: implications in cardiac hypertrophy. J Pharmacol Exp Ther. 1998;286:489–496. [PubMed] [Google Scholar]
- Devic E, Xiang Y, Gould D, Kobilka B. Beta-adrenergic receptor subtype-specific signaling in cardiac myocytes from β1 and β2 adrenoceptor knockout mice. Mol Pharmacol. 2001;60:577–583. [PubMed] [Google Scholar]
- Duka I, Duka A, Kintsurashvili E, Johns C, Gavras I, Gavras H. Mechanisms mediating the vasoactive effects of the B1 receptors of bradykinin. Hypertension. 2003;42:1021–1025. doi: 10.1161/01.HYP.0000097550.98865.35. [DOI] [PubMed] [Google Scholar]
- Duka I, Gavras I, Johns C, Handy DE, Gavras H. Role of the postsynaptic α2-adrenergic receptor subtypes in catecholamine-induced vasoconstriction. Gen Pharmacol The Vascular System. 2000;34:101–106. doi: 10.1016/s0306-3623(00)00051-3. [DOI] [PubMed] [Google Scholar]
- Duka I, Kintsurashvili E, Gavras I, Johns C, Bresnahan M, Gavras H. Vasoactive Potential of the B1 Bradykinin Receptor in Normotension and Hypertension. Circ Res. 2001;88:275–281. doi: 10.1161/01.res.88.3.275. [DOI] [PubMed] [Google Scholar]
- Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- Endoh M, Hiramoto T, Ishihata A, Takanashi M, Inui J. Myocardial α1-adrenoceptors mediate positive inotropic effect and changes in phosphatidylinositol metabolism. Species differences in receptor distribution and the intracellular coupling process in mammalian ventricular myocardium. Circ Res. 1991;68:1179–1190. doi: 10.1161/01.res.68.5.1179. [DOI] [PubMed] [Google Scholar]
- Gavras H, Handy D, Gavras I. Alpha-adrenergic Receptors in Hypertension. In: Laragh JH, Brenner BM, editors. Hypertension: Pathophysiology, Diagnosis, and Management. Raven; New York: 1995. pp. 853–861. [Google Scholar]
- Gavras I, Manolis AJ, Gavras H. The α2 -adrenergic receptors in hypertension and heart failure: experimental and clinical studies. J Hypertens. 2001;19:2115–2124. doi: 10.1097/00004872-200112000-00001. [DOI] [PubMed] [Google Scholar]
- Gisbert R, Noguera MA, Ivorra MD, D'Ocon P. Functional evidence of a constitutively active population of α1D-adrenoceptors in rat aorta. J Pharmacol Exp Ther. 2000;295:810–817. [PubMed] [Google Scholar]
- Gitlin L, Karelsky S, Andino R. Short interfering RNA confers intracellular antiviral immunity in human cells. Nature. 2002;418:430–434. doi: 10.1038/nature00873. [DOI] [PubMed] [Google Scholar]
- Handy DE, Johns C, Bresnahan MR, Tavares A, Bursztyn M, Gavras H. Expression of α2-adrenergic receptors in normal and atherosclerotic rabbit aorta. Hypertension. 1998;32:311–317. doi: 10.1161/01.hyp.32.2.311. [DOI] [PubMed] [Google Scholar]
- Hosoda C, Tanoue A, Shibano M, Tanaka Y, Hiroyama M, Koshimizu TA, Cotecchia S, Kitamura T, Tsujimoto G, Koike K. Correlation between vasoconstrictor roles and mRNA expression of alpha1-adrenoceptor subtypes in blood vessels of genetically engineered mice. Br J Pharmacol. 2005;146:456–466. doi: 10.1038/sj.bjp.0706325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrometz SL, Edelmann SE, McCune DF, Olges JR, Hadley RW, Perez DM, Piascik MT. Expression of multiple alpha1-adrenoceptors on vascular smooth muscle: correlation with the regulation of contraction. J Pharmacol Exp Ther. 1999;290:452–463. [PubMed] [Google Scholar]
- Hutchinson DS, Evans BA, Summers RJ. β1-Adrenoceptors compensate for β3-adrenoceptors in ileum from β3-adrenoceptor knockout mice. Br J Pharmacol. 2001;132:433–442. doi: 10.1038/sj.bjp.0703828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda U, Tsuruya Y, Yaginuma T. α1-adrenergic stimulation is coupled to cardiac myocyte hypertrophy. Am J Physiol. 1991;260:H953–956. doi: 10.1152/ajpheart.1991.260.3.H953. [DOI] [PubMed] [Google Scholar]
- Jeffries WB, Yang E, Pettinger WA. Renal α1-adrenergic receptor response coupling in spontaneously hypertensive rats. Hypertension. 1988;12:80–88. doi: 10.1161/01.hyp.12.1.80. [DOI] [PubMed] [Google Scholar]
- Kenny BA, Chalmers DH, Philpott PC, Naylor AM. Characterization of an alpha 1D-adrenoceptor mediating the contractile response of rat aorta to noradrenaline. Br J Pharmacol. 1995;115:981–986. doi: 10.1111/j.1476-5381.1995.tb15907.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kintsurashvili E, Gavras I, Johns C, Gavras H. Effects of Antisense oligodeoxynucleotide targeting of the α2B-adrenergic receptor messenger RNA in the central nervous system. Hypertension. 2001;38:1075–1080. doi: 10.1161/hy1101.093426. [DOI] [PubMed] [Google Scholar]
- Kintsurashvili E, Johns C, Ignjacev I, Gavras I, Gavras H. Central α2B-adrenergic receptor antisense in plasmid vector prolongs reversal of salt-dependent hypertension. J Hypertens. 2003;21:961–967. doi: 10.1097/00004872-200305000-00021. [DOI] [PubMed] [Google Scholar]
- Kurz T, Yamada KA, DaTorre SD, Corr P. Alpha1-adrenergic system and arrhythmias in ischaemic heart disease. Eur Heart J. 1991;12(Suppl F):88–98. doi: 10.1093/eurheartj/12.suppl_f.88. [DOI] [PubMed] [Google Scholar]
- Lefkowitz RJ, Caron MG. Adrenergic receptors. Models for the study of receptors coupled to guanine nucleotide regulatory proteins. J Biol Chem. 1988;263:4993–4996. [PubMed] [Google Scholar]
- Makaritsis KP, Johns C, Gavras I, Gavras H. Role of α2-adrenergic receptor subtypes in the acute hypertensive response to hypertonic saline infusion in anephric mice. Hypertension. 2000;35:609–613. doi: 10.1161/01.hyp.35.2.609. [DOI] [PubMed] [Google Scholar]
- McManus MT, Sharp PA. Gene silencing in mammals by small interfering RNAs. Nat Rev Genet. 2002;3:737–747. doi: 10.1038/nrg908. [DOI] [PubMed] [Google Scholar]
- Phillips MI. Gene therapy for hypertension: the preclinical data. Hypertension. 2001;38:543–548. doi: 10.1161/hy09t1.092927. [DOI] [PubMed] [Google Scholar]
- Piascik MT, Guarino RD, Smith MS, Soltis EE, Saussy DL, Jr, Perez DM. The specific contribution of the novel α-1D adrenoceptor to the contraction of vascular smooth muscle. J Pharmacol Exp Ther. 1995;275:1583–1589. [PubMed] [Google Scholar]
- Rohrer DK, Chruscinski A, Schauble EH, Bernstein D, Kobilka BK. Cardiovascular and metabolic alterations in mice lacking both β1- and β2-adrenergic receptors. J Biol Chem. 1999;274:16701–16708. doi: 10.1074/jbc.274.24.16701. [DOI] [PubMed] [Google Scholar]
- Ruffolo RR, Jr, Nichols AJ, Oriowo MA. Interaction of vascular α-1 adrenoceptors with multiple signal transduction pathways. Blood Vessels. 1991;28:122–128. doi: 10.1159/000158851. [DOI] [PubMed] [Google Scholar]
- Ross R. The smooth muscle cell. II Growth of smooth muscle in culture and formation of elastic fibers. J Cell Biol. 1971;50:172–186. doi: 10.1083/jcb.50.1.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenouda SM, Johns C, Kintsurashvili E, Gavras I, Gavras H. Long-Term Inhibition of the Central α2B-Adrenergic Receptor Gene via Recombinant Adeno-associated Virus-Delivered Antisense DNA in Hypertensive Rats. Am J Hypertens. 2006 doi: 10.1016/j.amjhyper.2006.04.001. (in press) [DOI] [PubMed] [Google Scholar]
- Tanoue A, Nasa Y, Koshimizu T, Shinoura H, Oshikawa S, Kawai T, Sunada S, Takeo S, Tsujimoto G. The α1D-adrenergic receptor directly regulates arterial blood pressure via vasoconstriction. J Clin Invest. 2002;109:765–75. doi: 10.1172/JCI14001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trendelenburg AU, Norenberg W, Hein L, Meyer A, Starke K. Alpha2-adrenoceptor-mediated inhibition of cultured sympathetic neurons: changes in alpha2A/D-adrenoceptor-deficient mice. Naunyn Schmiedebergs Arch Pharmacol. 2001;363:110–119. doi: 10.1007/s002100000331. [DOI] [PubMed] [Google Scholar]
- Villalobos-Molina R, Ibarra M. Vascular α1D-adrenoceptors: are they related to hypertension? Arch Med Res. 1999;30:347–352. doi: 10.1016/s0188-0128(99)00047-0. [DOI] [PubMed] [Google Scholar]
- Young MA, Vatner DE, Knight DR, Graham RM, Homcy CJ, Vatner SF. Alpha-adrenergic vasoconstriction and receptor subtypes in large coronary arteries of calves. Am J Physiol. 1988;255:H1452–1459. doi: 10.1152/ajpheart.1988.255.6.H1452. [DOI] [PubMed] [Google Scholar]