

Abstract
CD4+CD25+FOXP3+ regulatory T-cells (Tregs) form an important arm of the immune system responsible for suppressing untoward immune responses. Tregs can be thymically derived or peripherally induced, even from CD4+CD25−FOXP3− T-cells. FOXP3 expression and in vitro suppressive activity are considered unique hallmarks of this dedicated and stable lineage of regulatory cells. Here we show that virtually all human CD4+CD25−FOXP3− T-cells and CD8+CD25−FOXP3− T-cells attain a transient FOXP3+CD25+ state during activation. In this state of activation, these cells possess the classic phenotype of Tregs, in that they express similar markers and inhibit in vitro proliferation of autologous CD4+CD25− T-cells. This state is characterized by suppressed IFN-γ production and robust TNF-α and IL-10 production. Interestingly, the great majority of the activated cells eventually downregulate FOXP3 expression, with a concomitant drop in suppressive ability. Our results show that, in humans, FOXP3 expression and Treg functionality are not exclusive features of a stable or unique lineage of T-cells, but may also be a transient state attained by almost all T-cells. These results warrant caution in interpreting human studies using FOXP3 and suppressive activity as readouts and suggest that attempts to induce “Tregs” may paradoxically result in induction of effector T-cells, unless stability is confirmed.
Introduction
CD4+CD25+/high regulatory T (Treg) cells are an important arm of the immune system that downregulate potentially harmful effector immune responses [1]. They have been shown to play a role in autoimmune disorders, infections, tumors, asthma, allergy and transplantation and hence their modulation in these diseases is thought to be of great potential benefit [2]. Further elucidation of this subset has been hampered by the lack of a specific surface marker to isolate and study these cells. Markers identified as being expressed on Tregs are also expressed by activated T-cells. Hence, the discovery of the transcription factor FOXP3 as a marker that is expressed in Tregs but not on activated T-cells held major significance. Predominantly through murine studies it was shown that FOXP3 is necessary and sufficient for the development and function of Tregs [3; 4; 5; 6; 7]. Transduction of FOXP3 in human CD4+CD25− T-cells appeared to confer regulatory properties to those cells [7]. FOXP3 regulates T-cell activation by interacting with NF-AT or NF-κB and consequently repressing IL-2 secretion [8; 9].
CD4+CD25+/high Tregs can be subdivided into natural Tregs and induced Tregs. Natural CD4+CD25+FOXP3+ Tregs are thought to arise in the thymus and suppress harmful immune responses in the periphery [1]. While FOXP3 expression is thought to be a unique feature of natural Tregs in mice [3; 4; 5], several human studies and some murine studies have suggested that CD4+CD25− T-cells may give rise to induced CD4+CD25+FOXP3+ regulatory T-cells following stimulation [10; 11; 12; 13]. However, due to the lack of antibodies against intranuclear FOXP3 at that time, it had been repeatedly suggested that such cells merely represent an expansion of contaminating populations of natural Tregs [14; 15; 16; 17]. This continues to be a controversy in the field, with some studies reporting no upregulation of FOXP3 expression [7] and others proposing that induced FOXP3+ T-cells may not be regulatory in function [18]. However, these studies did not directly evaluate suppressor function. Also, it is currently unclear if the induced and natural Tregs represent separate lineages or if they belong to the same lineage but just differ in their location and timing of their origin. In either case, it is generally believed that FOXP3-expressing T-cells, induced or natural, are a stable population of T-cells with immune regulatory functions. Thus, the presence of FOXP3 and the ability to suppress effector T-cell responses in-vitro have been used as the hallmarks for the detection and quantification of this population [15]. This approach has been widely used recently in human disease settings where presence or absence of FOXP3+ T-cells at the disease site or in the blood (with or without the presence of concomitant suppressive activity) is interpreted as evidence for involvement of Tregs in the disease pathogenesis/modulation [19; 20; 21; 22; 23].
In this study, we evaluated the immune biology of adaptively induced FOXP3+ T-cells by using polychromatic flow cytometry and recently developed robust anti-FOXP3 antibodies including one that recognizes a specific spliced isoform of human FOXP3 [24]. Using CFSE staining of highly purified T-cell populations, we tracked their dynamics and function following activation with different stimuli. Using this approach, we conclusively show that virtually all activated CD4+ and CD8+ T-cells transiently upregulate FOXP3 and show transient suppressive activity. We further show that this transient regulatory state and certain effector functions are differentially regulated, suggesting that this state might be a general immune mechanism of fine-tuning an ongoing immune response.
Materials and Methods
Cell preparation and bead sorting
PBMC were isolated from fresh buffy coats from healthy blood donors using Ficoll Hypaque density gradient. “Untouched” CD4+ T-cells and CD8+ T-cells were negatively selected using negative selection kits and AutoMacs (DEPLETE program) from Miltenyi Biotech. Total CD3+ T-cells were negatively selected using MagCellect negative selection kits from R&D systems. All of these were negatively selected to greater than 85% purity. CD25+ T-cells were depleted from the purified CD4+ and CD8+ T-cells using CD25 microbeads and AutoMacs (DEPLETES program) to greater than 95% purity. CD45RO microbeads were used for CD45RO depletion (depletion greater than 95%). CD3 microbeads were used to deplete T-cells from PBMC to enrich for antigen-presenting cells (APC). The CD3+ T-cell-depleted population was irradiated at 3000 rads before being used as APC’s. Aliquots of CD4+CD25− T-cells and CD3-depleted cells were frozen for use at later time points as responders and APC’s, respectively.
CFSE-based proliferation assays
T-cell proliferation was detected and quantified by flow cytometric proliferation assays, utilizing the green fluorescent dye, 5 (and 6)-carboxyfluorescien diacetate succinimidyl ester (CFSE), as described previously [25; 26]. Briefly, cells were first suspended at 1 X 106/ml in PBS and incubated at 37oC for 7 min with 0.25 μM CFSE, followed by serum quenching and PBS washes. These cells were subjected to the indicated culture conditions (in human serum-containing media), followed by evaluation of their proliferative responses and expression of various markers. Importantly, no exogenous cytokines (such as IL-2) were added to any of the cultures.
FOXP3 Staining
Human FOXP3 staining kits from eBiosciences were used to stain for intracellular FOXP3. PCH101 anti-FOXP3-PE or anti-FOXP3-AlexaFluor700 were used to stain for total FOXP3, while FJK16S anti-FOXP3-PE was used to stain for full-length FOXP3.
Flow cytometric staining, antibodies, sorting and data analysis
Flow cytometric sorting of dividing vs. non-dividing populations by CFSE was performed on a FACSVantage (with FACSDiva upgrade) to greater than 95% purity, as described previously [25; 26]. Multicolor phenotyping panels were set up using different combinations of CTLA-4-PECY5, CCR7-PECY7, CD4-PECY5.5 (Caltag), CD8-Pacific Blue, CD25-APC or APCCY7, CD62L-PE or PECY5, CD27-APCCY7, CD28-PECY5 or Biotin+ strepavidin Qdot 655 (Invitrogen), CD127 (IL-7R) APC (R&D systems), CD45RA-FITC and CD45RO-APC (all antibodies from BD Biosciences unless indicated otherwise). Flow cytometric data was acquired on a 4-Laser, 17-color custom BD LSRII using FACSDiva software. Linear uncompensated data was then transferred as FCS 3.0 files and analysed after compensation and transformation using FlowJo version 6.4.1 (TreeStar), as recommended [27; 28]. Gating and analysis were performed as described [25; 26]. Cut-offs for positive populations were determined by using either fluorescence minus one (FMO) staining for polychromatic flow cytometry, no stimulus background staining or isotype control staining, as appropriate [28; 29]. Samples from experiments involving readouts at different days were run with the same appropriate instrument and compensation settings to ensure comparability. The proliferation platform in FlowJo was used to back-calculate the precursor frequency of the dividing cells by CFSE dilution.
Antigens
Anti-CD3 (BD Biosciences) was used at a concentration of 1 μg/ml/million cells. Anti-CD28 (BD Biosciences) was used at 1 μg/ml/million cells. Whole CMV lysates (50 μg/ml, Microbix Biosystems), tetanus toxoid (20 μg/ml; Accurate Chemical and Scientific) were used as antigens to stimulate PBMC.
3H-thymidine-based suppression assays
Suppression assays were performed in duplicate in 96-well plates. 50,000 CD4+CD25− T cells (Responders) were cultured with 50,000 irradiated APC and anti-CD3 in a total volume of 200 μl/well as previously described [11]. Varying ratios of induced or ex vivo-purified Tregs were added to the cultures. The cultures were pulsed with 3H-thymidine on day 4 and harvested on day 5 to determine their proliferation, as previously described [10; 25; 26].
Quantitative Real Time PCR
Total RNA was isolated using the QIAamp RNA mini kit (Qiagen, Valencia, CA). Random hexamers were used to convert mRNA into cDNA using SuperScript First-Strand Synthesis system from Invitrogen. Isoforms of FOXP3 were then amplified using 5’-GCCCTTGGACAAGGACCCGATG-3’ as the sense and 5’-CATTTGCCAGCAGTGGGTAGGA-3’ as the antisense primers (Invitrogen), allowing for detection of FOXP3 isoforms [30]. Relative expression of total FOXP3 was determined by normalizing to β-actin expression in SYBR-Green-based real time PCR reactions, as described previously [25; 26]. IFN-γ, IL-10, IL-4 and TNF-α messages were also quantified using published protocols and primers [26].
Results
All CD4+CD25− T-cells upregulate FOXP3 transiently following activation
First, we negatively selected “untouched” human CD3+CD25− (or CD4+CD25−) T-cells and stained them with CFSE. These cells were stimulated either in a mixed lymphocyte reaction using allogeneic, T-cell-depleted antigen-presenting cells (APC) or with anti-CD3 and autologous APC to stimulate the formation of induced FOXP3+ T-cells, as previously described [10; 11]. Following activation, these cells were monitored longitudinally for CFSE dilution and FOXP3/CD25 expression (to determine their proliferation and activation). In the absence of stimulation, the CD4+ T-cells did not show significant proliferation and remained CD25− and FOXP3− (Fig. 1a and 1b). In contrast, stimulated cells showed distinct upregulation of FOXP3 and CD25 as early as 2 days post-stimulation with peak FOXP3 expression at 4–6 days, depending on the stimulus. Of note, virtually all dividing cells (CFSElow) and CD25+ (activated) cells expressed FOXP3 at earlier time points (Fig. 1a, 1b and 1c). This was clearly visible in allostimulated cells at the 6-day time point. A similar pattern was noted following stimulation of PBMC with nominal antigens, such as tetanus toxoid (TT) or cytomegalovirus (CMV) lysate (Fig. 1d). Importantly, FOXP3 expression by all activated cells was a transient phenomenon, lasting for 48–72 hours, followed by downregulation and plateauing around day 10–14 of culture (Fig. 1a, 1c). While the kinetics varied according to the stimulus and geometry of culture (flask versus tube), all stimuli invariably resulted in the same pattern of transient upregulation followed by downregulation.
Figure 1. CD4+CD25− T-cells upregulate FOXP3 transiently following activation.
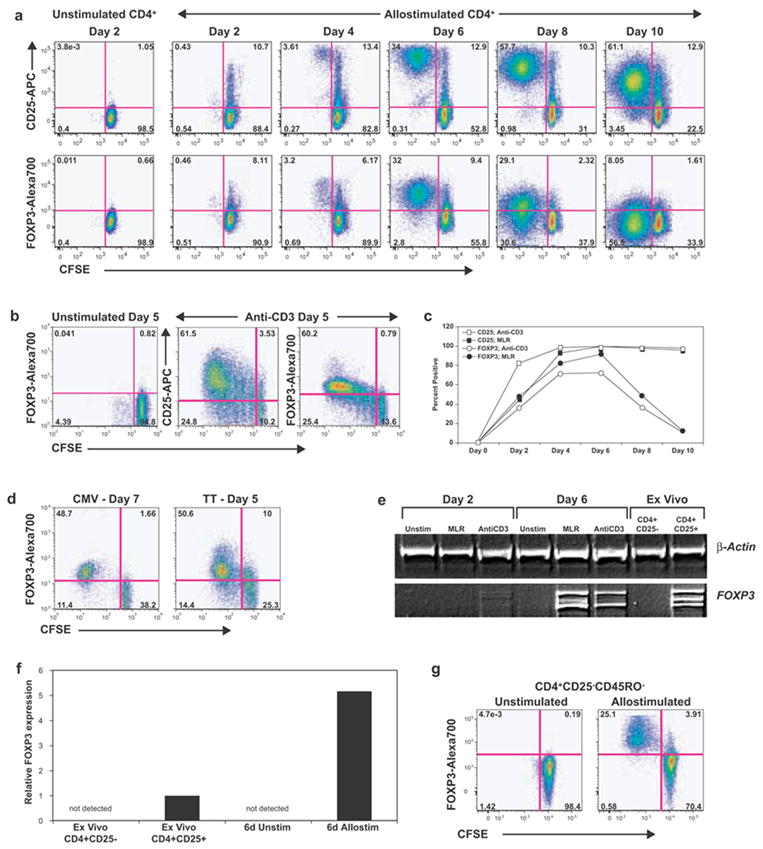
Bead sorted ‘untouched’ CD3+CD25− T-cells (a, c) or CD4+CD25− T-cells (b, e and f) were stained with CFSE and activated with either allogeneic T-cell-depleted PBMC (a) or with anti-CD3 antibody and autologous APC (b,c). CD25 and FOXP3 expression and CFSE dilution were monitored over time. a, Color-coded contour plots of gated CD4+ T-cells from allostimulated cultures are shown, with CFSE on the X-axis and CD25 (top row) or FOXP3 (bottom row) on the Y-axis. The vertical lines separate dividing (CFSElow) from non-dividing (CFSEhigh) cells. b, Anti-CD3 stimulated CD4+CD25− T-cells at five days of culture. c, Percentage of dividing cells that expressed CD25 (squares) or FOXP3 (circles) are shown at various time points following allogeneic (filled symbols) or anti-CD3 (open symbols) stimulation d, CFSE-stained PBMC stimulated with either CMV lysate (day 7) or tetanus toxoid (day 5) are shown. Data are gated for CD4+ T-cells. e, RT-PCR analysis of CD4+ T-cells for FOXP3 expression is shown. Primers were picked such that full-length isoform and the shorter alternatively spliced form could be detected as different bands in agarose gels. Cells were obtained from 2-day or 6-day cultures or purified ex vivo, as indicated. f, Total FOXP3 expression by quantitative real time PCR analysis. FOXP3 levels were first normalized to β-actin and then normalized to levels seen in ex vivo-purified CD4+CD25+ T-cells (assigned a value of 1.0). g, FOXP3 expression in allostimulated CD45RO-depleted CD4+CD25− naïve T-cells at 6 days of culture. All results are representative of at least 2 independent replicates performed on different donors (multiple replicates for a and b). Position of the positive gates for FOXP3 and CD25 in all graphs were determined based on the unstimulated control at the same time point and was also confirmed by either FMO (fluorescence minus one) staining (for polychromatic flow cytometry) or isotype control staining.
The expression of FOXP3 was confirmed with RT-PCR assays. Ex vivo-purified or unstimulated CD4+CD25− T-cells showed no detectable FOXP3 (Fig. 1e). In contrast, cells from activated cultures showed distinct upregulation of FOXP3, expressing both human isoforms [30]. Real time quantification showed that the levels of FOXP3 in activated cells were higher than ex vivo-purified CD4+CD25+ Tregs (Fig. 1f). As stated earlier and as shown in Fig. 1e, on day 6, FOXP3 expression had passed its peak in anti-CD3-stimulated cells, while it was at its peak in allostimulated cells. We also confirmed that FOXP3 message was downregulated at later time points (data not shown).
A significant portion of ex vivo-purified CD25− T-cells is made up of antigen-experienced, memory T-cells and it is possible that the induced FOXP3+ T-cells might predominantly arise from these antigen experienced populations. We next addressed whether FOXP3+ T-cells could be generated from naïve T-cells. We depleted the initial CD4+CD25− T-cells of CD45RO+ cells using microbeads, yielding >95% CD45RA+CD62Lhigh naïve T-cells [31]. Following anti-CD3 or allostimulation, we again observed a similar expression pattern of FOXP3 and CD25 over time (Fig.1g). Thus, virtually all activated/proliferating T-cells express FOXP3, even when originating from naïve T-cells.
It has been suggested that a minute population of contaminating FOXP3+ or CD25+ T-cells may account for the FOXP3+CD25+ population following stimulation [14; 15]. However, we did not observe appreciable FOXP3 expression in the initial or unstimulated population of CD4+CD25− T-cells, either by flow cytometry or by molecular assays. Even with liberal gating, FOXP3+ cells were always less than 1%. Moreover, the use of CFSE in our system allowed us to “back-calculate” the percentage of cells from the initial population that responded to the stimulus and expressed FOXP3. Following anti-CD3 stimulation, at least 40% of the original cells had divided and expressed FOXP3, making it impossible to explain this population merely as an expansion of contaminating FOXP3+ cells.
Induced FOXP3+ T-cells show transient in vitro suppressive ability
FOXP3+ Tregs suppress the in vitro proliferation of autologous CD4+CD25− effector T-cells [10; 11]. We asked whether the transient expression of FOXP3 in activated T-cells also imparted a transient regulatory functionality. For this, we used two approaches. First, we confirmed previous results by utilizing CD25 magnetic microbeads to obtain CD25+ T-cells from early cell cultures (at this time point, CD25 expression correlated with high FOXP3 expression). We tested their ability to suppress the proliferation of freshly purified, autologous CD4+CD25− T-cells in standard 3H-thymidine-based suppression assays (Fig. 2a). Similar to ex vivo-purified CD4+CD25+ Tregs, the CD25+ T-cells from anti-CD3 or allostimulated cultures showed robust suppressive activity as well as a relatively anergic phenotype (Fig. 2a). The induced Tregs being highly enriched in FOXP3+ T-cells appeared to have higher suppressive activity than bead-sorted CD25+ natural Tregs. However, this could be explained by the fact that the natural Treg population had contaminating CD25+FOXP3− effector T-cells (data not shown).
Figure 2. Induced “Tregs” show transient in vitro suppressive ability.
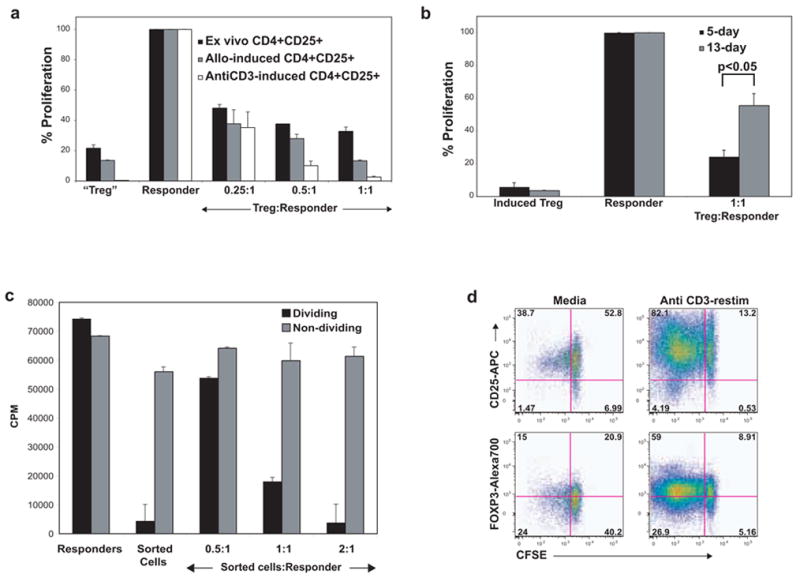
Bead-sorted, ‘untouched’ CD4+CD25− T-cells were activated with either anti-CD3 or allostimulation. a, CD4+CD25+ T-cells were sorted from PBMC ex vivo (black bars) or at 7 days of culture (as indicated) and tested for the ability to suppress the anti-CD3-mediated proliferation of freshly thawed, autologous responder CD4+CD25− T-cells in the presence of irradiated APC. The counts per minute (CPM) from a 5-day 3H-thymidine based assay were obtained and then normalized to the anti-CD3-induced proliferation of responder cells (assigned a value of 100; CPM were in the 50,000 to 80,000 range in most experiments on various donors). The sorted CD4+CD25+ T-cells were anergic and were able to suppress the responder cells at increasing ratios. In this experiment, the proportion of FOXP3+ cells was highest in anti-CD3-activated cells followed by allostimulated cells (data not shown). b, CFSElow dividing cells were flow sorted on day 5 (black bars) or day 13 (gray bars) from an anti-CD3-activated CD4+CD25− T-cell culture and evaluated for suppressive ability. c, CFSElow dividing cells (black bars) and CFSEhigh non-dividing cells (gray bars) were flow sorted on day 5 from an anti-CD3-activated CD4+CD25− T-cell culture and evaluated for suppressive ability. CPM values show that the non-dividing cells were not anergic and did not suppress. d, CFSEhigh (non-dividing) cells from day 13 of an anti-CD3-activated culture were flow sorted and were re-cultured with fresh autologous APC in media alone or anti-CD3 for a further 6 days. Robust proliferation and upregulation of CD25 and FOXP3 are shown.
In the second approach, we addressed whether this suppressive potential diminished over time, corresponding with downregulation of FOXP3. For this, we could not use a CD25-based sorting technique as CD25 expression decreases over time. Thus, we flow-sorted the CFSElow (dividing) and CFSEhigh (non-dividing) cells from stimulated cultures either on day 5 or day 13, which correspond to peak and plateaued FOXP3 expression, respectively (day 10 and day 13 FOXP3 expression was not significantly different). In suppression assays, the dividing cells from day 5 cultures robustly inhibited in vitro proliferation of CD4+CD25− T-cells (Fig. 2b, 2c). Interestingly, suppressive ability was significantly diminished in dividing cells sorted on day 13 (p<0.05), corresponding to downregulated FOXP3 expression. Non-dividing cells from the day 5 culture did not show either the anergic or the suppressive phenotype of 5-day dividing cells (Fig. 2c).
FOXP3+ induced Tregs may serve to limit local immune responses and tissue damage [15; 32]. In our culture system, activated T-cells accumulate higher quantities of FOXP3 as they divide, with cells in later divisions showing preferential FOXP3 and CD25 expression, suggesting that these cells may inhibit the division of other cells in the same culture (especially, the non-dividing, CFSEhighCD4+ cells). In order to test this hypothesis, we flow-sorted dividing and non-dividing cells from a 13-day anti-CD3-activated culture (Fig. 2d). On re-stimulation with anti-CD3 for an additional 6 days, the non-dividing cells robustly upregulated CD25, expressed FOXP3 and went through multiple rounds of division, suggesting that the non-dividing cells may have been suppressed in the prior culture. Re-stimulation of the CFSElow populations resulted in a re-upregulation of FOXP3 and CD25 (not shown), suggesting that T-cells may potentially upregulate FOXP3 and CD25 during multiple activation cycles, attaining a transient regulatory phenotype each time.
Transiently induced Tregs share immunophenotypic similarities with natural Tregs
We used 8-color flow cytometric panels to evaluate the immunophenotype of the transiently induced Tregs (Fig. 3a). The anti-CD3 or allo-induced FOXP3+ T-cells exhibited a CTLA-4 (CD152)high, CD25high, CCR7+, CD27+, CD62L+, CD28+ phenotype (Fig. 3a), similar to that of natural Tregs in humans [33; 34; 35]. Thus, transient FOXP3 expression by activated T-cells corresponds to not only transient suppressor (“Treg”) functionality but also immunophenotypic features similar to natural Tregs. These results suggest that, in humans, natural Tregs, induced Tregs or transient Tregs may be similar or highly related populations with the major difference being the location or timing of their origin.
Figure 3. Phenotypic analysis of transient FOXP3+ regulatory T cells.
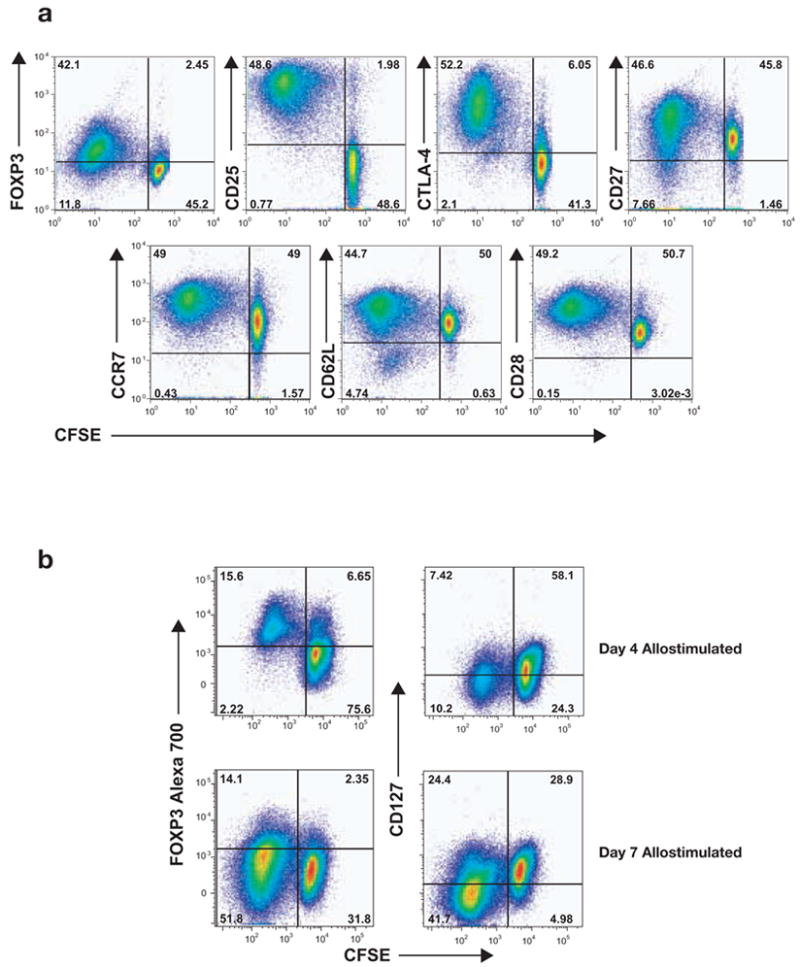
a, Allostimulated, CFSE-stained CD4+CD25− T-cells were phenotyped for FOXP3, CD25, CTLA-4, CD27, CD62L, CCR7 and CD28 expression at day 6 of culture. Dividing CFSElow cells were positive for most of the markers shown, similar to the phenotype of ex vivo-purified Tregs. b, CD127 (IL7-R) expression was monitored on allostimulated T-cells, revealing CD127 downregulation with the induction of FOXP3. CD127 remained low at a point where FOXP3 was downregulated.
It has been recently shown that low CD127 (IL-7 receptor) expression may be a specific marker for natural FOXP3+ Tregs [17; 36]. Thus, we evaluated CD127 status on transient Tregs (Fig. 3b). We observed that the induced Tregs downregulated CD127 and were CD127low (but not negative) when they were FOXP3+. This is strikingly similar to the CD127low phenotype of natural Tregs. They continued to remain CD127low even as they became FOXP3−. Hence, even CD127 does not appear to differentiate natural Tregs from transiently induced FOXP3+ Tregs.
FOXP3 in humans exists as at least two isoforms, in contrast to mice where only one isoform has been described [30]. To see whether there were differences in isoform expression in natural vs. induced/transient Tregs, we used commercially available antibodies to quantify the relative amounts of FOXP3 in human T-cell populations. We observed expression of both the full-length as well as the Δ2 isoform in ex vivo-sorted natural Tregs and induced Tregs arising from various sorted subsets (Fig. 4). We also observed downregulation of both isoforms (data not shown).
Figure 4. All sub-populations of Tregs express both isoforms of FOXP3.
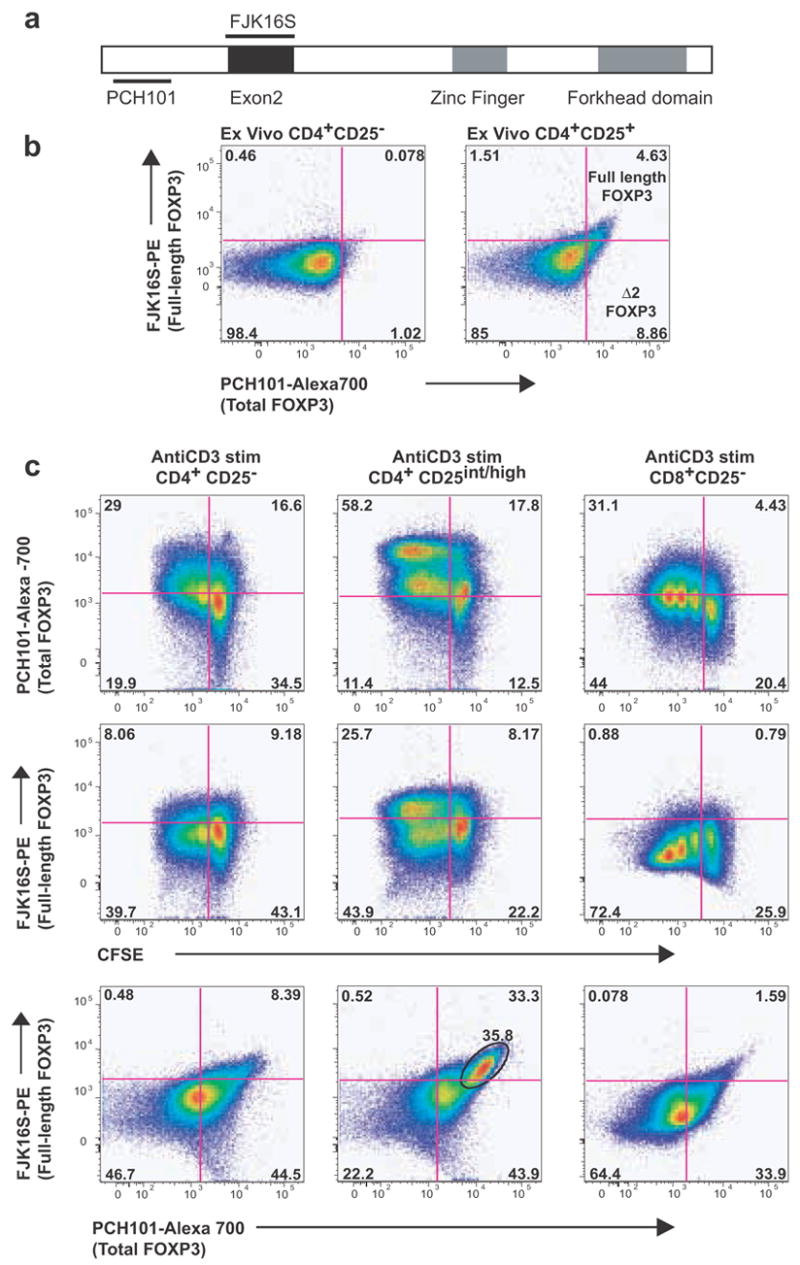
Commercially available antibodies were used to quantify total (PCH101) and full-length FOXP3 (FJK16S) in various Treg subpopulations. Spliced (Δ2) FOXP3 was detected by using a combination of both the antibodies (PCH101+/FJK16S−). a, Regions of the FOXP3 molecule that are targeted by the two antibodies are diagrammed. b, Using CD25 microbeads, we purified two subsets of CD4+ T-cells ex vivo: a subset enriched in CD25high/intermediate T-cells and a CD25− subset. CD4+CD25high/intermediate T-cells expressed both isoforms ex vivo, with a larger subset expressing the D2 isoform. c, Bead-sorted CD4+CD25− T-cells, CD4+CD25high/intermediate T-cells or CD8+CD25− T-cells were CFSE-stained and activated with anti-CD3. Following 4 days of anti-CD3 activation, CD4+CD25− T-cells induced predominantly the Δ2 isoform with a small subset expressing the full-length isoform. Activated CD4+CD25high/intermediate T-cells appeared to harbor a relatively larger subset expressing the full-length isoform. CD8+CD25− T-cells also upregulated both the isoforms with the Δ2 isoform predominating.
Differential dynamics of effector functions and FOXP3 in transient Tregs
As FOXP3 expression occurs during activation, we wanted to determine the relationship between transient Treg function and T-cell effector functions. Thus, we quantified the longitudinal expression of specific effector cytokines following activation, in flow-sorted CFSElowCD25+ cells from allostimulated T-cell cultures (Fig. 5). Interestingly, IFN-γ message was observed in the initial stages of activation, whereas FOXP3 and IL-10 were co-expressed at later time points. IFN-γ expression was also lowest when FOXP3 and IL-10 expression were highest. Of note, TNF-α also showed high levels of expression at this time, suggesting that different effector functions were differentially regulated during activation. IL-2 and IL-4 messages were either low or undetectable at the peak of FOXP3 expression (not shown). Similar dynamics were observed even when bulk populations from allo- or anti-CD3-stimulated cultures were used instead of flow-sorted, activated cells.
Figure 5. Effector functions are differentially regulated during Treg induction.
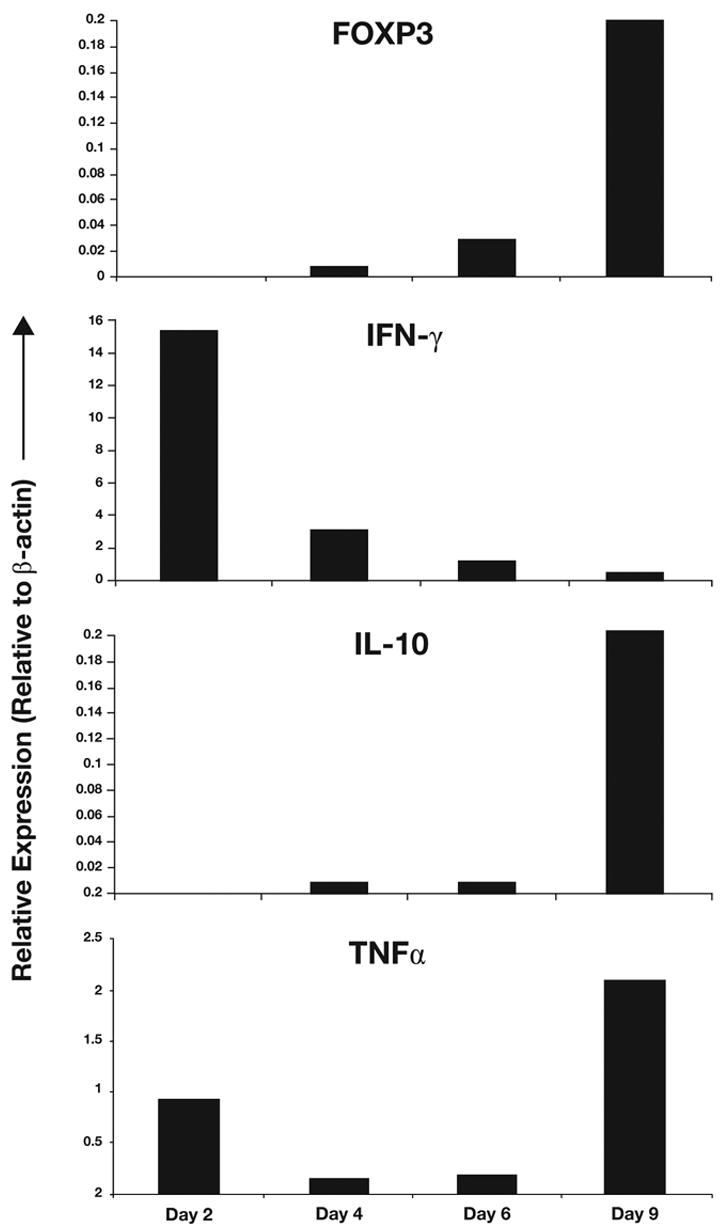
Allostimulated CD4+CD25− T-cell cultures were flow sorted for CD4+, CFSEhigh, CD25+ fraction (Day 2), CD4+, CFSElow, CD25+, dividing fraction (Days 4, 6 and 9) to obtain highly pure induced Treg populations at different stages of their formation. FOXP3, IFN-γ, TNF-α and IL-10 messages were quantified by real time PCR and normalized to β-actin. Presence of specific product was confirmed by dissociation curve and gel analysis.
CD8+CD25− T-cells also show transient FOXP3 upregulation and suppressor activity following activation
While CD4+FOXP3+ Tregs have been extensively evaluated, much less is known about the biology of CD8+FOXP3+ T-cells. Human CD8+ T-cells have been found to express FOXP3 [18; 24; 37; 38; 39]. We thus wanted to determine if activated CD8+ T-cells also show kinetics of FOXP3 expression and suppressive activity similar to CD4+ T-cells. Ex vivo, we did not detect appreciable FOXP3 expression on either CD8+CD25− or CD8+CD25+ T-cells. Starting with either CD3+CD25− T-cells or purified CD8+CD25− T-cells, we observed distinct FOXP3 upregulation in response to allo- or anti-CD3 stimulation (Fig. 6a and data not shown). Similar to CD4+ T-cells, virtually all the responding CD8+ T-cells expressed FOXP3, followed by eventual downregulation. Bead-sorted CD8+CD25+ T-cells from early cultures showed robust in vitro suppression of autologous CD4+CD25− T-cells, comparable to that of CD4+ Tregs (Fig. 6b). Similarly, flow-sorted dividing CD8+ T-cells from early cultures also showed robust in vitro suppression, which was partially reversed at later time points (Fig. 6c; p<0.05). Activated CD8+ T-cells also expressed both isoforms of FOXP3 (Fig. 4). Thus, the property of inducing transient FOXP3+ Tregs is not restricted to CD4+ T-cells, but may be a state attained by virtually all activated T-cells.
Figure 6. CD8+CD25− T-cells show transient FOXP3 upregulation and suppressor activity following activation.
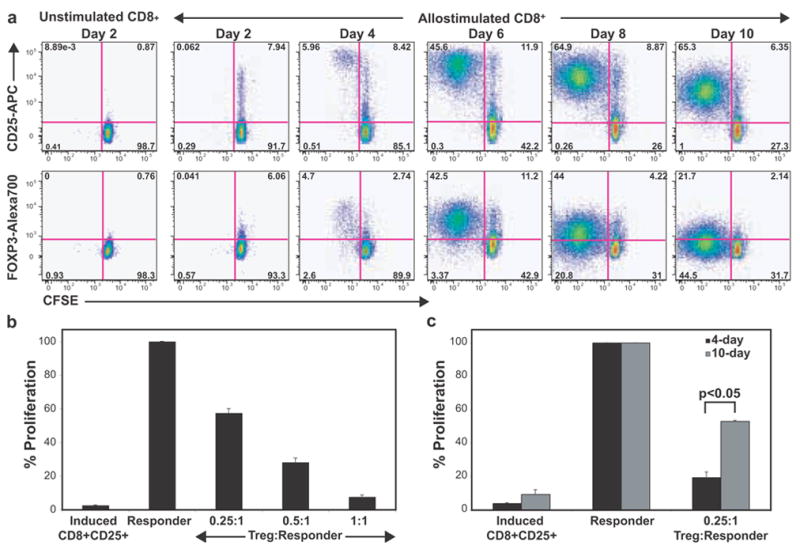
a, Gated CD8+ T-cells from an allostimulated CD3+CD25− T-cell culture are shown. Transient upregulation of FOXP3 and CD25 on CFSElow (dividing) cells is depicted. b, CD25+ T-cells were sorted from 5-day cultures of anti-CD3-stimulated CD8+CD25− T-cells and tested for their ability to suppress autologous CD4+CD25− T-cells in 3H-thymidine based suppression assays. Proliferation data was normalized to responder proliferation (100%), which ranged between 50,000 to 80,000 CPM in various experiments. c, Suppressive ability of flow-sorted CFSElow (dividing) cells from day 4 and day 10 of anti-CD3-activated CD8+CD25− T-cell cultures.
Discussion
It is known that CD25+FOXP3+ T-cells with regulatory properties can be induced following activation [10; 11; 12; 13]. For example, using PCR-based analysis, it has been demonstrated that CD4+CD25− T-cells can generate FOXP3+ regulatory T-cells [11]. However, it was widely proposed that these cells arose from a contaminating population of natural Tregs [14; 15; 16]. Moreover, the proportion of activated T-cells that become FOXP3+ and the stability and significance of such expression are poorly understood. In general, FOXP3+ T-cells are thought to be a unique subset of stable, regulatory T-cells.
In this study, we have demonstrated that virtually all activated CD4+CD25−FOXP3− T-cells attain a transient phase of FOXP3 expression and regulatory activity. We also show that these FOXP3+ cells must arise from CD4+CD25− T-cells and that this phenomenon occurs regardless of the stimulus used to activate these cells. While previous studies have shown that FOXP3+ T-cells can be induced from CD4+CD25−FOXP3− T-cells, it is thought that only a small subset of approximately 10–20% of the activated T-cells express FOXP3. Recent studies using anti-FOXP3 antibodies to detect FOXP3 expression have used either PBMC [18; 24] or PBMC depleted of CD25+ T-cells [18; 40] to study activation-induced FOXP3 expression. In those studies, cells evaluated for FOXP3 also included non-activated T-cells in the culture. The use of CFSE in our assay system clearly shows that, even in anti-CD3-stimulated cultures, not all T-cells undergo activation and proliferation. This allowed us to accurately gate on activated T-cell populations, demonstrating that all activated T-cells, rather than a minor subset, undergo this change. Transient FOXP3 expression is better visualized following allostimulation compared to anti-CD3 stimulation as only a percentage of the responding cells are activated by the allostimulus. The use of the CFSE-based approach also allowed us to visualize the non-dividing (unstimulated) cells as an internal control for CD25 and FOXP3 expression. Depending on the geometry of the culture (flask versus tubes) and the nature of the stimulus (anti-CD3 or antiCD3+antiCD28 versus MLR versus antigen), the kinetics of activation and consequently FOXP3 expression were different. Thus, if only a single time point is evaluated, one would reach a restricted conclusion about the number of cells that express FOXP3. Moreover, the actual day of peak expression is less important than the overall phenomenon of transient upregulation followed by downregulation. Our results support the conclusion that induction of a FOXP3+ Treg state is not a rare or unusual event, but rather the norm.
Importantly, most of the activated FOXP3+ cells downregulate FOXP3 and by day 10–14, only a subset of that population remains FOXP3+. We show that regulatory activity decreases at later time points when FOXP3 expression has plateaued. These results suggest that FOXP3 expression correlates with suppressive activity and that suppressive ability is a transient property of all activated CD4+ T-cell populations. It is possible that FOXP3 expression could be coincident with suppressive activity, rather than mechanistically involved in this function. However, regardless of its direct involvement, FOXP3 expression and regulatory function appear to be a transient phenomenon in all T-cells. Even when starting with naïve CD4+ T-cells, the same pattern is observed, suggesting that activation might be a way of producing Tregs in the periphery where every dividing T-cell has the potential to a become effector/memory T-cell and/or a Treg. Presumably, with multiple rounds of activation, a higher proportion of cells may be sustained as Tregs, as might happen in the setting of chronic infections, such as HCV and HIV [41; 42; 43; 44]. More importantly, our results also suggest that mere depletion of circulating Tregs may not be adequate therapy for tumors and infections. Not only would newer Tregs be produced during sustained antigenic stimulation, but the therapeutic approach may also carry the risk of eliminating potential effector T-cells. In these situations, tilting the balance from Treg sustenance toward effector T-cell induction may be a plausible immunotherapeutic intervention. In contrast to chronic viral infections, several autoimmune diseases are characterized by a functional deficit in CD4+ and/or CD8+ Tregs. From our studies, one might presume that the activation of autoreactive T-cells must also result in the generation of FOXP3+, transient Tregs. It is plausible that sustained autoreactivity, leading to autoimmunity, may be a defect in the sustenance of induced autoreactive Tregs in the periphery, rather than merely a global deficit in thymic Treg generation. Factors that promote such functional sustenance will be of great interest in future therapeutic strategies.
It has long been known that natural Tregs show an activated T-cell phenotype and efforts are still underway to identify a specific marker for Tregs that is not expressed by activated T-cells. Every putative “specific” Treg marker proposed thus far eventually turned out to be expressed by activated T-cells as well. We show here that induced Tregs also show an immunophenotype that is similar to that of natural Tregs. We propose that the similarities between Tregs and activated T-cells may, in fact, not be a coincidence. A major subset of circulating human Tregs may simply reflect the long-term sustenance of a subpopulation of activated cells. We found no obvious differences in the suppressive activity or phenotype between natural and induced Tregs, suggesting that these may be related populations with the location and timing of origin being the main difference between them.
In this study, we also evaluated effector functions in the context of FOXP3 expression, by flow-sorting pure populations of activated cells at multiple time points post-stimulation. IFN-γ expression preceded and inversely correlated with FOXP3 expression. FOXP3 expression coincided with peak IL-10 production and TNF-α production in these cultures. Thus, we show that effector function and regulatory function can occur in the same T-cell population. These results suggest that therapeutic attempts to increase Tregs may paradoxically increase potentially harmful effector responses. We speculate that this could be one of the plausible explanations for the results of the recent clinical trial using an anti-CD28 superagonist TGN1412, which attempted to expand Tregs but resulted in severe multiorgan failure [45; 46; 47]. Prior studies has shown increased Treg induction by this agent [48; 49]. However, based on our results, these may have potentially been transient Tregs, rather than a stable Treg population, a notion supported by recent studies using this agent in a humanized mouse system [50]. This underscores the importance of understanding the biology of induced Tregs and its relationship with effector functionality.
CD8+ T-cells have been shown to possess regulatory activity distinct from that of natural CD4+CD25+FOXP3+Tregs [39; 51]. Though a definitive CD8+ counterpart of the natural CD4+CD25+ Tregs has not been described, FOXP3 expression has been detected in CD8+ T-cells under various conditions [18; 24; 37; 38; 39]. One of our key findings is that CD8+ T-cells also show similar dynamics, where all activated CD8+ T-cells (in the presence or absence of CD4+ T-cells) express FOXP3 and exhibit regulatory activity transiently. It remains to be seen if the induced CD8+FOXP3+ Tregs persist in the periphery for any significant length of time compared to the CD4+FOXP3+ Tregs.
These findings strongly suggest that activation-induced FOXP3 expression and regulatory activity may be a broad T-cell phenomenon. Thus, upon T-cell activation, a FOXP3+ state may be coincident with or proximal to effector functionality. Presumably, such a mechanism might allow T-cells with the highest affinity (those that presumably generate the first or the strongest response following an immune stimulus) to effectively suppress lower affinity responses in their milieu [52]. It id currently unclear whether the attainment of such a FOXP3+ transient Treg state is an essential step toward attaining fully regulated effector functionality.
In summary, we have shown that virtually all activated CD4+ and CD8+ T-cells transiently upregulate FOXP3 and acquire suppressive properties. Thus, during an active immune process, most of the FOXP3+ cells could, in fact, represent effector T-cells that can downregulate FOXP3 and lose regulatory activity. Thus, neither FOXP3 expression nor in vitro suppressive ability is an indication of stable Treg function and these readouts should not be used to designate a cell as a permanent Treg. These observations warrant the re-evaluation of prior data relating to the role of Tregs in various human diseases.
Acknowledgments
We thank Drs. Ellen Vitetta, Mihail Firan and Mr. Ethan Baughman for review and discussions and Ms. Bonnie Darnell for assistance with flow sorting.
Footnotes
This work was supported by grants (to N.J.K.) from the NIH and National MS Society (NMSS). N.J.K. is a Harry Weaver Neuroscience Scholar of the NMSS.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sakaguchi S. Naturally arising Foxp3-expressing CD25+CD4+ regulatory T cells in immunological tolerance to self and non-self. Nat Immunol. 2005;6:345–52. doi: 10.1038/ni1178. [DOI] [PubMed] [Google Scholar]
- 2.Fehervari Z, Sakaguchi S. CD4+ regulatory cells as a potential immunotherapy. Philos Trans R Soc Lond B Biol Sci. 2005;360:1647–61. doi: 10.1098/rstb.2005.1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4:330–6. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 4.Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–61. doi: 10.1126/science.1079490. [DOI] [PubMed] [Google Scholar]
- 5.Fontenot JD, Rasmussen JP, Williams LM, Dooley JL, Farr AG, Rudensky AY. Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity. 2005;22:329–41. doi: 10.1016/j.immuni.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 6.Khattri R, Cox T, Yasayko SA, Ramsdell F. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat Immunol. 2003;4:337–42. doi: 10.1038/ni909. [DOI] [PubMed] [Google Scholar]
- 7.Yagi H, Nomura T, Nakamura K, Yamazaki S, Kitawaki T, Hori S, Maeda M, Onodera M, Uchiyama T, Fujii S, Sakaguchi S. Crucial role of FOXP3 in the development and function of human CD25+CD4+ regulatory T cells. Int Immunol. 2004;16:1643–56. doi: 10.1093/intimm/dxh165. [DOI] [PubMed] [Google Scholar]
- 8.Bettelli E, Dastrange M, Oukka M. Foxp3 interacts with nuclear factor of activated T cells and NF-kappa B to repress cytokine gene expression and effector functions of T helper cells. Proc Natl Acad Sci U S A. 2005;102:5138–43. doi: 10.1073/pnas.0501675102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schubert LA, Jeffery E, Zhang Y, Ramsdell F, Ziegler SF. Scurfin (FOXP3) acts as a repressor of transcription and regulates T cell activation. J Biol Chem. 2001;276:37672–9. doi: 10.1074/jbc.M104521200. [DOI] [PubMed] [Google Scholar]
- 10.Walker MR, Carson BD, Nepom GT, Ziegler SF, Buckner JH. De novo generation of antigen-specific CD4+CD25+ regulatory T cells from human CD4+CD25- cells. Proc Natl Acad Sci U S A. 2005;102:4103–8. doi: 10.1073/pnas.0407691102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Walker MR, Kasprowicz DJ, Gersuk VH, Benard A, Van Landeghen M, Buckner JH, Ziegler SF. Induction of FoxP3 and acquisition of T regulatory activity by stimulated human CD4+CD25- T cells. J Clin Invest. 2003;112:1437–43. doi: 10.1172/JCI19441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, McGrady G, Wahl SM. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. 2003;198:1875–86. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kretschmer K, Apostolou I, Hawiger D, Khazaie K, Nussenzweig MC, von Boehmer H. Inducing and expanding regulatory T cell populations by foreign antigen. Nat Immunol. 2005;6:1219–27. doi: 10.1038/ni1265. [DOI] [PubMed] [Google Scholar]
- 14.Fehervari Z, Sakaguchi S. CD4+ Tregs and immune control. J Clin Invest. 2004;114:1209–17. doi: 10.1172/JCI23395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fontenot JD, Rudensky AY. A well adapted regulatory contrivance: regulatory T cell development and the forkhead family transcription factor Foxp3. Nat Immunol. 2005;6:331–7. doi: 10.1038/ni1179. [DOI] [PubMed] [Google Scholar]
- 16.Shevach EM. Regulatory/suppressor T cells in health and disease. Arthritis Rheum. 2004;50:2721–4. doi: 10.1002/art.20500. [DOI] [PubMed] [Google Scholar]
- 17.Liu W, Putnam AL, Xu-Yu Z, Szot GL, Lee MR, Zhu S, Gottlieb PA, Kapranov P, Gingeras TR, de St Groth BF, Clayberger C, Soper DM, Ziegler SF, Bluestone JA. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J Exp Med. 2006 doi: 10.1084/jem.20060772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gavin MA, Torgerson TR, Houston E, Deroos P, Ho WY, Stray-Pedersen A, Ocheltree EL, Greenberg PD, Ochs HD, Rudensky AY. Single-cell analysis of normal and FOXP3-mutant human T cells: FOXP3 expression without regulatory T cell development. Proc Natl Acad Sci U S A. 2006;103:6659–64. doi: 10.1073/pnas.0509484103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lan RY, Cheng C, Lian ZX, Tsuneyama K, Yang GX, Moritoki Y, Chuang YH, Nakamura T, Saito S, Shimoda S, Tanaka A, Bowlus CL, Takano Y, Ansari AA, Coppel RL, Gershwin ME. Liver-targeted and peripheral blood alterations of regulatory T cells in primary biliary cirrhosis. Hepatology. 2006;43:729–37. doi: 10.1002/hep.21123. [DOI] [PubMed] [Google Scholar]
- 20.Campanelli AP, Roselino AM, Cavassani KA, Pereira MS, Mortara RA, Brodskyn CI, Goncalves HS, Belkaid Y, Barral-Netto M, Barral A, Silva JS. CD4+CD25+ T cells in skin lesions of patients with cutaneous leishmaniasis exhibit phenotypic and functional characteristics of natural regulatory T cells. J Infect Dis. 2006;193:1313–22. doi: 10.1086/502980. [DOI] [PubMed] [Google Scholar]
- 21.Epple HJ, Loddenkemper C, Kunkel D, Troeger H, Maul J, Moos V, Berg E, Ullrich R, Schulzke JD, Stein H, Duchmann R, Zeitz M, Schneider T. Mucosal but not peripheral FOXP3+ regulatory T cells are highly increased in untreated HIV infection and normalize after suppressive HAART. Blood. 2006 doi: 10.1182/blood-2006-04-016923. [DOI] [PubMed] [Google Scholar]
- 22.Cao D, Borjesson O, Larsson P, Rudin A, Gunnarsson I, Klareskog L, Malmstrom V, Trollmo C. FOXP3 identifies regulatory CD25bright CD4+ T cells in rheumatic joints. Scand J Immunol. 2006;63:444–52. doi: 10.1111/j.1365-3083.2006.001755.x. [DOI] [PubMed] [Google Scholar]
- 23.Xu D, Fu J, Jin L, Zhang H, Zhou C, Zou Z, Zhao JM, Zhang B, Shi M, Ding X, Tang Z, Fu YX, Wang FS. Circulating and Liver Resident CD4+CD25+ Regulatory T Cells Actively Influence the Antiviral Immune Response and Disease Progression in Patients with Hepatitis B. J Immunol. 2006;177:739–47. doi: 10.4049/jimmunol.177.1.739. [DOI] [PubMed] [Google Scholar]
- 24.Roncador G, Brown PJ, Maestre L, Hue S, Martinez-Torrecuadrada JL, Ling KL, Pratap S, Toms C, Fox BC, Cerundolo V, Powrie F, Banham AH. Analysis of FOXP3 protein expression in human CD4+CD25+ regulatory T cells at the single-cell level. Eur J Immunol. 2005;35:1681–91. doi: 10.1002/eji.200526189. [DOI] [PubMed] [Google Scholar]
- 25.Karandikar NJ, Crawford MP, Yan X, Ratts RB, Brenchley JM, Ambrozak DR, Lovett-Racke AE, Frohman EM, Stastny P, Douek DC, Koup RA, Racke MK. Glatiramer acetate (Copaxone) therapy induces CD8(+) T cell responses in patients with multiple sclerosis. J Clin Invest. 2002;109:641–649. doi: 10.1172/JCI14380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Crawford MP, Yan SX, Ortega SB, Mehta RS, Hewitt RE, Price DA, Stastny P, Douek DC, Koup RA, Racke MK, Karandikar NJ. High Prevalence of Autoreactive Neuroantigen-Specific CD8+ T Cells in Multiple Sclerosis Revealed by Novel Flow Cytometric Assay. Blood. 2004;103:4222–4231. doi: 10.1182/blood-2003-11-4025. [DOI] [PubMed] [Google Scholar]
- 27.Parks DR, Roederer M, Moore WA. A new "Logicle" display method avoids deceptive effects of logarithmic scaling for low signals and compensated data. Cytometry A. 2006 doi: 10.1002/cyto.a.20258. [DOI] [PubMed] [Google Scholar]
- 28.Herzenberg LA, Tung J, Moore WA, Herzenberg LA, Parks DR. Interpreting flow cytometry data: a guide for the perplexed. Nat Immunol. 2006;7:681–5. doi: 10.1038/ni0706-681. [DOI] [PubMed] [Google Scholar]
- 29.Perfetto SP, Chattopadhyay PK, Roederer M. Seventeen-colour flow cytometry: unravelling the immune system. Nat Rev Immunol. 2004;4:648–55. doi: 10.1038/nri1416. [DOI] [PubMed] [Google Scholar]
- 30.Allan SE, Passerini L, Bacchetta R, Crellin N, Dai M, Orban PC, Ziegler SF, Roncarolo MG, Levings MK. The role of 2 FOXP3 isoforms in the generation of human CD4+ Tregs. J Clin Invest. 2005;115:3276–84. doi: 10.1172/JCI24685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.De Rosa SC, Herzenberg LA, Herzenberg LA, Roederer M. 11-color, 13-parameter flow cytometry: identification of human naive T cells by phenotype, function, and T-cell receptor diversity. Nat Med. 2001;7:245–8. doi: 10.1038/84701. [DOI] [PubMed] [Google Scholar]
- 32.Ziegler SF. FOXP3: Of Mice and Men. Annu Rev Immunol. 2006;24:209–26. doi: 10.1146/annurev.immunol.24.021605.090547. [DOI] [PubMed] [Google Scholar]
- 33.Seddiki N, Santner-Nanan B, Tangye SG, Alexander SI, Solomon M, Lee S, Nanan R, de Saint Groth BF. Persistence of naive CD45RA+ regulatory T cells in adult life. Blood. 2006;107:2830–8. doi: 10.1182/blood-2005-06-2403. [DOI] [PubMed] [Google Scholar]
- 34.Wei S, Kryczek I, Zou W. Regulatory T cell compartmentalization and trafficking. Blood. 2006 doi: 10.1182/blood-2006-01-0177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Valmori D, Merlo A, Souleimanian NE, Hesdorffer CS, Ayyoub M. A peripheral circulating compartment of natural naive CD4 Tregs. J Clin Invest. 2005;115:1953–62. doi: 10.1172/JCI23963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Seddiki N, Santner-Nanan B, Martinson J, Zaunders J, Sasson S, Landay A, Solomon M, Selby W, Alexander SI, Nanan R, Kelleher A, de St Groth BF. Expression of interleukin (IL)-2 and IL-7 receptors discriminates between human regulatory and activated T cells. J Exp Med. 2006 doi: 10.1084/jem.20060468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morgan ME, van Bilsen JH, Bakker AM, Heemskerk B, Schilham MW, Hartgers FC, Elferink BG, van der Zanden L, de Vries RR, Huizinga TW, Ottenhoff TH, Toes RE. Expression of FOXP3 mRNA is not confined to CD4+CD25+ T regulatory cells in humans. Hum Immunol. 2005;66:13–20. doi: 10.1016/j.humimm.2004.05.016. [DOI] [PubMed] [Google Scholar]
- 38.Bisikirska B, Colgan J, Luban J, Bluestone JA, Herold KC. TCR stimulation with modified anti-CD3 mAb expands CD8+ T cell population and induces CD8+CD25+ Tregs. J Clin Invest. 2005;115:2904–13. doi: 10.1172/JCI23961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Manavalan JS, Kim-Schulze S, Scotto L, Naiyer AJ, Vlad G, Colombo PC, Marboe C, Mancini D, Cortesini R, Suciu-Foca N. Alloantigen specific CD8+CD28- FOXP3+ T suppressor cells induce ILT3+ ILT4+ tolerogenic endothelial cells, inhibiting alloreactivity. Int Immunol. 2004;16:1055–68. doi: 10.1093/intimm/dxh107. [DOI] [PubMed] [Google Scholar]
- 40.Mantel PY, Ouaked N, Ruckert B, Karagiannidis C, Welz R, Blaser K, Schmidt-Weber CB. Molecular mechanisms underlying FOXP3 induction in human T cells. J Immunol. 2006;176:3593–602. doi: 10.4049/jimmunol.176.6.3593. [DOI] [PubMed] [Google Scholar]
- 41.Boettler T, Spangenberg HC, Neumann-Haefelin C, Panther E, Urbani S, Ferrari C, Blum HE, von Weizsacker F, Thimme R. T Cells with a CD4+CD25+ Regulatory Phenotype Suppress In Vitro Proliferation of Virus-Specific CD8+ T Cells during Chronic Hepatitis C Virus Infection. J Virol. 2005;79:7860–7. doi: 10.1128/JVI.79.12.7860-7867.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rushbrook SM, Ward SM, Unitt E, Vowler SL, Lucas M, Klenerman P, Alexander GJ. Regulatory T Cells Suppress In Vitro Proliferation of Virus-Specific CD8+ T Cells during Persistent Hepatitis C Virus Infection. J Virol. 2005;79:7852–9. doi: 10.1128/JVI.79.12.7852-7859.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kinter AL, Hennessey M, Bell A, Kern S, Lin Y, Daucher M, Planta M, McGlaughlin M, Jackson R, Ziegler SF, Fauci AS. CD25(+)CD4(+) regulatory T cells from the peripheral blood of asymptomatic HIV-infected individuals regulate CD4(+) and CD8(+) HIV-specific T cell immune responses in vitro and are associated with favorable clinical markers of disease status. J Exp Med. 2004;200:331–43. doi: 10.1084/jem.20032069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Andersson J, Boasso A, Nilsson J, Zhang R, Shire NJ, Lindback S, Shearer GM, Chougnet CA. Cutting edge: The prevalence of regulatory T cells in lymphoid tissue is correlated with viral load in HIV-infected patients. J Immunol. 2005;174:3143–7. doi: 10.4049/jimmunol.174.6.3143. [DOI] [PubMed] [Google Scholar]
- 45.Can super-antibody drugs be tamed? Nature. 2006;440:855–6. doi: 10.1038/440855a. [DOI] [PubMed] [Google Scholar]
- 46.Wadman M. London's disastrous drug trial has serious side effects for research. Nature. 2006;440:388–9. doi: 10.1038/440388a. [DOI] [PubMed] [Google Scholar]
- 47.Beyersdorf N, Hanke T, Kerkau T, Hunig T. CD28 superagonists put a break on autoimmunity by preferentially activating CD4+CD25+ regulatory T cells. Autoimmun Rev. 2006;5:40–5. doi: 10.1016/j.autrev.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 48.Beyersdorf N, Gaupp S, Balbach K, Schmidt J, Toyka KV, Lin CH, Hanke T, Hunig T, Kerkau T, Gold R. Selective targeting of regulatory T cells with CD28 superagonists allows effective therapy of experimental autoimmune encephalomyelitis. J Exp Med. 2005;202:445–55. doi: 10.1084/jem.20051060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lin CH, Hunig T. Efficient expansion of regulatory T cells in vitro and in vivo with a CD28 superagonist. Eur J Immunol. 2003;33:626–38. doi: 10.1002/eji.200323570. [DOI] [PubMed] [Google Scholar]
- 50.Legrand N, Cupedo T, van Lent AU, Ebeli MJ, Weijer K, Hanke T, Spits H. Transient accumulation of human mature thymocytes and regulatory T cells with CD28 superagonist in "human immune system" Rag2(−/−)gammac(−/−) mice. Blood. 2006;108:238–45. doi: 10.1182/blood-2006-01-0190. [DOI] [PubMed] [Google Scholar]
- 51.Sarantopoulos S, Lu L, Cantor H. Qa-1 restriction of CD8+ suppressor T cells. J Clin Invest. 2004;114:1218–21. doi: 10.1172/JCI23152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mallone R, Kochik SA, Reijonen H, Carson B, Ziegler SF, Kwok WW, Nepom GT. Functional avidity directs T-cell fate in autoreactive CD4+ T cells. Blood. 2005;106:2798–805. doi: 10.1182/blood-2004-12-4848. [DOI] [PMC free article] [PubMed] [Google Scholar]