

Abstract
Medulloblastoma, the most common malignant brain tumor of childhood, is believed to derive from immature granule neuron precursors (GNPs) that normally proliferate in the external granule layer before exiting the cell cycle and migrating to their mature location in the inner granule layer. In this study, we examined the expression of D type cyclins in GNPs during cerebellar development and showed that GNPs in early development expressed only cyclin D1, whereas later GNPs expressed both cyclins D1 and D2. Coinciding with the period of cyclin D1-only expression, Ccnd1−/− mice showed reduced proliferation of GNPs and impaired growth of the cerebellum. Interestingly, removal of cyclin D1 was sufficient to drastically reduce the incidence of medulloblastoma in Ptch1+/− mice, despite the fact that these tumors showed upregulation of both cyclins D1 and D2. We showed that cyclin D1 has an earlier role in tumorigenesis: in the absence of cyclin D1, the incidence and overall volume of ‘pre-neoplastic’ lesions were significantly decreased. We propose a model that links a role of cyclin D1 in normal GNP proliferation with its early role in tumorigenesis.
Keywords: Cyclin D1, Cerebellar development, Medulloblastoma, Ptch1, Mouse
INTRODUCTION
During cerebellar development, granule neuron precursors (GNPs) migrate from the rhombic lip to the outer layer of the cerebellum to form the external granule layer (EGL), where they undergo a postnatal proliferative burst before exiting the cell cycle and migrating inward to form the mature inner granule layer. Proliferation occurs in the outer EGL, and, as post-mitotic cells begin to differentiate, they move inwards in the EGL before migrating to their mature location in the inner granule layer (IGL). Alterations in these tightly regulated events can lead to cerebellar dysfunction and ataxia or to medulloblastoma, the most prevalent malignant brain tumor of childhood (Gilbertson, 2004).
The conserved hedgehog signaling pathway plays critical roles in regulating the proliferation and differentiation of cerebellar GNPs (Wechsler-Reya and Scott, 2001). In vitro studies have shown that sonic hedgehog (Shh), which is secreted by the Purkinje neurons of the cerebellum, can sustain GNP proliferation and maintain their undifferentiated state (Wechsler-Reya and Scott, 1999). Shh acts by binding to its receptor patched (Ptch1) on target cells. In the absence of Shh, Ptch1 inhibits the signaling activity of smoothened (Smo), leading to the production of repressor forms of the Gli family of transcription factors. Binding of Shh to Ptch1 blocks the inhibitory activity of Ptch1 on Smo, resulting in the production of activator forms of the Gli transcription factors and increased expression of downstream genes (Stecca and Ruiz i Altaba, 2005).
Deregulation of the hedgehog pathway has been linked to the development of a number of cancers, including medulloblastoma (Goodrich et al., 1997; Hahn et al., 1996). Mice that are heterozygous for Ptch1 exhibit spontaneous development of medulloblastoma (Goodrich et al., 1997). These tumors exhibit increased Shh signaling (Goodrich et al., 1997), and treatment of mice with medulloblastomas with inhibitors of Shh signaling is sufficient to cause tumor regression (Berman et al., 2002; Romer et al., 2004). Interestingly, recent studies revealed the presence of pre-neoplastic lesions in young adult Ptch1+/− cerebella. These pre-neoplastic lesions exhibit an expression profile intermediate between those of normal proliferating GNPs and medulloblastomas (Oliver et al., 2005), suggesting that medulloblastomas in Ptch1+/− mice probably develop from these lesions. In addition, recent studies of human medulloblastomas have showed that only tumor cells positive for the stem cell marker CD133 are capable of forming tumors in mouse xenografts (Singh et al., 2004), and these tumor initiating cells exhibit other characteristics of neural stem cells, such as self-renewal, proliferation and the capacity to differentiate (Singh et al., 2003). These observations support a tumor stem cell model for the development of medulloblastomas.
Cyclins D1 and D2, but not D3, are upregulated in response to Shh signaling in cerebellar cells (Kenney and Rowitch, 2000). Both cyclins D1 and D2 are expressed in proliferating GNPs, and their joint removal results in an exceptionally hypoplastic cerebellum (Ciemerych et al., 2002), indicating that cyclins D1 and D2 play crucial roles in the rapid postnatal expansion of the GNP population. Interestingly, although removal of either cyclin D1 or D2 alone does not affect in vitro proliferation of postnatal day 4 GNPs in response to Shh signaling (Kenney and Rowitch, 2000), removal of cyclin D2 alone is sufficient to cause mild cerebellar hypoplasia (Huard et al., 1999), owing to both decreased proliferation and increased apoptosis of GNPs. This result is somewhat surprising as the D-type cyclins have very similar biochemical functions and, in general, specific effects of individual mutations are typically seen only in tissues where the other D-type cyclins are not expressed at high levels (Ciemerych et al., 2002).
The cerebellar phenotype of Ccnd2−/− mice could be due to a requirement for two D-type cyclins concurrently in the proliferation of GNPs, or it could be due to differences in the expression patterns of these two cyclins in the developing cerebellum. In this report, we show that early postnatal GNPs expressed only cyclin D1 while later GNPs expressed both cyclins D1 and D2. Consistent with this expression data, Ccnd1−/− mice had decreased early GNP proliferation and early ataxia as a consequence of a delay in acquiring normal cerebellar function. Interestingly, removal of cyclin D1 resulted in a significant decrease in the incidence of medulloblastoma in Ptch1+/− mice, as well as a decrease in the incidence and size of pre-neoplastic lesions. We suggest that cyclin D1 plays a crucial role in the proliferation of early GNPs and in the progression of the pre-neoplastic lesions to medulloblastomas.
MATERIALS AND METHODS
Mice
Ccnd1+/− mice were bred to obtain Ccnd1+/+ and Ccnd1−/− mice (Sicinski et al., 1995). For the righting reflex, five litters were tested daily beginning on P6. Mice were turned on their backs and observed to determine whether they could immediately right themselves. Ptch1+/−;Ccnd1+/− mice were bred to obtain Ptch1+/− (Goodrich et al., 1997) and Ptch1+/−;Ccnd1−/− mice. All mice were maintained on a mixed C57Bl/6 × 129Sv background. Mice used for the tumor study represent six generations beginning with the F2 generation. Mice were observed daily, and mice older than 35 weeks or demonstrating abnormal neurological symptoms were euthanized and brains were dissected under a dissecting microscope to examine for tumors. All mice were housed in a facility accredited by the Association for Assessment and Accreditation of Laboratory Animal Care and maintained in accordance with NIH Guidelines. The Animal Care and Use Committee at The University of Chicago approved all procedures.
Immunohistochemistry
Antibodies used: BrdU (BD Biosciences B44 1:800), cyclin D1 (Santa Cruz sc-450 1:400), cyclin D2 (Lab Vision Ab-4 1:500), NeuN (Chemicon MAB377 1:40,000), calbindin (Swant 1:6000) and phosphohistone H3 (Upstate 1:16,000). Samples were fixed, dehydrated and embedded in paraffin. Sections (6 μm) were rehydrated, and endogenous peroxidases were blocked by incubation in 3% H2O2, 10% methanol for 30 minutes. Antigen retrieval for all antibodies was performed by boiling in 0.01 M citric acid (pH 6) for 10 minutes in the microwave. The primary antibody was detected using the Vector Elite ABC kit (Vector Laboratories). Slides were lightly counterstained with Hematoxylin and Eosin. Sections incubated without primary antibody served as a negative control. For comparison of staining patterns within a single cerebellum, serial sections were examined. At least three pups from different litters were analyzed.
BrdU incorporation and statistic analysis
To monitor S-phase, mice were injected with BrdU (Sigma) (100 mg/kg body weight) 1 hour prior to euthanasia. For statistical analysis of tumor incidence, the Chi-squared test was used. For comparison of proliferation and number of pre-neoplastic lesions, the Student’s t-test was used.
All developmental sections examined for size, staining patterns and proliferation were midline sagittal sections of Ccnd1+/+ and Ccnd1−/−littermates. The midline was identified by the absence of cerebellar peduncles and fastigial nucleus. Multiple sections have been examined but only a single midline section was used to calculate the area. The area was determined by taking pictures with a Zeiss digital camera with a known pixel/μm ratio. The area in pixels was determined in Adobe Photoshop and converted to μm2. For quantification of proliferation, the number of BrdU and H3-P positive EGL cells per mm of EGL was counted. BrdU-positive cells were counted on midline sections in the same location in the anterior at P0 and at P6. As there were many fewer H3-P positive cells at P0, the H3-P positive cells over the entire length of the midline sections were counted to minimize variations. Statistical analysis used the paired two-tailed Student’s t-test.
RESULTS
Cyclin D1 and cyclin D2 exhibit distinct expression pattern in developing cerebellar GNPs
To determine to what extent cyclin D1 and D2 expression overlap in the developing EGL, we examined their localization during late embryonic and postnatal development when GNPs are proliferating. The earliest time point we examined was E16.5. At this point, cyclin D1 expression was primarily confined to the anteriormost 50% of the EGL (Fig. 1A,C) with only a few cells expressing cyclin D1 in the posterior (Fig. 1D). By P0, high cyclin D1 expression extended further posteriorly, with strong expression throughout the anterior 75% of the EGL (Fig. 1E–H), and almost all nuclei in the anterior outer EGL were positive for cyclin D1 expression (Fig. 1G). In addition, cyclin D1 also stained some cells deeper within the cerebellum and in the posterior at both E16.5 and P0 (Fig. 1D,H). By P6, cyclin D1 expression was detected throughout the entire length of the EGL and was limited to the outer half of the EGL (Fig. 1I,J). Scattered cyclin D1-positive cells were also found within the inner granule layer at this time, particularly directly adjacent to Purkinje cells (data not shown). Based on their location, these are likely to be glial cells, but we did not confirm their identity. By P12, cyclin D1 was almost entirely absent from the remaining EGL of wild-type cerebellum (Fig. 1K,L). With the exception of background endothelial and erythrocyte staining (also seen in the absence of primary antibody), this antibody showed no staining in Ccnd1−/−samples, confirming its specificity.
Fig. 1. Cyclin D1 (brown) is expressed at high levels in the EGL during early cerebellar development.
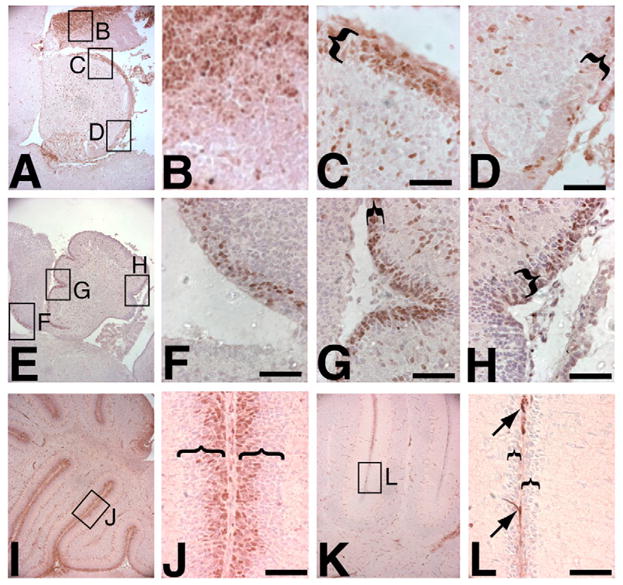
(A–D) The E16.5 cerebellum (A, with high-power views of boxed areas shown in B–D) expresses cyclin D1 strongly in the ventricular region (B) and anterior EGL (C), with fewer positive cells in the posterior EGL (D). (E–H) The P0 cerebellum (E, with high-power views of boxed areas shown in F–H) shows some remaining ventricular expression (F), and strong expression in the EGL extending much further posteriorly with enlarged anterior (G) and posterior (H) views of the EGL, although the extreme posterior still has fewer positive cells (H). (I,J) P6 cerebellum (I, with high-power of view of boxed area in J) shows strong staining in the outer EGL throughout the entire AP axis. (K,L) At P12, only background staining of blood vessels and meninges is seen (arrows) (K, with high-power of view of boxed area in L). Brackets indicate the width of the EGL. Nuclei are lightly stained purple with Hematoxylin and Eosin. Scale bars: 50 μm.
The EGL expression of cyclin D2 was in striking contrast to that of D1 at E16.5 and P0, even though similar strong nuclear staining of both cyclins D1 and D2 were detected in the ventricular region of the midbrain (Fig. 1A,B,E,F; Fig. 2A,B,E,F) and in some cells deeper within the inner cerebellum (Fig. 1C,G; Fig. 2C,G arrowheads). As shown in Fig. 2C, very few cells with nuclear cyclin D2 staining were observed in the EGL at E16.5 (Fig. 2C,D, arrow), consistent with reports of absent cyclin D2 mRNA in these cells during embryonic proliferation (Ross et al., 1996). Similarly, very few cyclin D2 positive nuclei were detected in the EGL at P0 (Fig. 2G,H). At P6, cyclin D2 was detected throughout the outer zone of the EGL in an area that was broader than that of cyclin D1 (Fig. 2I,J), although staining was not uniform: a subpopulation of cells, generally towards the middle of the EGL, expressed much stronger cyclin D2 levels. Furthermore, unlike cyclin D1, nuclear cyclin D2 staining was still present in some of the EGL cells by P12 (Fig. 2K,L, arrows). These immunohistochemistry results were further supported by western blot analysis of total cerebellar extracts, which showed very low levels of cyclin D2 at P0 and much higher levels at P6, while high levels of cyclin D1 were present at both time points (see Fig. S1 in the supplementary material).
Fig. 2. Cyclin D2 (brown) is not expressed at high levels in the EGL until later in cerebellar development.

(A–H) The E16.5 (A, with high-power views of boxed areas shown in B–D) and P0 (E, with high-power views of boxed areas shown in F–H) EGL show little nuclear cyclin D2 staining. Enlarged views of the ventricular region (B,F), anterior EGL (C,G) and posterior EGL (D,H) are shown. Only a few nuclei in the posterior EGL (arrow in D) are weakly positive, in comparison with strong staining of the ventricular zone of the midbrain and of cells deeper within the cerebellum (arrowheads in C,G). (I,J) Strong cyclin D2 expression is seen in the outer half to two-thirds of the EGL at P6 (I, with high-power of view of boxed area in J). (K,L) Scattered EGL cells are positive (arrows) at P12 (K, with high-power of view of boxed area in L). Brackets indicate the width of the EGL. Nuclei are lightly stained purple with Hematoxylin and Eosin. Scale bars: 50 μm.
To determine whether cyclin D2 was upregulated to compensate for the loss of cyclin D1, we evaluated the levels of cyclin D2 at P0 in Ccnd1−/− cerebella. Comparison of Ccnd1−/− and wild-type siblings showed no difference in the cyclin D2 staining pattern (see Fig. S2 in the supplementary material). As Ccnd1−/−;Ccnd2−/− mice show severe cerebellar hypoplasia with only slight upregulation of cyclin D3 (Ciemerych et al., 2002), it was unlikely that cyclin D3 would be significantly upregulated in Ccnd1−/− cerebella. Consistent with this, we were unable to detect cyclin D3 in either wild-type or Ccnd1−/− GNPs (see Fig. S2 in the supplementary material). Therefore, the absence of cyclin D1 does not lead to an upregulation of cyclins D2 or D3 in the EGL.
Ccnd1−/− mouse cerebellar phenotype
Given the absence of high levels of D-type cyclins in the EGL during early cerebellar development in Ccnd1−/− mice, we wondered whether they might have a cerebellar phenotype. Ccnd1−/− mice exhibit several symptoms such as early lethality, decreased size and a hindleg clasping reflex that have long been hypothesized to reflect a neurological defect (Sicinski et al., 1995), but no specific histological deficiency has been reported. To test if Ccnd1−/− mice might have a mild ataxia related to their cerebellar development, we examined the righting reflex of Ccnd1−/− pups (Aruga et al., 1998; Crawley, 1999). Wild-type pups could right themselves by P8, but Ccnd1−/− pups were unable to turn over until at least P14, and in some pups the reflex was delayed until as late as P18. After this point, the Ccnd1−/− adults displayed no obvious ataxia, suggesting that the cerebellar functional defect had been corrected or compensated for.
Examination of the Ccnd1−/− cerebellum indicated no gross abnormalities in foliation or organization of cellular layers at any age (data not shown). However, cerebellar cross sections did appear smaller than normal, so the sizes of Ccnd1−/− cerebella were compared with those of their littermates. At early time points the cerebellum was too small to be accurately dissected and weighed, so we estimated the size by measuring the area of a midline histological section. At P0 we found no significant difference in the cross-sectional area of the midline cerebellum (Fig. 3A,D); however, by P6 the cerebellum was reduced in size to 68% of that of wild-type siblings (Fig. 3B,E,G). This relative size difference persisted but did not increase from P6 to P12 (Fig. 3C,F,G). By contrast, the total brain weight of Ccnd1−/− brains (including the cerebellum) was 84% and 80% of the Ccnd1+/+ littermate weight at P6 and P12, respectively. Therefore, the cerebellum was more severely affected than other brain regions. In addition, the difference in the cerebellar sizes differed in its timing and severity from that of the total body weight of Ccnd1−/− mice, which continued to fall behind that of their littermates from 72% at P6 to 53% at P12 (Fig. 3H).
Fig. 3. Ccnd1−/− cerebella show a postnatal size defect.
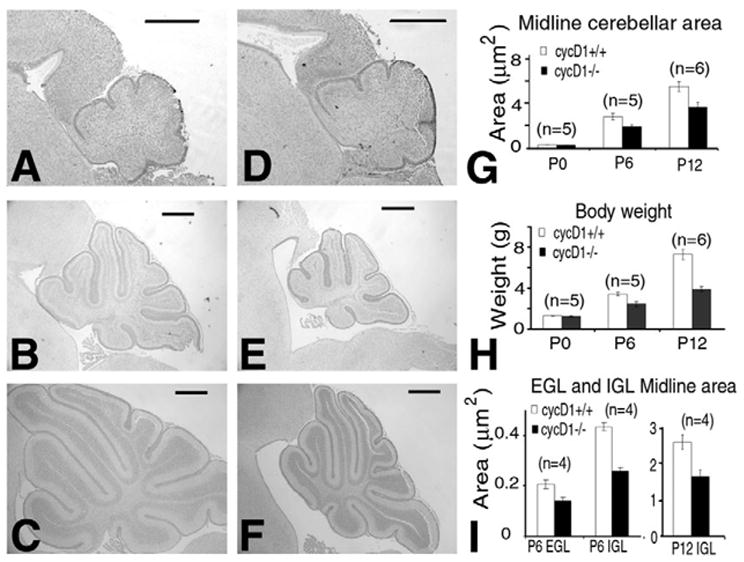
The midline cross-sections of wild-type littermates (A–C) are shown next to Ccnd1−/− (cycD1 on figure) pups (D–F). At P0 there is no difference in cerebellar size (A,D), but by P6 Ccnd1−/− cerebella are markedly smaller (B,E). The relative size difference is maintained but not increased by P12 (C,F). (G) Quantification of the relative cerebellar midline area. (H) Total body weight of Ccnd1−/− pups continued to fall further behind wild-type littermates from P6 to P12. (I) The size of the granule cell population was similarly decreased at P6 and P12. Scale bars: 500 μm. The number of littermates analyzed at each time point is indicated in each chart.
Much of the normal postnatal size increase of the cerebellum is due to an increase in the granule cell population, so the normal sized cerebellum at birth suggested that the Ccnd1−/− cerebellar size defect was probably due to the impaired postnatal proliferation of GNPs. In Ccnd1−/− cerebella, the thickness of the EGL was not altered, but the entire surface area was decreased. As the nuclear density was similar to wild type, we measured the granule neuron population by determining the cross-sectional area of the EGL and IGL in midline histological sections. This confirmed a decrease in the EGL and IGL at P6 (69% and 61% respectively) and the IGL at P12 (63%) in Ccnd1−/− cerebella relative to wild-type littermates (Fig. 3I). Therefore, the phenotypically crucial period for cyclin D1 in cerebellar growth is between P0 and P6, corresponding to the developmental phase when cyclin D1 is the only highly expressed D type cyclin in GNPs.
Removal of cyclin D1 affects early granule neuron precursor proliferation
The size defect of Ccnd2−/− cerebella was reported to be due to both decreased entry into S phase and increased apoptosis of GNPs (Huard et al., 1999). To determine if increased apoptosis could contribute to the decreased granule neuron population in Ccnd1−/−cerebella, we performed TUNEL assays on both P0 and P6 cerebella. Both wild-type and Ccnd1−/− cerebella showed very low levels of apoptosis, and no significant difference was seen between them (see Fig. S3 in the supplementary material). Therefore, there is no evidence that increased apoptosis significantly contributes to the smaller granule neuron population in Ccnd1−/− mice, although we cannot absolutely exclude this possibility.
We then examined whether Ccnd1−/− GNPs had decreased proliferation. As the D-type cyclins are known to be involved in the transition through G1 (Sherr and Roberts, 2004), we expected this phase of the cell cycle to be lengthened and the percentage of cells in S phase to be decreased. To identify cells in S-phase, pups were injected with BrdU 1 hour prior to sacrifice. Midline sections were labeled with anti-BrdU or anti-phosphohistone H3 (H3-P) antibodies to measure S-phase or M phase cells, respectively. BrdU incorporation and H3-P staining were observed throughout the entire anteroposterior axis of the midline EGL at E16.5 and P0, except in the innermost one to two cell layers (Fig. 4A,B,E,F; data not shown). By P6 when the thickness of the EGL had greatly increased, both markers were found only in the outer half of the EGL (Fig. 4C,D,G,H; data not shown), consistent with known proliferation and migration patterns.
Fig. 4. Proliferation is decreased in Ccnd1−/− cerebella during early development.

(A–H) Representative BrdU (A–D) and H3-P (E–H) immunohistochemistry of cerebella from P0 (A,B,E,F) and P6 (C,D,G,H) pups sacrificed 1 hour after BrdU injection. (I) Quantification of BrdU incorporation in the anterior EGL of wild-type and Ccnd1−/− (cycD1 on figure) siblings at P0 (n=7) and P6 (n=4). *P≤0.03 when comparing between +/+ and −/− littermates. (J) Quantification of H3-P positive nuclei in the EGL at P0 (n=8, P=0.07) and P6 (n=4, P=0.22) shows a trend of decreased mitotic cells in P0 but not in P6 GNPs in the mutant.
The pattern of BrdU incorporation was roughly similar between Ccnd1−/− and wild-type littermates except that BrdU incorporation was consistently decreased in Ccnd1−/− cerebella at P0 (Fig. 4A,B,I). Quantification of S phase cells in the same anterior region of the EGL at P0 revealed that Ccnd1−/− cerebella exhibit a significant decrease in BrdU incorporation (Fig. 4I; P0+/+=427 cells/mm, P0−/−=363 cells/mm; P=0.028). Interestingly, this difference was not observed at P6, when both cyclins D1 and D2 are expressed at high levels (Fig. 4C,D,I). There was similarly a trend in decreased mitosis in the Ccnd1−/− EGL at P0 (Fig. 4E–F,J), although this was not statistically significant (P=0.07) probably due to the small number of mitotic cells as a consequence of the very short length of mitosis. This modest (about 15%) decrease in BrdU incorporation could account for the decrease in the granule cell population over 6 days of rapid proliferation. For example, if early GNPs proliferate at a rate of 15 hours/cycle (Yoshioka et al., 1985), there could be 6×24/15 (i.e. 9.6) rounds of GNP proliferation between P0 and P6. If each round of cell proliferation is decreased by 15%, this could lead to the number of cells at P6 being decreased to as little as (1–0.15)9.6×100 (i.e. 21%) of the normal number. Therefore, a small decrease in proliferation over the course of multiple cell cycles can have a profound effect on the cerebellar size. Taken together, these results indicate that at P0, when cyclin D1 is the only highly expressed D-type cyclin, GNPs are impaired in their ability to progress into the cell cycle in the absence of cyclin D1. The observed defect in the G1/S transition of Ccnd1−/− GNPs corresponds precisely to the appearance of the cerebellar size defect seen between P0 and P6.
Delayed Purkinje cell development in the Ccnd1−/− cerebellum
Purkinje cell progenitors complete proliferation by E14.5, so the normal size of Ccnd1−/− P0 cerebella suggested that Purkinje cell numbers were not drastically affected; however, other mice with granule neuron defects show abnormal Purkinje cell maturation (Goldowitz and Hamre, 1998). To determine whether Purkinje cell dendrite development was altered in Ccnd1−/− cerebella, we examined sections from pups at different developmental stages. At P6, Purkinje cell dendrites have not extended very far, and there is little difference between wild-type and Ccnd1−/− pups (data not shown). At P9, when dendrites have begun to extend but have not developed the more complex branches characteristic of adult cerebella, staining with calbindin showed stunted dendrites in Ccnd1−/− pups (Fig. 5A,B). We further examined the dendrites by confocal microscopy and found that, at P9, Ccnd1−/− Purkinje cells had simplified disorganized dendritic trees (data not shown). Interestingly, adult Ccnd1−/− Purkinje cells looked normal (Fig. 5C,D); therefore, the timing of the Purkinje cell abnormality corresponds to the period of ataxia for Ccnd1−/− pups. As Purkinje cells did not express cyclin D1 at any of the postnatal time points examined, it is unlikely the observed Purkinje cell defect represents a cell-autonomous event; more likely it represents a response to abnormalities in other cell types such as the GNPs.
Fig. 5. Purkinje cell dendritic development is abnormal in Ccnd1−/− mice at P9.

Comparison of wild-type (A,C) and Ccnd1−/− (B,D) Purkinje cells stained with anti-calbindin antibody shows that at P9 (A,B), Ccnd1−/− Purkinje cell dendrites are abnormally oriented and stunted, but by 4 weeks (C,D) they look normal. Scale bars: 50 μm in A,B; 100 μm in C,D.
Comparison of Ccnd1−/− pups with siblings at P6, P12 and P15 showed similar decreases in EGL thickness (see Fig. S4 in the supplementary material), suggesting that GNPs were not accumulating in the EGL due to migration abnormalities. Therefore, we did not expect to see abnormalities in the radial glia along which they migrate. Consistent with this, staining with GFAP showed a normal density and orientation for these cells (data not shown). We conclude that the granule neuron proliferation defect is more likely to be due to a cell-intrinsic effect of cyclin D1 than to abnormal signals from the other cells such as the Purkinje cells or glial cells.
Decreased medulloblastoma incidence in Ptch1+/−;Ccnd1−/− mice
Ptch1+/− mice, in which the Ptch1 allele has been replaced with the lacZ gene, have been reported to show a 15–20% incidence of spontaneous medulloblastoma (Goodrich et al., 1997; Wetmore et al., 2000). Transcriptional profiling has shown that cyclin D1 is among the genes most robustly activated by Shh signaling in GNPs (Lee et al., 2003; Oliver et al., 2003), the cell from which the tumors are believed to arise, and in vitro cyclin D1 protein levels are more rapidly upregulated relative to other D-type cyclins in GNPs in response to Shh (Kenney and Rowitch, 2000). The observation that cyclin D1 plays a crucial role in normal early GNP proliferation prompted us to determine whether loss of cyclin D1 might decrease medulloblastoma incidence in Ptch1+/−mice.
Ptch1+/−;Ccnd1+/− mice were crossed to generate both Ptch1+/−;Ccnd1+/+ and Ptch1+/−;Ccnd1−/− mice. As had been previously reported (Sicinski et al., 1995), Ccnd1−/− pups showed high rates of early lethality with an especially steep decline in survival during the first 5 weeks of life, with 54% of Ptch1+/−;Ccnd1−/− pups surviving to 5 weeks. After this period, the rate of mortality was very low, allowing us to examine medulloblastoma development in surviving adults. The peak incidence of symptomatic medulloblastoma in Ptch1+/− mice occurs at 16–24 weeks of age (Wetmore et al., 2000), and a similar incidence of tumors (7/23) was found in asymptomatic mice in this age range (Goodrich et al., 1997). All the Ptch1+/−;Ccnd1−/− mice included in our analysis (Fig. 6H) survived within or beyond this time point.
Fig. 6. Medulloblastoma development is significantly decreased in Ptch1+/−;Ccnd1−/− mice compared with Ptch1+/− mice, despite upregulation of both cyclins D1 and D2 in tumors.
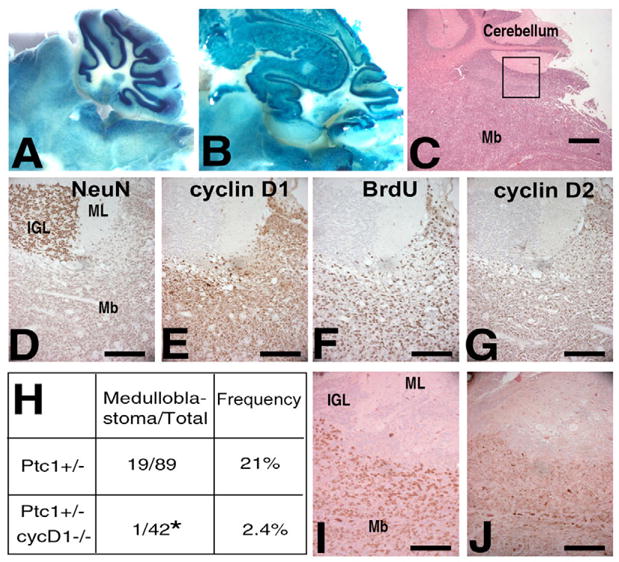
Tumors were identified by staining with X-gal (A,B): in the normal cerebellum (A), the staining is strongest in the Purkinje cell layer and inner granule layer, but medulloblastomas (B) were easily identified by their disruption of this pattern resulting in staining throughout the tumor, including at the surface of the brain. (C) Medulloblastomas were also identified histologically. (D–G) Immunohistochemistry of Ptch1+/−medulloblastomas. (D) In contrast to mature granule cells of the IGL, tumor cells are only weakly positive for the postmitotic marker NeuN. Expression of both cyclins D1 (E) and D2 (G), as well as BrdU incorporation (F) in the tumor but not in adjacent IGL or molecular layers. (H) Ptch1+/−;Ccnd1−/− (cycD1 on figure) mice develop significantly fewer medulloblastomas than do Ptch1+/−mice P=0.0105. (I,J) The medulloblastoma that developed in Ptch1+/−;Ccnd1−/− background showed similar patterns of BrdU incorporation (I) and cyclin D2 (J) expression. Mb, medulloblastoma; ML, molecular layer; IGL, inner granule layer. Scale bars: 500 μm in C; 100 μm in D–G,I,J.
Adult mice were monitored and maintained until they showed symptoms of illness (decreased weight, lethargy, difficulty moving, change in the shape of their head) or reached well beyond the window of normal tumor development. All brains were examined either histologically or grossly for tumors. Brains were stained with X-gal and the cerebella were assessed under the dissecting scope or after histological sectioning (Fig. 6A–C). Out of 89 Ptch1+/−;Ccnd1+/+ brains inspected, 19 were found to have medulloblastomas (21%) (Fig. 6H). We did not find any tumors in asymptomatic mice, suggesting that by this age tumors are large enough to produce unmistakable symptoms. By contrast, only one of 42 (2.4%) Ptch1+/−;Ccnd1−/− mice developed a medulloblastoma (Fig. 6H). In total another five of the 42 adult Ptch1+/−;Ccnd1−/− mice showed rapid weight loss and lethargy and were sacrificed, but none of these had a medulloblastoma. One Ptch1+/−;Ccnd1+/+ and 3 Ptch1+/−;Ccnd1−/− mice were found dead in their cages despite no previous symptoms, and their brains could not be examined because of advanced autolysis.
To exclude the possibility that we did not find tumors in adult Ptch1+/−;Ccnd1−/− mice because pups with tumors were selected against during earlier development, we examined eight Ptch1+/−;Ccnd1−/− cerebella from mice 2.5–3.0 weeks old by staining the brain for β-gal activity and comparing them with Ptch1+/−;Ccnd1+/+ cerebella of the same age. Although we expected only about half of the Ptch1+/−;Ccnd1−/− mice of this age to survive to adulthood, we could find no major abnormalities in cerebellar patterns of β-galactosidase activity. In fact, there appeared to be fewer regions of abnormal surface staining than in Ptch1+/−;Ccnd1+/+ cerebella. In conclusion, our results show that although not absolutely essential, cyclin D1 plays an important role in Shh signaling induced medulloblastomas.
Medulloblastomas from Ptch1+/− mice express both cyclins D1 and D2
As the biochemical activities of the D-type cyclins appear similar, it has been hypothesized that the requirement for a specific cyclin in a specific tissue type is due largely to the regulation of its expression. In mouse breast tumor models, the inhibition of tumorigenesis in Ccnd1−/− tissue has been linked to the ability of an oncogenic signaling pathway to upregulate cyclin D1 specifically (Yu et al., 2001). To determine if the requirement for cyclin D1 in medulloblastomas in Ptch1+/− mice could be explained by it being the only D-type cyclin expressed, we assessed the levels of both cyclins D1 and D2 by immunohistochemistry. We found that both cyclins D1 and D2 were highly expressed in the eight Ptch1+/−;Ccnd1+/+ tumors examined (Fig. 6E,G). All tumors had a large fraction of cells expressing cyclins D1 (39±12.6%) and D2 (42±9.2%). BrdU staining showed that these tumors were also highly proliferative, with an average of 25% of cells showing BrdU incorporation (Fig. 6F). Interestingly, the single Ptch1+/−;Ccnd1−/− tumor that developed had a similar percentage of cyclin D2 positive cells (40%) (Fig. 6J) and was highly proliferative (33% BrdU positive) (Fig. 6I). Thus, the absence of cyclin D1 did not impact tumor cell proliferation at this stage. As there was no further increase in the level of cyclin D2 in the Ptch1+/−;Ccnd1−/− medulloblastoma, these observations indicate that cyclin D1 is not required for medulloblastoma cell proliferation and suggest that it is more likely to play a key role at an earlier stage in medulloblastoma development.
Decreased ‘pre-neoplastic’ lesions in Ptch1+/−;Ccnd1−/− mice
Adult Ptch1+/− mice have been reported to maintain ectopic clusters of cells at the cerebellar surface, and these have been suggested to represent pre-neoplastic lesions (Goodrich et al., 1997; Kim et al., 2003; Oliver et al., 2005). To determine if removal of cyclin D1 affected the incidence of these pre-neoplastic lesions, we dissected 3-week-old Ptch1+/−;Ccnd1+/+ and Ptch1+/−;Ccnd1−/− cerebella, and examined sagittal sections every 200 μm for ectopic lesions. As expected, in the five wild-type cerebella examined the EGL was entirely absent, and no ectopic clusters of cells were observed. By contrast, all Ptch1+/−;Ccnd1+/+ and some Ptch1+/−;Ccnd1−/− mice had ectopic clusters of cells above the molecular layer. We divided these clusters into two types based on cell proliferation: one that was proliferative (Fig. 7A–E) and the other that was not proliferative (Fig. 7F–J), as determined by staining for BrdU or H3-P. Similar to mature granule neurons in the IGL, all cells in the non-proliferative clusters expressed high levels of the post-mitotic neural marker NeuN (Fig. 7F,G), and these clusters did not express cyclin D1 or D2 (Fig. 7I,J). As we observed similar small ectopic clusters of non-proliferative granule neurons in adult (35-week-old) Ptch1+/−;Ccnd1+/+ mice that did not develop medulloblastomas, these non-proliferative clusters are unlikely to represent pre-neoplastic lesions. Interestingly, no significant difference in the incidence of these non-proliferative lesions was observed between adult Ptch1+/−;Ccnd1+/+ and Ptch1+/−;Ccnd1−/− mice (Fig. 7K).
Fig. 7. Ptch1+/−;Ccnd1−/− mice develop fewer pre-neoplastic lesions.
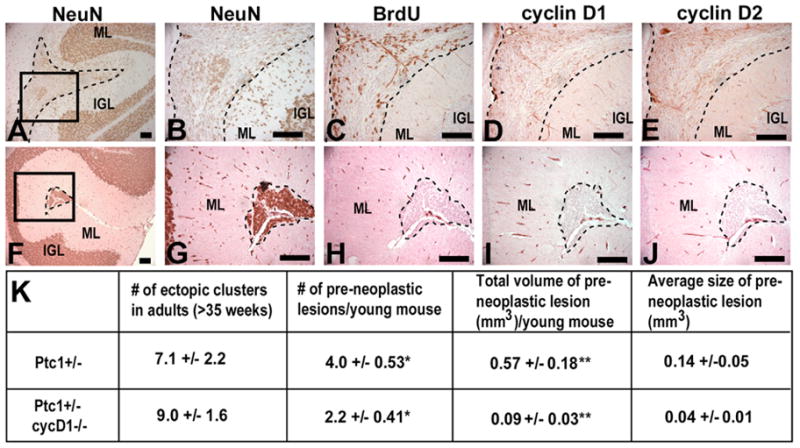
(A–E) Pre-neoplastic lesions (indicated by broken lines) express both cyclins D1 (D) and D2 (E) and incorporate BrdU (C). (A,B) Some cells in these lesions, particularly in BrdU-negative regions, are weakly NeuN positive. The density of NeuN positive cells is increased in the molecular layer below the lesion (A,B) when compared with below the mature cluster (F,G). (F–J) Clusters of mature ectopic granule cells (indicated by broken lines) do not incorporate BrdU (H) or express cyclins D1 (I) or D2 (J), and all cells are strongly positive for the post-mitotic neural marker NeuN (F,G). Scale bars: 100 μm. (K) There is a significant decrease in the number of pre-neoplastic lesions in Ptch1+/−;Ccnd1−/− (cycD1 on figure) mice when compared with Ptch1+/− mice (*P=0.021), and a smaller total volume of preneoplastic lesions (**P=0.020), but no significant difference in the number of mature ectopic clusters seen in older adults. ML, molecular layer; IGL, inner granule layer.
By contrast, proliferative lesions were only detected in young mice, and they correspond to the previously described ‘pre-neoplastic’ lesions in terms of their histology, proliferation and gene expression properties as described below (Oliver et al., 2005). These lesions expressed both cyclins D1 and D2 (Fig. 7D,E), and were only weakly positive for NeuN (Fig. 7A,B), similar to medulloblastomas (Fig. 6D). Unlike the normal EGL (Fig. 4C,D), there was no organization into outer proliferative and inner post-mitotic layers. In some proliferative lesions, BrdU-positive cells were found relatively evenly throughout the entire collection of cells (not shown), while in others (Fig. 7C) there were areas of abnormal organization with BrdU-positive cells at both the inner and outer edges of the cluster. In these cases, comparison of serial sections showed that non-proliferative regions were more likely to express NeuN (Fig. 7B). There was also an increased number of strongly NeuN-positive cells in the molecular layer below the proliferative lesions (compare Fig. 7B and 7G), suggesting that some cells from the lesion were differentiating and migrating out of these clusters. Additionally, TUNEL staining revealed apoptotic cells within these lesions that were not detected in the normal mature granule layer (see Fig. S3 in the supplementary material).
Each section was examined for proliferative pre-neoplastic lesions. If a lesion appeared in the same folia in two adjacent sections, it was deemed to represent a single lesion, whereas if a lesion appeared distinctly anterior or posterior to a lesion in a neighboring section it probably represented a different lesion. The size of individual lesions varied widely, with some small lesions appearing in only two consecutive sections (therefore extending less than 600 μm in the mediolateral direction), while other large lesions spanned up to 11 sequential sections (up to 2400 μm). The volume of a lesion was estimated based on its average cross-sectional area and its mediolateral span. Ptch1+/−;Ccnd1−/− pups were found to have significantly fewer of these pre-neoplastic lesions (Fig. 7K). The average number of independent lesions was approximately half that of Ptch1+/−;Ccnd1+/+ mice, while the total volume of lesions per mouse was sixfold smaller. The average size of an individual lesion was also one quarter of that in Ptch1+/−;Ccnd1+/+ mice. Therefore, we conclude that cyclin D1 contributes to the growth of pre-neoplastic lesions in young Ptch1+/− mice and that the decrease in the number of these cells probably accounts for the decreased incidence of medulloblastoma in Ptch1+/−;Ccnd1−/− mice.
DISCUSSION
Early and late GNPs have different D type cyclin expression
Our results demonstrate that the expression of D-type cyclins in GNPs changes over time, possibly indicative of different stages of GNPs as they progress towards differentiation. Specifically, our analysis of the expression of cyclins D1 and D2 revealed that GNPs at P0 expressed high levels of only cyclin D1, while at P6 both cyclins D1 and D2 were expressed. Consistent with these observations, the absence of cyclin D1 impaired the proliferation of P0 GNPs but not P6 GNPs. We have not examined time points between P0 and P6 to determine exactly when cyclin D2 began to appear during cerebellar development, but it was reported that cyclin D2 mRNA was absent from the embryonic cerebellum from E14.5 to E18.5, and was detected postnatally as early as P2 (Ross et al., 1996) and that Ccnd2−/− cerebella began to show phenotypes after P3 (Huard et al., 1999). These reports are consistent with our cyclin D2 expression data and our suggestion that cyclin D2 can compensate for the absence of cyclin D1 at P6.
Although both early and late GNPs can proliferate, it is possible that they have distinct capacities to be transformed. For example, early GNPs potentially behave more like stem cells and are therefore more susceptible to transformation, while later GNPs have a more limited proliferation potential because they are more committed to differentiation. In support of this idea, it was reported that there was a gradual decrease in the ability of irradiation to increase the incidence of medulloblastoma in Ptch1+/− pups as they aged from P1 to P10: irradiation at P1 or P4 increased medulloblastoma incidence to 81% or 51%, respectively, while by P10 irradiation had no effect (Pazzaglia et al., 2002; Pazzaglia et al., 2006). This is somewhat surprising given that there are many more GNPs at P4 than at P1, so this difference probably reflects a greater susceptibility of early GNPs to transformation.
An early role of cyclin D1 in medulloblastoma development
We propose a model that explains a role for cyclin D1 in medulloblastoma development. Recently a pre-neoplastic stage of medulloblastoma was found to exhibit gene expression patterns intermediate between those of GNPs and tumor cells (Oliver et al., 2005), suggesting that medulloblastomas are derived from these lesions. The pre-neoplastic cells lacked expression of the wild-type Ptch1 allele, suggesting that loss of Ptch1 expression is a rate-limiting step in the transition from normal GNPs to pre-neoplastic lesions. We suggest that genetic or epigenetic changes that activate Shh signaling in either early or late GNPs could result in pre-neoplastic lesions with cells that have the capacity to not only proliferate but also differentiate or die. However, lesions derived from early GNPs also contain a population of cells that behave more like stem cells and continue to renew, allowing them to acquire additional genetic and epigenetic changes, which may lead to malignant medulloblastoma. By contrast, transformation of late GNPs would be less likely as these cells have a decreased proliferation potential and would require re-activation of self-renewal pathways. This model predicts that removal of cyclin D1, by limiting the proliferation of early GNPs, would reduce the number of lesions arising from early GNPs. This is consistent with our findings of an approximate halving in the number of independent pre-neoplastic lesions. In addition, Ccnd1−/− early GNPs may be intrinsically less competent for tumorigenesis, owing to their impaired ability to proliferate. In support of this, we saw a more dramatic decrease in the total volume of pre-neoplastic cells and in the incidence of medulloblastomas in Ptch1+/−;Ccnd1−/− mice.
Interestingly, while multiple pre-neoplastic lesions per mouse were observed in young mice, only one or two lesions were observed by 6–8 weeks of age (Oliver et al., 2005). These results suggest that the majority of pre-neoplastic lesions will not progress to tumors. As we detected both apoptosis and differentiation within these pre-neoplastic lesions, it is likely that the abnormal cells in most of these lesions either differentiate or undergo apoptosis, resulting in the disappearance of the lesions. In addition to pre-neoplastic lesions, we found that both young and adult Ptch1+/− mice had non-proliferative clusters of cells, which were abnormally localized, morphologically similar to mature granule neurons and strongly NeuN-positive. We speculate that pre-neoplastic lesions might transition into these non-proliferative clusters. In support of this, the pre-neoplastic lesions often exhibited areas of non-proliferation with NeuN-positive cells.
This specific requirement for cyclin D1 during early tumorigenesis in the Ptch1+/− medulloblastoma model contrasts with its role as described in other tumor models (Robles et al., 1998; Yu et al., 2001), which revealed an important role for cyclin D1 only when cyclin D1 was the only D-type cyclin upregulated in the tumor. Furthermore, in the breast tumor model, continued cyclin D1/cdk4 kinase activity is required for tumor cell proliferation (Yu et al., 2006). By contrast, it does not seem likely that cyclin D1 is required for proliferation of medulloblastoma cells as medulloblastomas that developed in Ptch1+/−;Ccnd1+/+ mice showed upregulation of both cyclins D1 and D2, and the one Ptch1+/−;Ccnd1−/− medulloblastoma that developed did not show a decreased level of proliferation.
Other cell cycle regulators in GNPs and medulloblastomas
During granule cell development, a number of cell cycle regulatory proteins, including cyclin D2 and the cell cycle inhibitors p18 and p27, are expressed in at least a partially overlapping manner, and deletion of any one is often sufficient to create a mild cerebellar phenotype. The Cip/Kip family member p27 was reported to be the only member of the Cip/Kip family of Cdk inhibitors expressed in granule cell precursors, and it is detected in the inner two-thirds of the EGL and in the IGL at P7 (Miyazawa et al., 2000). In addition to p27, at least one member of the Ink family of Cdk inhibitors, p18ink4c, which inhibits cyclin D/cdk4 kinase activity, is expressed in the pool of proliferating granule cell precursors by P5, although it is absent from early EGL cells at E15.5 (Zindy et al., 2003). Given the intriguing possibility from our results that medulloblastoma development may involve early GNPs that are present at P0, it will be interesting to define more precisely the spatiotemporal expression of other cell cycle regulators and the signaling pathways that regulate them in the GNPs at this stage. In addition, as cyclin D2 is not expressed in early GNPs and removal of cyclin D2 affects later GNP proliferation and apoptosis, it will be interesting to determine if removal of cyclin D2 affects the incidence of medulloblastomas and the number or size of pre-neoplastic lesions.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Footnotes
Supplementary material
Supplementary material for this article is available at http://dev.biologists.org/cgi/content/full/133/19/3929/DC1
This work is supported by grants from the American Cancer Society, National Institute of Health and the Fletcher Scholar award from the Cancer Research Foundation to W.D. J.P. was supported by the Medical Scientist National Research Service Award Grant and W.D. is a Scholar of the Leukemia and Lymphoma Society.
References
- Aruga J, Minowa O, Yaginuma H, Kuno J, Nagai T, Noda T, Mikoshiba K. Mouse Zic1 is involved in cerebellar development. J Neurosci. 1998;18:284–293. doi: 10.1523/JNEUROSCI.18-01-00284.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman DM, Karhadkar SS, Hallahan AR, Pritchard JI, Eberhart CG, Watkins DN, Chen JK, Cooper MK, Taipale J, Olson JM, et al. Medulloblastoma growth inhibition by hedgehog pathway blockade. Science. 2002;297:1559–1561. doi: 10.1126/science.1073733. [DOI] [PubMed] [Google Scholar]
- Ciemerych MA, Kenney AM, Sicinska E, Kalaszczynska I, Bronson RT, Rowitch DH, Gardner H, Sicinski P. Development of mice expressing a single D-type cyclin. Genes Dev. 2002;16:3277–3289. doi: 10.1101/gad.1023602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawley JN. Behavioral phenotyping of transgenic and knockout mice: experimental design and evaluation of general health, sensory functions, motor abilities, and specific behavioral tests. Brain Res. 1999;835:18–26. doi: 10.1016/s0006-8993(98)01258-x. [DOI] [PubMed] [Google Scholar]
- Gilbertson RJ. Medulloblastoma: signalling a change in treatment. Lancet Oncol. 2004;5:209–218. doi: 10.1016/S1470-2045(04)01424-X. [DOI] [PubMed] [Google Scholar]
- Goldowitz D, Hamre K. The cells and molecules that make a cerebellum. Trends Neurosci. 1998;21:375–382. doi: 10.1016/s0166-2236(98)01313-7. [DOI] [PubMed] [Google Scholar]
- Goodrich LV, Milenkovic L, Higgins KM, Scott MP. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science. 1997;277:1109–1113. doi: 10.1126/science.277.5329.1109. [DOI] [PubMed] [Google Scholar]
- Hahn H, Wicking C, Zaphiropoulous PG, Gailani MR, Shanley S, Chidambaram A, Vorechovsky I, Holmberg E, Unden AB, Gillies S, et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell. 1996;85:841–851. doi: 10.1016/s0092-8674(00)81268-4. [DOI] [PubMed] [Google Scholar]
- Huard JM, Forster CC, Carter ML, Sicinski P, Ross ME. Cerebellar histogenesis is disturbed in mice lacking cyclin D2. Development. 1999;126:1927–1935. doi: 10.1242/dev.126.9.1927. [DOI] [PubMed] [Google Scholar]
- Kenney AM, Rowitch DH. Sonic hedgehog promotes G(1) cyclin expression and sustained cell cycle progression in mammalian neuronal precursors. Mol Cell Biol. 2000;20:9055–9067. doi: 10.1128/mcb.20.23.9055-9067.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Nelson AL, Algon SA, Graves O, Sturla LM, Goumnerova LC, Rowitch DH, Segal RA, Pomeroy SL. Medulloblastoma tumorigenesis diverges from cerebellar granule cell differentiation in patched heterozygous mice. Dev Biol. 2003;263:50–66. doi: 10.1016/s0012-1606(03)00434-2. [DOI] [PubMed] [Google Scholar]
- Lee Y, Miller HL, Jensen P, Hernan R, Connelly M, Wetmore C, Zindy F, Roussel MF, Curran T, Gilbertson RJ, et al. A molecular fingerprint for medulloblastoma. Cancer Res. 2003;63:5428–5437. [PubMed] [Google Scholar]
- Miyazawa K, Himi T, Garcia V, Yamagishi H, Sato S, Ishizaki Y. A role for p27/Kip1 in the control of cerebellar granule cell precursor proliferation. J Neurosci. 2000;20:5756–5763. doi: 10.1523/JNEUROSCI.20-15-05756.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver TG, Grasfeder LL, Carroll AL, Kaiser C, Gillingham CL, Lin SM, Wickramasinghe R, Scott MP, Wechsler-Reya RJ. Transcriptional profiling of the Sonic hedgehog response: a critical role for N-myc in proliferation of neuronal precursors. Proc Natl Acad Sci USA. 2003;100:7331–7336. doi: 10.1073/pnas.0832317100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver TG, Read TA, Kessler JD, Mehmeti A, Wells JF, Huynh TT, Lin SM, Wechsler-Reya RJ. Loss of patched and disruption of granule cell development in a pre-neoplastic stage of medulloblastoma. Development. 2005;132:2425–2439. doi: 10.1242/dev.01793. [DOI] [PubMed] [Google Scholar]
- Pazzaglia S, Mancuso M, Atkinson MJ, Tanori M, Rebessi S, Majo VD, Covelli V, Hahn H, Saran A. High incidence of medulloblastoma following X-ray-irradiation of newborn Ptc1 heterozygous mice. Oncogene. 2002;21:7580–7584. doi: 10.1038/sj.onc.1205973. [DOI] [PubMed] [Google Scholar]
- Pazzaglia S, Tanori M, Mancuso M, Rebessi S, Leonardi S, Di Majo V, Covelli V, Atkinson MJ, Hahn H, Saran A. Linking DNA damage to medulloblastoma tumorigenesis in patched heterozygous knockout mice. Oncogene. 2006;25:1165–1173. doi: 10.1038/sj.onc.1209032. [DOI] [PubMed] [Google Scholar]
- Robles AI, Rodriguez-Puebla ML, Glick AB, Trempus C, Hansen L, Sicinski P, Tennant RW, Weinberg RA, Yuspa SH, Conti CJ. Reduced skin tumor development in cyclin D1-deficient mice highlights the oncogenic ras pathway in vivo. Genes Dev. 1998;12:2469–2474. doi: 10.1101/gad.12.16.2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romer JT, Kimura H, Magdaleno S, Sasai K, Fuller C, Baines H, Connelly M, Stewart CF, Gould S, Rubin LL, et al. Suppression of the Shh pathway using a small molecule inhibitor eliminates medulloblastoma in Ptc1(+/−)p53(−/−) mice. Cancer Cell. 2004;6:229–240. doi: 10.1016/j.ccr.2004.08.019. [DOI] [PubMed] [Google Scholar]
- Ross ME, Carter ML, Lee JH. MN20, a D2 cyclin, is transiently expressed in selected neural populations during embryogenesis. J Neurosci. 1996;16:210–219. doi: 10.1523/JNEUROSCI.16-01-00210.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherr CJ, Roberts JM. Living with or without cyclins and cyclin-dependent kinases. Genes Dev. 2004;18:2699–2711. doi: 10.1101/gad.1256504. [DOI] [PubMed] [Google Scholar]
- Sicinski P, Donaher JL, Parker SB, Li T, Fazeli A, Gardner H, Haslam SZ, Bronson RT, Elledge SJ, Weinberg RA. Cyclin D1 provides a link between development and oncogenesis in the retina and breast. Cell. 1995;82:621–630. doi: 10.1016/0092-8674(95)90034-9. [DOI] [PubMed] [Google Scholar]
- Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, Dirks PB. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63:5821–5828. [PubMed] [Google Scholar]
- Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- Stecca B, Ruiz i Altaba A. Brain as a paradigm of organ growth: Hedgehog-Gli signaling in neural stem cells and brain tumors. J Neurobiol. 2005;64:476–490. doi: 10.1002/neu.20160. [DOI] [PubMed] [Google Scholar]
- Wechsler-Reya R, Scott MP. The developmental biology of brain tumors. Annu Rev Neurosci. 2001;24:385–428. doi: 10.1146/annurev.neuro.24.1.385. [DOI] [PubMed] [Google Scholar]
- Wechsler-Reya RJ, Scott MP. Control of neuronal precursor proliferation in the cerebellum by Sonic Hedgehog. Neuron. 1999;22:103–114. doi: 10.1016/s0896-6273(00)80682-0. [DOI] [PubMed] [Google Scholar]
- Wetmore C, Eberhart DE, Curran T. The normal patched allele is expressed in medulloblastomas from mice with heterozygous germ-line mutation of patched. Cancer Res. 2000;60:2239–2246. [PubMed] [Google Scholar]
- Yoshioka H, Mino M, Morikawa Y, Kasubuchi Y, Kusunoki T. Changes in cell proliferation kinetics in the mouse cerebellum after total asphyxia. Pediatrics. 1985;76:965–969. [PubMed] [Google Scholar]
- Yu Q, Geng Y, Sicinski P. Specific protection against breast cancers by cyclin D1 ablation. Nature. 2001;411:1017–1021. doi: 10.1038/35082500. [DOI] [PubMed] [Google Scholar]
- Yu Q, Sicinska E, Geng Y, Ahnstrom M, Zagozdzon A, Kong Y, Gardner H, Kiyokawa H, Harris LN, Stal O, et al. Requirement for CDK4 kinase function in breast cancer. Cancer Cell. 2006;9:23–32. doi: 10.1016/j.ccr.2005.12.012. [DOI] [PubMed] [Google Scholar]
- Zindy F, Nilsson LM, Nguyen L, Meunier C, Smeyne RJ, Rehg JE, Eberhart C, Sherr CJ, Roussel MF. Hemangiosarcomas, medulloblastomas, and other tumors in Ink4c/p53-null mice. Cancer Res. 2003;63:5420–5427. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.