

Abstract
A number of reports indicate the potential for redox signalling via extracellular signal-regulated protein kinases (ERK) during neuronal injury. We have previously found that sustained ERK activation contributes to toxicity elicited by 6-hydroxydopamine (6-OHDA) in the B65 neuronal cell line. To determine whether reactive oxygen species (ROS) play a role in mediating ERK activation and 6-OHDA toxicity, we examined the effects of catalase, superoxide dismutase (SOD1), and metalloporphyrin antioxidants (‘SOD mimetics’) on 6-OHDA-treated cells. We found that catalase and metalloporphyrin antioxidants not only conferred protection against 6-OHDA but also inhibited development of sustained ERK phosphorylation in both differentiated and undifferentiated B65 cells. However, exogenously added SOD1 and heat-inactivated catalase had no effect on either toxicity or sustained ERK phosphorylation. This correlation between antioxidant protection and inhibition of 6-OHDA-induced sustained ERK phosphorylation suggests that redox regulation of ERK signalling cascades may contribute to neuronal toxicity.
Keywords: 6-hydroxydopamine, mitogen activated protein kinase, Parkinson’s disease, redox signalling
1. Introduction
6-hydroxydopamine (6-OHDA) is a redox active neurotoxin (Cohen and Heikkila 1974), commonly used to produce a Parkinsonian pattern of neuronal loss in rodents (Ungerstedt 1968). Exact mechanisms by which 6-OHDA elicits its neurotoxic effects have yet to be fully elucidated, although many studies implicate a role for oxidative mediators (Glinka and Youdim 1995; Asanuma et al 1998).
Recently, it has been appreciated that reactive oxygen species (ROS) can serve as modulators of signal transduction pathways (reviewed in Suzuki et al 1997). Thus, one possible molecular mechanism by which oxidants may contribute to neuronal death is through their ability to influence critical molecules within intracellular signalling cascades. Several recent studies indicate that activation of the extracellular signal-regulated protein kinase (ERK) branch of the mitogen-activated protein (MAP) kinase superfamily may play a pathologic role in neurons exposed to increased oxidative stress (Ohhashi et al 1999; Stanciu et al 2000; Kulich and Chu 2001). We have previously reported that the neurotoxin 6-OHDA elicits sustained ERK-phosphorylation and cytotoxicity in B65 cells, which could be attenuated by the MEK inhibitor PD98059 (Kulich and Chu 2001). In the present study we investigated the potential role of ROS in 6-OHDA-mediated sustained ERK activation and cytotoxicity.
2. Materials and methods
2.1 Cell culture
Chemical reagents (except where specified) were purchased from Sigma, St. Louis, MO, USA. B65 cells, a gift from Dr David Schubert of the Salk Institute (Schubert et al 1974), were plated at 280 cells/mm2, and grown as described previously (Kulich and Chu 2001). For differentiation studies, cells were transferred to DH2 differentiation media, DMEM containing 2% FCS, 10 mM HEPES, 5 mM butyrate, and 5 μM UO126, 24 h after plating and maintained for 7 days. For toxicity and ERK phosphorylation studies, the media was changed to DH2, minus UO126, 30 min prior to addition of 6-OHDA or vehicle.
2.2 Toxicity assays
Cell injury was determined using two independent methods: metabolism of the tetrazolium salt [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulphophenyl)-2H-tetrazolium, inner salt] (MTS assay); and lactate dehydrogenase (LDH) release, as described previously (Kulich and Chu 2001). The antioxidant reagents were diluted in DH10 (Kulich and Chu 2001), and added 30 min prior to the addition of 6-OHDA. Heat-inactivation (5 min, 100°) of beef liver catalase (Roche Molecular Biochemicals, Indianapolis, IN, USA) and bovine liver Cu/Zn superoxide dismutase (SOD1) (Alexis Biochemicals, 260,000 U/ml) resulted in > 90% loss of activity as confirmed by in vitro assays for catalase (Aebi 1984) and SOD activity (Fattman et al 2001). In studies utilizing Mn-tetrakis-(N-ethyl-2-pyridyl) porphyrin (MnTE-2-PyP) (Aeol 10113, gift of Incara Pharmaceuticals, Research Triangle Park, NC, USA) and Mn-tetrakis-(4-benzoic acid) porphyrin (MnTBAP) (Alexis Biochemicals, San Diego, CA, USA), only the LDH assay was performed because the metalloporphyrin compounds interfere with tetrazolium salt-based assays.
2.3 Cell lysates, immunoblotting and immunocytochemistry
Cell lysis and immunoblots for phospho-ERK (Cell Signalling, Beverly, MA, USA) and total ERK (Upstate Biotechnology, Lake Placid, NY, USA) were performed following 18 h of exposure to 6-OHDA as previously described (Chu et al 1997; Kulich and Chu 2001). B65 cells, fixed in 3% paraformaldehyde on glass coverslips, were stained with antibodies against nestin and neurofilament (Chemicon, Temecula, CA, USA) 1 : 4000 and 1 : 2000, respectively, followed by Alexa 488 goat anti-mouse (Molecular Probes, Eugene, OR, USA). Following nuclear counterstaining with propidium iodide, cells were imaged using the Zeiss LSM510 laser scanning microscope. Phase contrast microscopy was performed using the Olympus CR2 microscope.
3. Results
3.1 Effect of catalase and SOD on 6-OHDA toxicity
6-OHDA is a dopamine analogue that readily undergoes non-enzymatic oxidation producing hydrogen peroxide, superoxide, and hydroxyl radical at physiologic pH (Cohen and Heikkila 1974). In order to characterize the contribution of hydrogen peroxide and superoxide to cytotoxicity, B65 cells were exposed to 6-OHDA in the presence of either catalase or SOD. Preincubation of cells with catalase-containing media conferred significant protection from cell injury as determined by metabolism of MTS (figure 1) and LDH release (figure 2), and this effect could be blocked by prior heat-inactivation of catalase (figure 1). Conversely, SOD did not confer cell injury protection (figure 2).
Figure 1.
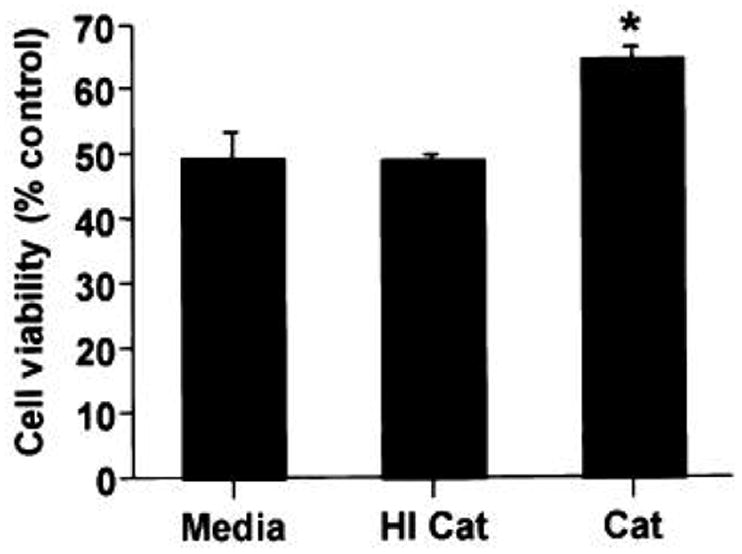
Influence of catalase on 6-OHDA-mediated cell injury. B65 cells were exposed to 6-OHDA for 20 h. Thirty minutes prior to the addition of 500 μM 6-OHDA, media was replaced with fresh media with or without catalase (Cat, 30 U/ml) or heat inactivated catalase (HI Cat). Cell injury was determined by measuring MTS metabolism, normalized to that of vehicle (0.05% ascorbate) treated cells, and plotted as percent control. Each value represents the mean of triplicate wells ± SEM. *P < 0.05 by two-tailed Student’s t-test compared to 6-OHDA-treated cells lacking catalase. Representative plots are illustrated.
Figure 2.
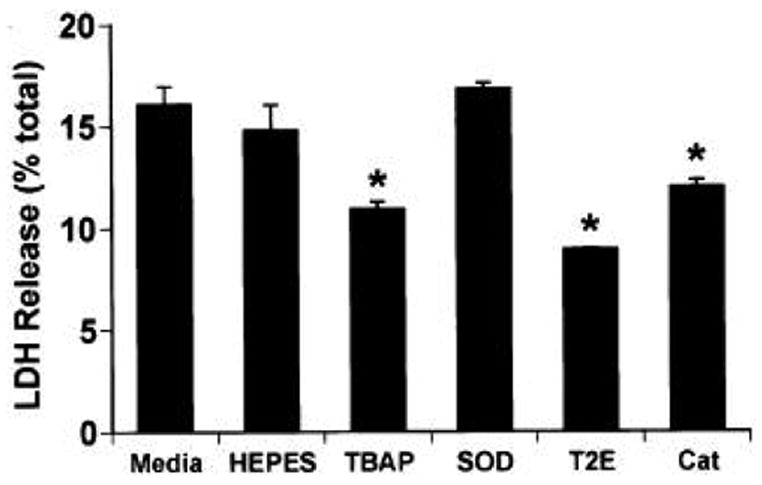
Metalloporphyrins also inhibit 6-OHDA cell injury. B65 cells were exposed to 500 μM 6-OHDA for 20 h. Thirty minutes prior to the addition of 6-OHDA or vehicle, media was replaced with fresh media alone (Media), HEPES (the diluent for the antioxidants) or media containing SOD (150 U/ml), catalase (30 U/ml, Cat), MnTBAP (100 μM, TBAP), or MnTE-2-PyP (T2E, 50 μM). Cell injury was determined by determining the percent of LDH released into the media. The baseline percent release from ascorbate (the vehicle for 6-OHDA) treated cells was 11.7 ± 0.4%. *P < 0.05 by two-tailed Student’s t-test as compared to cells exposed to 6-OHDA in the absence of catalytic antioxidants. Specific activities of Mn-TBAP and MnTE-2-PyP were 60,000 and 3,000,000 U/mmol respectively, and of catalase, 260,000 U/ml and of SOD, 4,600 U/ml. A representative plot is illustrated.
3.2 Metalloporphyrins confer protection from 6-OHDA toxicity
Metalloporphyrins are catalytic antioxidants which have been shown to be protective in a variety of in vitro models of oxidative stress (reviewed in Patel and Day 1999). Unlike large proteins such as catalase and SOD, metal-loporphyrins provide a mechanism to deliver intracellular catalytic antioxidant activity (Li et al 2001). Pre-incubation of cells in media containing either the benzoic acid-substituted metalloporphoryn (MnTBAP) or the pyridine-substituted analogue (MnTE-2-PyP) resulted in significant protection from cell injury (figure 2).
3.3 Effect of catalase, SOD, and metalloporphyrins on 6-OHDA-induced ERK activation
We have previously found that 6-OHDA elicits a sustained pattern of ERK activation in B65 cells (Kulich and Chu 2001). Furthermore, inhibition of 6-OHDA-mediated ERK phosphorylation by the MEK1/2 inhibitor PD98059 confers significant protection from cytotoxicity (Kulich and Chu 2001), suggesting that ERK activation may play a detrimental role in neuronal cells exposed to 6-OHDA.
We examined the effects of catalase and SOD on 6-OHDA-mediated sustained ERK phosphorylation. Catalase, but not SOD, was capable of attenuating 6-OHDA-mediated sustained ERK phosphorylation (figure 3). Similarly, cytoprotective doses of MnTE-2-PyP attenuated 6-OHDA-mediated sustained ERK phosphorylation (figure 3). These results indicate a correlation between the ability of an antioxidant treatment to inhibit 6-OHDA-elicited ERK phosphorylation and its ability to protect against 6-OHDA toxicity.
Figure 3.
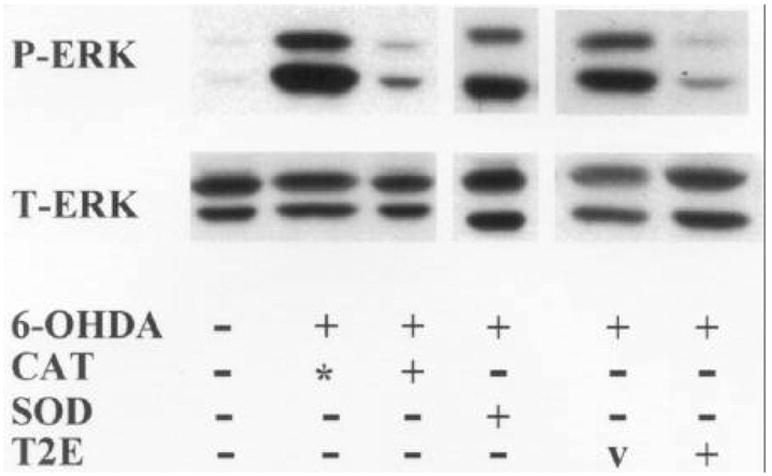
Effects of catalytic antioxidants on 6-OHDA-induced ERK phosphorylation. B65 cells were exposed to 500 μM 6-OHDA (+) or vehicle (–) for 20 h in the presence of catalase (CAT, 30 U/ml), heat-inactivated catalase (*), SOD (100 U/ml), MnTE-2-PyP (T2E, 50 μM) or the diluent for the mimetic (v, HEPES). Equal amounts of protein from cell lysates (50 μg) were subjected to immunoblot analysis using an antibody specific for activated dually phosphorylated ERK (top, P-ERK). The blots were then stripped and reprobed with antibody against total ERK (bottom, T-ERK). Representative experiments are shown.
3.4 Catalase and metalloporphyrins protect differentiated B65 from 6-OHDA toxicity
Although B65 cells are CNS derived and have been reported to express neuron-specific markers (Schubert et al 1974), when assessed under the assay conditions utilized above, they remain proliferative. However, when cultivated under lowered serum concentrations in the presence of butyrate and UO126, the cells undergo a morphologic conversion from small bipolar cells to larger, complex cells with extensive process formation (figure 4A, B). Immunocytochemical stains for neuronal markers, such as nestin and neurofilament, confirm that this morphologic transformation is accompanied by appropriate expression of neuronal markers (figure 4C, D). In analogy to undifferentiated B65 cells, treatment of differentiated B65 cells with either catalase or MnTE-2-PyP conferred protection from 6-OHDA cytotoxicity (figure 5A, B). Moreover, both catalase and MnTE-2-PyP were capable of inhibiting sustained ERK phosphorylation in differentiated B65 cells (figure 5C).
Figure 4.
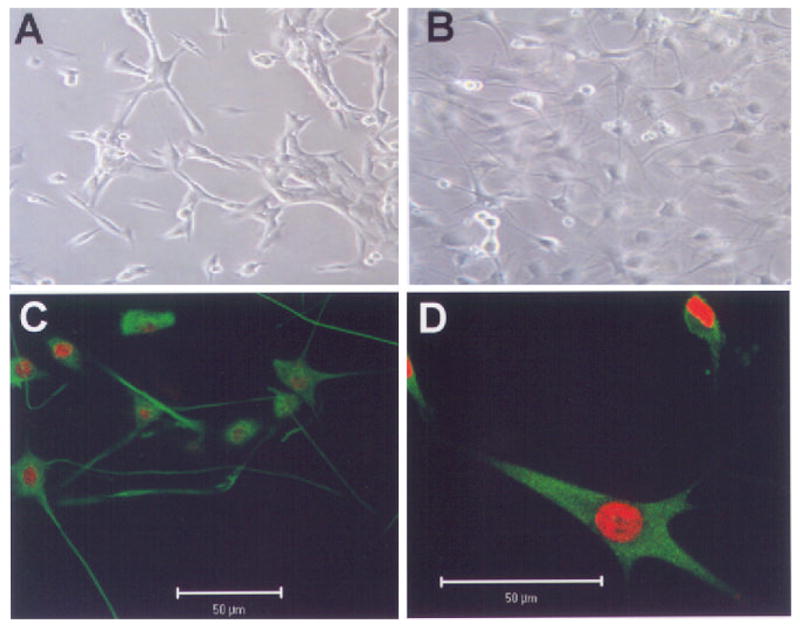
Differentiation of B65 cells. Phase contrast microscopy of (A) undifferentiated, and (B) differentiated B65 cells. Immunocytochemical stains (green) of differentiated B65 cells stained for (C) nestin, and (D) 70 kDa neurofilament. Nuclei were counterstained with propidium iodide (red).
Figure 5.
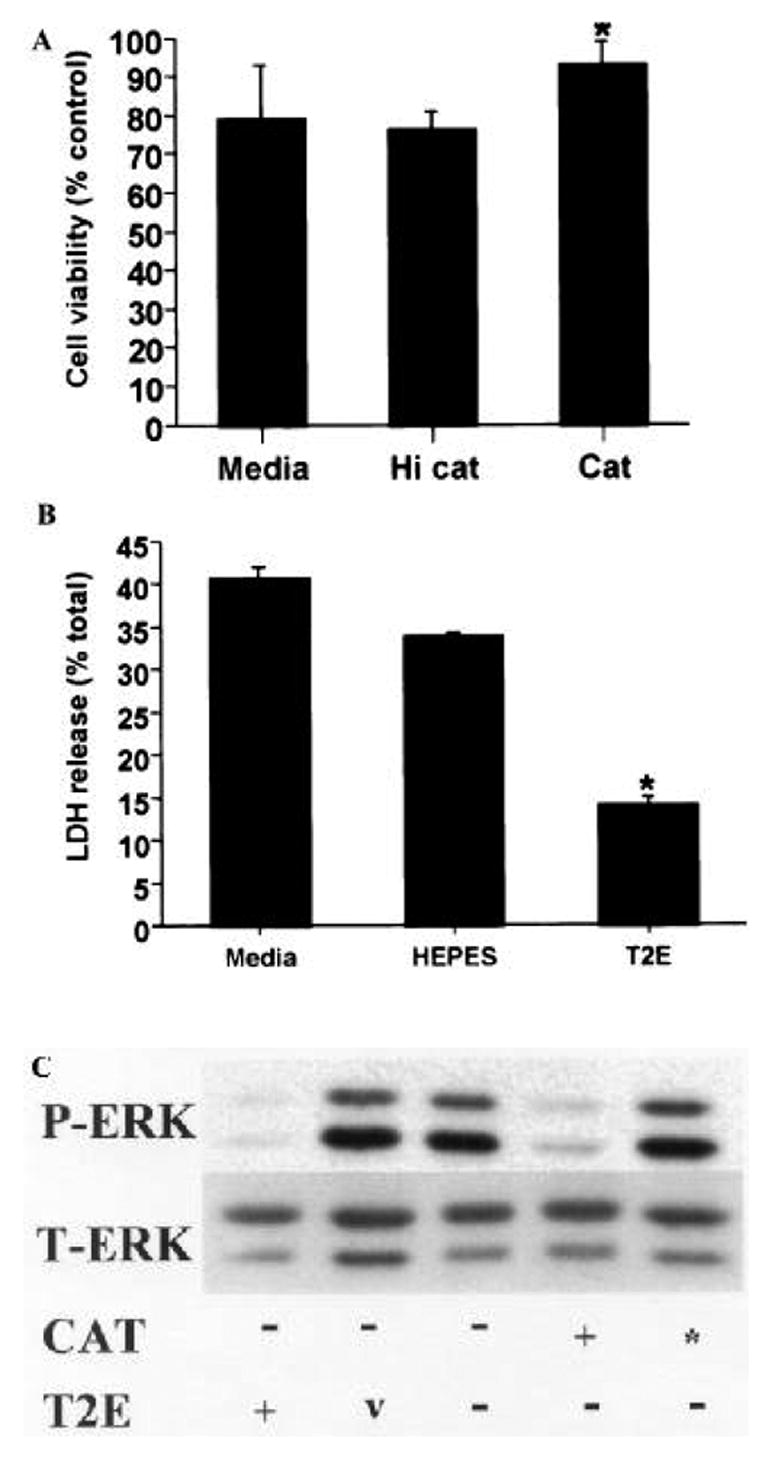
Cytotoxic doses of 6-OHDA induce ERK phosphorylation in differentiated B65 cells. Differentiated B65 cells were exposed to 500 μM 6-OHDA or vehicle for 20 h in media alone or media containing (A) catalase (Cat, 30 U/ml) or heat inactivated catalase (HI Cat), or (B) MnTE-2-PyP (T2E, 50 μM) or mimetic diluent (HEPES) and then assessed for cell injury as in figures 1 and 2, respectively. LDH release of vehicle treated cells (no 6-OHDA) was 15 ± 0.8%. *P < 0.05 by two-tailed Student’s t-test compared to 6-OHDA treated cells lacking antioxidant (Media). Representative plots are illustrated. (C) Equal protein (10 μg) from cell lysates prepared following exposure of differentiated B65 cells to 500 μM 6-OHDA for 20 h in media containing catalase (30 U/ml), heat inactivated catalase (*), MnTE-2-PyP (50 μM) or mimetic diluent (v) was subjected to immunoblot analysis for P-ERK (top) and T-ERK (bottom) as in figure 3.
4. Discussion
Recently, we have demonstrated that 6-OHDA produces dose-dependent cytotoxicity in B65 cells that is associated with sustained ERK phosphorylation (Kulich and Chu 2001). The current studies demonstrate that catalase- and metalloporphyrin-sensitive reactions are involved in both toxicity and sustained ERK phosphorylation, and implicate ROS-mediated sustained ERK activation as a key contributor to 6-OHDA toxicity in B65 cells.
6-OHDA toxicity in B65 cells was inhibited by catalase and metalloporphyrins, but not by SOD. In fact, SOD dose curves indicate a trend towards increased injury as assessed by the MTS assay (not shown), similar to that reported in SY5Y cells (a different tyrosine hydroxylase expressing cell line) (Tiffany-Castiglioni et al 1982). The observation that extracellular application of SOD, a non-cell permeable macromolecule, may be detrimental, but that cell permeable metalloporphyrin (SOD mimetics) (Day et al 1997) are protective, suggests that compartmentalization of superoxide scavengers may be an important determinant of 6-OHDA toxicity. An alternative explanation would be that metalloporphyrins protect by providing a source of intracellular catalase activity, as metalloporphyrins also possess low levels of catalase activity and have been shown to protect against H2O2 toxicity (Antunes and Cadenas 2000). Although the in vitro catalase activity of the mimetic dosage used in this study is similar to that of our heat-inactivated catalase preparation (data not shown), we cannot rule out the possibility that this low-level catalase activity, when introduced into the intracellular compartment, is sufficient for protection from 6-OHDA toxicity. Additional studies using methodologies capable of specifically elevating intracellular SOD and catalase activities will be necessary to distinguish these various possibilities.
Both catalase and metalloporhyrins were capable of inhibiting 6-OHDA-induced ERK phosphorylation. Interestingly, treatment of B65 cells with equivalent cytotoxic doses of hydrogen peroxide did not result in sustained ERK phosphorylation (Kulich and Chu 2001). These results are similar to those observed with zymosan-activated serum treatment of alveolar macrophages, where hydrogen peroxide appears to be necessary but not sufficient for ERK activation (Torres and Forman 1999), support a role for a secondary mediator, such as intracellular superoxide and/or 6-OHDA oxidation products, in sustained ERK activation.
We have observed a correlation between the ability of specific antioxidant treatments to protect B65 cells from 6-OHDA toxicity and the ability to inhibit sustained ERK activation. These observations are consistent with our previous data demonstrating that the compound PD98059, which inhibits the MEK1/2 kinases that activate ERK, protects B65 cells from 6-OHDA toxicity. However, recent studies have questioned the specificity of PD98059, since it is capable of inhibiting MEK 5 and kinase suppressor of ras (Cavanaugh et al 2001; Wang and Studzinski 2001). Studies utilizing dominant negative forms of MEK will be necessary to further investigate the potential role of ERK activation in neuronal cell death.
While we have shown that 6-OHDA-induced sustained ERK activation occurs via catalase- and metalloporphyrin-sensitive mechanisms, the molecular site of action of 6-OHDA associated ROS remains to be defined. Possible ROS-sensitive targets which may be important in regulating sustained ERK activation include upstream activators of the ERK pathway (Nishida et al 2000), protein phosphatases (Runden et al 1998), and proteasomal function (Hashimoto et al 2000). The contribution of these processes to 6-OHDA-mediated sustained ERK activation is currently under investigation.
The results of this study demonstrate that both sustained ERK activation and cytotoxicity induced by the neurotoxin 6-OHDA, which is commonly used in Parkinsonian models, is mediated by catalase- and metalloporphyrin-sensitive events. Since dopamine itself undergoes similar redox cycling (Hastings et al 1996; Luo et al 1998), it is possible that abnormal ERK activation can contribute to neuronal death and dysfunction in Parkinson’s disease and other neurodegenerative diseases with overlapping histopathological features (Chu et al 2000). In support of this hypothesis, we have observed discrete granular cytosolic accumulations of phospho-ERK and increased ERK activity in substantia nigral tissue from patients with Parkinson’s disease and dementia with Lewy bodies as well as in this cell culture model (Zhu et al 2002). Taken together, these observations suggest that identifying the redox-sensitive signalling proteins that are affected by 6-OHDA to yield sustained ERK activation in B65 cells will be important for understanding the molecular pathogenesis of neuronal loss in Parkinson’s disease and related neurodegenerative diseases.
Acknowledgments
The authors would like to thank Manisha Patel for providing us with MnTE-2-PyP, and Amy Sartori and Lisa Schaefer for technical assistance. This work was supported by grants from the NIH (RO1 NS40817), the American Federation for Aging Research, and the Rockefeller Brothers Fund/Charles E Culpeper Scholarship. SMK was supported in part by the University of Pittsburgh Pathology Post-doctoral Research Training Program.
Abbreviations used
- ERK
Extracelluar signal-regulated protein kinase
- LDH
lactate dehydrogenase
- MnTBAP
Mn-tetrakis-(4-benzoic acid) porphyrin
- MTS
metabolism of the tetrazolium salt [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulphophenyl)-2H-tetrazolium, inner salt]
- 6-OHDA
6-hydroxydopamine
- ROS
reactive oxygen species
- SOD
superoxide dismutase
References
- Aebi H. Catalase in vitro. Methods Enzymol. 1984;105:121–126. doi: 10.1016/s0076-6879(84)05016-3. [DOI] [PubMed] [Google Scholar]
- Antunes F, Cadenas E. Estimation of H2O2 gradients across biomembranes. FEBS Lett. 2000;475:121–126. doi: 10.1016/s0014-5793(00)01638-0. [DOI] [PubMed] [Google Scholar]
- Asanuma M, Hirata H, Cadet JL. Attenuation of 6-hydroxydopamine-induced dopaminergic nigrostriatal lesions in superoxide dismutase transgenic mice. Neuroscience. 1998;85:907–917. doi: 10.1016/s0306-4522(97)00665-9. [DOI] [PubMed] [Google Scholar]
- Cavanaugh JE, Ham J, Hetman M, Poser S, Yan C, Xia Z. Differential regulation of mitogen-activated protein kinases ERK1/2 and ERK5 by neurotrophins, neuronal activity, and cAMP in neurons. J Neurosci. 2001;21:434–443. doi: 10.1523/JNEUROSCI.21-02-00434.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu CT, Caruso JL, Cummings TJ, Ervin J, Rosenberg C, Hulette CM. Ubiquitin immunochemistry as a diagnostic aid for community pathologists evaluating patients who have dementia. Modern Pathol. 2000;13:420–426. doi: 10.1038/modpathol.3880072. [DOI] [PubMed] [Google Scholar]
- Chu CT, Everiss KD, Batra S, Wikstrand CJ, Kung H-J, Bigner DD. Receptor dimerization is not a factor in the signalling activity of a transforming variant epidermal growth factor receptor (EGFRvIII) Biochem J. 1997;324:855–861. doi: 10.1042/bj3240855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen G, Heikkila RE. The generation of hydrogen peroxide, superoxide radical, and hydroxyl radical by 6-hydroxydopamine, dialuric acid, and related cytotoxic agents. J Biol Chem. 1974;249:2447–2452. [PubMed] [Google Scholar]
- Day BJ, Fridovich I, Crapo JD. Manganic porphyrins possess catalase activity and protect endothelial cells against hydrogen peroxide-mediated injury. Arch Biochem Biophys. 1997;347:256–262. doi: 10.1006/abbi.1997.0341. [DOI] [PubMed] [Google Scholar]
- Fattman CL, Chu CT, Kulich SM, Enghild JJ, Oury TD. Altered expression of extracellular superoxide dismutase in mouse lung after bleomycin treatment. Free Radical Biol Med. 2001;31:1198–1207. doi: 10.1016/s0891-5849(01)00699-2. [DOI] [PubMed] [Google Scholar]
- Glinka YY, Youdim MB. Inhibition of mitochondrial complexes I and IV by 6-hydroxydopamine. Eur J Pharmacol. 1995;292:329–332. doi: 10.1016/0926-6917(95)90040-3. [DOI] [PubMed] [Google Scholar]
- Hashimoto K, Guroff G, Katagiri Y. Delayed and sustained activation of p42/p44 mitogen-activated protein kinase induced by proteasome inhibitors through p21(ras) in PC12 cells. J Neurochem. 2000;74:92–98. doi: 10.1046/j.1471-4159.2000.0740092.x. [DOI] [PubMed] [Google Scholar]
- Hastings TG, Lewis DA, Zigmond MJ. Role of oxidation in the neurotoxic effects of intrastriatal dopamine injections. Proc Natl Acad Sci USA. 1996;93:1956–1961. doi: 10.1073/pnas.93.5.1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulich SM, Chu CT. Sustained extracellular signal-regulated kinase activation by 6-hydroxydopamine: Implications for Parkinson’s disease. J Neurochem. 2001;77:1058–1066. doi: 10.1046/j.1471-4159.2001.00304.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li QY, Pedersen C, Day BJ, Patel M. Dependence of excitotoxic neurodegeneration on mitochondrial aconitase inactivation. J Neurochem. 2001;78:746–755. doi: 10.1046/j.1471-4159.2001.00457.x. [DOI] [PubMed] [Google Scholar]
- Luo Y, Umegaki H, Xiantao W, Abe R, Roth GS. Dopamine induces apoptosis through an oxidation-involved SAPK/JNK activation pathway. J Biol Chem. 1998;273:3756–3764. doi: 10.1074/jbc.273.6.3756. [DOI] [PubMed] [Google Scholar]
- Nishida M, Maruyama Y, Tanaka R, Kontani K, Nagao T, Kurose H. G alpha(i) and G alpha(o) are target proteins of reactive oxygen species. Nature. 2000;408:492–495. doi: 10.1038/35044120. London. [DOI] [PubMed] [Google Scholar]
- Oh-hashi K, Maruyama W, Yi H, Takahashi T, Naoi M, Isobe K. Mitogen-activated protein kinase pathway mediates peroxynitrite-induced apoptosis in human dopaminergic neuroblastoma SH-SY5Y cells. Biochem Biophys Res Commun. 1999;263:504–509. doi: 10.1006/bbrc.1999.1237. [DOI] [PubMed] [Google Scholar]
- Patel M, Day BJ. Metalloporphyrin class of therapeutic catalytic antioxidants. Trends Pharmacol Sci. 1999;20:359–364. doi: 10.1016/s0165-6147(99)01336-x. [DOI] [PubMed] [Google Scholar]
- Runden E, Seglen PO, Haug FM, Ottersen OP, Wieloch T, Shamloo M, Laake JH. Regional selective neuronal degeneration after protein phosphatase inhibition in hippocampal slice cultures: evidence for a MAP kinase-dependent mechanism. J Neurosci. 1998;18:7296–7305. doi: 10.1523/JNEUROSCI.18-18-07296.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert D, Heinemann S, Carlisle W, Tarikas H, Kimes B, Patrick J, Steinback JH. Clonal cell lines from the rat central nervous system. Nature. 1974;249:224–227. doi: 10.1038/249224a0. London. [DOI] [PubMed] [Google Scholar]
- Stanciu M, Wang Y, Kentor R, Burke N, Watkins S, Kress G, Reynolds I, Klann E, Angiolieri M, Johnson J, DeFranco DB. Persistent activation of ERK contributes to glutamate-induced oxidative toxicity in a neuronal cell line and primary cortical neuron cultures. J Biol Chem. 2000;275:12200–12206. doi: 10.1074/jbc.275.16.12200. [DOI] [PubMed] [Google Scholar]
- Suzuki YJ, Forman HJ, Sevanian A. Oxidants as stimulators of signal transduction. Free Radical Biol Med. 1997;22:269–285. doi: 10.1016/s0891-5849(96)00275-4. [DOI] [PubMed] [Google Scholar]
- Tiffany-Castiglioni E, Saneto RP, Proctor PH, Perez-Polo JR. Participation of active oxygen species in 6-hydro-xydopamine toxicity to a human neuroblastoma cell line. Biochem Pharmacol. 1982;31:181–188. doi: 10.1016/0006-2952(82)90208-8. [DOI] [PubMed] [Google Scholar]
- Torres M, Forman HJ. Activation of several MAP kinases upon stimulation of rat alveolar macrophages: role of the NADPH oxidase. Arch Biochem Biophys. 1999;366:231–239. doi: 10.1006/abbi.1999.1225. [DOI] [PubMed] [Google Scholar]
- Ungerstedt U. 6-Hydroxy-dopamine induced degeneration of central monoamine neurons. Eur J Pharmacol. 1968;5:107–110. doi: 10.1016/0014-2999(68)90164-7. [DOI] [PubMed] [Google Scholar]
- Wang X, Studzinski GP. Phosphorylation of raf-1 by kinase suppressor of ras is inhibited by “MEK-specific” inhibitors PD 098059 and U0126 in differentiating HL60 cells. Exp Cell Res. 2001;268:294–300. doi: 10.1006/excr.2001.5292. [DOI] [PubMed] [Google Scholar]
- Zhu J-H, Kulich SM, Oury TD, Chu CT. Cytoplasmic aggregates of phosphorylated extracellular signal-regulated kinase in Lewy body diseases. Am J Pathol. 2002;161:2087–2098. doi: 10.1016/S0002-9440(10)64487-2. [DOI] [PMC free article] [PubMed] [Google Scholar]