

Abstract
Although the toxin 6-hydroxydopamine (6-OHDA) is utilized extensively in animal models of Parkinson's disease, the underlying mechanism of its toxic effects on dopaminergic neurons is not completely understood. We examined the effects of 6-OHDA on the CNS-derived tyrosine hydroxylase expressing B65 cell line, with particular attention to the regulation of the extracellular signal-regulated protein kinases (ERK). 6-OHDA elicited a dose-dependent cytotoxicity in B65 cells. Toxic doses of 6-OHDA also elicited a biphasic pattern of ERK phosphorylation with a prominent sustained phase, a pattern that differed from that observed with hydrogen peroxide (H2O2) treatment. 6-OHDA-elicited ERK phosphorylation was blocked by PD98059, an inhibitor of the upstream mitogen activated protein kinase kinase (MEK) that phosphorylates and activates ERK. PD98059 also conferred protection against 6-OHDA cytotoxicity, but did not affect H2O2 toxicity in B65 cells. These results suggest that ERK activation plays a direct mechanistic role in 6-OHDA toxicity, rather than representing a protective compensatory response, and raise the possibility that abnormal patterns of ERK activation may contribute to dopaminergic neuronal cell death.
Keywords: dopaminergic cell death, mitogen-activated protein kinases, oxidative stress
Parkinson's disease, defined clinically by the triad of tremor, bradykinesia, and rigidity, is a common neurodegenerative disease. Although Parkinson's disease may affect younger individuals with specific genetic susceptibilities, the majority of cases occur sporadically in the aging population and lack a well-defined etiology (Moghal et al. 1994). The neuropathologic findings of Parkinson's disease are characterized by selective neuronal death affecting the locus ceruleus, dorsal motor nucleus of vagus, nucleus basalis of Meynert and, most importantly, the dopaminergic neurons of the substantia nigra pars compacta (SNpc) (reviewed in (Lang and Lozano 1998). It is the loss of the striatal projections from the SNpc neurons that is believed to result in the majority of symptoms in Parkinson's disease. The mechanisms leading to neuronal cell death in Parkinson's disease remain unclear.
Insight into potential mechanisms contributing to neurodegeneration in Parkinson's disease has been gained from animal models of Parkinson's disease. One such model involves the neurotoxin 6-hydroxydopamine (6-OHDA). 6-OHDA is a dopamine analog that readily undergoes non-enzymatic oxidation producing hydrogen peroxide (H2O2), superoxide, and hydroxyl radical at physiologic pH (Cohen and Heikkila 1974). Moreover, intrastriatal injections of 6-OHDA result in a parkinsonian pattern of neuronal loss in rats (Ungerstedt 1968). Although used as an exogenous neurotoxin in this model, there is evidence suggesting that 6-OHDA can be formed from dopamine in vivo. The neurons within the SNpc contain increased iron in Parkinson's disease (Hirsch et al. 1991). In addition, the activities of catalase and glutathione peroxidase, the major enzymes responsible for the elimination of H2O2, are reduced in Parkinson's disease brains (Ambani et al. 1975; Kish et al. 1985). In the presence of free ferric iron and H2O2, the major product of dopamine oxidation has been reported to be 6-OHDA (Jameson and Linert 2000). Indeed, 6-OHDA is elevated in the urine of Parkinson's disease patients treated with levodopa (Andrew et al. 1993), suggesting that 6-OHDA may play a role in parkinsonian disease progression.
Although many lines of data support the role of oxidative mediators in 6-OHDA-induced neurotoxicity, the exact mechanisms by which 6-OHDA elicits its neurotoxic effects have yet to be fully elucidated. Mice that overexpress superoxide dismutase show protection from intraventricular injections of 6-OHDA (Asanuma et al. 1998). 6-OHDA can inhibit mitochondrial complex I activity (Glinka and Youdim 1995), potentially contributing to increased mitochondrial superoxide generation. In addition, other non-mitochondrial mechanisms may also contribute to 6-OHDA toxicity. Activation of c-Jun N-terminal kinase (JNK) occurs in response to 6-OHDA in the dopaminergic MN9D cell line (Choi et al. 1999). JNK is a member of the mitogen-activated protein (MAP) kinase family, protein serine/ threonine kinases that play critical roles in cellular responses to a wide range of extracellular stimuli. JNK, p38 MAP kinase, and extracellular signal-regulated kinases (ERK) comprise the three major classes of MAP kinases. The dynamic balance between branches of the MAP kinase family is believed to regulate neuronal decisions to live or die in response to stressors (Xia et al. 1995). In particular, ERK activation may play a pivotal role.
Many neuroprotective\neurotrophic factors activate receptor tyrosine kinases. A major mechanism by which these kinases transmit signals is through ERK, which undergoes a dual phosphorylation event leading to its activation (reviewed in Segal and Greenberg 1996). ERK activation appears to antagonize apoptotic pathways in some cell systems (Xia et al. 1995; Holmström et al. 1998). Several recent studies, however, indicate that ERK activation may also play a pathologic role in neurons exposed to increased oxidative stress (Oh-hashi et al. 1999; Stanciu et al. 2000). The effects of 6-OHDA upon ERK signaling pathways are unknown.
This study was designed to investigate the role of ERK activation in neuronal cell responses to 6-OHDA. The rat CNS-derived B65 cell line exhibits electrically excitable membranes that can produce regenerative action potentials, expresses tyrosine hydroxylase and neuronal markers, binds α-neurotoxin, and extends neurites in response to low serum or dibutyral cAMP (Schubert et al. 1974). 6-OHDA elicited sustained ERK phosphorylation in B65 cells that could be inhibited by the MEK inhibitor PD98059. PD98059 also conferred significant protection against 6-OHDA cytotoxicity. These effects appeared specific to 6-OHDA, as they could not be recapitulated with comparable doses of H2O2. These results suggest that abnormal patterns of ERK activation may contribute to mechanisms of dopaminergic neuron death relevant to Parkinson's disease.
Materials and methods
Chemical reagents (except where specified) were purchased from Sigma (St Louis, MO, USA).
Cell culture
Cell culture reagents were purchased from BioWhittaker (Walkerville, MD, USA). Cell culture plates were purchased from Corning (Acton, MD, USA). B65 cells were the kind gift of Dr David Schubert of the Salk Institute (Schubert et al. 1974). B65 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10 mM HEPES, 2 mM L-glutamine, and 10% heat-inactivated fetal calf serum (FCS; DH10). Cells were plated at a density of 140 cells/mm2 for all experiments and grown for 2 days prior to use at 37°C in a humidified incubator with 5% CO2.
Toxicity assays
6-OHDA was prepared fresh for each assay in ice-cold 0.05% (wt/vol) ascorbate. H2O2 was diluted in water. The concentration of the H2O2 stock solution was confirmed by measuring the absorbance at 240 nm using the extinction coefficient of 0.0437 cm/mM−1. Cell injury in response to 6-OHDA and H2O2 was determined using two independent methodologies:
Lactate dehydrogenase (LDH) release assay
Release of LDH into the culture medium was determined using the Sigma 340-UV LDH detection system. Briefly, B65 cells, grown in a 96-well plate format, were exposed to the varying concentrations of 6-OHDA, H2O2, or vehicle for 18–20 h. Following visual inspection and gentle centrifugation (5 min, 225 g), aliquots were obtained from the supernatant. An equal volume of 2% (v/v) Triton X-100 was added to the wells and cells were lysed using vigorous pipetting. LDH activities in supernatant and lysate were determined by measuring the decrease in absorbance at 340 nm associated with reduction of pyruvate in a linear velocity reaction (Wroblewski and LaDue 1955). Levels of LDH released into the supernatant were expressed as percent of total LDH activity.
MTS assay
The CellTiter 96 Aqueous non-radioactive cell proliferation assay (Promega, Madison, WI, USA), which measures the conversion of the tetrazolium salt [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt] (MTS), was used as follows: Twenty microliters of a 1 : 20 mixture of PMS (phanazine methsulfate): MTS was added to each well of a 96-well plate containing treated cells in a final volume of 100 μL of media. The A490 was determined immediately following addition of PMS : MTS, and again following a 4-h incubation of the cells at 37°C, with the change in absorbance representing enzymatic conversion of tetrazolium to formazan. For experiments utilizing 6-OHDA, each concentration of 6-OHDA utilized had its own background control (media plus 6-OHDA in the absence of cells), since superoxide radicals generated by 6-OHDA may react directly with tetrazolium dyes such as MTS (Sutherland and Learmonth 1997). Using these controls, we determined that little to no further superoxide was generated by 6-OHDA following overnight incubations. Assay conditions were thus selected to minimize this non-enzymatic contribution. All data are expressed as a percentage of control values obtained from cells treated with vehicle alone.
MEK inhibitor experiments
PD98059 (Cell Signaling, Beverly, MA, USA) was prepared as a 50-mM stock in 100% dimethylsulfoxide (DMSO). Thirty minutes prior to the addition of 6-OHDA or H2O2, cells were exposed to fresh DH10 media containing: (a) 50 μM PD98059, (b) 0.1% DMSO (concentration of vehicle present in PD98059 treated cells), or (c) no additional additives.
Cell lysates and immunoblotting
Cell lysates were obtained at various time points using lysis buffer containing 0.1% Triton X-100 and a protease/phosphatase inhibitor cocktail as previously described (Chu et al. 1997). For pulse-chase experiments, the pulse of 6-OHDA was terminated by aspiration of the media containing 6-OHDA, washing once with the same volume of fresh DH10, and then growing the cells in fresh DH10. Equal amounts of protein, as determined by Coomassie Plus Protein Assay (Pierce, Rockford, IL, USA), were subjected to electrophoresis through 5–15% polyacrylamide gels under reducing conditions (Chu and Pizzo 1993), and transferred to Immobilon-P membranes (Millipore, Bedford, MA, USA). The membranes were blocked for 2 h with 5% non-fat dry milk in 20 μM potassium phosphate, 150 mM potassium chloride, pH 7.4 containing 0.3% (w/v) Tween-20 (PBST) and then probed overnight at room temperature (25°C) with an activation-specific antiphospho-ERK antibody, which only recognizes ERK that is phosphorylated on both Y202 and T204 (Cell Signaling, Beverly, MA, USA; diluted 1 : 2000 in PBST), followed by incubation with HRP-conjugated sheep–anti-mouse IgG (Amersham, Piscataway, NJ, USA). Antibody detection was carried out using an ECL detection kit (Amersham, Piscataway, NJ, USA). Blots were then stripped as previously described (Chu et al. 1997), and reprobed with antitotal ERK kinase (Upstate Biotechnology, Lake Placid, NY, USA) (1 : 10 000, 1 h, room temperature, 1% non-fat milk in PBST) followed by incubation with HRP-conjugated donkey anti-rabbit IgG (Amersham, Piscataway, NJ, USA) and ECL detection. Specificity of the antisera were confirmed by substitution of non-immune antisera for the primary antibodies, and by inclusion of control lanes containing lysates from a fibroblast-line treated with known activators of ERK phosphorylation (Chu et al. 1997). Densitometric analysis of blots was performed using the electrophoresis documentation and analysis system 120 (Kodak, Rochester, NY, USA).
Results
6-OHDA toxicity in B65 cells
Exposure of B65 cells to 6-OHDA resulted in dose-dependent cell injury. Incubation of B65 cells overnight in the presence of 6-OHDA resulted in cell injury as detected by either increased amounts of LDH released from the cells or decreased ability of the cells to metabolize the tetrazolium dye MTS (Fig. 1a). H2O2, a potential byproduct of 6-OHDA metabolism (Cohen and Heikkila 1974), also exhibited a dose-dependent toxicity in B65 cells (Fig. 1b). The EC50 for 6-OHDA and H2O2 under these conditions were 573 ± 41 and 880 ± 35 μM, respectively (mean ± SEM, 10 independent experiments for each reagent). For subsequent experiments, the concentrations of 6-OHDA and H2O2 utilized were based upon the dose–response curves for the cell passage used.
Fig. 1.

Cytotoxicity of 6-OHDA and H2O2 in B65 cells. B65 cells were exposed to (a) 6-OHDA or (b) H2O2 for 18–20 h. Cell injury was determined by measuring LDH activity released into the media (○) or by altered MTS metabolism (●). Values were normalized to total LDH activity or the A490 of vehicle treated control cells, respectively, as described in the Materials and methods section. Each value represents the mean of triplicate wells ± SE. Each experiment was performed independently at least three times. Representative plots are illustrated.
ERK phosphorylation elicited by 6-OHDA
Immunoblot analysis of cell lystates from B65 cells following an overnight exposure to 6-OHDA revealed strong induction of ERK phosphorylation affecting both the ERK1 (p44) and ERK2 (p42) isoforms (Fig. 2). The level of ERK activation was dose-dependent, with sublethal doses showing little ERK phosphorylation and lethal doses showing the greatest amount of ERK phosphorylation. This pattern of ERK phosphorylation did not appear to be a general response to cell injury or H2O2 generation since similar effective concentrations of H2O2 were not effective at eliciting ERK phosphorylation following overnight incubation (Fig. 2). Although 6-OHDA has not been reported to bind any known dopamine receptors, D2 dopamine receptor stimulation may result in ERK activation (Cai et al. 2000). Potential ligation of dopamine receptors by 6-OHDA did not contribute to these observations, however, as concentrations of up to 1 mM dopamine failed to elicit ERK phosphorylation.
Fig. 2.
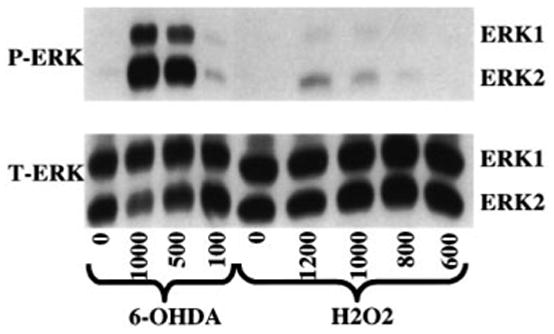
Dose–response study of ERK activation in B65 cells. Cells were treated with the indicated μM concentrations of 6-hydroxydopamine (6-OHDA), H2O2 (H2O2) or vehicle (0.05% ascorbate for 6-OHDA, water for H2O2) for 18 h. The cells were then lysed and equal amounts of protein (20 μg) were subjected to immunoblot analysis using an antibody specific for activated dually phosphorylated ERK (top, P-ERK). The blots were then stripped and reprobed with antibody against total ERK (bottom, T-ERK). Blots depicted are representative of eight independent experiments.
Kinetics of 6-OHDA elicited ERK phosphorylation
In an attempt to better define the nature of 6-OHDA-mediated ERK activation in B65 cells, an analysis of the kinetics of ERK phosphorylation was performed. Although the kinetics of ERK phosphorylation following 6-OHDA treatment were slightly variable with different passages of B65 cells, a biphasic response with an intense early phosphorylation by 15 min, a decrease towards baseline levels at 60–90 min, followed by a second, sustained phase of phosphorylation was consistently observed (Fig. 3). Separate studies indicate that the initial phase of ERK activation may occur as early as 5 min after application of 6-OHDA. This phosphorylation response was not observed with vehicle-treated cells (Fig. 3c) or with cells treated with fresh media alone, indicating that this observation was not simply an effect of fresh media. In contrast to 6-OHDA, H2O2, gave a unimodal response that was not sustained (Fig. 3). The sustained phase of 6-OHDA-mediated ERK phosphorylation peaked within 2–4 h of treatment and, unlike that observed following H2O2 treatment, remained substantially elevated above that observed in the vehicle control throughout the time course of the experiment (Figs 3c and d).
Fig. 3.
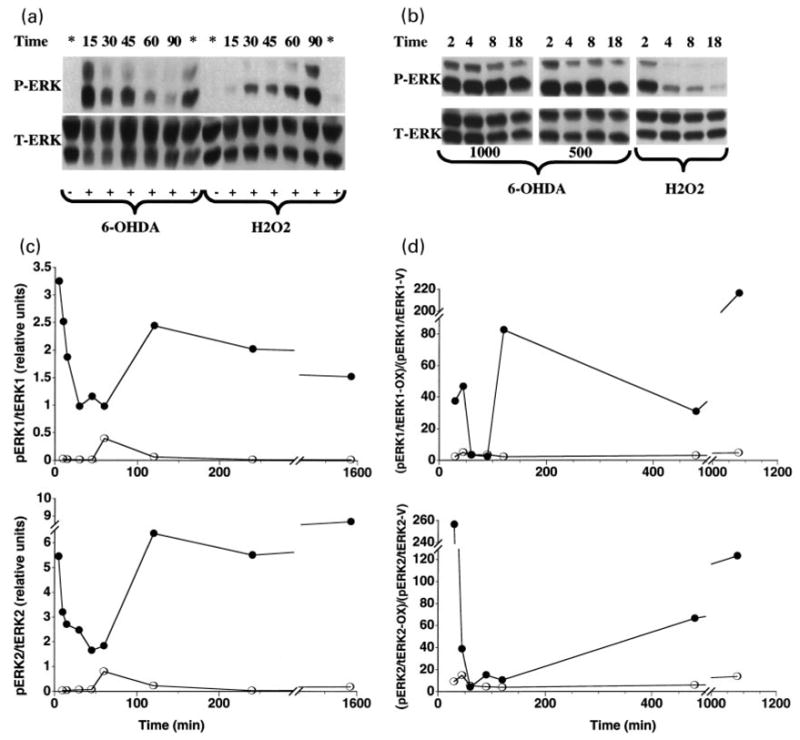
Kinetics of ERK activation in B65 cells in response to 6-OHDA and H2O2. (a) Cells treated with 500 μM 6-hydroxydopamine (6-OHDA) or 1 mM H2O2 (H2O2) for the indicated times (minutes) were lysed and equal amounts of protein (50 μg) were subjected to immunoblot analysis using antibody against the activated form of ERK (top, P-ERK). The blots were then stripped and reprobed with antibody against total ERK (bottom, T-ERK). Lanes designated with * were harvested following an overnight (16 h) incubation. Lanes designated with – were treated with the appropriate vehicle (0.05% ascorbate for 6-OHDA, water for H2O2). (b) Cells were treated with either 1000 or 500 μM 6-OHDA or 1 mM H2O2 for the indicated time (hours) then subjected to immunoblot analysis as in (a). (c) Immunoblots from cells treated with 1000 mM 6-OHDA were subjected to densitometric analysis, and the data for the ERK1 (top panel) and ERK2 (bottom panel) isoforms from a representative time-course is shown. Closed circles (●) represent the relative band intensity of phosphorylated ERK/total ERK elicited by 6-OHDA; open circles (○) represent the phosphorylated ERK/total ERK ratio observed in vehicle-treated cells. Cells treated with only fresh DH10 exhibited a pattern of ERK phosphorylation similar to that seen with the vehicle controls. (d) Immunoblots from cells treated with 500 μM 6-OHDA and 1 mM H2O2 were subjected to densitometric analysis, and the data for the ERK1 (top panel) and ERK2 (bottom panel) isoforms from a representative time-course is shown. Closed circles represent the relative band intensity elicited by 6-OHDA while open circles represent the relative band intensity elicited by H2O2. Data are expressed as the ratio of phosphorylated ERK to total ERK elicited by the oxidizing agents (pERK/tERK-OX) normalized to the ratio of phosphorylated ERK to total ERK elicited in vehicle treated cells (pERK/tERK-V). Data shown in panels A–D are representative compilations of 10 independent time-course experiments.
The continued presence of phosphorylated ERK elicited by 2–26 h exposures to 6-OHDA contrasts strikingly with growth factor receptor-mediated ERK phosphorylation, for which down-regulating systems rapidly terminate the response even in the continued presence of ligand. Because 6-OHDA undergoes fairly rapid auto-oxidation at physiologic pH (Cohen and Heikkila 1974), we wished to define the minimum duration of 6-OHDA exposure that would elicit sustained ERK phosphorylation. Pulse-chase experiments showed that in order to elicit sustained ERK phosphorylation, B65 cells required the presence of 6-OHDA for at least 2–4 h (Fig. 4a) since shorter exposures did not result in sustained ERK activation. Similarly, a 2-h pulse with 6-OHDA resulted in cytotoxicity, although a 4-h incubation was necessary to elicit dose–response curves identical to longer treatments (Fig. 4b). These experiments suggest that the pulse length that commits the cells to death correlates with the time it takes to achieve induction of the sustained phase of 6-OHDA elicited ERK-phosphorylation.
Fig. 4.
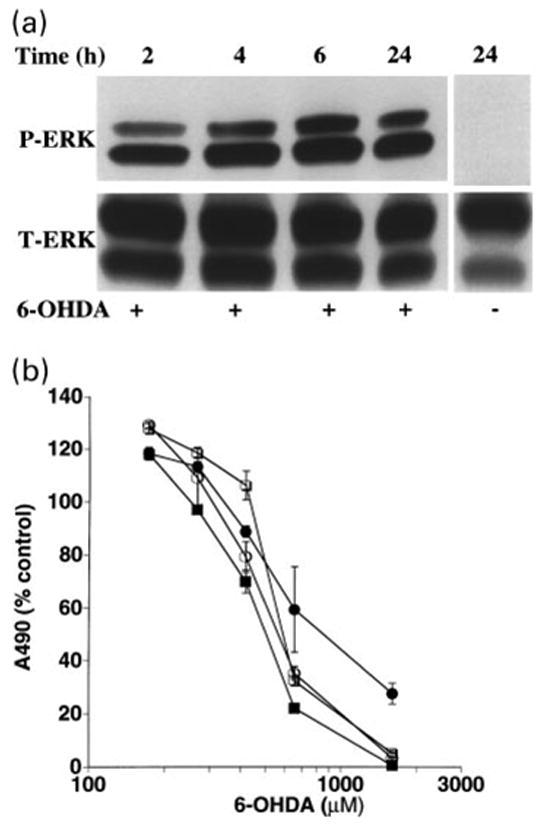
Pulse-chase studies comparing ERK phosphorylation and cytotoxicity. (a) Cells were pulsed with 1 mM 6-hydroxydopamine (6-OHDA) for the indicated times (hours) at which time 6-OHDA was removed. The cells were washed with DH10 and then incubated in fresh DH10 for a total incubation time of 24 h. Immunoblot analysis of cell lysates was performed as described above. Cells treated with vehicle did not exhibit increased phosphorylation at any time point (24 h time point shown as an example). Data shown are representative of four independent experiments. (b) Cells were pulsed with various concentrations of 6-OHDA for the indicated times (2 h: ●, 4 h: ○, 6 h: ■, 22 h: □), washed and placed in fresh DH10 for a total incubation time of 22 h. Cell injury was determined using the MTS viability assay. Each value represents the mean of triplicate wells ± SEM. The experiment was performed independently three times, and a representative plot is illustrated.
Inhibition of 6-OHDA-mediated ERK phosphorylation by PD98059
Sustained ERK phosphorylation secondary to 6-OHDA treatment could result from a variety of different mechanisms. Activation of upstream signaling molecules, inhibition of dephosphorylation, and inhibition of protein degradation have been previously observed as mediators of sustained ERK phosphorylation in different cellular contexts (Traverse et al. 1992; Runden et al. 1998; Hashimoto et al. 2000). In an attempt to better define the role of upstream activators of ERK in 6-OHDA-mediated sustained ERK phosphorylation, B65 cells were exposed to PD98059, an inhibitor of MEK 1/2, a dual potential protein kinase that is capable of directly activating ERK. As illustrated in Fig. 5, the presence of 50 μM PD98059 substantially inhibited 6-OHDA-mediated sustained ERK phosphorylation, suggesting that 6-OHDA affects the signal transduction machinery upstream of MEK 1/2. The presence of PD98059 also inhibited the H2O2-induced activation of ERK (data not shown).
Fig. 5.
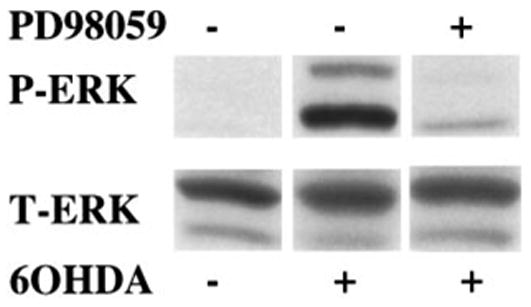
Effect of the MEK inhibitor PD98059 upon 6-OHDA mediated ERK phosphorylation. B65 cells treated for 20 h with 500 mM 6-hydroxydopamine (6-OHDA) in the presence (+) or absence (−) of 50 μM PD98059 were lysed and equal amounts of protein (50 μg) were subjected to immunoblot analysis using antibody against the activated form of ERK (top, P-ERK). The blots were then stripped and reprobed with antibody against total ERK (bottom, T-ERK).
Inhibition of 6-OHDA-mediated cytotoxicity by PD98059
Although traditionally associated with responses such as differentiation or proliferation, recent reports have implicated sustained ERK activation in oxidative toxicity to primary cortical neurons and the hippocampal neuronal cell line HT22 (Stanciu et al. 2000). In addition, compensatory up-regulation of neurotrophic factors or their receptors is a hypothetical possibility. In order to assess the role of ERK phosphorylation in 6-OHDA-mediated toxicity, B65 cells were exposed to 6-OHDA in the presence of PD98059. The presence of 50 μM PD98059 resulted in decreased B65 injury in response to 6-OHDA as determined by both MTS and LDH release assays (Figs 6a and b). PD98059 had no effect on H2O2-mediated cell injury as determined by MTS (Fig. 6c) or LDH release assays (data not shown). These results support a role for sustained ERK phosphorylation in 6-OHDA cytotoxicity in this tyrosine hydroxylase-expressing CNS neuronal cell line.
Fig. 6.
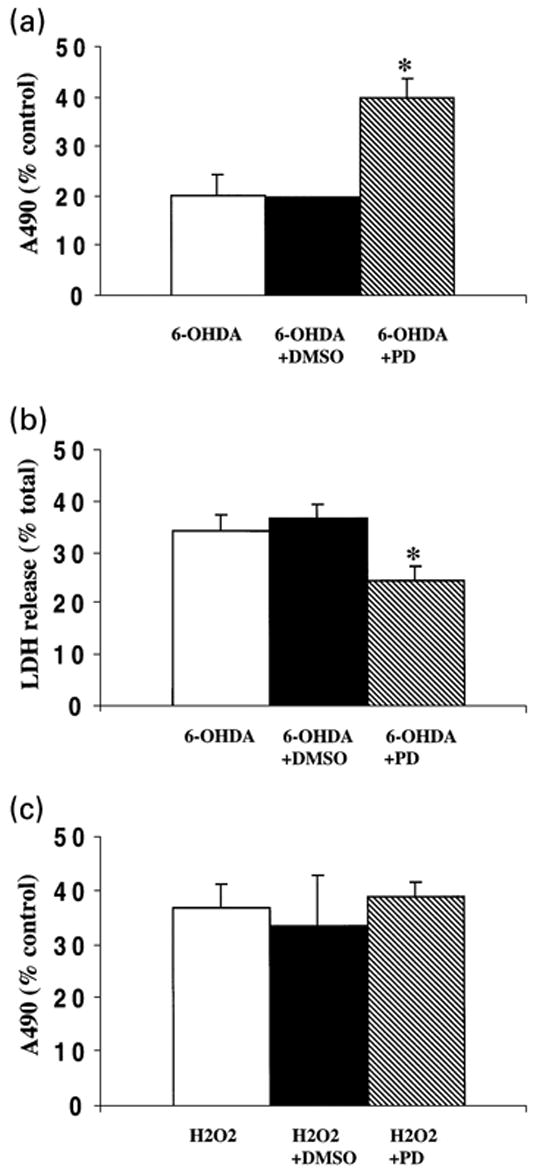
Effect of PD98059 on 6-OHDA-mediated cytotoxicity. Following overnight exposure of B65 cells to 655 mM 6-OHDA in the absence (white bar) or presence (hatched bar) of 50 mM PD98059, cytotoxicity was determined utilizing MTS (a) and LDH release (b) assays. Cells treated with 6-OHDA in the presence of 0.1% DMSO, the vehicle in which the inhibitor was dissolved (black bar), showed values similar to those lacking DMSO. PD98059 treatment had no effect upon the A490 or total LDH values of ascorbate-treated control wells against which the other treatment conditions were normalized (A490 values: 2.26 ± 0.06, untreated; 2.23 ± 0.08 PD98059 treated; total LDH values: 129 ± 27 IU/mL, untreated; 132 ± 23 IU/mL, PD 98059 treated). MTS data is a compilation of five independent experiments and LDH release data is a compilation of three independent experiments. Data are expressed as the mean ± SEM. Analysis by two-tailed Student's t-tests yielded p-values of <0.05 (*). (c) Following overnight exposure of B65 cells to 1 mM H2O2 in the absence (white bar) or presence (hatched bar) of 50 mM PD98059, cytotoxicity was determined utilizing a MTS assay. Cells treated with H2O2 in the presence of 0.1% DMSO (black bar), showed values similar to those lacking DMSO. MTS data is a compilation of five independent experiments, and is expressed as the mean ± SEM (p = 0.75, two-tailed Student's t-test).
Discussion
Although the toxin 6-OHDA is utilized extensively in animal models of Parkinson's disease, the underlying mechanism of its toxic effects on dopaminergic neurons is not completely understood. As MAP kinase signaling cascades play important roles in neuronal survival and differentiation, this study was designed to investigate the effects of 6-OHDA upon activation of the ERK branch of the MAP kinase superfamily. A CNS-derived neuronal cell line was selected to generate a uniform population of tyrosine hydroxylase-expressing cells for cytotoxicity and immunoblot analysis. Using this model, two significant findings regarding potential mechanisms underlying 6-OHDA-mediated toxicity were observed. First, 6-OHDA, but not H2O2, elicits a dose-dependent sustained activation of ERK. Second, PD98059, inhibits 6-OHDA-elicited ERK phosphorylation, and confers protection from 6-OHDA-mediated toxicity, but not H2O2 toxicity. These results suggest a specific detrimental role of ERK phosphorylation in 6-OHDA-mediated toxicity.
ERK isoforms are activated by neurotrophic factors such as brain-derived neurotrophic factor or glial cell line-derived neurotrophic factor, both of which are neuroprotective for dopaminergic neurons (Frim et al. 1994; Choi-Lundberg et al. 1997). While it has been well documented that ERK activation leads to favorable responses such as differentiation and neuroprotection, recent evidence has also implicated ERK in detrimental responses to oxidative stress. In neuroblastoma SH-SY5Y cells, sequential activation of both p38 and ERK contributes to peroxynitrite-induced apoptosis (Oh-hashi et al. 1999). An abnormal immunohistochemical staining pattern for phosphorylated ERK has also been observed in susceptible neurons from the brains of patients with Alzheimer's disease (Perry et al. 1999). In several non-neuronal cell types, apoptosis and inhibition of DNA synthesis are associated with ERK activation (Tombes et al. 1998; Bhat and Zhang 1999; Ishikawa and Kitamura 1999). In addition, recent studies in cell lines and primary cortical neurons suggest ERK activation may play a direct role in neuronal cytotoxicity (Stanciu et al. 2000). Although kinase inhibitor studies must always be interpreted with caution as the compound may have effects on unrecognized signaling pathways, PD98059 exhibits strong in vitro selectivity for inhibiting MEK 1/2 (Alessi et al. 1995). Since the development of sustained ERK phosphorylation correlates with commitment to death (Fig. 4), and PD98059, which effectively blocks 6-OHDA-mediated ERK phosphorylation (Fig. 5) confers protection (Fig. 6), this study offers additional support for the concept that sustained ERK phosphorylation can contribute to oxidative neuronal injury.
Two general patterns of ERK activation have been observed, transient and sustained (reviewed in Marshall 1995). The divergent roles of ERK phosphorylation patterns are well illustrated in PC12 cells, where transient ERK activation by epidermal growth factor leads to cell proliferation but sustained ERK activation by nerve growth factor leads to nuclear translocation of ERK and differentiation (Traverse et al. 1992). It is important to point out that, in these physiologic patterns of ERK activation, the transient component peaks at 2 min and returns to baseline by ~60 min, whereas the `sustained' component refers to elevation of ERK phosphorylation for 1.5–3 h. Much longer periods of ERK activation, as seen in this study, may prove detrimental, particularly in the setting of neuronal responses to oxidative stressors. For example, elevated ERK phosphorylation was reported in cortical neurons and a hippocampal cell line for 3–15 h in response to oxidative glutamate toxicity associated with glutathione depletion (Stanciu et al. 2000). Sustained ERK phosphorylation secondary to phosphatase inhibition appears to selectively target certain susceptible neuronal populations (Runden et al. 1998). This report is the first to demonstrate an association between the redox cycling 6-OHDA and sustained ERK activation that is not down-regulated >20 h following removal of the neurotoxin (Fig. 4a).
A second interesting observation is that B65 cells demonstrate a biphasic response to 6-OHDA (Fig. 3) with an initial robust ERK activation followed by a return towards baseline and a subsequent delayed, sustained ERK activation. The decrease in ERK phosphorylation at ~60–90 min is most prominent with lower doses of 6-OHDA, and this period of time may be associated with refractoriness to ERK activation by neurotrophic factors (CTC, unpublished observations). The previous reports of detrimental sustained ERK phosphorylation have not reported this early activation phase. Additional studies to define the contributions of these phases of ERK phosphorylation to the detrimental effect observed in B65 cells are currently being pursued.
While the concentrations of 6-OHDA utilized in the present study are higher than the doses reported in recent reports of 6-OHDA-mediated toxicity in primary cultures (Eggert et al. 1999; Lotharius et al. 1999), it is difficult to make direct comparisons between the experimental systems. Although highly enriched for neurons, primary cultures contain additional cell populations that may influence the responsiveness of the target neuron population, which often comprises only 5% of the cells (Lotharius et al. 1999). In addition, there are differences in the length of exposure to the toxin and the methodology used to assess cell injury. Finally, our preliminary studies utilizing differentiated B65 cells suggest that cell cycle status may also influence susceptibility to toxins.
Metabolism of 6-OHDA is complex and may generate several different reactive species. H2O2 and superoxide have both been shown to modulate the activities of protein kinases and protein phosphatases in multiple systems (reviewed in Suzuki et al. 1997; Klann and Thiels 1999). Although the kinetics of ERK phosphorylation in B65 differs between 6-OHDA, H2O2 (Fig. 3) and xanthine\xanthine oxidase-treated cells (unpublished observation), we cannot rule out a potential role for H2O2 and superoxide in 6-OHDA-mediated ERK activation based on the present studies. It is possible that intracellularly generated superoxide or interactions between H2O2 and superoxide are important. Another potential 6-OHDA metabolite, dopamine quinone, is an electron-deficient intermediate of dopamine metabolism that is capable of covalently modifying free or protein bound thiol groups. Modification of cys118 of ras has been associated with direct ras activation (Lander et al. 1997). Activation of a signaling molecule such as ras that lies upstream of ERK would be consistent with our findings that PD98059, an inhibitor of MEK 1/2, is capable of attenuating 6-OHDA-mediated sustained ERK activation. Additional studies will be required to further evaluate the potential contributions of these metabolic intermediates and to better define which members of the signal transduction cascade are responsible for 6-OHDA-mediated ERK activation.
Oxidative stress has been postulated to play a role in many neurodegenerative diseases (reviewed in Beal 1995). Our study lends further support to other studies indicating an association between oxidative stressors and sustained ERK activation in neuronal cells (Runden et al. 1998; Stanciu et al. 2000). While the relevance of these in vitro observations to in vivo situations is unclear, recent reports have described an abnormal pattern of activated ERK staining in pathologic states linked to increased oxidative stress. Increased levels of both total and activated ERK has been observed within atherosclerotic lesions of cholesterol-fed rabbits (Hu et al. 2000). An abnormal punctate staining pattern of activated ERK has also been described in susceptible neurons in the brains of patients with Alzheimer's disease (Perry et al. 1999). While the status of ERK activation in patients with Parkinson's disease is currently unknown, the involvement of ERK in 6-OHDA toxicity suggests that abnormal ERK activation could contribute to mechanisms of neuronal cell death relevant to Parkinson's disease.
Only recently has it been appreciated that oxidative species such as H2O2 and superoxide may play important roles in modulating signal transduction pathways (reviewed in Suzuki et al. 1997). This study links the redox cycling neurotoxin 6-OHDA, which is commonly used in parkinsonian models, to an unusual biphasic pattern of sustained ERK phosphorylation. As dopamine itself can also undergo redox cycling, and high doses of dopamine can contribute to oxidative toxicity both in vivo and in vitro (Hastings et al. 1996; Luo et al. 1998), similar mechanisms may contribute to enhanced susceptibility of dopaminergic neurons. As 6-OHDA can be identified in the urine of both control and Parkinson's disease patients, and it is elevated in patients that have received levodopa (Andrew et al. 1993), it is possible that these patterns of abnormal ERK activation can contribute to ongoing neuron loss and progression of Parkinson's disease. Identification of the oxidative mechanisms involved and the members of the signal transduction cascade that are affected by 6-OHDA to yield sustained ERK activation would be important for understanding the molecular pathogenesis of neuronal loss relevant to Parkinson's disease and related neurodegenerative diseases.
Acknowledgments
We thank Dr Tim Oury for helpful discussions. Dr Chu is a Charles E. Culpeper Scholar in Medical Science, and this work was supported in part by the Rockefeller Brothers Fund, grant RO1 NS40817 from the National Institutes of Health, and grants from the Scaife Family Foundation of Pittsburgh, the American Parkinson Disease Association, and the National Parkinson Foundation.
Abbreviations used
- DH10
DMEM media with 10 mM HEPES, 2 mm L-glutamine, 10% fetal calf serum
- ECL
enhanced chemiluminescence
- ERK
extracellular signal-regulated protein kinase
- HRP
horseradish peroxidase
- JNK
c-Jun N-terminal kinase/stress activated protein kinase
- LDH
lactate dehydrogenase
- MAP
mitogen activated protein
- MEK
kinase that phosphorylates and activates ERK (also known as MAP kinase kinase)
- MTS
[3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt]
- 6-OHDA
6-hydroxydopamine
- PBST
phosphate buffered saline with 0.3% (w/v) Tween-20
- PMS
phanazine methsulfate
- SNpc
substantia nigra pars compacta
References
- Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J Biol Chem. 1995;270:27489–27494. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- Ambani LM, Van Woert MH, Murphy S. Brain peroxidase and catalase in Parkinson disease. Arch Neurol. 1975;32:114–118. doi: 10.1001/archneur.1975.00490440064010. [DOI] [PubMed] [Google Scholar]
- Andrew R, Watson DG, Best SA, Midgley JM, Wenlong H, Petty RKH. The determination of hydroxydopamines and other trace amines in the urine of Parkinsonian patients and normal controls. Neurochem Res. 1993;18:1175–1177. doi: 10.1007/BF00978370. [DOI] [PubMed] [Google Scholar]
- Asanuma M, Hirata H, Cadet JL. Attenuation of 6-hydroxydopamine-induced dopaminergic nigrostriatal lesions in superoxide dismutase transgenic mice. Neuroscience. 1998;85:907–917. doi: 10.1016/s0306-4522(97)00665-9. [DOI] [PubMed] [Google Scholar]
- Beal MF. Aging, energy, and oxidative stress in neurodegenerative diseases. Ann Neurol. 1995;38:357–366. doi: 10.1002/ana.410380304. [DOI] [PubMed] [Google Scholar]
- Bhat NR, Zhang P. H2O2 activation of multiple mitogen-activated protein kinases in an oligodendrocyte cell line: role of extracellular signal-regulated kinase in hydrogen peroxide-induced cell death. J Neurochem. 1999;72:112–119. doi: 10.1046/j.1471-4159.1999.0720112.x. [DOI] [PubMed] [Google Scholar]
- Cai G, Zhen X, Uryu K, Friedman E. Activation of extracellular signal-regulated protein kinases is associated with a sensitized locomotor response to D2 dopamine receptor stimulation in unilateral 6-hydroxydopamine-lesioned rats. J Neurosci. 2000;20:1849–1857. doi: 10.1523/JNEUROSCI.20-05-01849.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi WS, Yoon SY, Oh TH, Choi EJ, O'Malley KL, Oh YJ. Two distinct mechanisms are involved in 6-hydroxydopamine- and MPP+-induced dopaminergic neuronal cell death: role of caspases, ROS, and JNK. J Neurosci Res. 1999;57:86–94. doi: 10.1002/(SICI)1097-4547(19990701)57:1<86::AID-JNR9>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Choi-Lundberg D, Lin Q, Chang Y, Chiang Y, Hay C, Mohajeri H, Davidson B, Bohn M. Dopaminergic neurons protected from degeneration by GDNF gene therapy. Science. 1997;275:838–841. doi: 10.1126/science.275.5301.838. [DOI] [PubMed] [Google Scholar]
- Chu CT, Pizzo SV. Receptor-mediated antigen delivery into macrophages: Complexing antigen to α2-macroglobulin enhances presentation to T-cells. J Immunol. 1993;150:48–58. [PubMed] [Google Scholar]
- Chu CT, Everiss KD, Batra S, Wikstrand CJ, Kung HJ, Bigner DD. Receptor dimerization is not a factor in the signalling activity of a transforming variant epidermal growth factor receptor (EGFRvIII) Biochem J. 1997;324:855–861. doi: 10.1042/bj3240855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen G, Heikkila RE. The generation of hydrogen peroxide, superoxide radical, and hydroxyl radical by 6-hydroxydopamine, dialuric acid, and related cytotoxic agents. J Biol Chem. 1974;249:2447–2452. [PubMed] [Google Scholar]
- Eggert K, Schlegel J, Oertel W, Wurz C, Krieg JC, Vedder H. Glial cell line-derived neurotrophic factor protects dopaminergic neurons from 6-hydroxydopamine toxicity in vitro. Neurosci Lett. 1999;269:178–182. doi: 10.1016/s0304-3940(99)00443-7. [DOI] [PubMed] [Google Scholar]
- Frim D, Uhler T, Galpern W, Beal M, Breakefield X, Isacson O. Implanted fibroblasts genetically engineered to produce brain-derived neurotrophic factor prevent 1-methyl-4-phenylpyridinium toxicity to dopaminergic neurons in the rat. Proc Natl Acad Sci USA. 1994;91:5104–5108. doi: 10.1073/pnas.91.11.5104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glinka YY, Youdim MB. Inhibition of mitochondrial complexes I and IV by 6-hydroxydopamine. Eur J Pharmacol. 1995;292:329–332. doi: 10.1016/0926-6917(95)90040-3. [DOI] [PubMed] [Google Scholar]
- Hashimoto K, Guroff G, Katagiri Y. Delayed and sustained activation of p42/p44 mitogen-activated protein kinase induced by proteasome inhibitors through p21 (ras) in PC12 cells. J Neurochem. 2000;74:92–98. doi: 10.1046/j.1471-4159.2000.0740092.x. [DOI] [PubMed] [Google Scholar]
- Hastings TG, Lewis DA, Zigmond MJ. Role of oxidation in the neurotoxic effects of intrastriatal dopamine injections. Proc Natl Acad Sci USA. 1996;93:1956–1961. doi: 10.1073/pnas.93.5.1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch EC, Brandel JP, Galle P, Javoy-Agid F, Agid Y. Iron and aluminum increase in the substantia nigra of patients with Parkinson's disease: an X-ray microanalysis. J Neurochem. 1991;56:446–451. doi: 10.1111/j.1471-4159.1991.tb08170.x. [DOI] [PubMed] [Google Scholar]
- Holmström TH, Chow SC, Elo I, Coffey ET, Orrenius S, Sistonen L, Eriksson JE. Suppression of Fas/APO-1-mediated apoptosis by mitogen-activated kinase signaling. J Immunol. 1998;160:2626–2636. [PubMed] [Google Scholar]
- Hu Y, Dietrich H, Metzler B, Wick G, Xu Q. Hyperexpression and activation of extracellular signal-regulated kinases (ERK1/2) in atherosclerotic lesions of cholesterol-fed rabbits. Arterioscler Thromb Vasc Biol. 2000;20:18–26. doi: 10.1161/01.atv.20.1.18. [DOI] [PubMed] [Google Scholar]
- Ishikawa Y, Kitamura M. Dual potential of extracellular signal-regulated kinase for the control of cell survival. Biochem Biophys Res Commun. 1999;264:696–701. doi: 10.1006/bbrc.1999.1542. [DOI] [PubMed] [Google Scholar]
- Jameson GNL, Linert W. 6-Hydroxydopamine, dopamine, and ferritin: a cycle of reactions sustaining parkinson's disease? In: Poli G, Cadenas E, Packer L, editors. Free Radicals in Brain Pathophysiology. Marcel Dekker, Inc.; New York: 2000. pp. 247–272. [Google Scholar]
- Kish SJ, Morito C, Hornykiewicz O. Glutathione peroxidase activity in Parkinson's disease brain. Neurosci Lett. 1985;58:343–346. doi: 10.1016/0304-3940(85)90078-3. [DOI] [PubMed] [Google Scholar]
- Klann E, Thiels E. Modulation of protein kinases and protein phosphatases by reactive oxygen species: implications for hippocampal synaptic plasticity. Prog Neuropsychopharmacol Biol Psychiatry. 1999;23:359–376. doi: 10.1016/s0278-5846(99)00002-0. [DOI] [PubMed] [Google Scholar]
- Lander HM, Hajjar DP, Hempstead BL, Mirza UA, Chait BT, Campbell S, Quilliam LA. A molecular redox switch on p21 (ras). Structural basis for the nitric oxide-p21 (ras) interaction. J Biol Chem. 1997;272:4323–4326. doi: 10.1074/jbc.272.7.4323. [DOI] [PubMed] [Google Scholar]
- Lang AE, Lozano AM. Parkinson's disease. First of two parts. N Engl J Med. 1998;339:1044–1053. doi: 10.1056/NEJM199810083391506. [DOI] [PubMed] [Google Scholar]
- Lotharius J, Dugan LL, O'Malley KL. Distinct mechanisms underlie neurotoxin-mediated cell death in cultured dopaminergic neurons. J Neurosci. 1999;19:1284–1293. doi: 10.1523/JNEUROSCI.19-04-01284.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y, Umegaki H, Xiantao W, Abe R, Roth GS. Dopamine induces apoptosis through an oxidation-involved SAPK/JNK activation pathway. J Biol Chem. 1998;273:3756–3764. doi: 10.1074/jbc.273.6.3756. [DOI] [PubMed] [Google Scholar]
- Marshall CJ. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell. 1995;80:179–185. doi: 10.1016/0092-8674(95)90401-8. [DOI] [PubMed] [Google Scholar]
- Moghal S, Rajput AH, D'Arcy C, Rajput R. Prevalence of movement disorders in elderly community residents. Neuroepidemiology. 1994;13:175–178. doi: 10.1159/000110376. [DOI] [PubMed] [Google Scholar]
- Oh-hashi K, Maruyama W, Yi H, Takahashi T, Naoi M, Isobe K. Mitogen-activated protein kinase pathway mediates peroxynitrite-induced apoptosis in human dopaminergic neuroblastoma SH-SY5Y cells. Biochem Biophys Res Commun. 1999;263:504–509. doi: 10.1006/bbrc.1999.1237. [DOI] [PubMed] [Google Scholar]
- Perry G, Roder H, Nunomura A, Takeda A, Friedlich AL, Zhu X, Raina AK, Holbrook N, Siedlak SL, Harris PL, Smith MA. Activation of neuronal extracellular receptor kinase (ERK) in Alzheimer disease links oxidative stress to abnormal phosphorylation. Neuroreport. 1999;10:2411–2415. doi: 10.1097/00001756-199908020-00035. [DOI] [PubMed] [Google Scholar]
- Runden E, Seglen PO, Haug FM, Ottersen OP, Wieloch T, Shamloo M, Laake JH. Regional selective neuronal degeneration after protein phosphatase inhibition in hippocampal slice cultures: evidence for a MAP kinase-dependent mechanism. J Neurosci. 1998;18:7296–7305. doi: 10.1523/JNEUROSCI.18-18-07296.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert D, Heinemann S, Carlisle W, Tarikas H, Kimes B, Patrick J, Steinback JH. Clonal cell lines from the rat central nervous system. Nature. 1974;249:224–227. doi: 10.1038/249224a0. [DOI] [PubMed] [Google Scholar]
- Segal RA, Greenberg ME. Intracellular signaling pathways activated by neurotrophic factors. Ann Rev Neurosci. 1996;19:463–489. doi: 10.1146/annurev.ne.19.030196.002335. [DOI] [PubMed] [Google Scholar]
- Stanciu M, Wang Y, Kentor R, Burke N, Watkins S, Kress G, Reynolds I, Klann E, Angiolieri M, Johnson J, Defranco DB. Persistent activation of ERK contributes to glutamate-induced oxidative toxicity in a neuronal cell line and primary cortical neuron cultures. J Biol Chem. 2000;275:12200–12206. doi: 10.1074/jbc.275.16.12200. [DOI] [PubMed] [Google Scholar]
- Sutherland MW, Learmonth BA. The tetrazolium dyes MTS and XTT provide new quantitative assays for superoxide and superoxide dismutase. Free Radic Res. 1997;27:283–289. doi: 10.3109/10715769709065766. [DOI] [PubMed] [Google Scholar]
- Suzuki YJ, Forman HJ, Sevanian A. Oxidants as stimulators of signal transduction. Free Radic Biol Med. 1997;22:269–285. doi: 10.1016/s0891-5849(96)00275-4. [DOI] [PubMed] [Google Scholar]
- Tombes RM, Auer KL, Mikkelsen R, Valerie K, Wymann MP, Marshall CJ, Mcmahon M, Dent P. The mitogen-activated protein (MAP) kinase cascade can either stimulate or inhibit DNA synthesis in primary cultures of rat hepatocytes depending upon whether its activation is acute/phasic or chronic. Biochem J. 1998;330:1451–1460. doi: 10.1042/bj3301451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traverse S, Gomez N, Paterson H, Marshall C, Cohen P. Sustained activation of the mitogen-activated protein (MAP) kinase cascade may be required for differentiation of PC12 cells. Comparison of the effects of nerve growth factor and epidermal growth factor. Biochem J. 1992;288:351–355. doi: 10.1042/bj2880351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungerstedt U. 6-Hydroxy-dopamine induced degeneration of central monoamine neurons. Eur J Pharmacol. 1968;5:107–110. doi: 10.1016/0014-2999(68)90164-7. [DOI] [PubMed] [Google Scholar]
- Wroblewski F, Ladue J. Lactic dehydrogenase activity in blood. Proc Soc Exp Biol Med. 1955;90:210. doi: 10.3181/00379727-90-21985. [DOI] [PubMed] [Google Scholar]
- Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]