

Abstract
No3 (nuclear opacity 3) is a novel congenital nuclear cataract in mice. Microsatellite mapping placed the No3 locus on chromosome 1 between D1Mit480 (32cM) and D1Mit7 (41cM), a region containing seven crystallin genes; Cryba2 and the Cryga-Crygf cluster. Although polymorphic variants were observed, no candidate mutations were found for six of the genes. However, DNA walking identified a murine endogenous retrovirus (IAPLTR1: ERVK) insertion in exon 3 of Cryge, disrupting the coding sequence for γE-crystallin. Recombinant protein for the mutant γE was completely insoluble. The No3 cataract is mild compared with the effects of similar mutations of γE. Quantitative RT-PCR showed that γE/F mRNA levels are reduced in No3, suggesting that the relatively mild phenotype results from suppression of γE levels due to ERVK insertion. However, the severity of cataract is also strain dependent suggesting that genetic background modifiers also play a role in the development of opacity.
Keywords: cataract, crystallin, endogenous retrovirus
Introduction
As the major macromolecular components of the ocular lens, crystallins are centrally important to the transparency and refractive power of the tissue [1; 2]. While there may be many different initial insults and upstream triggering events, loss of the structural integrity and organization of crystallins is thought to play a large part in the light scattering that causes opacity in cataract [2].
Many crystallin-related inherited cataracts are known in human and mouse [3; 4]. The most frequently observed class of mutation, perhaps because it has the greatest severity, results from truncations or frame shift mutations that severely disrupt protein structure, preventing normal folding and leading to immediate formation of insoluble aggregates and perhaps to amyloid [5]. Such cataracts are exemplified by Elo [6], Cat2t [7]and Nop [5] in mice. A second kind of mutation involves more subtle mutations that allow normal three-dimensional folding but apparently reduce thermodynamic stability or compromise intermolecular interactions (with other proteins or with water). This second class, exemplified by the temperature sensitive Opj mutation in mouse γS-crystallin [8] and several surface mutations in human γD-crystallin [9; 10; 11; 12; 13; 14], may be better models for processes in “normal” human age-related cataract in which proteins that were correctly folded and soluble lose their integrity and undergo phase separation or aggregation [15; 16]. In addition, it has become clear that identical mutations in the same crystallin gene can produce diagnostically different forms of cataract in different human lineages [10; 11; 12], suggesting that genetic background is important even in apparently simple mutational models.
Here we describe a novel murine cataract, No3 (nuclear opacity 3) which produces mild nuclear opacity that is highly dependent upon the genetic background of the carrier mice. Unlike most other murine crystallin mutations that have been catalogued, No3 results from insertion of a full-length endogenous retroviral element into the coding sequence of a crystallin gene. This produces a frame-shifted and truncated γE-crystallin that is superficially similar to the mutations in the severe Elo and Cat2t cataracts, but with significant difference in the reported phenotype.
Results
Identification and Inheritance
The founder animal, designated iPMS-8, resulted from chemical mutagenesis of male (C3H/He x 102/El)F1 mice [17] and was said to have “nuclear opacities”. When crossed with a normal animal it produced offspring like itself, indicating autosomal dominant inheritance. The cataract was given the name “nuclear opacity 3” and gene symbol No3 (figure 1).
Figure 1. The appearance of the No3 opacity on the C3H/HeH background.
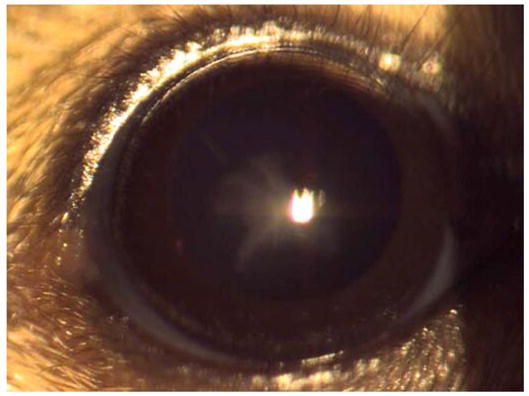
A mild nuclear opacity is visible in affected animals.
Crosses of affected mice with normal animals yielded offspring with mild diffuse opacities in the lens nucleus. When the normal mate was from the C3H/HeH strain the ratio of affected to normal offspring was not significantly different from 1:1 (χ2=0.48, p>0.3). However, when affected animals were crossed with C57BL/6 there was a severe shortage of affected animals (χ2= 59.8, p<.001) (Table 1). The probable explanation of this shortage was incomplete penetrance. There may have been some misclassification of No3/+ as normal offspring in the C3H/HeH crosses also as the phenotype was sometimes very mild. When two affected animals were crossed together some offspring showed a more severe phenotype, and were suspected to be homozygotes. When these animals were crossed to C3H/HeH, almost all offspring showed the mild phenotype of No3/+, confirming the parents to be No3/No3. Crosses of No3/No3 animals together yielded almost all offspring with the more severe phenotype of No3/No3.
Table 1.
Breeding behavior of No3/+ and No3/No3 mice.
| Genotype of parents | Offspring | Percent cataract | ||
|---|---|---|---|---|
| Severe opacity | Mild opacity | +/+ | ||
| No3/+ x +/+ (C3H/HeH) | 0 | 71 | 63 | 53.0 |
| No3/+ x +/+ (C57BL/6) | 0 | 92 | 231 | 28.5 |
| No3/+ x No3/+ | 13 | 14 | 16 | 62.8 |
| No3/No3 + +/+ | 0 | 54 | 1 | 98.2 |
| No3/No3 x No3/No3 | 58 | 8 | 1 | 98.5 |
Histological examination of eyes from new born and adult No3/No3 mice showed no obvious deviations from normal lens size or organization (not shown).
Mapping
Preliminary results with markers covering the whole genome showed significant linkage with the marker D1Mit46 (χ2= 10.7, p< .01) (data not shown). The same animals were then typed for several more markers on Chromosome 1. A few animals gave anomalous results suggesting misclassification for No3 and these were excluded. Results from the remaining 41 mice gave the following recombination percentages: D1Mit3 – 17.6 ±6.2 – D1Mit213 – 12.2± 5.1 – (D1Mit282, D1Mit480, No3) – 2.4 ± 2.4 – (D1Mit7, D1Mit181, D1Mit46) – 4.1 ± 3.4 – D1Mit80 – 21.9 + 6.5 – D1Mit227. These results place the No3 locus between D1Mit480 at 32cM and D1Mit7 at 41cM, thus making both the Cryg cluster at 32cM and Cryba2 at 40.8cM possible candidate genes (fig 2).
Figure 2. Schematic of the candidate genes for No3 on mouse chromosome 1.

The mapped interval contains genes for seven crystallins: Cryba2 and the six genes of the Cryga-Crygf cluster. Not to scale.
Candidate Gene Analysis
Prior to the availability of the complete mouse genome sequence, the CrybA2 gene from No3 was examined by PCR amplification of genomic DNA using primer sequences based on the published mouse cDNA sequence. No coding sequence mutations were found. The structure of the CrybA2 gene will be described separately. In addition, a cDNA library was constructed from adult No3 mouse eyes. Since the No3 background contained the rd gene, this provided an example of a retinal degeneration model to add to the NEIBank database of eye-expressed genes (http://neibank/nei.nih.gov). It also provided full length cDNA sequences for βA2, γB, γC and γD-crystallins from No3/No3. These all appeared to be normal, matching the sequences for C57BL/6, with one exception. The predicted amino acid sequence of γD from No3 has 52A (counting from the initiator codon), instead of 52T found in C57BL/6. However this difference is also seen in another mouse strain (GenBank accession NP_031802) and may therefore represent a common polymorphism.
No cDNA clones were observed for γA, γE or γF-crystallins in this EST analysis. These were addressed by PCR amplification of exons from genomic DNA using primers based on C57BL/6 genome sequence and by RT-PCR of RNA extracted from adult No3 and FVBN mouse eyes. All three genes were successfully amplified from the control DNA, but only γA and γF were obtained from No3. Exons and flanking regions were sequenced from the γA and γF amplimers. Several apparent polymorphisms, including a 29bp insertion of a TA-rich sequence in intron 2 were observed in Cryga, but none that were plausible candidates for the No3 mutation. For Crygf, three sequence differences with C57BL/6 were observed in exon 3, although all three were identical with the 129SV/j genomic sequence. Two of these were silent, but the third resulted in a predicted difference of G165S compared with the amino acid sequence of C57BL/6. This also appeared to be a strain related polymorphism that might contribute to background susceptibility but was not thought to be a likely candidate for the No3 mutation. γA and γF coding sequences were confirmed by RT-PCR of No3 eye RNA. Again, no product was obtained for γE using sequence specific primers.
The Cryge gene was then amplified in sections. Exons 1 and 2 and flanking sequences were amplified in one piece and sequenced (figure 3). Sections of intron 2 and 3′ flanking sequence were also amplified and sequenced. One apparent polymorphism was identified in exon 2 that would lead to an A>T amino acid change at position 51 in the mature protein (figure 4c) relative the C57BL/6 sequence, however T at this position has also been observed in γE sequences from other strains (for example [18])and the A/T polymorphism appears in both γE and γF of different strains in GenBank.
Figure 3. PCR and Gene Walking strategy for Cryge in No3.
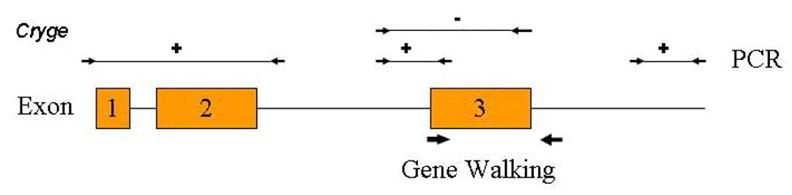
Schematic of wild type Cryge structure with exons shown as boxes (not to scale). Small arrow pairs indicate regions amplified directly from genomic DNA, with +/− indicating success or failure. Thick arrows indicate the regions examined by Gene Walking using nested gene specific and degenerate primers.
Figure 4. Retroviral insertion in Cryge.
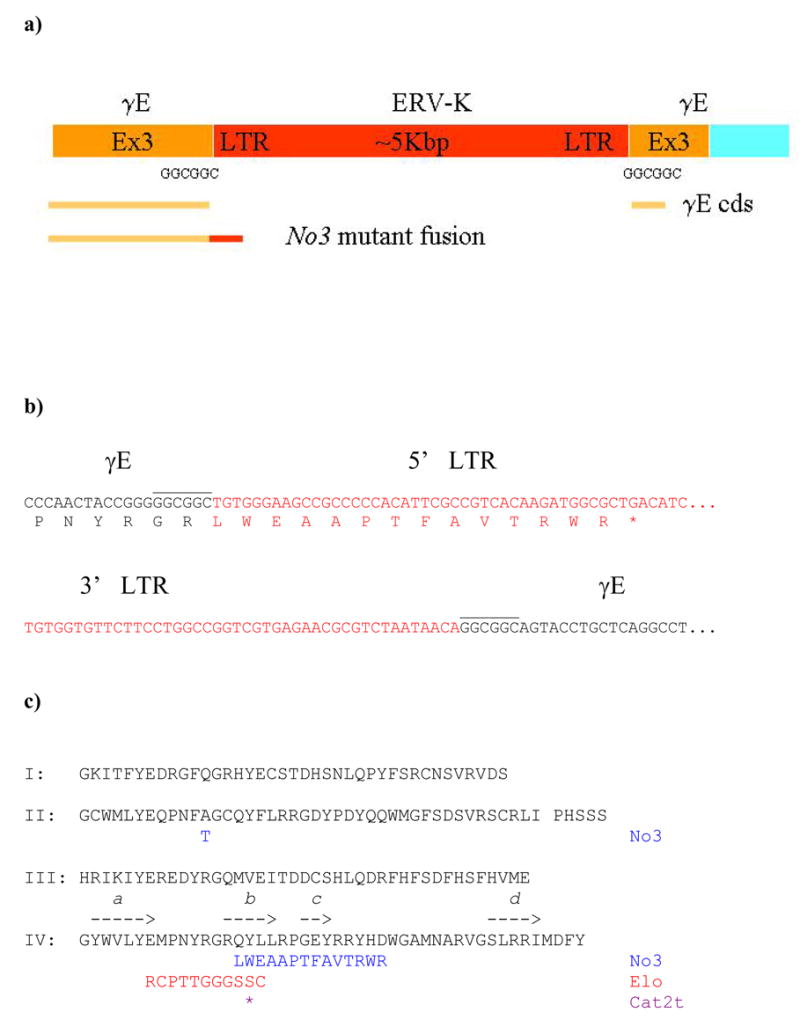
a) Schematic of exon 3 of Cryge in No3. The interrupted ORF sequence of exon 3 is shown in yellow (3′ UTR in blue) and the ERV-K insert in red (not to scale). The direct repeat GGCGGC sequences are indicated. Yellow lines indicate the (broken) ORF of exon 3 and below that, the yellow and red line shows the ORF of the read through transcript into the LTR.
b) Sequence at the 5′ (above) and 3′ (below) ends of the ERV-K insertion. Sequence for Cryge exon 3 is in black and LTR sequence is in red. The read through ORF producing the mutant γE sequence is shown translated below the 5′ sequence.
c) Sequence comparison of C57BL/6 wild type, No3, Elo and Cat2tγE crystallins. The complete wild type sequence is shown arranged in four lines corresponding to the four structural motifs (I 1–39; II 40–81; Connecting peptide between domains 82–86; III 87–127; IV 128–173). Above the sequence for the fourth motif, arrows show the positions of the four β-strands. Below the wild type sequence are shown the C-terminal truncations of the different mutants. Also shown is the A51T polymorphism in the second motif of γE from No3.
No PCR product was obtained for Cryge exon 3. This suggested the presence of a large deletion, insertion or rearrangement in exon 3. To map the unknown sequence of exon 3, gene walking was used. This is a procedure that uses known, specific primers (in this case taken from known intron 2 and 3′ flanking sequence) in successive amplifications with overlapping degenerate primers [19]. Products were successfully obtained for both sense and antisense directions, mapping the site of the disruption in exon 3 sequence. Sequence analysis showed that exon 3 was interrupted by a murine endogenous retrovirus (ERV-K), with intact LTR sequences at both ends and a duplicated GGCGGC hexamer at the insertion site (figure 4a).
The complete ERV-K insertion was cloned by PCR from No3 genomic DNA and sequenced (GenBank Accession DQ864756). The insertion is a full-length ERV-K element, 5kbp in length, flanked by identical LTRs of 354bp and containing a full ORF for a gag/pol gene. The closest matches of this new element in the C57BL/6 genome are two ERV-K elements on mouse chromosome 16. Either of these may be the progenitor of the new element, or it could be derived from a different element in the C3H/HeH strain. Genotyping for wild type and No3Cryge in No3/No3, No3/+ and +/+ littermates (from No3/+ matings), from parental C3H and 102 strains (from both MRC and IMG colonies) and from FVBN mice showed that this is a de novo mutation in No3 (figure 5a).
Figure 5. The ERV insertion is a de novo mutation in No3.
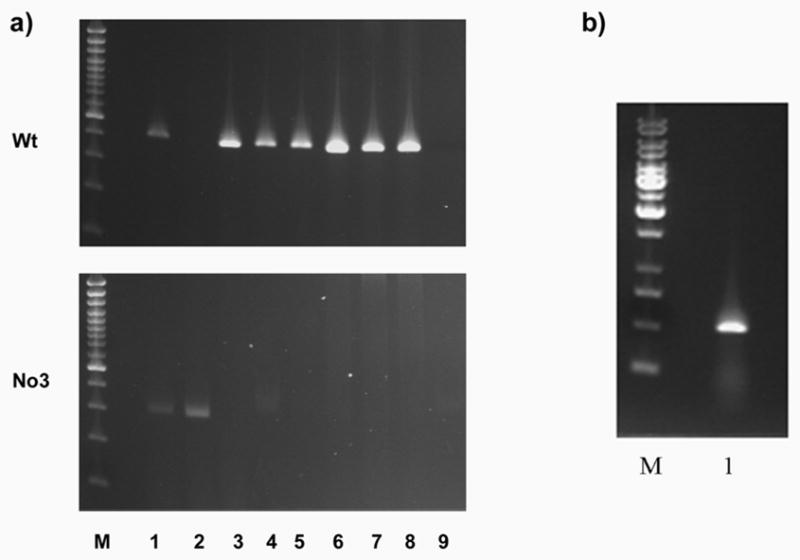
a) Genomic PCR of Wt Cryge third exon (upper panel) and No3 ERV insertion (lower panel) from No3 and other mouse strains. M: size markers; 1:No3/+; 2: No3/No3; 3: +/+ littermate (from No3/+ mating); 4: C3H (IMG, Germany); 5: 102 (IMG, Germany); 6: 102 (MRC, UK); 7: C3H (MRC, UK); 8: FVBN; 9: No DNA control. The Wt allele is missing only from No3/No3 and the mutant allele is present only in No3/No3 and No3/+.
b) The mutant γE-No3 transcript is expressed. RT-PCR of No3 eye RNA with intron crossing primers for the complete mutant coding sequence.
The retroviral insertion occurs after codon 142 of the ORF (figure 4b, c). Read through into LTR sequence provides 14 more in frame codons before a stop codon. γcrystallins consist of two domains, each made from two repeated structural motifs [20; 21]. In No3, γE has a normal N-terminal domain and a normal third motif. However, the fourth motif is truncated and would not be able to complete the C-terminal domain. This would prevent normal formation of intra-domain β-sheets and could also expose hydrophobic core residues of the C-terminal domain. RT-PCR from No3 eye RNA amplified the complete coding sequence (crossing 2 intron positions), demonstrating that the mutant mRNA is expressed (fig 6a).
Figure 6. Mutant γE-No3 is insoluble.
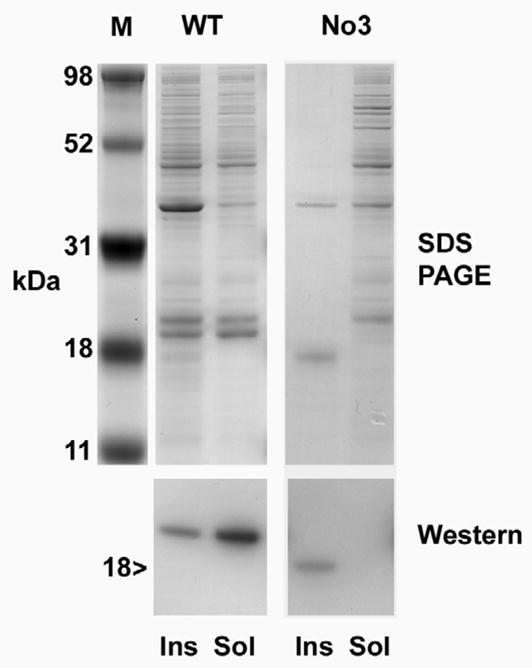
Expression of recombinant WT γE and γE-No3. Upper panel shows SDS PAGE, lower panel shows Western blot using antiserum to γ-crystallin. M: Size markers. Ins and Sol designate insoluble and soluble fractions of whole cell lysate from E.coli expressing either WT or No3 recombinant protein. Recombinant γE-No3 is found only in the insoluble fraction.
Recombinant protein
To examine the effects of the mutation on the protein, the complete γE-No3 coding sequence was amplified from RT-PCR product and cloned into pET31b for expression in E.coli (fig. 6a, b). The WT coding sequence of γE from FVBN lens (with the 51T polymorphism common to No3) was also cloned and expressed. For the WT sequence, much of the recombinant protein was found in the soluble fraction, while some was also retained in inclusion bodies, as is typical for γ-crystallin expression in this system (figure 6b). In contrast, although γE-No3 expressed well, western blotting showed that all of the expressed protein was in the insoluble fraction (fig. 6b). The identity of the insoluble band was confirmed by mass spectroscopy of tryptic digests which identified peptides covering more than half of the protein sequence for the mutant γE and by electrospray mass spectroscopy which identified a major species with relative mass 18806 Da, in agreement with the predicted size for the mature mutant protein. Attempts were made to denature and refold the insoluble protein but no soluble product was obtained.
Examination of γE-No3 transcript levels
RT-PCR was used to give a semi-quantitative comparison of expression levels for γA and γE-crystallin mRNAs in eyes of No3/No3, No3/+ and +/+ mice (from No3/+ x No3/+ matings) (figure 7a). Levels for γA were similar in all three genotypes. Levels for γE appeared lower in No3/No3 mice. Furthermore, although the PCR primers were designed to exploit the limited sequence differences between γE and γF, it was not possible to verify that only γE was being amplified.
Figure 7. Suppression of γE/F transcript levels in No3.
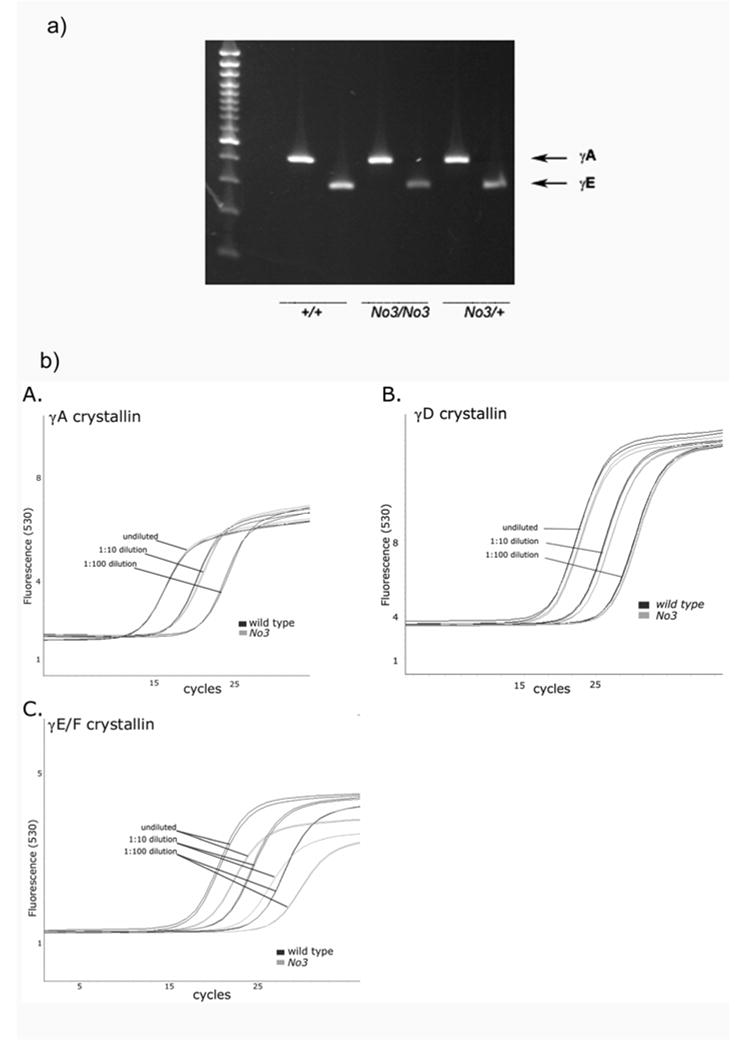
a) RT-PCR of transcripts for γA (upper band) and γE (lower) crystallins in +/+, No3/No3 and No3/+ lenses (from No3/+ x No3/+ matings).
b) QPCR of transcripts for γA, γD and γE/F crystallins in No3/No3 and +/+ lenses A: Tc curves for amplification of (A) γA, (B) γD and (C) γE/F RNA from wild type and No3/No3 lens at three dilutions of template. The shift in cycle number of the crossing point for the No3 samples indicates a reduction in target γE/F RNA levels.
To address this issue more quantitatively, quantitative RT-PCR (Q-PCR) was used (figure 7b). It was determined that transcripts for both γA and γD were present in equivalent amounts in wild type and No3/No3 mice. Again, this method was unable to reliably distinguish between the very similar γE and γF transcripts. However, using γA as a reference, Q-PCR showed that the sum of combined γE and γF transcripts detected in No3/No3 eye was reduced to approximately 50% of the wild type levels. This suggests that there is a substantial drop in the level of γE mRNA in No3.
Discussion
Several models of inherited cataract in mice have been created by chemical or radiation mutagenesis [3; 22; 23]. Many of these are associated with single base changes or small insertion/deletions that modify the coding sequence of one of the crystallins, the major soluble proteins of the eye lens. No3 is a nuclear cataract observed in a chemical mutagenesis study. The causative mutation was localized to the region of mouse chromosome 1 that contains the genes for βA2-crystallin and the cluster of six genes for the γA-F-crystallins. Since mutations in murine γ-crystallin genes often produce severe phenotypes, this raised the interesting possibility that No3 could be the first example of a mutation in βA2-crystallin. However, this proved not to be the case and, surprisingly, considering the reported severity of similar mutations [6; 7], No3 was found to be associated with a fourth motif truncation mutation of γE-crystallin. In the generally similar Elo and Cat2t truncation mutants, lens fiber cell necrosis and microophthalmia have been reported [6; 7], while the No3/No3 lens appears superficially normal in organization and only exhibits a nuclear opacity. Furthermore, the severity of this opacity is variable and strongly strain-dependent.
It was also surprising to find that the cause of the mutation was a probably spontaneous insertion of a full-length murine endogenous retrovirus, rather than chemical mutagenesis. Endogenous retroviruses (ERV) make up a large fraction of the mammalian genome [24; 25]. While the majority of the ERV elements are incomplete and apparently inactive, some full-length copies exist and may give rise to daughter elements that can insert elsewhere in the genome. In mice, the major class of actively transposing ERV elements is known as intracisternal A particles (IAP), and those with long terminal repeats also carry the designation for LTR. The frequency of IAP/ERV insertion varies with mouse strain; the most susceptible strain being C3H/He [24]. The 5kbp insertion in the Cryge gene in No3 belongs to the ERV-K class of IAPLTR1 transposons [24; 25; 26], with a perfectly conserved 354bp LTR at both ends and a characteristic direct repeat (GGCGGC) at the insertion site. Genotyping confirmed that this insertion is unique to No3 and was not present in parental strains.
The ERV insertion into exon 3 of Cryge disrupts the coding sequence of γE-crystallin approximately half way through the fourth and final “Greek-key” like motif of the protein at codon 142, after the first β-strand of the motif but eliminating the final three β-strands needed to complete the structure of the C-terminal domain [20; 21] (figure 4). The open reading frame continues into the ERV LTR sequence for a further 14 codons. Thus the γE polypeptide in No3 is longer than the truncated mutants of Elo and Cat2t (which are also fourth motif truncations of γE) and is potentially able to form an additional β-strand. The No3 mutant protein would not be able fold correctly and would be expected to be entirely insoluble. However, since the severity of the No3 cataract was so different from that reported for Elo and Cat2t, we considered the remote possibility that the apparent differences in the effects of the mutant proteins might be due to altered folding, or even formation of multimers, that might permit at least partial solubility for No3.
To test the solubility issue, recombinant γE-No3 was expressed in E.coli. The protein expressed well, but, in contrast to wild type FVBN γE, was undetectable in the soluble fraction. Attempts were made to denature and refold the insoluble protein but no solubilization was achieved. This suggests that the No3 mutant γE is indeed completely insoluble and no more functional in the lens than its counterparts in the other cataract models. There is still the possibility that the mild phenotype in No3 is due to a difference in the aggregation properties of the mutant γE (perhaps a reduced tendency for amyloid formation). However other more likely explanations for the mild phenotype are available.
Quantitation of transcript levels in No3 shows that expression of the mutant Cryge gene is suppressed. Retroposons, particularly those with LTR sequences can suppress expression of nearby genes, either transcriptionally or post-transcriptionally [24; 25]. Quantitative RT-PCR of crystallin gene transcripts from +/+ and No3/No3 littermates showed that levels of mRNA for γA and γD were similar but that the combined level of mRNA for the highly similar γE and γF-crystallins (indistinguishable in this assay) was significantly reduced. Since Crygf (the γF gene) is normal in sequence and is also the most distantly located member of the γ-crystallin gene cluster from the mutated Cryge (figure 2), it is unlikely that its level of mRNA is significantly affected. This suggests that the level of mRNA for γE is very low in the No3/No3 lens. This may act to offset the severity of the No3 mutation at the protein level by reducing the amount of insoluble, aggregated protein in the lens fiber cells.
Opacity due to No3 may be the result of insolubilization of the mutant protein and/or to loss-of-function of γE-crystallin. Indeed, a possible functional interaction for γE-crystallin with the major lens membrane protein MIP/APQ0 has recently been reported [27]. No3/No3 mice may be close to a “knock out” or “knock down” of γE that could be used to further investigate its role and to shed some light on the functional basis for the loss of γE expression as part of a suite of molecular changes in the evolution of the human lens [28; 29].
The severity of cataract associated with the No3 mutation is also affected by differences in genetic background. This is illustrated by the incomplete penetrance of No3 in C57BL/6 matings, which suggests that another genetic variant in the C3H/HeH strain is required for formation of an obvious opacity. In other words the No3 mutation alone is not sufficient for severe opacification of the lens but combines with some modifier that itself does not produce cataract in isolation. For example, it is possible that a coding sequence polymorphism exists in C3H/HeH that compromises the solubility or function of another lens protein but is ameliorated by normal levels of wild type γE, perhaps by direct protein-protein interaction. Loss of γE, whether by insolubilization or suppression, might then lead to loss of function or increased insolubilization of the partner protein, increasing the severity of opacity due to mutant γE alone. In strains that lack the modifier, such as C57Bl/6, the effects of the No3 mutation on γE would have a milder phenotype than in the C3H/HeH parental strain.
One possible modifier for lens phenotypes has recently been observed in mice of the 129 strain families. This involves a truncation and loss of the major lens cytoskeleton protein Bfsp2/CP49 [30]. No3/No3 mice on the original background were genotyped for the known CP49 mutation but only wild type alleles were observed, suggesting that at least one other genetic variant capable of modifying lens function or severity of cataract exists in laboratory mouse strains and awaits discovery.
Many simple mutations in crystallins have been associated with severe inherited cataract in both humans and mice. However, these results show that in some cases the crystallin mutation may be just the final step in a multifactorial process, with the phenotype and severity of the cataract dependent also on genetic background and environment. Such a multi-step progression towards cataract may be similar to what happens in human age related cataract, as accumulating post-translational modifications in the aging lens interact with polymorphic variant sequences, eventually producing opacity that is dependent upon both genetic and lifestyle differences.
Materials and Methods
Animals
The original animal was found at the Institute of Mammalian Genetics, Neuherberg, Germany, among the offspring in an experiment in which male mice of genotype (C3H/He x 102/El)F1 were treated with the mutagen iso-propyl methane sulphonate, and their offspring were scored for eye defects [17]. The mutant was transferred to the Medical Research Council, Harwell, Oxfordshire, UK, and given the name “nuclear opacity 3” and gene symbol No3. All studies were performed under the guidance issued by the Medical Research Council and Home Office licenses 30/1517 and 30/2049.
Phenotype Screen
Mice aged 4–6 weeks were scored for cataracts by slit-lamp examination (Zeiss 30SL/M) at 20x magnification with illumination at a 25–30° angle from the direction of observation. Dilatation of the pupil was achieved with 1% atropine sulphate (Schering-Plough) applied at least 10 minutes before examination.
Mapping
No3/+ mice were crossed to C57BL/6 and affected offspring were backcrossed to C3H/HeH. Backcross offspring were scored for cataract, and were then typed for microsatellite markers spaced at ~20cM intervals throughout the genome. When linkage was found the same animals were typed for additional markers in the region of interest. DNA was extracted from tail tissue and PCR conditions were as described [31].
Expressed Sequence Tag Analysis
Twenty eyes from adult No3/No3 mice were extracted with RNAzol (Tel-Test Inc., Friendswood, TX), yielding 1133μg of total RNA. 11.3μg of twice selected polyA+ RNA cDNA was prepared by oligo(dT) cellulose affinity chromatography of which 5μg was used to synthesize cDNA using the Superscript II system (Invitrogen, Carlsbad CA). The cDNA was cloned into the Sal I-Not I sites of the pCMVSport6 vector (Invitrogen), essentially as described before [32; 33]. The unamplified library was cloned as two sublibraries, designated ob and oc, that were combined for subsequent sequence analysis. Library construction was carried out at Bioserve Biotechnologies (Laurel, MD).
Clones were randomly selected for sequencing at the NIH Intramural Sequencing Center, as described before [32]. Just over 2800 clones were sequenced, with average read length of 597nt. Sequence data from the expressed sequence tag (EST) analysis of all libraries were processed for quality and to remove vector and other non-cDNA sequences as described previously [34]. Insert sequences were analyzed using GRIST [35] to group and identify sequences. 2500 high quality reads were obtained after trimming and removal of mitochondrial and other contaminant sequences. Data are available through GenBank and at http://neibank.nei.nih.gov/cgi-bin/showDataTable.cgi?lib=NbLib0074.
PCR methods
Primers were designed from C57BL/6 genome sequence to amplify specific candidate genes from genomic DNA from No3/No3 and wild type FVBN mice using the Fast Start High Fidelity PCR system (Roche, Indianapolis IN). Primers for Cryga: MGA1: ACACTGACCATTTGCTGTCAACAAC; MGA2: GATTATTTATTGCATATATGGGCTG. For Cryge and Crygf, a common sense primer: MGEDF1: TCCCATCCGACCTGCCAACACCAGC; and specific antisense primers: MGE2: TTATTACTGTCCAGATGGAGAAAAT; MGF2: ATTAACTTCCAAATGATGAAAGGGC; were used. Several more primer sets were then used to amplify exons 1 and 2 of Cryge from No3, intron 2 sequences and 3′ flanking sequences.
The DNA Walking SpeedUp Premix Kit (Seegene Inc., Seoul Korea) was used to amplify unknown sequence in exon 3 from both 5′ and 3′ sides using gene specific primers and degenerate primers from the kit. Nested 5′ primers: MGEI2AS.1500: CCCTCTAGATTCTTGGTAATCCGAG; MGEX3.S2: AGCTCAGGTTTTCTGACGTCCTGCTG; MGEX3.S: TCTGACGTCCTGCTGTTCTCTGGAC. Nested 3′ primers: NAS173311: TCGTCAGGTCGGGATTAATACCTCTC; NAS173264 : GGGCACAGAAATGATAAGTGACATCAATC; NAS173234: ATCGCCTGAAGCTAGTGACCCTGGCCAC.
Primers MGEX3.S and NAS173234 were also used to amplify the complete ERV insertion using the Expand Long range PCR kit (Roche) for subsequent cloning using the Expand Cloning kit (Roche). To produce a subclone lacking LTR sequence primers LTR1: GTGGTGTTCTTCCTGGCCGGTCGTGAG; LTR2: TCTGCGGCAAAGCTTTATTCTTACATC; were used.
Genotyping for No3/No3 was performed using MGEX3.S and GENXho1: CTCGAGTCATGCGCAGATTATTTTGTTTAC. The wild type allele was genotyped using MGEX3.S and WTEX3AS: GCCCTTCAATTGAGGGTGAAAGGAACAG. Mice were also genotyped for a known Cp49 mutation found in mouse 129 strains as described [30].
Genomic DNA sequence
PCR derived fragments were sequenced using the Beckman CEQ 2000 system (Beckman Coulter, Fullerton CA). The ERV insertion was partially sequenced in house and completed with primer walking at Agencourt (Beverly MA).
Recombinant protein
The coding sequence of No3 mutant mouse γE was cloned by PCR from cDNA template generated by RT-PCR from No3 eye RNA using primers GEN1: CATATGGGGAAGATCACCTTCTATGAGG and GENXho1: CTCGAGTCATGCGCAGATTATTTTGTTTAC. WT was cloned in a similar, substituting GEWTX: CTCGAGTCAATAGAAATCCATGATTCTCCT as 3′ Xho-containing primer. Clones were confirmed by sequencing and cloned into Nde I/Xho I sites of pET 31b (Novagen). Each plasmid was transformed into E.coli BL21(DE3) pLysS (Novagen). Induction yielded high expression of both proteins.
For protein isolation, the pellet was thawed, sonicated and centrifuged at 15000g for 20 minutes at 4°C. Soluble and insoluble fractions were examined by SDS PAGE and western blotting. The antibody used was a polyclonal against purified recombinant human γD-crystallin [9] raised in rabbits at Spring Valley Labs (Woodbine, MD).
Mass Spectroscopy
The recombinant protein was also confirmed by mass spectroscopy. For tryptic peptide mapping, whole cell lysate, ~50 μg, was separated by SDS-PAGE. The over-expressed recombinant protein band was excised from the stained gel and digested with trypsin (1:100) (Promega) according to manufacturer’s protocol. Peptides were extracted, reduced, alkylated and dried. Extracts were resuspended, applied to zip-tip columns (Millipore), spotted in the presence of cyano-hydroxycinnamic acid (Sigma) and analyzed using MALDI (Voyager, Applied Biosystems) in reflector mode, delayed extraction mode with 68% grid voltage and a 700 nsec delay.
Protein was also sized using Electrospray liquid chromatography – mass spectrometry. The LC-MS system consisted of an Agilent (Palo Alto, CA) binary pump, degasser, autosampler, and an HP1100 Mass Selective Detector (MSD). Data were acquired on the HP ChemStation data system. The instrument was calibrated with a standard Agilent ES tuning mix. The mass spectrometer was scanned from m/z 500 to 2000 every four seconds. Nitrogen was used to assist nebulization and desolvation. Solvent A of the mobile phase consisted of 5% (v/v) acetic acid, solvent B consisted of 100% acetonitrile. After an initial wash phase of 25 min at 5% solvent B, chromatographic separation was done using a gradient from 5 to 95 % solvent B within 25 min at 40°C on a Zorbax SB-C3 reversed phase column (150 mm × 2.1 mm ID) and a C3 guard column at a flow rate of 0.2 ml/min. The injection volume was 100 μL equaling approximately 25 pmol of protein.
Refolding
Inclusion bodies containing γE-No3 were purified from E. coli whole cell lysate by centrifugation. The recombinant protein was dissolved in guanidinium, 8M and DTT 4 mM. The dissolved protein was then added drop-wise to refolding buffer: Tris100mM, L-arginine 400 mM, EDTA 3 mM, oxidized glutathione 0.5 mM and reduced glutathione, 5 mM. The suspension was gently stirred overnight at 4 C, dialyzed in Tris, 50 mM.
Q-PCR
The mRNA from equal quantities of wild type and No3 mouse total RNA was converted to cDNA via reverse transcription as per protocol using the Roche Transcriptor First Strand cDNA Synthesis Kit. Ten-fold dilution series of the cDNAs were made for four orders of magnitude.
Quantitative PCR was performed using a Roche LightCycler and Universal ProbeLibrary hybridization probes (Roche). Using the ProbeFinder software from Roche (available at the Assay Design Center, http://www.universalprobelibrary.com) we designed primer and probe pairs to assay transcripts from Cryga, Crygd, Cryge, Crygf, and also from Alas and Gapdh genes (serving as low and high expressing “housekeeping” genes). Due to the high degree of sequence similarity, the assays could not distinguish between Cryge and Crygf. Amplification and detection of the transcripts was performed following the protocols of the LightCycler TaqMan Master Reagent kit (Roche).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wistow G, Piatigorsky J. Lens crystallins: evolution and expression of proteins for a highly specialized tissue. Ann Rev Biochem. 1988;57:479–504. doi: 10.1146/annurev.bi.57.070188.002403. [DOI] [PubMed] [Google Scholar]
- 2.Harding JJ, Crabbe MJC. The lens: Development, proteins, metabolism and cataract. In: Davson H, editor. The Eye. Academic Press; New York: 1984. pp. 207–492. [Google Scholar]
- 3.Graw J. Congenital hereditary cataracts. Int J Dev Biol. 2004;48:1031–44. doi: 10.1387/ijdb.041854jg. [DOI] [PubMed] [Google Scholar]
- 4.Hejtmancik JF, Smaoui N. Molecular genetics of cataract. Dev Ophthalmol. 2003;37:67–82. doi: 10.1159/000072039. [DOI] [PubMed] [Google Scholar]
- 5.Sandilands A, Hutcheson AM, Long HA, Prescott AR, Vrensen G, Loster J, Klopp N, Lutz RB, Graw J, Masaki S, Dobson CM, MacPhee CE, Quinlan RA. Altered aggregation properties of mutant gamma-crystallins cause inherited cataract. Embo J. 2002;21:6005–14. doi: 10.1093/emboj/cdf609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cartier M, Breitman ML, Tsui LC. A frameshift mutation in the gammaE-crystallin gene of the Elo mouse. Nature Genetics. 1992;2:42–45. doi: 10.1038/ng0992-42. [DOI] [PubMed] [Google Scholar]
- 7.Klopp N, Favor J, Loster J, Lutz RB, Neuhauser-Klaus A, Prescott A, Pretsch W, Quinlan RA, Sandilands A, Vrensen GF, Graw J. Three murine cataract mutants (Cat2) are defective in different gamma-crystallin genes. Genomics. 1998;52:152–158. doi: 10.1006/geno.1998.5417. [DOI] [PubMed] [Google Scholar]
- 8.Sinha D, Wyatt MK, Sarra R, Jaworski C, Slingsby C, Thaung C, Pannell L, Robison WG, Favor J, Lyon M, Wistow G. A temperature-sensitive mutation of Crygs in the murine Opj cataract. J Biol Chem. 2001;276:9308–15. doi: 10.1074/jbc.M010583200. [DOI] [PubMed] [Google Scholar]
- 9.Evans P, Wyatt K, Wistow GJ, Bateman OA, Wallace BA, Slingsby C. The P23T Cataract Mutation Causes Loss of Solubility of Folded gammaD-Crystallin. J Mol Biol. 2004;343:435–44. doi: 10.1016/j.jmb.2004.08.050. [DOI] [PubMed] [Google Scholar]
- 10.Mackay DS, Andley UP, Shiels A. A missense mutation in the gammaD crystallin gene (CRYGD) associated with autosomal dominant “coral-like” cataract linked to chromosome 2q. Mol Vis. 2004;10:155–62. [PubMed] [Google Scholar]
- 11.Nandrot E, Slingsby C, Basak A, Cherif-Chefchaouni M, Benazzouz B, Hajaji Y, Boutayeb S, Gribouval O, Arbogast L, Berraho A, Abitbol M, Hilal L. Gamma-D crystallin gene (CRYGD) mutation causes autosomal dominant congenital cerulean cataracts. J Med Genet. 2003;40:262–7. doi: 10.1136/jmg.40.4.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shentu X, Yao K, Xu W, Zheng S, Hu S, Gong X. Special fasciculiform cataract caused by a mutation in the gammaD-crystallin gene. Mol Vis. 2004;10:233–9. [PubMed] [Google Scholar]
- 13.Xu WZ, Zheng S, Xu SJ, Huang W, Yao K, Zhang SZ. Localization and screening of autosomal dominant coralliform cataract associated gene. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2004;21:19–22. [PubMed] [Google Scholar]
- 14.Stephan DA, Gillanders E, Vanderveen D, Freas-Lutz D, Wistow G, Baxevanis AD, Robbins CM, VanAuken A, Quesenberry MI, Bailey-Wilson J, Juo SH, Trent JM, Smith L, Brownstein MJ. Progressive juvenile-onset punctate cataracts caused by mutation of the gammaD-crystallin gene. Proc Natl Acad Sci U S A. 1999;96:1008–12. doi: 10.1073/pnas.96.3.1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Benedek GB, Pande J, Thurston GM, Clark JI. Theoretical and experimental basis for the inhibition of cataract. Prog Retin Eye Res. 1999;18:391–402. doi: 10.1016/s1350-9462(98)00023-8. [DOI] [PubMed] [Google Scholar]
- 16.Clark JI, Clark JM. Lens cytoplasmic phase separation. Int Rev Cytol. 2000;192:171–87. doi: 10.1016/s0074-7696(08)60526-4. [DOI] [PubMed] [Google Scholar]
- 17.Favor J, Neuhauser-Klaus A. Saturation mutagenesis for dominant eye morphological defects in the mouse Mus musculus. Mamm Genome. 2000;11:520–5. doi: 10.1007/s003350010099. [DOI] [PubMed] [Google Scholar]
- 18.Graw J, Coban L, Liebstein A, Werner T. Murine gamma E-crystallin is distinct from murine gamma 2-crystallin. Gene. 1991;104:265–70. doi: 10.1016/0378-1119(91)90260-i. [DOI] [PubMed] [Google Scholar]
- 19.Hwang IT, Kim YJ, Kim SH, Kwak CI, Gu YY, Chun JY. Annealing control primer system for improving specificity of PCR amplification. Biotechniques. 2003;35:1180–4. doi: 10.2144/03356st03. [DOI] [PubMed] [Google Scholar]
- 20.Blundell T, Lindley P, Miller L, Moss D, Slingsby C, Tickle I, Turnell B, Wistow G. The molecular structure and stability of the eye lens: x-ray analysis of gamma-crystallin II. Nature. 1981;289:771–7. doi: 10.1038/289771a0. [DOI] [PubMed] [Google Scholar]
- 21.Wistow G, Turnell B, Summers L, Slingsby C, Moss D, Miller L, Lindley P, Blundell T. X-ray analysis of the eye lens protein gamma-II crystallin at 1.9 A resolution. J Mol Biol. 1983;170:175–202. doi: 10.1016/s0022-2836(83)80232-0. [DOI] [PubMed] [Google Scholar]
- 22.Everett CA, Glenister PH, Taylor DM, Lyon MF, Kratochvilova-Loester J, Favor J. Mapping of six dominant cataract genes in the mouse. Genomics. 1994;20:429–434. doi: 10.1006/geno.1994.1197. [DOI] [PubMed] [Google Scholar]
- 23.Graw J, Neuhauser-Klaus A, Klopp N, Selby PB, Loster J, Favor J. Genetic and allelic heterogeneity of Cryg mutations in eight distinct forms of dominant cataract in the mouse. Invest Ophthalmol Vis Sci. 2004;45:1202–13. doi: 10.1167/iovs.03-0811. [DOI] [PubMed] [Google Scholar]
- 24.Maksakova IA, Romanish MT, Gagnier L, Dunn CA, van de Lagemaat LN, Mager DL. Retroviral elements and their hosts: insertional mutagenesis in the mouse germ line. PLoS Genet. 2006;2:e2. doi: 10.1371/journal.pgen.0020002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Druker R, Whitelaw E. Retrotransposon-derived elements in the mammalian genome: a potential source of disease. J Inherit Metab Dis. 2004;27:319–30. doi: 10.1023/B:BOLI.0000031096.81518.66. [DOI] [PubMed] [Google Scholar]
- 26.Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 2005;110:462–7. doi: 10.1159/000084979. [DOI] [PubMed] [Google Scholar]
- 27.Fan J, Fariss RN, Purkiss AG, Slingsby C, Sandilands A, Quinlan R, Wistow G, Chepelinsky AB. Specific interaction between lens MIP/Aquaporin-0 and two members of the gamma-crystallin family. Mol Vis. 2005;11:76–87. [PubMed] [Google Scholar]
- 28.Brakenhoff RH, Aarts HJ, Reek FH, Lubsen NH, Schoenmakers JG. Human gamma-crystallin genes. A gene family on its way to extinction. Journal of Molecular Biology. 1990;216:519–532. doi: 10.1016/0022-2836(90)90380-5. [DOI] [PubMed] [Google Scholar]
- 29.Wistow G. The NEIBank project for ocular genomics: data-mining gene expression in human and rodent eye tissues. Prog Retin Eye Res. 2006;25:43–77. doi: 10.1016/j.preteyeres.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 30.Sandilands A, Wang X, Hutcheson AM, James J, Prescott AR, Wegener A, Pekny M, Gong X, Quinlan RA. Bfsp2 mutation found in mouse 129 strains causes the loss of CP49′ and induces vimentin-dependent changes in the lens fibre cell cytoskeleton. Exp Eye Res. 2004;78:875–89. doi: 10.1016/j.exer.2003.09.028. [DOI] [PubMed] [Google Scholar]
- 31.Arkell RM, Cadman M, Marsland T, Southwell A, Thaung C, Davies JR, Clay T, Beechey CV, Evans EP, Strivens MA, Brown SD, Denny P. Genetic, physical, and phenotypic characterization of the Del(13)Svea36H mouse. Mamm Genome. 2001;12:687–94. doi: 10.1007/s00335-001-2066-2. [DOI] [PubMed] [Google Scholar]
- 32.Wistow G, Bernstein SL, Wyatt MK, Behal A, Touchman JW, Bouffard G, Smith D, Peterson K. Expressed sequence tag analysis of adult human lens for the NEIBank Project: over 2000 non-redundant transcripts, novel genes and splice variants. Mol Vis. 2002;8:171–84. [PubMed] [Google Scholar]
- 33.Wistow G, Bernstein SL, Wyatt MK, Ray S, Behal A, Touchman JW, Bouffard G, Smith D, Peterson K. Expressed sequence tag analysis of human retina for the NEIBank Project: retbindin, an abundant, novel retinal cDNA and alternative splicing of other retina-preferred gene transcripts. Mol Vis. 2002;8:196–204. [PubMed] [Google Scholar]
- 34.Bouffard GG, Iyer LM, Idol JR, Braden VV, Cunningham AF, Weintraub LA, Mohr-Tidwell RM, Peluso DC, Fulton RS, Leckie MP, Green ED. A collection of 1814 human chromosome 7-specific STSs. Genome Res. 1997;7:59–64. doi: 10.1101/gr.7.1.59. [DOI] [PubMed] [Google Scholar]
- 35.Wistow G, Bernstein SL, Touchman JW, Bouffard G, Wyatt MK, Peterson K, Behal A, Gao J, Buchoff P, Smith D. Grouping and identification of sequence tags (GRIST): bioinformatics tools for the NEIBank database. Mol Vis. 2002;8:164–70. [PubMed] [Google Scholar]