

Abstract
The receptor for advanced glycation end products (RAGE) is a member of the immunoglobulin superfamily of cell surface proteins that has been implicated as a progression factor in a number of pathologic conditions from chronic inflammation to cancer to Alzheimer’s disease. In such conditions, RAGE acts to facilitate pathogenic processes. Its secreted isoform, soluble RAGE or sRAGE, has the ability to prevent RAGE signaling by acting as a decoy. sRAGE has been used successfully in animal models of a range of diseases to antagonize RAGE-mediated pathologic processes. In humans, sRAGE results from alternative splicing of RAGE mRNA. This study was aimed to determine whether the same holds true for mouse sRAGE and, in addition, to biochemically characterize mouse sRAGE. The biochemical characteristics examined include glycosylation and disulfide patterns. In addition, sRAGE was found to bind heparin, which may mediate its distribution in the extracellular matrix and cell surfaces of tissues. Finally, our data indicated that sRAGE in the mouse is likely produced by carboxyl-terminal truncation, in contrast to the alternative splicing mechanism reported in humans.
The receptor for advanced glycation end products (RAGE)1 is a member of the immunoglobulin superfamily of cell surface receptors (1). It is composed of three extracellular immunoglobulin-like domains, a single-pass transmembrane domain, and a short highly charged cytoplasmic domain that is essential for RAGE-mediated signaling (2). It binds multiple families of ligands, all of which can be associated with pathology, namely advanced glycation end products, S100/calgranulins, amphoterin/HMGB1, and amyloid fibrils (reviewed in Ref. 3). Upon ligand binding in humans, a cell type-specific signaling cascade is initiated, in most cases culminating in the activation of the immune/inflammatory-associated transcription factor NF-κB (2, 4–6). Notably, the human RAGE promoter contains two NF-κB-responsive elements (7) that act to form a positive feedback loop such that RAGE is up-regulated where its ligands are present. In effect, RAGE and its ligands show sustained colocalization. This is thought to explain the observation that RAGE is expressed at low levels in healthy adults, except at certain pathological sites where it is up-regulated, including diabetic vasculature, sites of chronic inflammation, Alzheimer’s brain tissue, and some tumors (4, 8–11).
RAGE has a secreted isoform called soluble RAGE or sRAGE. Because this isoform lacks a transmembrane domain, it is secreted and acts as a decoy receptor. Indeed, sRAGE has been administered in a number of cell culture and animal models of RAGE-mediated disorders where it successfully prevented or reversed RAGE signaling effects such as diabetic atherosclerosis and impaired wound healing (8, 12, 13), colitis (4), amyloid-β penetration of the blood-brain barrier (14), and tumor cell migration and invasion (9).
Although murine sRAGE has been used in many studies, it has not been fully characterized. We sought to biochemically characterize mouse sRAGE and describe the mechanism of its synthesis. Malherbe et al. (15) and Yonekura et al. (16) found that in humans, sRAGE is produced by alternative splicing of RAGE mRNA. The resultant frameshift occurs upstream of the transmembrane domain-encoding sequence, thus eliminating this domain and causing human RAGE and sRAGE to have distinct carboxyl-terminal amino acid sequences. Here, we report data that suggest that mouse sRAGE is produced by carboxyl-terminal truncation rather than alternative splicing. We describe the glycosylation and disulfide patterns of murine sRAGE and also report a novel purification strategy for sRAGE from mouse lungs that includes heparin affinity chromatography, a characteristic of sRAGE that could have implications on the extracellular localization of sRAGE.
EXPERIMENTAL PROCEDURES
sRAGE Purification from Mouse Lungs
Frozen mouse lungs from Pel-Freez Biologicals (Rogers, AZ) were used as starting material. Lungs were homogenized in homogenization buffer (50 mM potassium phosphate, pH 7.4, 0.3 M potassium bromide, 3 mM diethylenetriamine-pentacetic acid, 0.5 mM phenylmethylsulfonyl fluoride) (10 ml/g tissue) followed by brief sonication and centrifugation (20,000 × g, 20 min, 4 °C). Polyethyleneimine (0.01% final) (ICN Biomedicals, Aurora, OH) was added to the supernatant to precipitate nucleic acids (17), and then the homogenate was centrifuged again as above.
Concanavalin A Batch Extraction
Concanavalin A-Sepharose 4B (Sigma) (~100 ml) was added to the cleared homogenate and gently stirred overnight at 4 °C. Matrix with bound protein was collected using a scinted glass funnel and packed into a column (2.5 × 20 cm). The column was washed with HEPES buffer (50 mM HEPES, pH 7.0, 0.25 M sodium chloride) until A280 reached 0. Bound protein was batch eluted with 50 mM HEPES, pH 7.0, 0.25 M sodium chloride, 200 mM methyl α-mannopyranoside at a flow rate of 10 ml/min.
Heparin-Sepharose Chromatography
Bulk eluate from the concanavalin A batch extraction was dialyzed into Buffer A (50 mM Tris, pH 7.5, 50 mM sodium chloride) and concentrated to ~50 ml using an Amicon concentrator (10,000 Da cutoff). The sample was then applied to a column packed with Affi-Gel heparin (Bio-Rad) (~80-ml column material, 2.5 × 20-cm column) and washed with Buffer A until A280 reached 0. Heparin-binding proteins were eluted with a 0.6%/min gradient of Buffer B (50 mM Tris, pH 7.5, 1.0 M sodium chloride) into 130 fractions at a flow rate of 5 ml/min. Fractions containing sRAGE were identified by Western blotting using an antibody against mouse RAGE/sRAGE as previously described (18). sRAGE-containing fractions were pooled and dialyzed in Buffer A.
Anion-exchange Chromatography
Pooled fractions from heparin-Sepharose chromatography were concentrated to ~50 ml as detailed above and applied to a 1-ml Mono Q column (Amersham Biosciences). The column was washed with Buffer A until A280 reached 0. Proteins were eluted with Buffer B using the following elution profile: 0.5%/min Buffer B for the first 60 ml; 1%/min Buffer B for the next 30 ml; and, thereafter, 100% Buffer B at a flow rate of 1 ml/min. 95 fractions were collected, and sRAGE-containing fractions were identified as described above and pooled. All of the chromatography was performed on a Pharmacia fast protein liquid chromatography system.
Amino-terminal Sequencing
Amino-terminal sequencing by Edman degradation was done as described previously (19) using an Applied Biosystems Model 477A sequencer with on-line phenylthiodantoin analysis using an Applied Biosystems Model 120A HPLC system.
Removal of Amino-terminal Blocking Group of sRAGE
Purified sRAGE was deblocked as previously described (19) by incubation with Pfu pyroglutamate aminopeptidase (Takara Bio Inc., Shiga, Japan).
PCR and RT-PCR
While attempting to clone an alternatively spliced mouse RAGE cDNA that could encode sRAGE, RT-PCR or PCR was performed with mouse lung RNA isolated as previously described (20, 21) or a mouse lung cDNA library (Spring Bioscience, Fremont, CA). Fig. 1 shows the approximate sites along the mouse RAGE cDNA2 to which primers were targeted. RT-PCR was performed using EZ rTth RNA PCR kit (Applied Biosystems, Branchburn, NJ). Conditions were as follows: RT step at 60.0 °C for 30 min and 94.0 °C for 2 min; 40 cycles of 94.0 °C for 15 s, 60.0 °C for 30 s, and 70.0 °C for 1 min 30 s; and a final extension step at 65.0 °C for 7 min. PCR was performed using Taq DNA polymerase (Fisher Scientific) and PCR nucleotide mixture (Roche Applied Science). Cycling conditions were as follows: denaturation at 94 °C for 5 min; 35 cycles of 94 °C for 30 s, 58.0 °C for 30 s, and 72.0 °C for 2 min; and a final extension step at 72.0 °C for 8 min.
Fig. 1. Primers used in attempt to detect an alternatively spliced form of RAGE mRNA.
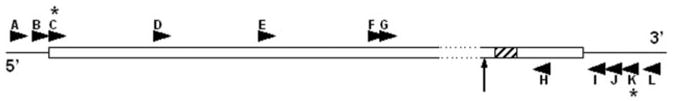
The mouse RAGE cDNA sequence is represented by the rectangle, whereas 5′- and 3′-untranslated regions are represented by horizontal lines (GenBank™ accession number AF030001.1 for nucleotides 74101–77630). Each arrowhead indicates the position of one primer sequence and the direction it was designed to amplify along the cDNA. Primer sequences are shown in Table I. The dashed outline represents the sequence homologous to that spliced out in the human sRAGE cDNA (15). The diagonally striped sequence encodes the transmembrane domain of RAGE. The vertical arrow indicates the position at which the frameshift occurs in human sRAGE. Thus, after this codon, the amino acid sequences of human RAGE and sRAGE became distinct from one another. Asterisks indicate the primers used to successfully amplify and clone full-length mouse RAGE.
Protease Digestion of sRAGE for Liquid Chromatography Tandem Mass Spectrometry (LC-MS/MS)
For each reaction, 5 μg of purified sRAGE was lyophilized and then reduced (30-min incubation at room temperature in the dark with 8 M urea and 5 mM dithiothreitol) and alkylated (30-min incubation in the dark at room temperature with 10 mM iodoacetamide). Protein was then diluted in 100 mM Tris, pH 7.5, to dilute the urea and incubated with 0.1 μg of trypsin (Promega, Madison, WI) or 0.1 μg of thermolysine (Sigma) with 2 mM calcium chloride or 0.1 μg of chymotrypsin (Sigma) for 4 h at 37 °C.
LC-MS/MS
The LC-MS/MS analyses were performed using a Micromass Q-TOF Ultima Global mass spectrometer (Micromass/Waters, Manchester, United Kingdom) connected to an LC-Packings UltiMate nanoLC system (LC-Packings, Amsterdam, The Netherlands). A nano-spray ion source was used to hold the packed PicoFrit™ columns (New Objective, Inc., Woburn, MA) and apply capillary voltage through a Valco union. The PicoFrit columns (75 μm, inner diameter × 10 cm) were packed with Zorbax SB C18 3.5-μm reverse phase column material (Agilent, Palo Alto, CA) using a high pressure column loader (Proxeon Biosystems, Odense, Denmark). The column was developed at a flow rate of 200 nl/min and linear gradients from 0.02% heptafluoro-butyric acid/0.5% acetic acid in water to 0.02% heptafluorobutyric acid/0.5% acetic acid in 75% acetonitrile/24.5% water. After data acquisition, the individual MS/MS spectra acquired for each of the precursors within a single LC run were combined, smoothed, deisotoped, and centroided using the Micromass Masslynx data processing software and output as a single Mascot-searchable peak list. The peak list files were used to query the Swiss-Prot data base using the Mascot program (22).
Matrix-assisted Laser Desorption Ionization MALDI-MS
Buffer salts were removed from sRAGE using zip-tips (Millipore Corporation, Danvers, MA), and MALDI-MS was performed using a Micromass Q-TOF Ultima Global mass spectrometer operated in MALDI-MS mode. Spectra were recorded in linear mode (mass determination) or reflector mode (glycosylation studies).
Disulfide Characterization
A tryptic map of mouse RAGE was generated by GPMAW (General Protein/Mass Analysis for Windows) software (Lighthouse data, Odense, Denmark). Approximately 400 μg of native purified sRAGE was incubated with 8 μg of trypsin overnight at room temperature. The reaction was then analyzed by SDS-PAGE to confirm reaction completion. After trypsinization of nonreduced sRAGE, the peptides were separated by gel filtration (Superdex peptide HR10/30 column (Amersham Biosciences)) in 20 mM sodium phosphate, pH 7.2, 250 mM sodium chloride. Each peak from gel filtration was further separated by reverse-phase (RP)-HPLC on an Aquapore RP-300 column (Solvent A, 0.1% trifluoroacetic acid; Solvent B, 90% acetonitrile, 0.08% trifluoroacetic acid; elution profile, 2% solvent B/min linear gradient at 200 μl/min). Both chromatography steps were performed using a Pharmacia SMART system (Amersham Biosciences). Peaks from RP-HPLC were subjected to 3–6 cycles of Edman degradation, and these residues were used to identify the corresponding peptide on the tryptic map. If two cysteine-containing peptides were identified from a single RP-HPLC peak with an ~1:1 intensity, this was taken as evidence of their linkage by a disulfide bond. For confirmation, each of these peaks was subjected to MALDI-MS to verify that the mass corresponded to the combined mass of the two peptides. Finally, protein from each peak was reduced with 5 mM dithiothreitol and again analyzed by MALDI-MS to see whether two distinct masses resulted after reduction. If so, this result would confirm that the two peptides are linked by a disulfide bond.
Deglycosylation of Mouse sRAGE
100 μg of purified sRAGE was boiled for 5 min in denaturation buffer (50 mM sodium phosphate, pH 7.5, 0.1% SDS, 50 mM 2-mercaptoethanol) and cooled. Triton X-100 was added (0.5% final concentration) followed by the addition of 9 units of PNGase F (Calbiochem), and the reaction was incubated at 37 °C for 3 h. For O-deglycosylation, 100 μg of sRAGE in 50 mM sodium acetate, pH 5.0, was incubated with 0.1 unit of neuraminidase (Roche Applied Science) at 37 °C for 18 h. Following this incubation, 1.5 milliunits of O-glycosidase (Roche Applied Science) was added with reaction buffer (50 mM sodium phosphate, pH 7.5), and the reaction was incubated at 37 °C for 18 h. Where indicated, sRAGE underwent PNGase F treatment followed by neuraminidase treatment and O-glycosidase treatment as detailed above. Reactions were stopped by the addition of SDS sample buffer. One microgram of sRAGE from each reaction was subjected to SDS-PAGE and Western blotting as described previously (18, 23).
N-linked Glycan Characterization
A portion of each of the RP-HPLC fractions determined to contain the peptides with Asn2 and Asn27 (potential N-glycosylation sites) was subjected to MALDI-MS before and after deglycosylation by PNGase F. Glycan components were identified from MALDI-MS analysis using GlycoMod on Expasy (us.expasy.org/tools/glycomod/).
RESULTS
Three-step Purification of sRAGE from Mouse Lungs
Mouse lungs were used as starting material for sRAGE purification, because sRAGE is found most abundantly in mouse lungs compared with other tissues (18). Subsequent to homogenization, lung protein was subjected to batch extraction with concanavalin A-Sepharose followed by Affi-Gel heparin affinity chromatography and finally Mono Q ion-exchange chromatography. Fig. 2 shows the stepwise purification of mouse sRAGE.
Fig. 2. Three-step purification of sRAGE from mouse lungs.
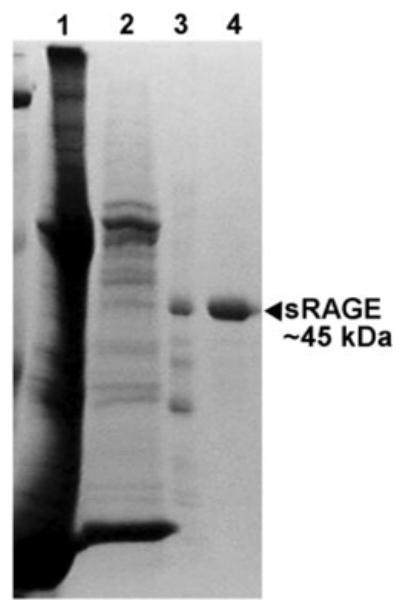
Mouse lungs were homogenized as described under “Experimental Procedures.” The homogenate was then subjected to three successive chromatography steps: 1) concanavalin A-Sepharose; 2) Affi-Gel heparin; and 3) Mono Q-Sepharose. Proteins (26 μl of each sample) were separated after reduction and denaturation in a 5–15% gradient acrylamide gel, and after electrophoresis, the gel was stained with Coomassie Brilliant Blue. Lane 1, lung homogenate; lane 2, concanavalin A eluate; lane 3, Affi-Gel heparin eluate; lane 4, Mono Q eluate (purified mouse sRAGE).
Amino-terminal Blockage of Mouse sRAGE
We were unable to determine the amino-terminal sequence of the native purified mouse sRAGE using Edman degradation, so we hypothesized that the amino terminus of sRAGE was blocked. After intracellular removal of the signal peptide (Met-Pro-Ala … Ala-Gly-Gly, the first 23 residues of the mouse RAGE polypeptide),3 the amino-terminal sequence becomes Gln1-Asn-Ile-Thr-Ala-Arg … (see Fig. 3). When Gln is the amino-terminal amino acid residue, it may spontaneously undergo deamidation and cyclization to form the cyclic amino acid pyroglutamate, which is unreactive in Edman degradation. To determine whether pyroglutamate is the blocking group at the amino terminus of mouse sRAGE, the purified protein was incubated with Pfu pyroglutamate aminopeptidase and then subjected to Edman degradation. The resulting amino-terminal sequence was determined to begin with Xaa-Ile-Thr-Ala-Arg … This sequence corresponds to an unidentified amino acid residue (Xaa) followed by the third residue after the signal peptide (Ile3), and so on. Notably, Asn2 was not identified as the amino-terminal amino acid residue after treatment with Pfu, because it was glycosylated (see below). Thus, these studies indicate that, after removal of the sRAGE signal peptide, the amino-terminal Gln is modified to form pyroglutamate (depicted in Fig. 4B).
Fig. 3. Mass spectroscopy sequencing highlights for mouse sRAGE.

Mouse sRAGE was proteolyzed with trypsin, chymotrypsin, or thermolysine, and resultant peptides were subjected to LC-MS/MS for sequencing. Depicted here is the composite sequencing of all three peptide sets. Numbers indicate amino acid positions relative to the first residue after the removal of the signal peptide. The portion of the sequence bordered with a dashed line indicates the signal peptide. The arrow indicates the residue at which our sequence data terminated (proposed carboxyl-terminus of sRAGE). Diamonds indicate N-glycosylation sites.
Fig. 4. Disulfide pattern of mouse sRAGE.
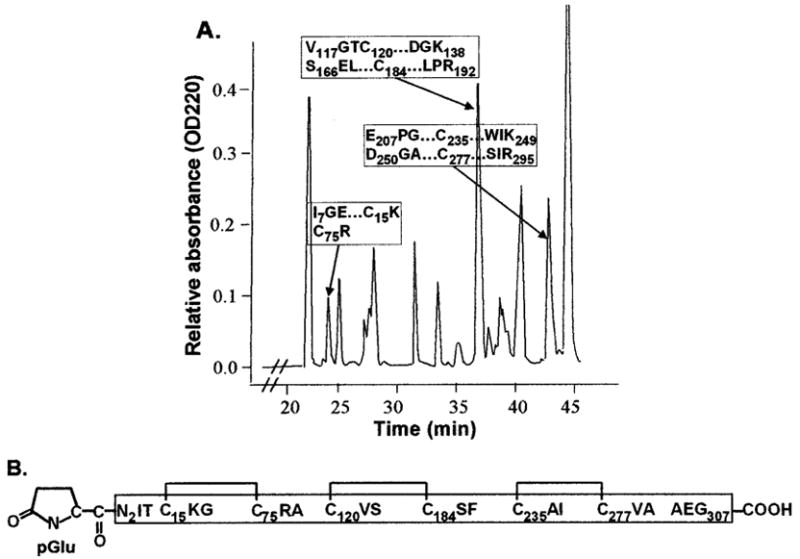
A, a representative RP-HPLC trace is shown. Native mouse sRAGE was trypsinized, and the resulting peptides were separated by RP-HPLC. Edman degradation was used to determine which peaks contained the indicated pairs of cysteine-containing peptides, and MALDI-MS analysis was used to confirm that the pair of the peptides in each of these peaks is joined by a disulfide bond (see “Experimental Procedures”). The disulfide pattern within sRAGE is shown in B with horizontal brackets indicating each pair of cysteine residues joined by a disulfide bond. pGlu, pyroglutamate, the amino-terminal blocking group on sRAGE.
Mouse sRAGE Sequence
Data from others (15, 16) show that human sRAGE is a product of alternative splicing of RAGE mRNA. The alternative splice site is located just upstream of the transmembrane domain and results in a frame-shift so that a transmembrane domain is no longer encoded for sRAGE. Thus, human RAGE and sRAGE have distinct carboxyl-terminal sequences. To see whether mouse sRAGE is also produced by alternative splicing, we attempted to detect an alternative RAGE transcript. We used either mouse lung RNA or a mouse lung cDNA library as a template in RT-PCR or PCR. Primers were targeted to each end of the RAGE transcript as well as various internal sites including sequences directly flanking the region homologous to the sequence spliced out in humans (Fig. 1 and Table I) in attempt to detect a smaller amplicon that would indicate alternative splicing analogous to that seen in humans. We found no evidence of an alternative RAGE transcript. However, we were able to amplify mouse membrane RAGE (Fig. 1 and Table I, primers are indicated by an asterisk). Furthermore, we also were able to amplify and clone sRAGE from a human cDNA library whose sequence matched that found by Malherbe et al. (15), indicating that our failure to find an alternative mouse RAGE transcript is not due to failure of our primers to amplify RAGE cDNA.
Table I.
Primer sequences used in attempt to detect alternatively spliced mouse RAGE transcript
| Primera | Sequence (5′ . . . . 3′) |
|---|---|
| Ab | CTGGCATTCTCTGACAGAACGAGATG |
| Bb | AGAAGATGGAGNCTGGGAAGGAA |
| Cc | ATGCCAGCGGGGACAGCAGCTA |
| D | CAAGTCCAACTACCGAG |
| E | AAGAGACCAGGAGACACCCT |
| F | TCCAGAGGGCATTCAGCTGTT |
| G | CATTCAGCTGTTGGTTGAGCCTG |
| H | GACTGATTCAGCTCTGCACGTTC |
| I | TTTACTCAGCATGGATCATGTGGGC |
| J | CGTATCAAATGTTTACTCAGCAT |
| Kb,c | GTGCCACTCTGACCAGCCTGGTATG |
| Lb | GTCTCTTTCCATCCCTCACTACCTC |
Primer designations and their relative positions are indicated in Fig. 1.
Primers were designed using untranslated sequence. Untranslated sequences flanking mouse RAGE can be found in the GenBank™ data base under GenBank™ accession number AF030001.1 (nucleotides 74101–77630).
Primers used to successfully amplify mouse membrane RAGE.
To investigate the mouse sRAGE amino acid sequence, we used LC-MS/MS. Purified mouse sRAGE was digested with trypsin, chymotrypsin, or thermolysine, and peptides were analyzed by LC-MS/MS. We chose to use three proteases so that peptides would overlap, and thus, the sequence data from one digestion would complement the data from the others. Fig. 3 shows highlights from our sequencing data. 81.8% of the sequence between residues Gln1 and Gly307 was covered by the LC-MS/MS analyses. Notably, the carboxyl-terminal end of mouse sRAGE determined from our sequence data is homologous to the point at which the human sRAGE amino acid sequence becomes distinct from that of human membrane RAGE as determined by Yonekura et al. (Fig. 1, vertical arrow) (16).
To determine whether the mass of sRAGE corroborates this putative carboxyl terminus (Gly307), sRAGE was deglycosylated and subjected to MALDI-MS. If mouse sRAGE indeed ends at the last amino acid residue detected in our LC-MS/MS analysis (… Pro-Ala-Glu-Gly307), its expected (calculated) size would be ~32.736 kDa after the removal of the signal peptide. We found that after deglycosylation, mass spectroscopy size determination of mouse sRAGE yielded ~32.854 kDa (Table II). Such a small discrepancy supports the hypothesis that mouse sRAGE is truncated and does not have a distinct amino acid sequence from mouse membrane-bound RAGE.
Table II.
Mouse sRAGE masses determined by indicated methods
| Description | Method | Mass |
|---|---|---|
| Da | ||
| Calculation based on amino acid sequencea | 32736.28 | |
| Reduced denatured | SDS-PAGE | ~45000 |
| Native | MALDI-MS | 36033.00 |
| Deglycosylated | MALDI-MS | 34165.03b |
| 32854.15 |
Amino acid residues 1–307 (see Fig. 3).
A result of incomplete deglycosylation of sRAGE.
Disulfide Pattern of Mouse sRAGE
To further characterize mouse sRAGE, we investigated its disulfide pattern. Trypsin was used for this analysis because the tryptic digest map predicted that there would be no more than one cysteine in any single peptide. Tryptic peptides from purified native mouse sRAGE were subjected to gel filtration. Peak fractions from gel filtration were subsequently subjected to RP-HPLC to further separate peptides. Fractions from RP-HPLC were then sequenced using Edman degradation. Two peptides were presumed to be linked by a disulfide bond when the sequence analysis of one RP-HPLC peak revealed two distinct amino-terminal peptide sequences and both peptides were identified by the tryptic map to contain cysteine residues. To confirm that the peptides were linked by a disulfide bond, a portion of the RP-HPLC fraction of interest was subjected to MALDI-MS to verify that the mass corresponded to two peptides. Another portion of each fraction of interest was then reduced and analyzed by MALDI-MS to determine whether reduction would result in two masses that matched the predicted masses of the relevant peptides. Mouse sRAGE has six cysteines, and using this method, we found that all six cysteines were engaged in disulfide bonds (Cys15 shares a disulfide bond with Cys75 as does Cys120 with Cys184, and Cys235 with Cys277, Fig. 4).
Glycosylation of Mouse sRAGE
The importance of RAGE glycosylation was shown by Srikrishna et al. (24) when they found a reduction in amphoterin-RAGE binding after RAGE deglycosylation. Presumably, the same is true for sRAGE because it has the same ligand specificity as RAGE. To determine whether mouse sRAGE has N-linked and O-linked carbohydrates, purified mouse sRAGE was digested with O-glycosidase, PNGase F, or a combination of the two enzymes. An analysis of sRAGE after deglycosylation by Western blotting (Fig. 5A) shows that mouse sRAGE is N-glycosylated but not O-glycosylated. To confirm this finding, sRAGE was subjected to amine monosaccharide analysis but no N-acetylgalactosamine was detected (not illustrated), which supports the conclusion that mouse sRAGE is not O-glycosylated. Mouse sRAGE contains two potential N-glycosylation sites (Fig. 3), and Fig. 5A suggests that both of them are used since two sRAGE bands appeared after deglycosylation (corresponding to partially and completely deglycosylated forms). In addition, mass spectroscopic size analysis of mouse sRAGE before and after PNGase F treatment (36,033 and 32,854 Da, respectively) (Table II) indicates a reduction in size of ~3.2 kDa, corresponding to two glycan groups. To further confirm this result and identify the components of these glycans, each of the two sRAGE tryptic peptides that were determined to contain one of the possible N-glycosylation sites by Edman degradation was analyzed by MALDI-MS in reflector mode before and after PNGase F treatment. MALDI-MS data (Table III) were used with the GlycoMod program to identify the components of each N-linked carbohydrate. Fig. 5B summarizes this data, illustrating that Asn2 probably has a di-antennary complex-type glycosylation with core fucosylation and Asn57 has a complex or hybrid-type glycan.
Fig. 5. sRAGE is N-glycosylated.
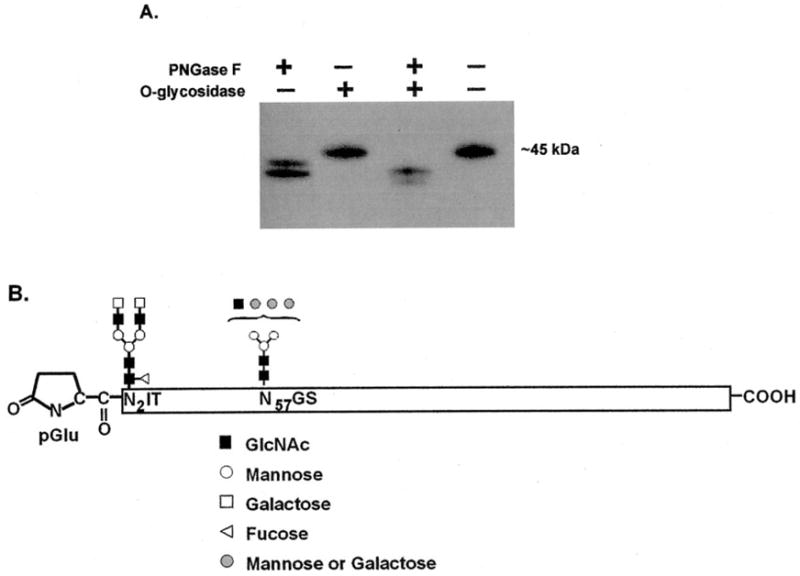
A, purified sRAGE was incubated with O-glycosidase and/or PNGase F and then subjected to Western blotting using an antibody targeted to mouse RAGE/sRAGE described previously (18). B, mouse sRAGE tryptic peptides that contained a possible N-glycosylation site were subjected to MALDI-MS for size determination before and after deglycosylation. Data from MALDI-MS (see Table III) were used in conjunction with the GlycoMod program to determine the composition of the N-linked sugar groups.
Table III.
MALDI-MS data for N-glycosylated peptides
| Peptide range | Glycosylated residue | Massa (glycosylated) | Massa after deglycosylation | Massa of N-glycanb |
|---|---|---|---|---|
| pGlu1-Arg6 | Asn2 | 2454.0029 | 686.3291 | 1768.7 |
| Val40-Arg74 | Asn57 | 5189.8867 | 3608.2102 | 1582.7 |
Masses are reported in Da.
Difference between peptide mass before and after deglycosylation by PNGase F treatment, plus 1 Da to account for the conversion of Asn to Asp resulting from PNGase F treatment.
DISCUSSION
Here we describe a novel purification strategy for sRAGE from frozen mouse lungs, one step of which is heparin affinity chromatography. Notably, in terms of lung physiology, sRAGE is not detectable in bronchoalveolar lining fluid or serum (18). This observation suggests that sRAGE is bound to the extracellular matrix (ECM) or cell surfaces, and our finding that sRAGE has heparin affinity suggests that sRAGE may be bound to heparan sulfate in the ECM or cell membranes. Localized in the ECM or cell surfaces, sRAGE may act as a decoy receptor by binding ligands and preventing them from reaching endothelial and inflammatory cell surface RAGE and activating potentially harmful RAGE signaling. It is interesting to consider the possibility that ligand binding could cause a conformational change in sRAGE that could alter its heparin affinity, perhaps allowing sRAGE-ligand complexes to be released from the surrounding ECM and enter the bloodstream to be cleared. It has been postulated that sRAGE-ligand complexes are eliminated from the blood via the spleen and/or liver (25). Further studies are needed to determine whether the loss of ECM affinity upon ligand binding is a component of sRAGE physiology. Incidentally, heparin affinity of bovine RAGE has also been demonstrated (10). The domain of RAGE/sRAGE involved in heparin binding has yet to be defined.
The data presented here suggest that mouse sRAGE is produced by a mechanism distinct from human sRAGE, which is produced by alternative splicing. We found no evidence for an alternative RAGE transcript in mouse lung RNA or cDNA. Because it appears to be a truncated form of RAGE, it is likely that RAGE is proteolyzed to produce sRAGE. Matrix metallo-proteinase-9 has been found to cause murine pulmonary epithelial cells to shed sRAGE into the culture medium (26), implicating this proteinase in sRAGE production from RAGE. Our finding may suggest that human sRAGE production could have two levels of regulation. In addition to alternative splicing, human sRAGE production may also be regulated by proteinases in a manner similar to that seen in mouse sRAGE.
Of the three immunoglobulin-like domains, the most amino-terminal one (V-like domain) has been shown to be involved in ligand binding (27). Because sRAGE is glycosylated in this domain, it is likely that glycosylation is important for the ability of sRAGE to bind its ligands, which would be in agreement with previous observations made by Srikrishna et al. (24) regarding bovine RAGE. Thus, a bacterial expression system for sRAGE should be used cautiously because a lack of glycosylation could decrease or inhibit ligand binding by sRAGE.
It is interesting to note that there is a discrepancy between the mass of mouse sRAGE as determined by SDS-PAGE (~45 kDa, Fig. 2) and mass spectroscopy of native sRAGE (~36 kDa, Table II). It appears that there is some residual structure of sRAGE after denaturation with SDS and reduction with dithiothreitol, making sRAGE appear larger in SDS-PAGE. Also interesting is the apparent size of mouse sRAGE in native PAGE. Under non-denaturing and non-reducing conditions, purified mouse sRAGE runs at ~200 kDa (data not illustrated). It has not yet been determined whether another protein is also contained in this high molecular mass band or whether it is solely sRAGE. To our knowledge, no evidence of sRAGE oligomerization has been reported to date. Schmidt et al. (28) reported that bovine RAGE binds to a lactoferrin-like peptide. When cross-linked, this binding results in a high molecular mass complex (~180 kDa) when analyzed by SDS-PAGE. Further studies will determine whether this type of interaction also occurs in the mouse and is responsible for the high molecular mass band we observe in native PAGE analysis.
In summary, these studies indicate that sRAGE has heparin affinity, which probably mediates its localization in tissues. Furthermore, no evidence was found to suggest that alternative splicing of RAGE mRNA contributes to sRAGE biosynthesis in the mouse as was found for human sRAGE. In contrast, the carboxyl-terminal proteolysis of RAGE appeared to be the likely mechanism of sRAGE formation in the mouse. Mechanisms regulating this proteolysis are probably important in regulatory balance between proinflammatory RAGE signaling and anti-inflammatory sequestration of RAGE ligands by sRAGE. These proteolytic pathways may also be important additional mechanisms of regulating sRAGE expression in humans.
Acknowledgments
We thank Ida Thøgersen, Toni Termin, Cheryl Fattman, and Rod Tan for excellent technical assistance.
The abbreviations used are
- RAGE
receptor for advanced glycation end products
- sRAGE
soluble RAGE
- RT-PCR
reverse transcription PCR
- LC-MS/MS
liquid chromatography tandem mass spectrometry
- MALDI-MS
matrix-assisted laser desorption-ionization mass spectrometry
- RP-HPLC
reverse-phase high performance liquid chromatography
- PNGase F
N-glycosidase F
- ECM
extracellular matrix
Footnotes
This work was supported by American Heart Association Grants 702359 (to T. D. O.) and 0415412U (to L. E. H.) and National Institutes of Health Grant ROI HL-063700.
Mouse RAGE cDNA sequence can be found in the GenBank™ database under GenBank™ accession number L33412.1.
The amino acid sequence of mouse RAGE can be found in the GenBank™ database under GenBank™ accession number 6671525.
References
- 1.Neeper M, Schmidt AM, Brett J, Yan SD, Wang F, Pan YC, Elliston K, Stern D, Shaw A. J Biol Chem. 1992;267:14998–15004. [PubMed] [Google Scholar]
- 2.Huttunen HJ, Fages C, Rauvala H. J Biol Chem. 1999;274:19919–19924. doi: 10.1074/jbc.274.28.19919. [DOI] [PubMed] [Google Scholar]
- 3.Schmidt AM, Yan SD, Yan SF, Stern DM. Biochim Biophys Acta. 2000;1498:99–111. doi: 10.1016/s0167-4889(00)00087-2. [DOI] [PubMed] [Google Scholar]
- 4.Hofmann MA, Drury S, Fu C, Qu W, Taguchi A, Lu Y, Avila C, Kambham N, Bierhaus A, Nawroth P, Neurath MF, Slattery T, Beach D, McClary J, Nagashima M, Morser J, Stern D, Schmidt AM. Cell. 1999;97:889–901. doi: 10.1016/s0092-8674(00)80801-6. [DOI] [PubMed] [Google Scholar]
- 5.Bierhaus A, Schiekofer S, Schwaninger M, Andrassy M, Humpert PM, Chen J, Hong M, Luther T, Henle T, Kloting I, Morcos M, Hofmann M, Tritschler H, Weigle B, Kasper M, Smith M, Perry G, Schmidt AM, Stern DM, Haring HU, Schleicher E, Nawroth PP. Diabetes. 2001;50:2792–2808. doi: 10.2337/diabetes.50.12.2792. [DOI] [PubMed] [Google Scholar]
- 6.Arumugam T, Simeone DM, Schmidt AM, Logsdon CD. J Biol Chem. 2004;279:5059–5065. doi: 10.1074/jbc.M310124200. [DOI] [PubMed] [Google Scholar]
- 7.Li J, Schmidt AM. J Biol Chem. 1997;272:16498–16506. doi: 10.1074/jbc.272.26.16498. [DOI] [PubMed] [Google Scholar]
- 8.Park L, Raman KG, Lee KJ, Lu Y, Ferran LJ, Jr, Chow WS, Stern D, Schmidt AM. Nat Med. 1998;4:1025–1031. doi: 10.1038/2012. [DOI] [PubMed] [Google Scholar]
- 9.Taguchi A, Blood DC, del Toro G, Canet A, Lee DC, Qu W, Tanji N, Lu Y, Lalla E, Fu C, Hofmann MA, Kislinger T, Ingram M, Lu A, Tanaka H, Hori O, Ogawa S, Stern DM, Schmidt AM. Nature. 2000;405:354–360. doi: 10.1038/35012626. [DOI] [PubMed] [Google Scholar]
- 10.Yan SD, Chen X, Fu J, Chen M, Zhu H, Roher A, Slattery T, Zhao L, Nagashima M, Morser J, Migheli A, Nawroth P, Stern D, Schmidt AM. Nature. 1996;382:685–691. doi: 10.1038/382685a0. [DOI] [PubMed] [Google Scholar]
- 11.Kuniyasu H, Oue N, Wakikawa A, Shigeishi H, Matsutani N, Kuraoka K, Ito R, Yokozaki H, Yasui W. J Pathol. 2002;196:163–170. doi: 10.1002/path.1031. [DOI] [PubMed] [Google Scholar]
- 12.Goova MT, Li J, Kislinger T, Qu W, Lu Y, Bucciarelli LG, Nowygrod S, Wolf BM, Caliste X, Yan SF, Stern DM, Schmidt AM. Am J Pathol. 2001;159:513–525. doi: 10.1016/S0002-9440(10)61723-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wear-Maggitti K, Lee J, Conejero A, Schmidt AM, Grant R, Breitbart A. Ann Plast Surg. 2004;52:519–521. doi: 10.1097/01.sap.0000122857.49274.8c. [DOI] [PubMed] [Google Scholar]
- 14.Deane R, Du Yan S, Submamaryan RK, LaRue B, Jovanovic S, Hogg E, Welch D, Manness L, Lin C, Yu J, Zhu H, Ghiso J, Frangione B, Stern A, Schmidt AM, Armstrong DL, Arnold B, Liliensiek B, Nawroth P, Hofman F, Kindy M, Stern D, Zlokovic B. Nat Med. 2003;9:907–913. doi: 10.1038/nm890. [DOI] [PubMed] [Google Scholar]
- 15.Malherbe P, Richards JG, Gaillard H, Thompson A, Diener C, Schuler A, Huber G. Brain Res Mol Brain Res. 1999;71:159–170. doi: 10.1016/s0169-328x(99)00174-6. [DOI] [PubMed] [Google Scholar]
- 16.Yonekura H, Yamamoto Y, Sakurai S, Petrova RG, Abedin MJ, Li H, Yasui K, Takeuchi M, Makita Z, Takasawa S, Okamoto H, Watanabe T, Yamamoto H. Biochem J. 2003;370:1097–1109. doi: 10.1042/BJ20021371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jendrisak J. In: Protein Purification: Micro to Macro: Proceedings of a Cetus-UCLA Symposium, Frisco, March 29–April 4, 1987. Burgess R, editor. A. R. Liss, Inc.; New York: 1987. pp. 75–97. [Google Scholar]
- 18.Hanford LE, Fattman CL, Shaefer LM, Enghild JJ, Valnickova Z, Oury TD. Am J Respir Cell Mol Biol. 2003;29:S77–81. [PubMed] [Google Scholar]
- 19.Petersen SV, Valnickova Z, Enghild JJ. Biochem J. 2003;374:199–206. doi: 10.1042/BJ20030313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oury TD, Schaefer LM, Fattman CL, Choi A, Weck KE, Watkins SC. Am J Physiol. 2002;283:L777–L784. doi: 10.1152/ajplung.00011.2002. [DOI] [PubMed] [Google Scholar]
- 21.Chomczynski P, Sacchi N. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 22.Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Electrophoresis. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 23.Fattman CL, Enghild JJ, Crapo JD, Schaefer LM, Valnickova Z, Oury TD. Biochem Biophys Res Commun. 2000;275:542–548. doi: 10.1006/bbrc.2000.3327. [DOI] [PubMed] [Google Scholar]
- 24.Srikrishna G, Huttunen HJ, Johansson L, Weigle B, Yamaguchi Y, Rauvala H, Freeze HH. J Neurochem. 2002;80:998–1008. doi: 10.1046/j.0022-3042.2002.00796.x. [DOI] [PubMed] [Google Scholar]
- 25.Renard C, Chappey O, Wautier MP, Nagashima M, Lundh E, Morser J, Zhao L, Schmidt AM, Scherrmann JM, Wautier JL. Mol Pharmacol. 1997;52:54–62. doi: 10.1124/mol.52.1.54. [DOI] [PubMed] [Google Scholar]
- 26.Devaux Y, Senior RM, Ray P. Am J Respir Crit Care Med. 2004;169:A456. [Google Scholar]
- 27.Kislinger T, Fu C, Huber B, Qu W, Taguchi A, Du Yan S, Hofmann M, Yan SF, Pischetsrieder M, Stern D, Schmidt AM. J Biol Chem. 1999;274:31740–31749. doi: 10.1074/jbc.274.44.31740. [DOI] [PubMed] [Google Scholar]
- 28.Schmidt AM, Mora R, Cao R, Yan SD, Brett J, Ramakrishnan R, Tsang TC, Simionescu M, Stern D. J Biol Chem. 1994;269:9882–9888. [PubMed] [Google Scholar]