

Abstract
In many cell types agonist-receptor activation leads to a rapid and transient release of Ca2+ from intracellular stores via activation of inositol 1,4,5 trisphosphate (InsP3) receptors (InsP3Rs). Stimulated cells activate store- or receptor-operated calcium channels localized in the plasma membrane, allowing entry of extracellular calcium into the cytoplasm, and thus replenishment of intracellular calcium stores. Calcium entry must be finely regulated in order to prevent an excessive intracellular calcium increase. Junctate, an integral calcium binding protein of endo(sarco)plasmic reticulum membrane, (a) induces and/or stabilizes peripheral couplings between the ER and the plasma membrane, and (b) forms a supramolecular complex with the InsP3R and the canonical transient receptor potential protein (TRPC) 3 calcium entry channel. The full-length protein modulates both agonist-induced and store depletion–induced calcium entry, whereas its NH2 terminus affects receptor-activated calcium entry. RNA interference to deplete cells of endogenous junctate, knocked down both agonist-activated calcium release from intracellular stores and calcium entry via TRPC3. These results demonstrate that junctate is a new protein involved in calcium homeostasis in eukaryotic cells.
Keywords: calcium homeostasis; IP3R; calcium entry channel; calcium binding protein; ER
Introduction
The overall importance of Ca2+ signaling in a variety biological functions is firmly established. Agonist-receptor interactions cause a rapid and transient release of Ca2+ from ER stores via inositol 1,4,5 trisphosphate (InsP3) receptor (InsP3R) activation, which in turn stimulates numerous cellular responses ranging from secretion to gene expression (Clapham, 1995; Berridge et al., 2000). Stimulation of InsP3R and/or depletion of intracellular calcium stores, activate a set of receptor- or store-operated calcium channels localized on the plasma membrane, allowing entry of extracellular calcium into the cytoplasm and thus replenishment of intracellular calcium stores (Irvine, 1990; Zhu et al., 1996; Kiselyov et al., 1998; Ma et al., 2000; Elliot, 2001; Putney et al., 2001). The canonical transient receptor potential proteins (TRPCs) constitute some of the calcium influx channels. Several mechanisms have been put forward to explain how calcium entry occurs (for reviews see Patterson et al., 1999; Elliot, 2001; Putney et al., 2001; Clapham, 2003). The “conformational coupling” model predicts that calcium entry occurs via a mechanism similar to that underlying excitation-contraction coupling of skeletal muscle (Irvine, 1990; Kiselyov et al., 2000). The latter event involves interaction between the ryanodine receptor in the sarcoplasmic reticulum membrane and the dihydropyridine receptor on the plasma membrane, but a number of accessory proteins are also involved in excitation-contraction coupling (Marty et al., 1994; Nakai et al., 1996; Franzini-Armstrong and Protasi, 1997). By analogy, the InsP3R might be localized vis a vis the plasma membrane in microdomains of the ER endowed with different protein components which could be involved in the mechanisms of calcium entry (Tang et al., 2001). As part of a comprehensive search of proteins involved in excitation-contraction coupling, we identified junctate, a 33-kD integral membrane calcium binding protein (21 moles of Ca2+ per mole of protein, K d 217 μM) of sarco(endo)plasmic reticulum membranes which is expressed in a variety of tissues (Dinchuk et al., 2000; Treves et al., 2000; Hong et al., 2001). The first 78 residues of junctate are identical to those of junctin; the latter protein is found in the sarcoplasmic reticulum of heart and skeletal muscle, and forms a quaternary complex with the ryanodine receptor, calsequestrin, and triadin (Zhang et al., 1997). Overexpression of junctate in COS-7 cells affects calcium release via InsP3R activation (Treves et al., 2000). In this work, we demonstrate that junctate is part of a supramolecular complex with InsP3Rs and TRPC3 and modulates calcium entry through receptor- and store-activated channels.
Results
The NH2-domain of junctate interacts with the InsP3R
The present work was undertaken in order to determine whether InsP3R and junctate interact in vivo. We found that junctate: (a) is an integral protein component of bovine cerebellum microsomal membranes (Fig. 1, top, lanes 1 and 2), a tissue fraction enriched in InsP3R; and (b) coimmunoprecipitates with InsP3R (Fig. 1, top, lane 6).
Figure 1.

Coimmunoprecipitation of InsP 3 R with antijunctate Ab. (Top) Proteins present in the total microsomal fraction (lane 1, 10 μg protein) or integral membrane proteins extracted with 100 mM Na2CO3 (lane 2, 10 μg protein) of bovine cerebella were stained with antijunctate Ab. Solubilized microsomal proteins were incubated in the absence (lanes 3 and 4) and in the presence of affinity-purified antijunctate Ab (lanes 5 and 6). Proteins complexing with junctate were coimmunoprecipitated with protein-A Sepharose beads and stained with anti-InsP3R Ab (lanes 3–6). Lanes 3 and 5, void; lanes 4 and 6, proteins bound to Protein–A Sepharose beads. (Bottom) Peptide pull-down experiments. (A) Triton X-100–solubilized rabbit brain microsomes stained with antibodies against type 1 InsP3R (lane 1, void; lane 2, last wash; lane 3, beads) and type 2 InsP3R (lane 4, void; lane 5, last wash; lane 6, beads). (B) Lung microsomes stained with type 3 InsP3R Ab (lane 1, void; lane 2, last wash; lane 3, beads). (C) Brain microsomes were incubated with a peptide corresponding to the scrambled sequence of the NH2-terminal domain of junctate and stained with anti-InsP3R Abs (lane 1, void; lane 2, last wash; lane 3, beads). (D) Brain microsomes were incubated with a GST fusion protein containing the COOH-terminal domain of junctate and stained with anti-InsP3R Ab (lane 1, void; lane 2, last wash; lane 3 glutathione-Sepharose beads).
Junctate is made up of a short NH2-terminal cytoplasmic domain, followed by a hydrophobic transmembrane domain and a lumenal COOH-terminal calcium-binding domain. To define the domain of junctate involved in the interaction with the InsP3R, we performed pull-down experiments with a synthetic biotinylated peptide corresponding to the cytoplasmic NH2-domain and with a fusion protein containing the COOH-terminal domain. The NH2-domain of junctate specifically binds to InsP3R types 1 and 2 in rabbit brain microsomes (Fig. 1 A, lanes 3 and 6, respectively), and to type 3 InsP3R present in rabbit lung microsomes. (Fig. 1 B, lane 3). The InsP3R did not interact either with (a) a peptide corresponding to the scrambled sequence of the NH2-terminal domain (Fig. 1 C, lane 3) or (b) a fusion protein encompassing the COOH-terminal domain of junctate (Fig. 1 D, lane 3).
Junctate affects agonist-induced calcium release and receptor-activated calcium entry
We investigated the role of junctate (a) on the regulation of InsP3-mediated agonist induced calcium release from intracellular stores and (b) on calcium entry elicited in response to InsP3R activation, i.e., Ca2+-influx mediating refilling of the intracellular calcium stores. ATP stimulation of the PLC/InsP3 pathway in COS-7 cells in the presence of nominally calcium-free Krebs-Ringer, caused an immediate rapid rise in the [Ca2+]i followed by a slow decline to resting levels (Fig. 2, A–D). COS-7 cells overexpressing full-length junctate (junctate-EGFP) exhibited a significant increase in peak ATP-induced Ca2+ release (mean ± SEM was 20% ± 2, n = 57; **P < 0.0004; Fig. 2 E). The transient increases in [Ca2+]i of cells transfected with either NH2-terminal (NH2-TM-EGFP) or COOH-terminal (EGFP-TM-COOH) domains of junctate were not significantly different from those observed in mock-transfected cells, excluding the possibility that the increases in calcium release observed in cells transfected with full-length junctate, is due to nonspecific cellular responses secondary to overexpression of recombinant proteins. Junctate overexpression did not affect the amount of InsP3R expressed, as determined by Western blot analysis (unpublished data). To establish if the effect of the different junctate constructs on calcium transients were due to increases of releasable calcium from ER, we determined the amount of calcium released from the stores by treating transfected COS-7 cells with the sarcoplasmic and ER Ca2+ATPase (SERCA) inhibitor Di-tert-butylhydroquinone (t-BuBHQ) in the presence of nominally calcium-free medium. Overexpression of either full-length junctate, or of its COOH-terminal calcium binding domain significantly increased the t-BuBHQ releasable Ca2+; on the other hand in cells overexpressing the NH2-terminal domain, the t-BuBHQ releasable-Ca2+ was not different from control COS-7 cells (Fig. 2 G). The ER-free [Ca2+] of mock-transfected cells and cells overexpressing junctate were not significantly different (the means ± SD were 456 ± 54 and 412 ± 27 μM, respectively; n = 47; t test, P = 0.497). These results indicate that overexpression of the COOH-terminal Ca2+-binding domain of junctate increases the releasable calcium present in the intracellular stores of COS-7 cells.
Figure 2.
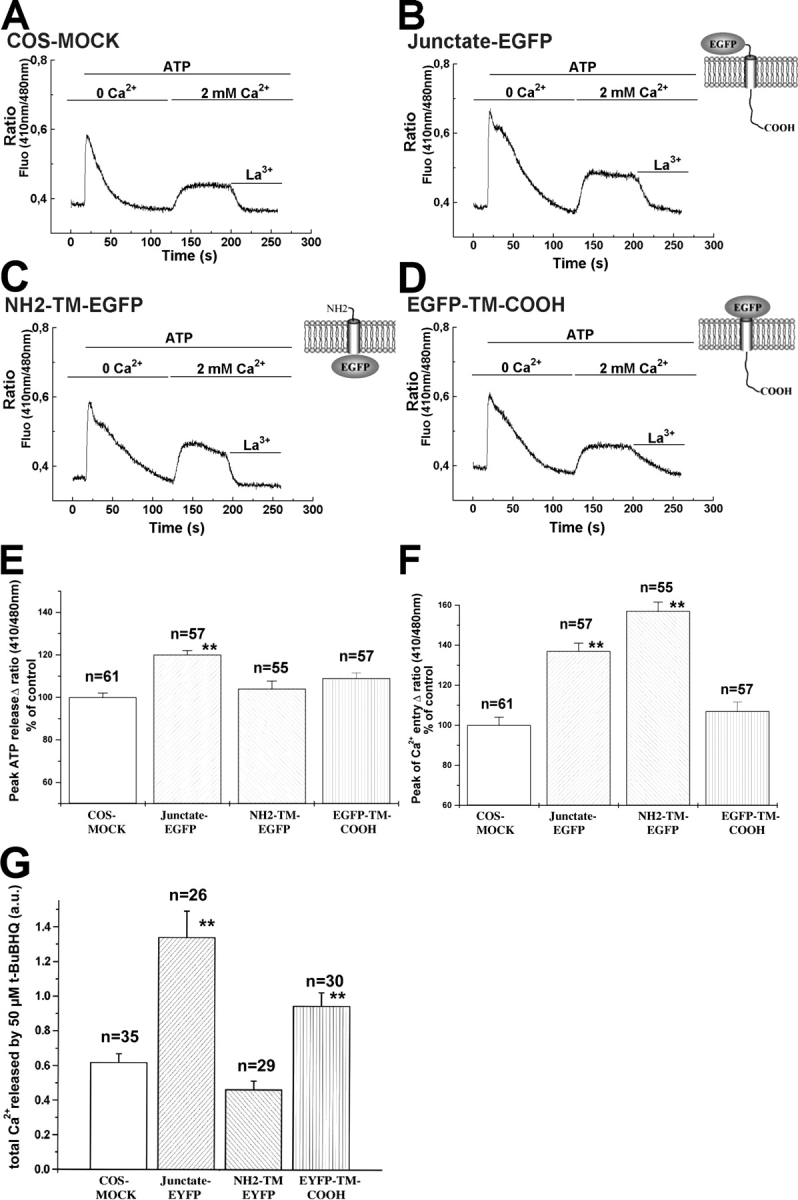
Junctate affects agonist-induced peak calcium release, receptor-activated calcium entry and t-BuBHQ-sensitive calcium pools. (A–D) Indo-1–loaded COS-7 cells were flushed with a solution containing 100 μM ATP in nominally Ca2+-free Krebs-Ringer; once the [Ca2+]i had returned to resting levels, calcium influx was stimulated by addition of 2 mM Ca2+ in the presence of 100 μM ATP in Krebs-Ringer. Each record is from a single cell. Calcium influx was blocked by exposing the cell to a solution containing 2 mM lanthanum. (E and F) Calcium released and calcium entry peak amplitudes are presented as a percentage of the same changes in mock-transfected cells (mean ± SEM; n = single cells from four different transfections). (G) COS-7 cells were loaded with 5 μM fura-2/AM and then perfused with 50 μM t-BuBHQ in nominally Ca2+-free Krebs-Ringer. Total amount of calcium released was measured by calculating the integral of the calcium transient, using the Origin computer program software. Values are mean ± SEM; n = number of cells from three different transfections; **, indicates statistically significant difference from mock-transfected cells. a.u., arbitrary units.
Cells expressing cDNA either for the full-length junctate or for the NH2-terminal cytoplasmic domain exhibited a larger calcium entry (P < 0.001) compared with mock-transfected COS-7 cells: the increases were 37% ± 4; n = 57 (mean ± SEM) for cells overexpressing the full-length junctate (Fig. 2 F) and 57% ± 4.5; n = 55 (mean ± SEM) for cells overexpressing the cytoplasmic domain (Fig. 2 F). Calcium entry in cells transfected with the COOH-terminal domain of junctate was not different from that of mock-transfected cells. The effect of junctate on calcium entry is not due to nonspecific leak of calcium through the plasma membrane induced by protein overexpression, because lanthanum or gadolinium, two cations that have been shown to inhibit store-operated Ca2+ entry (Kwan et al., 1990; Trebak et al., 2002), caused the [Ca2+]i to return to resting levels in all transfected cells we examined (Fig. 2, A–D, traces).
We also determined whether the potentiation of receptor activated calcium entry by junctate requires PLC activation and/or generation of diacylglycerol by studying the characteristics of calcium entry after application of U73122, which is a specific inhibitor of PLC (Smith et al., 1990; Fig. 3 A). The presence of 10 μM U73122 almost completely abolished calcium influx in both mock-transfected COS-7 cells and cells transfected with the full-length junctate-EGFP (Fig. 3, A and C). Under similar experimental conditions, the inactive analogue U73343 did not block calcium influx (not depicted).
Figure 3.

Junctate requires activation of PLC to modulate calcium entry. Cells were first stimulated with ATP; after the intracellular Ca2+ concentration returned to basal the cells were flushed for 40 s with the PLC-inhibitor U73122 and with 2-APB. Traces show the recordings of the calcium influx stimulated by addition of 2 mM calcium in the presence of ATP plus 10 μM of PLC-inhibitor U73122 or 75 μM of 2-APB.
Treatment of cells with 75 μM 2-aminoethyl diphenylborate (2-APB), a compound which has been reported to block the InsP3R and calcium entry pathway (Bootman et al., 2002), also blocked calcium influx into mock-transfected COS-7 cells and cells transfected with the full-length junctate-EGFP construct (Fig. 3, B and D), supporting the observation that the increased receptor activated calcium entry observed in junctate overexpressing cells relies on a PLC-dependent mechanism.
Junctate affects calcium entry stimulated by depletion of ER stores independently of receptor-coupled events
2-APB has been reported to block the calcium entry pathways activated either by receptor agonist-activation or by store-depletion (Bootman et al., 2002). Therefore, we examined the effect of junctate on calcium entry mediated by depletion of ER calcium stores. To ensure store depletion, transfected COS-7 cells were loaded with 4 μM indo-1/AM in the presence of 1 mM EGTA (Fig. 4). This procedure depletes intracellular calcium stores because addition of 50 μM of the SERCA inhibitor t-BuBHQ (Fig. 4) failed to induce a [Ca2+]i transient. No difference in peak amplitude calcium entry was observed between mock-transfected cells and cells overexpressing either the NH2- or the COOH-terminal domains of junctate. On the contrary, cells transfected with the full-length junctate construct exhibited a 51 ± 6% increase (mean ± SEM; n = 48; *P < 0.0001) of peak amplitude calcium entry compared with mock-transfected cells.
Figure 4.

Junctate modulates calcium entry mediated by depletion of the ER independently of receptor activation. (A–D) Transfected cells were loaded with 5 μM indo-1/AM in the presence of 1 mM EGTA. Cells were exposed to 50 μM t-BuBHQ in the presence of nominally Ca2+-free Krebs-Ringer. Calcium influx was stimulated by addition of 2 mM Ca2+. (E) Values are mean ± SEM; n = number of cells from three different transfections. *P < 0.0001.
Junctate induces ER proliferation and an increase in frequency and extent of peripheral couplings
COS-7 cells transfected only with GFP show diffuse fluorescence in the cytoplasm (Fig. 5 A), whereas junctate-GFP overexpressing COS-7 cells show an extensive GFP-positive network that pervades the entire cytoplasm from the nucleus to the cell periphery (Fig. 5, B and C). The channels responsible for calcium entry in COS-7 cells have not been identified. Therefore, we investigated the effect of junctate overexpression in T3-HEK293 cells, a cell line of human origin, which was transfected and stably expresses the TRPC3 calcium entry channel (Zhu et al., 1998). This cellular model has been exploited to test the conformational coupling hypothesis of calcium entry predicting that a peripheral membrane coupling between the ER membrane and the plasma membrane is involved in activation of calcium entry (Irvine, 1990; Kiselyov et al., 2000).
Figure 5.
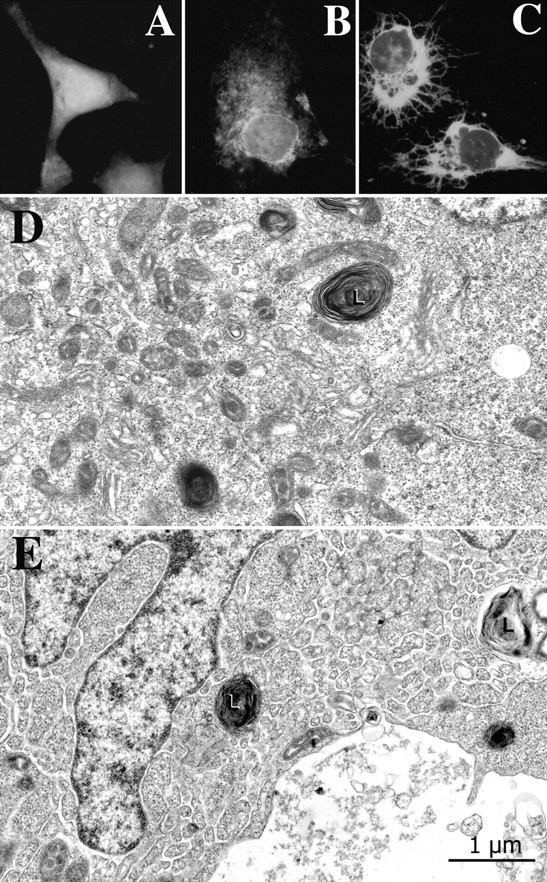
Junctate-positive network in junctate overexpressing T3-HEK293 cells. (A) An example of a COS-7 cell transfected with GFP. The cell shows an amorphous cytoplasmic fluorescence. (B and C) Examples of COS-7 cells transfected with the full-length junctate-GFP. An extensive GFP-positive network extends from the perinuclear region throughout the cytoplasm. Approximately 25% junctate-GFP–positive COS-7 cells had the spindle-like appearance as shown in C. (D) GFP-transfected T3-HEK293 cells have a fairly extensive amount of smooth ER near the cell center (left portion of the image) but little of it elsewhere (at right). L, lysosome. (E) This junctate-GFP overexpressing cell exhibits a very extensive ER network, continuous with the nuclear envelope and reaching out to the cell periphery. Such an extensive network was seen in a minority of cell profiles in sections from junctate-GFP expressing cells, but never in the control cells expressing GFP. Other cells from the same cultures show variable amounts of ER.
The ultrastructure of junctate-GFP overexpressing T3-HEK293 was compared with that of GFP-expressing T3-HEK293 cells (controls) and of T3-HEK293 cells which had been cotransfected with the reporter plasmid pEGFP and depleted of their endogenous junctate using RNA interference (RNAi). Transfected cells were sorted with FACS® on the basis of GFP expression. The underlying assumption was that T3-HEK293 cells taking in the reporter plasmid pEGFP were also taking in the RNAi construct. Control cells (Fig. 5 D) and cells transfected with pShag-1-RNAi-junctate construct have an active cell center with numerous Golgi system profiles and isolated ER vesicles. Few ER profiles belonging to the continuous intracellular network are seen elsewhere in the cell. By contrast, the cytoplasm of junctate overexpressing cells contains a very extensive, labyrinthine ER, sometimes filling the entire cell (Fig. 5 E). The ER profiles are quite clearly continuous with the nuclear envelope and reach all the way to the cell periphery. The relative extent of ER overdevelopment was roughly estimated by classifying sectioned cell profiles on the basis of their content of ER network, in areas other than the cell center, from almost nil to very extensive (Table I). Cells that were clearly in the process of breakdown were not included in the counts. Cells with moderate levels of ER may appear ER-free at one section level, but have ER profiles elsewhere. However, cells that have very extensive ER would show it at all section levels. Table I shows that junctate overexpressing cells are more highly represented than controls in the medium to extensive ER content categories and pShag-1-RNAi-junctate–treated cells have even less frequent ER profiles than the controls.
Table I. ER statistics.
| Cell type | No. of experiments |
Viable cell profiles with no ER |
Viable cell profiles with minor ER |
Viable cell profiles with medium ER |
Viable cell profiles with extensive ER |
|---|---|---|---|---|---|
| Sorted GFP-expressing T3-HEK293 cells |
1 (n = 134 cells) | 75% | 20% | 5% | 0 |
| Sorted GFP-junctate-expressing T3-HEK293 cells |
2 (n = 262 cells) | 21% | 40% | 33% | 6% |
| Sorted GFP-pShag-RNAi-junctate T3-HEK293 cells |
1 (n = 109 cells) | 71% | 29% | 0 | 0 |
The ER network reaches from the nuclear envelope to the cell periphery and forms junctional contacts (peripheral couplings) with the plasma membrane, in which the two apposed membranes run parallel to each other and are separated by a small gap of fairly uniform width (Fig. 6). The ER surface involved in the formation of these peripheral couplings is somewhat variable, but usually fairly small in control T3-HEK293 cells (Fig. 6 A), in which sectioned profiles of apposed ER are 218 ± 94 nm long (n = 165 measurements; see Table II). The ER profiles of peripheral couplings in junctate overexpressing cells are significantly longer and more variable in length (339 ± 192 nm; n = 126; P < 0.0001, Table II) due to the presence of some large areas of ER-plasma membrane junctional contact (Fig. 6, B–D). The profiles of pShag-1-RNAi-junctate–treated cells are significantly shorter than those in control cells (139 ± 51 nm; n = 15; P < 0.002; Table II). Note that in the latter case very few junctions were measured because they are very scarce. The frequency of peripheral couplings is also modulated by the expression levels of junctate: pShag-1-RNAi-junctate–treated cells show on the average 0.1 peripheral couplings/cell profile; control cells have 0.5 peripheral couplings/cell profile and junctate overexpressing cells have one peripheral coupling/cell profile (Table II). Thus, the contacts between the ER and the plasma membrane are dependent on the level of expression of junctate; more frequent and extended in junctate overexpressing cells and scarce and short when it is expressed at very low levels.
Figure 6.

Overexpression of junctate results in larger peripheral couplings. The ER forms junctions with the plasma membrane (peripheral couplings, between arrows). The area of close apposition is smaller in the control (A) than in junctate overexpressing (B–D) T3-HEK293 cells (see also Table II).
Table II. ER–plasma membrane junction (peripheral coupling) statistics.
| Cell type | Average length of junction (mean ± SD) | No. of junctions/cell profile |
|---|---|---|
| (A) Sorted GFP-expressing T3-HEK293 cells | 218 ± 94 nm (n = 165 junctions) | 0.5/cell (n = 112 cells) |
| (B) Sorted GFP-junctate–expressing T3-HEK293 cells | 339 ± 51 nm (n = 126 junctions)* | 1/cell (n = 100 cells)** |
| (C) Sorted GFP-pShag-RNAi-junctate T3-HEK293 cells | 139 ± 51 nm (n = 15 junctions)*** | 0.1/cell (n = 108 cells)**** |
t test: A versus B,
P < 0.0001; A versus B,
P < 0.007; A versus C,
P < 0.002; A versus C,
P < 0.009.
Junctate modulates calcium entry via TRPC3 channels
We performed a set of experiments to gain insight into the molecular and functional basis of the peripheral couplings in T3-HEK293 cells. Microsomal membrane fractions from T3-HEK293 cells contain a 33-kD integral membrane protein, which is identified as junctate because it is immunodecorated with antijunctate Ab (Fig. 7 A, lane 2). Because TRPC3 interacts with the InsP3R and our data shows that the InsP3R interacts with junctate, we investigated whether junctate forms a complex with the TRPC3 channel. The NH2-domain of junctate pulled down the TRPC3 channel (Fig. 7 A, lane 4), whereas the corresponding scrambled peptide was unable to do so (Fig. 7 A, lane 3). We then examined whether the InsP3R is also a component of the complex by quantifying the junctate-InsP3R complex in HEK293 (i.e., wild-type cells) and in T3-HEK293 cells, by using a peptide pull-down technique (Lund-Johansen et al., 2000; see Fig. 7 B for a schematic representation of the experiment). We found that when T3-HEK293 cell extracts were used, there was a 3.6-fold increase in the association of InsP3R to the beads coated with the peptide corresponding to the NH2 terminus of junctate (Fig. 7 C). The presence of the TRPC3 protein increased the geometric mean fluorescence intensity from a mean intensity of 386 for HEK293 cells to 776 for T3-HEK293 cells. In HEK293 cells, the geometric mean fluorescence level generated by the NH2 terminus of junctate is slightly higher compared with that obtained when the beads were coated with an unrelated biotinylated peptide (386 and 237, respectively). This result reflects binding via endogenous TRPC3 channels, which have been reported to be expressed endogenously at low levels in wild-type HEK293 cells (Zhu et al., 1998).
Figure 7.

A peptide encompassing the cytoplasmic domain of junctate complexes with the InsP3R via TRPC3 and inhibits TRPC3 current induced by carbachol in HEK293 cells stably expressing TRPC3. (A) Microsomal membrane fraction from T3-HEK293. Lane 1, Ponceau red staining; lane 2, antijunctate Ab immunostaining. Arrow indicates the band corresponding to junctate. The microsomal proteins of T3-HEK293 were solubilized, pulled down with Streptavidine beads coated either with a biotinylated peptide corresponding to the NH2 terminus of junctate (lane 4) or a biotinylated peptide corresponding to the scrambled sequence of the NH2- terminal domain of junctate (lane 3), and stained with anti–HA-tag Ab (lanes 3 and 4, respectively). (B) Model depicting the experimental approach used in C. (C) Peptide pull-down reactions analyzed by flow cytometry. Beads used in the pull down were coated with either an irrelevant peptide (gray lines, unshaded histograms) or junctate-derived peptide (shaded histograms). After incubation with cell lysates, beads were stained with goat anti-InsP3R type 3, followed by anti–goat IgG-FITC (x axis, log scale of geometric mean fluorescence intensity; y axis, number of events counted) and analyzed by flow cytometry. The geometric mean fluorescence intensity readout values are noted above each peak. (D) Immunoreactivity of total lysates prepared from HEK293 cells (lanes 1, 3, and 5) and T3-HEK293 cells (lanes 2, 4, and 6). Total lysates from 5 × 104 (lanes 1 and 2), 105 (lanes 3 and 4), and 1.5 × 105 cells (lanes 5 and 6) were stained with anti-InsP3R type 3 Ab. (E–G) Cells were clamped at −40 mV and stimulated for 40 s with 200 μM carbachol (CCh, black bar) 8 min after establishment of the whole cell configuration (t = 0 min). Recordings of TRPC3 currents in control cells (E) and peptide dialyzed cells (F). Histogram (G) showing mean ± SD of control and peptide-treated current densities of seven different sets of experiments performed with wild-type HEK293 and T3-HEK293 cells. *P < 0.02.
The increase in fluorescent signal could be due to a higher content of InsP3R in T3-HEK293 cells compared with that of HEK293, or to an increase in the association between TRPC3, InsP3R and junctate. To exclude the former possibility, we performed Western blot analysis of the total soluble extracts of HEK293 and T3-HEK293 cells with anti-type 3 InsP3R Ab. No appreciable changes in the quantity of InsP3R expression was found between HEK293 and T3-HEK293 (Fig. 7 D) supporting the hypothesis that the presence of TRPC3 channel protein, enforces the association of the supramolecular complex. When extracts were incubated with the unrelated biotinylated peptide the mean fluorescence intensity was the same for HEK293 and T3-HEK293 cells (237 and 236, respectively; Fig. 7 C, gray lines, unshaded histogram) and is due to nonspecific fluorescence of the FITC-conjugated anti–goat antibodies binding to the beads.
On the basis of these data, we would expect that intracellular perfusion in T3-HEK293 cells of the peptide encompassing the NH2 terminus of junctate would compete with the endogenous junctate and affect calcium influx via the TRPC3 channels. We performed patch clamp experiments in the whole cell configuration on T3-HEK293 cells. Bath application of 200 μM of the muscarinic agonist carbachol produced an inward current, previously characterized as being mostly carried by TRPC3 channels (Hurst et al., 1998). To test the effect of the NH2-domain of junctate, the corresponding peptide (5 μM) was introduced into the pipette solution and allowed to diffuse into the cytoplasm for 8 min after establishment of the whole cell configuration. Peptide dialysis produced a significant decrease (70%; P < 0.02; Fig. 7 G) of the peak inward current density induced by carbachol (Fig. 7 E). Fig. 7 (E and F) shows representative recordings of inward current induced by carbachol in control cells and in cells dialyzed with the junctate peptide, respectively. It is important to note that dialysis of peptide alone did not induce the appearance of any current (Fig. 7 F). Furthermore, dialysis with the corresponding scrambled peptide (10 μM) did not induce significant changes of the inward current amplitude (not depicted). Wild-type HEK293 cells showed negligible current densities (<0.05 pA/pF; Fig. 7 G).
Effect of junctate knockdown in T3-HEK293 cells
To investigate the role of endogenous junctate on [Ca2+]i of T3-HEK293 we cotransfected cells with the pShag-1 RNAi construct and the reporter plasmid p enhanced YFP (EYFP). Control cells were transfected with the pEYFP plasmid. From both groups, YFP-positive cells were sorted (Fig. 8 A). In RNAi-treated cells the mRNA encoding junctate was less abundant than in pEYFP-transfected control cells (Fig. 8 B). The presence of residual mRNA for junctate could be due either to the presence of a small subpopulation of cells which had been transfected only with the marker plasmid, or to the inability of the construct to fully eliminate the expression of junctate. The amount of β-actin transcript did not differ between cells transfected with the two constructs (Fig. 8 B, compare lanes 1 and 2 to lanes 3 and 4, top and bottom). Depletion of junctate in pShag-1-RNAi-junctate–treated cells was also confirmed at the protein level by Western blot analysis (Fig. 8 C). The RNAi construct we used specifically degraded the mRNA specific for junctate because (a) the level of β-actin mRNA was unchanged and (b) the expression of other key proteins involved in calcium signaling such as the InsP3R and the TRPC3 channel was not affected (Fig. 8 C). We also performed an additional control to ensure the specificity of the RNAi protocol. In mammalian cells RNAi has been reported to induce an interferon response, which leads to nonspecific mRNA degradation (Sledz et al., 2003). We tested for such a response in our cellular model by measuring the production of α and β interferons. We did not observe significant differences in the production of either interferon in RNAi-treated T3-HEK293 cells (unpublished data).
Figure 8.
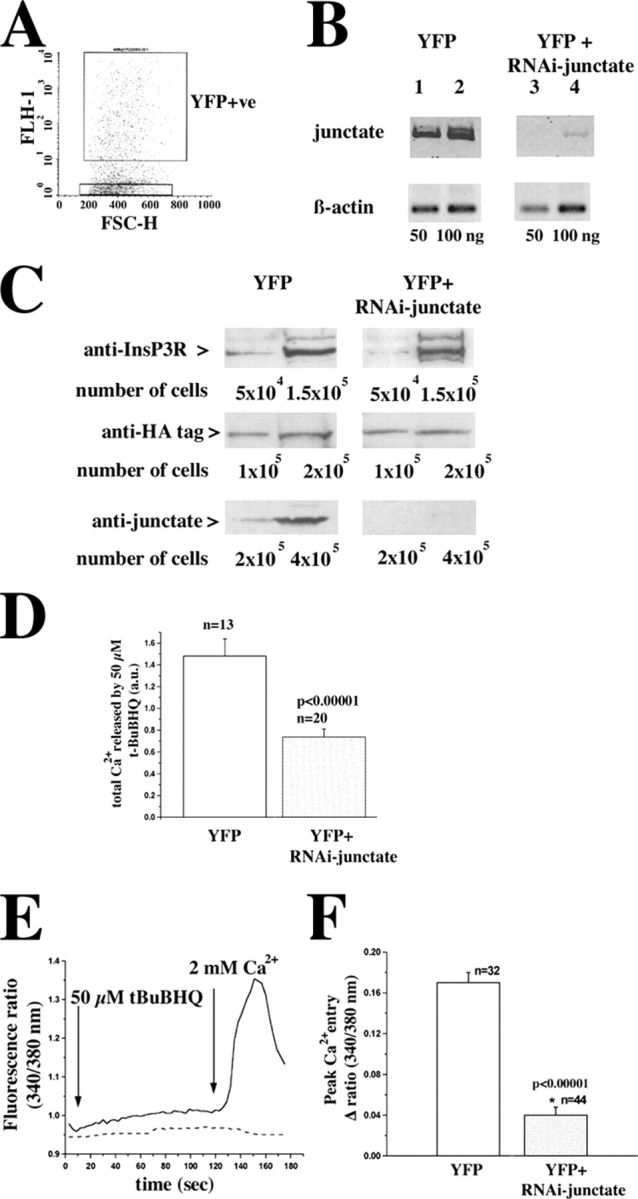
Transfection of T3-HEK293 cells with RNAi-junctate plasmid reduces the junctate transcript and protein level, affects intracellular store calcium content and diminishes store depletion-mediated calcium entry. (A) 60 h after transfection cells were sorted and YFP-positive cells were selected; the boxed scattergram represents a typical experiment in which ∼33% of all sorted cells were YFP positive. The purity of the YFP-positive population of cells was >90% as determined by reanalysis of the sorted cells by FACS® as well as fluorescence microscopy. (B) Total RNA was extracted from the YFP-positive cells. RNA was converted into cDNA and semiquantitative RT-PCR was performed on 50 and 100 ng RNA. The figure is representative of three different transfection experiments. (C) Total cellular protein extracts were prepared from the indicated number of YFP-positive cells as described in the methods section. Proteins were stained with affinity-purified antijunctate antibodies, anti-HA antibodies, and anti-InsP3R antibodies. (D) Calcium content of the stores was determined by perfusing T3-HEK393 transfected with 50 μM t-BuBHQ in nominally Ca2+-free Krebs-Ringer; mean ± SEM; n = cells from two transfections; a.u., arbitrary units. (E and F) YFP-positive T3-HEK293 cells were selected and intracellular Ca2+ measurements were performed. (E) pEYFP-transfected (continuous line) and in pEYFP+pShag-RNAi-junctate–transfected cells (dashed line). (F) T3-HEK293 cells transfected with pEYFP (unshaded histogram) and pEYFP+pShag-RNAi-junctate (shaded histogram). Values are mean ± SEM; n = number of cells from three different transfections.
Because in COS-7 cells junctate overexpression leads to alterations of intracellular calcium stores, we reasoned that depletion of junctate would similarly affect intracellular calcium pools. We assessed the releasable calcium of the stores, by measuring the integral of the calcium signal induced by 50 μM t-BuBHQ in both pEYFP-control and RNAi-treated cells in the presence of 0.5 mM EGTA in the external medium. Depletion of junctate induced a twofold decrease of the releasable calcium by t-BuBHQ (Fig. 8 D). Next, we determined whether knocking down junctate also affected calcium entry induced by store depletion (Fig. 8 E, continuous trace pEYFP-transfected cells; dashed trace RNAi-junctate–treated cells). Intracellular calcium stores were first equally and completely depleted by loading cells with fura-2/AM in the presence of EGTA. This maneuver was effective in depleting Ca2+ stores because t-BUBHQ induced a negligible calcium transient. pEYFP-transfected cells exhibited a large calcium entry transient (Fig. 8 E, continuous line; Fig. 8 F, unshaded box, mean peak calcium entry was 0.17 ± 0.01 fluorescence units), whereas in pEYFP /pShag-1-RNAi-junctate cotransfected cells, the transient was reduced near to zero (Fig. 8 E, dashed line; Fig. 8 F, shaded box, mean peak calcium entry was 0.04 ± 0.008 fluorescence units; P < 0.00001).
To further substantiate the role of junctate on calcium signaling, we also monitored the changes in [Ca2+]i induced by ATP in pShag-1-RNAi-junctate–transfected cells by studying the [Ca2+]i of single T3-HEK293 cells. Based on the presence YFP expressing cells, we measured Ca2+ fluxes in groups of 5–10 cells. Stimulation of cells with 100 μM ATP induced a rapid and transient increase in [Ca2+]i (Fig. 9, C and I) which was reduced in T3-HEK293 cells in which endogenous junctate had been knocked down. The decrease in the amplitude of calcium release is evident not only in all the cells cotransfected with the pEYFP, but also in two adjacent cells which apparently did not pick up the pEYFP plasmid (Fig. 9, G and I). This is probably due to the fact that transfection efficiency is lower with larger plasmids, thus it is likely that the cells which did not exhibit the YFP fluorescence were transfected with the smaller construct carrying the RNAi cassette. Once the calcium signal returned to basal level (Fig. 9, D and J) calcium entry was measured (Fig. 9, E and K). Cells transfected with pEYFP alone exhibited a larger agonist-activated calcium entry compared with cells transfected with pShag-1-RNAi-junctate (Fig. 9 N).
Figure 9.
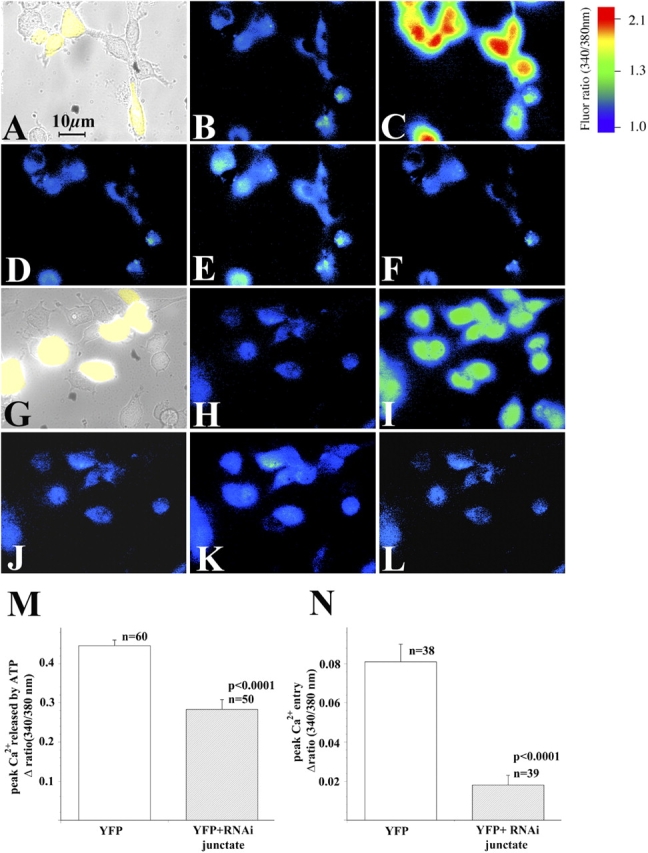
Intracellular calcium measurements of T3-HEK293 cells depleted of junctate by RNAi. Ca2+ measurements in fura-2 loaded T3-HEK293 cells were performed 60 h after transfection with pEYFP (A–F) and with pEYFP+pShag-RNAi-junctate plasmids (G–L). (B and H) Resting [Ca2+]i; (C and I) peak [Ca2+]i after addition of 100 μM ATP in nominally Ca2+-free Krebs-Ringer; (D and J) [Ca2+]i returned to resting level 25 s after addition of ATP; (E and K) calcium entry induced by replacement of EGTA-containing extracellular medium, to Krebs-Ringer containing 2 mM CaCl2, in the continuous presence of 100 μM ATP; (F and L) [Ca2+]i returned to resting levels by adding 0.5 mM La3+ to Krebs-Ringer containing 100 μM ATP and 2 mM Ca2+. Images were acquired at 340/380 nm and ratioed every 0.75 s. (M and N) Statistical analysis of mean (±SEM) peak [Ca2+]i induced by the addition of 100 μM ATP and refilling of calcium pools induced by the readdition of 2 mM CaCl2 in YFP-transfected T3-HEK293 cells (unshaded histograms) and YFP+RNAi-junctate–transfected cells (shaded histograms); n = number of cells analyzed. At least three different transfection experiments were performed and cells from two coverslips were analyzed per transfection experiment.
Discussion
In the present paper, we investigated the role of junctate in calcium homeostasis in eukaryotic cells. Our results suggest that junctate plays an important role both in calcium release after InsP3R activation and in calcium entry induced by agonist-receptor activation and store depletion, perhaps by facilitating and/or stabilizing connections between the ER and the plasma membrane.
Role of junctate in agonist-activated calcium release
The COOH-terminal domain of junctate is a moderate affinity, high capacity calcium binding domain. As expected, overexpression either of the full-length protein or of the lumenal COOH-domain induces an increase in the amount of releasable calcium from the stores sensitive to the SERCA inhibitor t-BuBHQ. However, although overexpression of full-length junctate also increases the amount of calcium released by ATP-mediated receptor activation, the COOH-terminal domain does not. Thus, the COOH domain acts similarly to calreticulin, a high capacity calcium binding protein uniformly distributed within the lumen of the ER. Overexpression of calreticulin increases the calcium content of ER calcium stores, but does not affect InsP3-mediated calcium release (Bastianutto et al., 1995; Xu et al., 2000).
The free calcium concentration in the endoplasmic reticulum lumen has been calculated to be ∼500–600 μM (Elliot, 2001). In the presence of physiological concentrations of KCl, junctate binds calcium with an affinity of 217 μM. Thus, under resting conditions the calcium binding sites of junctate are saturated by calcium. Our results clearly demonstrate that in vivo junctate interacts with the InsP3R and though we do not exactly know how many junctate binding sites there are in the InsP3R complex, the simplest stoichiometric ratio is one junctate binding site per InsP3R complex. This implies that under resting conditions one mole of the junctate-InsP3R complex binds at least 21 moles of calcium and these are adjacent to the inner leaflet of the ER membrane. Thus, the moderate affinity calcium binding sites of junctate are close to the lumenal mouth of the InsP3R and it is tempting to speculate that an interaction between junctate's NH2 terminus and InsP3R causes a local increase of releasable Ca2+ upon agonist-receptor activation. This hypothesis is in agreement with the results obtained by the junctate RNAi experiments: knocking down junctate induced a decrease in the amplitude of agonist-induced calcium release. Measurements of [Ca2+]ER during receptor-mediated stimulation of calcium release have shown that the amplitude of InsP3R-mediated calcium release declines to near zero below a lumenal calcium concentration of 100 μM (Barrero et al., 1997), and at such a free [Ca2+]ER only ∼10% of junctate's calcium binding sites would be occupied (Treves et al., 2000). Under these conditions, the junctate-associated calcium would be too low to drive further release of calcium via the InsP3R and would thus result in the decline in calcium release, which has been experimentally demonstrated.
These data suggest that the local releasable calcium adjacent to the lumenal mouth of the ER calcium release channel, as opposed to the global ER calcium content, is the major limiting factor influencing InsP3-induced calcium release events triggered by activation of surface receptors.
Role of junctate on calcium entry
The elucidation of store- and receptor-activated calcium entry has emerged as a major theme in the field of cellular signaling in a variety of cell types. According to the conformational coupling hypothesis the InsP3R localized next to the plasma membrane interacts with the calcium channel responsible for calcium entry (Irvine, 1990; Kiselyov et al., 2000). The membrane compartment containing the InsP3R and the calcium entry channel may be endowed with other protein components that could be involved in the calcium entry mechanism. The ultrastructural and functional data reported here not only support this model, but also identify junctate as a candidate directly involved both in the assembly of the supramolecular complex and in the modulation of its function. In T3-HEK293 cells a small amount of ER extends throughout the cytoplasm and approaches the plasma membrane, where it forms discrete, small contacts with the plasma membrane (peripheral couplings). In the presence of extra amounts of junctate, the ER of T3-HEK293 cells is much more extensive, and peripheral couplings are more frequent and larger. Considering the dynamics of the ER, one likely explanation is that the ER is continuously reaching toward the plasma membrane and retracting from it and that junctate either induces an association between the ER and the plasma membrane or stabilizes it once it is formed. Coimmunoprecipitation and pull-down assay are consistent with such a view and indicate that the molecular basis of the peripheral coupling in T3-HEK293 cells is a heteroligomeric complex formed by junctate, IP3R and TRPC3 channels.
In addition to an indirect role mediated by an increase in peripheral couplings, our results suggest a more direct effect of junctate on calcium entry and indicate that this role may differ depending on how entry is elicited in an activated cell. Modulation of receptor-activated calcium entry is affected by the NH2 terminus of junctate, i.e., the domain of the protein interacting with the InsP3R. This interaction is not sufficient per se to modulate calcium entry, because the PLC inhibitor U73122 blocked the modulatory effect. This suggests that junctate affects receptor-activated calcium entry by modulating the steps which link the ligand-bound InsP3R and the plasma membrane calcium channel. It is plausible that initiation of the retrograde signal for stimulation of the receptor-activated calcium entry requires ligand-bound InsP3R. Binding of InsP3 to its receptor unmasks the effect on calcium entry, which derives from the interaction between the InsP3R and the NH2-terminal domain of junctate. In contrast to receptor-activated calcium entry, modulation of calcium entry induced by store depletion requires the entire junctate molecule: the overexpression of either the NH2-terminal domain or the COOH-terminal domain does not modulate store-depletion activated calcium entry, whereas it is significantly diminished in cells in which endogenous junctate has been knocked down. The retrograde signal activating store-depletion activated calcium entry is probably initiated by the removal of the calcium bound to the calcium binding domain of junctate, which is adjacent to the lumenal mouth of the release channel. Such a signal would be sensed by the calcium entry channel via the ligand-free InsP3R.
In conclusion, our data demonstrate that junctate, a protein of sarco(endo)plasmic reticulum membranes, is a novel component of the supramolecular machinery involved in calcium entry.
Materials and methods
COS-7 and HEK293 cells were obtained from American Type Culture Collection. Peroxidase-conjugated anti–mouse, anti–rat IgG, and protein-A Sepharose were purchased from Sigma-Aldrich. Indo-1/AM, Fura-2/AM, Mag-Fura-2/AM, and the calcium calibration kit were purchased from Molecular Probes. ATP, EDTA-free protease inhibitor cocktail tablets, anti-HA antibodies, ECL reagents, and DNA modifying enzymes were purchased from Roche Molecular Biochemicals. 2,5-Di(t-butyl)-1,4-hydroquinone (t-BuBHQ), U73122, U73343, and 2-APB were purchased from Calbiochem. Streptavidin-conjugated Dynabeads were purchased from Dynal. The pSilencer kit was purchased from Ambion. The pGex plasmid, glutathione-Sepharose 4B, and nitrocellulose were purchased from Amersham Biosciences. The pEGFP and pEYFP plasmids were purchased from CLONTECH Laboratories, Inc. U6 RNA polymerase promoter pShag-1 vector was a gift of G. Hannon (Cold Spring Laboratories, Cold Spring Harbor, NY). Tissue culture reagents and Lipofectin were purchased from Invitrogen. Triton X-100, CaCl2, and La3+ were purchased from Merck. Goat anti-InsP3R Abs were purchased from Biosource International. Peroxidase-conjugated protein A was purchased from Fluka. Streptavidin was purchased from Southern Biotechnology Associates, Inc. Polystyrene latex beads were purchased from Interfacial Dynamics Corporation. FITC-conjugated anti–goat IgG was purchased from BD Biosciences. All other chemicals were reagent or highest available grade.
EGFP-junctate constructs
Junctate-EGFP, EGFP-junctate, and EYFP-junctate, were cloned into the MCS of the pEGFPN3, pEGFPC1, and pEYFPC1 plasmids, respectively. NH2-TM-EGFP and NH2-TM-EYFP, in which the first 85 aa of junctate, including the transmembrane domain, were cloned into the MCS of pEGFPN3 and pEYFPN3, respectively. EGFP-TM-COOH and EYFP-TM-COOH in which the transmembrane and COOH domain of junctate were cloned into the MCS of pEGFPC1 and pEYFPC1, respectively. For the latter four constructs cDNA was amplified by PCR using the following forward and reverse primers: 5′-AGAATTCACAAATGGCTGAAG-3′ and 5′-GGGATCCCTTTGCTTTGGCTAGA-3′ for the NH2-TM-EGFP(EYFP) constructs; and 5′-GGAATTCCACCATGAGGAAAGGCGGACTCTCA-3′ and 5′-GAAGCTTTTAGGATCCTGGTG-3′ for the EGFP (EYFP)-TM-COOH constructs. All DNA constructs were checked by sequencing.
Expression of recombinant junctate in COS-7 cells and intracellular calcium measurements
Expression of GFP-junctate and single cell intracellular calcium measurements in COS-7 cells were performed as described previously (Treves et al., 2000). Measurements were made on cells from four distinct transfections.
RNAi for junctate with small double-stranded RNA
The RNAi oligo (5′-GATCAAAAAAGTGTACCATTTCATGGAGGGACTGAATTCAAGCTTCAATTCAGTCCCTTCTCCATGAAATGGTACG-3′) was annealed with equimolar amounts of the complementary oligo missing nucleotides CG and GATC at the 5′ and 3′ ends, respectively. The annealed oligos were subcloned into the U6 RNA pol III promoter pShag-1 vector. The nucleotide sequence of the construct was verified by sequencing. RT-PCR on YFP-positive cells using junctate specific primers (5′-TCAAAAAGACTGCCCCTACC-3′ and 5′-GGACATGCAGGAATGTAACATGAC-3′ for forward and reverse, respectively) or human β-actin specific primers (5′-TGACGGGGTCACCCAC-3′ and 5′-CTAGAAGCATTGCGGT-3′ for forward and reverse, respectively). YFP-positive T3-HEK293 cells were sorted in a FACSVantageTM SE sorter (model 621, Becton Dickinson; 488 nm Enterprise laser, filter 530/30).
For single cell intracellular measurements, transfected T3-HEK293 cells were loaded with 5 μM fura-2/AM; YFP-positive cells were identified using an inverted microscope (model Axiovert S100 TV; Carl Zeiss MicroImaging, Inc.) using a 40× Plan-Neofluar objective (NA = 1.3) and filter (Carl Zeiss MicroImaging, Inc.) set N°44 (BP 475/40, FT 500, BP 530/50). Intracellular calcium measurements were obtained using a 40× plan-Neofluar oil-immersion objective, at 340 and 380 nm excitation and 510 nm emission (filters BP 340/ BP 380; FT 425; 500–530). Fluorescence values obtained at 340 and 380 nm were acquired with a multiformat CCD C4880-80 camera (Hamamatsu), ratioed and the [Ca2+]i was analyzed using an Openlab imaging system. Calcium calibration was performed following the instructions provided with the calcium calibration kit. The [Ca2+]ER was determined in cells loaded with 2 μM Mag-Fura-2/AM as described by Solovyova et al. (2002).
Subcellular fractionation of HEK293
Subcellular fractions from T3-HEK293 cells stably transfected with the HA-tagged TRPC channel protein (Zhu et al., 1998) were prepared as described previously (Treves et al., 2000).
Coimmunoprecipitation and pull-down experiments
Total bovine cerebellum microsomal proteins (2 mg/ml) or integral membrane proteins extracted from microsomes with 100 mM Na2CO3 as described previously (Treves et al., 2000) were solubilized in: 1% Triton X-100, 200 mM NaCl, and 50 mM Tris-HCl, pH 7.4, at RT for 60 min in the presence of a protease inhibitor cocktail. Insoluble material was removed by centrifugation. The solubilized fraction was diluted fivefold with 200 mM NaCl, and 50 mM Tris-HCl, pH 7.4. Affinity-purified polyclonal antijunctate antibodies (Treves et al., 2000) were incubated with 200 μg solubilized bovine cerebellum microsomes for 2 h at RT, followed by incubation with 30 μl protein-A Sepharose. After 1 h, the beads were washed three times with 200 mM NaCl, 50 mM Tris-HCl, pH 7.4; bound proteins were separated on a SDS-PAG and blotted onto nitrocellulose.
For the pull-down experiments, microsomal proteins from T3-HEK293 cells, rabbit brain, and lung microsomes were washed with 0.6 M KCl, solubilized as described above, and incubated for 60 min with Streptavidin beads coated with 10 μg of a biotinylated peptide corresponding either to the NH2 terminus of junctate or to its scrambled sequence, and GST–COOH junctate. The beads were washed three times with 0.1% Triton X-100, 200 mM NaCl, 1 mM EDTA, and 50 mM Tris-HCl, pH 7.4, and Western blots of bound protein were stained with antibodies. GST–COOH-terminal domain of junctate was prepared as described previously (Treves et al., 2000). Protein concentration was measured as described previously (Treves et al., 2000).
Electrophoresis and immunoblotting
SDS-PAGE, protein transfer onto nitrocellulose and immunostaining were performed as described previously (Treves et al., 2000). Blots were probed with a polyclonal anti-InsP3R, or monoclonal anti-InsP3R type 1, 2, and 3 antibodies (Sugiyama et al., 1994), with affinity-purified antijunctate Ab (Treves et al., 2000) and anti-HA antibodies.
Electrophysiology
Recordings were performed on T3-HEK293 cells stably expressing TRPC3, in the whole cell configuration of the patch clamp technique. The pipette solution contained (mM) CaCl2 2, CsCl 120, EGTACs 5, Hepes 10, MgCl2 2, pH 7.2. Cells were bathed in Ringer's solution containing (mM) NaCl 140, KCl 5.4, MgCl2 1, CaCl2 2, d-glucose 10, and Hepes 10, pH 7.4. Pipettes (5–7 MΩ were pulled from Kimax 51 glass (Kimball). Cells were clamped with an Axopatch 200B amplifier (Axon Instruments) and currents analyzed with pClamp 6.0. Current was filtered at 1 kHz. Holding potential was set at –40 mV.
Detection of peptide pull downs by flow cytometry
Streptavidin was covalently conjugated to ∼3.2-μm-diam carboxylate-modified polystyrene latex beads. The beads were then incubated in 150 mM NaCl, 10 mM Hepes, pH 7.4, for 30 min at RT with 100 μg/ml biotinylated peptide corresponding either to the NH2 terminus of junctate or an irrelevant peptide (biotin-SHIPGLRPSQQQQL). After washing, the beads were incubated with postnuclear lysates (150 mM NaCl, 50 mM Tris-HCl, pH 8.0, 6 mM EDTA, 1% NP-40, 0.5% deoxycholate; Boulay et al., 1999) from 2 × 105 HEK293 or T3-HEK293 cells for 60 min at RT. Beads were washed and stained with goat anti-InsP3R type 3 and subsequently with anti–goat IgG-FITC. Bead fluorescence was acquired on a FACScalibur cytometer (Becton Dickinson Immunocytometry Systems); geometric mean fluorescence intensity calculated using CellQuest software.
LM and EM analysis of GFP-junctate expressing cells
Transfected COS-7 cells were examined 48 h after transfection using an inverted microscope (model Diaphot 300; Nikon) equipped with a PlanApo × 100/1.4 oil-immersion objective under fluorescent light (excitation 480 nm; emission 510 nm) as described previously (Treves et al., 2000). FACS-sorted GFP-positive T3-HEK293 cells were fixed in 3% glutaraldehyde in PBS and pelleted. The cells were maintained in the fixative for several days. The pellets were fixed after in osmium tetraoxide, en-bloc “stained” in uranyl acetate, and embedded. Thin sections showed 1–200 profiles of cells/section cut in random orientations. The section were stained in uranyl acetate and lead containing solutions and examined in an electron microscope (model 410; Philips).
Statistical analysis
Statistical analysis was performed using the t test for unpaired samples; means were considered statistically significant when the P value was <0.05.
Acknowledgments
We would like to thank: Evgueni Voronkov and Verena Jäggin for expert technical assistance; Professors Ed Palmer, Gennaro DeLibero, and George Holländer for helpful suggestions; and Professor Alex Eberle for the biotinylated unrelated peptide.
This work was supported by grants from Ministero Universita' e Ricerca scientifica e Tecnologica 40%, F.I.R.B. RBAUO01ERMX, HPRN-CT-2002-00331 from the European Union, by the Dept. of Anaesthesia, Kantonsspital Basel and by National Institute of Health grants AR PO144650 to CFA and RO1 NS42183 to M.X. Zhu.
Abbreviations used in this paper: 2-APB, 2-aminoethyl diphenylborate; EYFP, enhanced YFP; InsP3, inositol 1,4,5 trisphosphate; InsP3R, InsP3 receptor; RNAi; RNA interference; SERCA, sarcoplasmic and ER Ca2+ATPase; t-BuBHQ, Di-tert-butylhydroquinone; TRPC, canonical transient receptor potential protein.
References
- Barrero, M.J., M. Montero, and J. Alvarez. 1997. Dynamics of [Ca2+] in the endoplasmic reticulum and cytoplasm of intact HeLa cells. A comparative study. J. Biol. Chem. 272:27694–27699. [DOI] [PubMed] [Google Scholar]
- Bastianutto, C., E. Clementi, F. Codazzi, P. Podini, F. De Giorgi, R. Rizzuto, J. Meldolesi, and T. Pozzan. 1995. Overexpression of calreticulin increases the Ca2+ capacity of rapidly exchanging Ca2+ stores and reveals aspects of their lumenal microenvironment and function. J. Cell Biol. 130:847–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge, M.J., P. Lipp, and M.D. Bootman. 2000. The versatility and universality of calcium signaling. Nat. Rev. Mol. Cell Biol. 1:11–21. [DOI] [PubMed] [Google Scholar]
- Bootman, M.D., T.J. Collins, L. MacKenzie, H.L. Roderick, M.J. Berridge, and C.M. Peppiat. 2002. 2-aminoethoxydiphenyl borate (2-APB) is a reliable blocker of store-operated Ca2+ entry but an inconsistent inhibitor of InP3-induced Ca2+ release. FASEB J. 16:1145–1150. [DOI] [PubMed] [Google Scholar]
- Boulay, G., D.M. Brown, N. Qin, M. Jiang, A. Dietrich, M.X. Zhu, Z. Chen, M. Birnbaumer, K. Mikoshiba, and L. Birnbaumer. 1999. Modulation of calcium entry by polypeptides of the IP3R that bind TRP: evidence for roles of TRP and IP3R in store depletion-activated Ca2+ entry. Proc. Natl. Acad. Sci. USA. 96:14955–14960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapham, D.E. 1995. Calcium signaling. Cell. 80:259–268. [DOI] [PubMed] [Google Scholar]
- Clapham, D.E. 2003. TRP channels as cellular sensors. Nature. 426:517–524. [DOI] [PubMed] [Google Scholar]
- Dinchuk, J.E., N.L. Henderson, T.C. Burn, R. Huber, S.P. Ho, J. Link, K.T. O'Neil, R.J. Focht, M.S. Scully, J.M. Hollis, et al. 2000. Aspartyl beta-hydroxylase (Asph) and an evolutionarily conserved isoform of Asph missing the catalytic domain share exons with junctin. J. Biol. Chem. 275:39543–39554. [DOI] [PubMed] [Google Scholar]
- Elliot, A.C. 2001. Recent developments in non-excitable cell calcium entry. Cell Calcium. 30:73–93. [DOI] [PubMed] [Google Scholar]
- Franzini-Armstrong, C., and F. Protasi. 1997. Ryanodine receptors of striated muscles: a complex channel capable of multiple interactions. Physiol. Rev. 77:699–729. [DOI] [PubMed] [Google Scholar]
- Hong, C.S., Y.G. Kwak, J.H. Ji, S.W. Chae, and D. Han Kim. 2001. Molecular cloning and characterization of mouse cardiac junctate isoforms. Biochem. Biophys. Res. Commun. 289:882–887. [DOI] [PubMed] [Google Scholar]
- Hurst, R.S., X. Zhu, G. Boulay, L. Birnbaumer, and E. Stefani. 1998. Ionic currents underlying HTRP3 mediated agonist-dependent Ca2+ influx in stably transfected HEK293 cells. FEBS Lett. 422:333–338. [DOI] [PubMed] [Google Scholar]
- Irvine, R.F. 1990. “Quantal” Ca2+ release and the control of Ca 2+ entry by inositol phosphates-a possible mechanism. FEBS Lett. 263:5–9. [DOI] [PubMed] [Google Scholar]
- Kiselyov, K., X. Xu, G. Mozhayeva, T. Kuo, I. Pessah, G. Mignery, X. Zhu, L. Birnbaumer, and S. Muallem. 1998. Functional interaction between InsP3 receptor and store-operated Htrp3 channels. Nature. 396:478–482. [DOI] [PubMed] [Google Scholar]
- Kiselyov, K.I., D.M. Shin, Y. Wang, I.N. Pessah, P.D. Allen, and S. Muallem. 2000. Gating of store-operated channels by conformational coupling to ryanodine receptors. Mol. Cell. 6:421–431. [DOI] [PubMed] [Google Scholar]
- Kwan, C.Y., H. Takemura, J.F. Obie, O. Thastrup, and J.W. Putney Jr. 1990. Effect of MeCh, thapsigargin, La3+ on plasmalemma and intracellular Ca2+ transport in lacrimal acinar cells. Am. J. Physiol. 258:C1006–C1015. [DOI] [PubMed] [Google Scholar]
- Lund-Johansen, F., K. Davis, J. Bishop, and R. de Waal Malefyt. 2000. Flow cytometric analysis of immunoprecipitates: high-throughput analysis of protein phosphorylation and protein-protein interactions. Cytometry. 39:250–259. [DOI] [PubMed] [Google Scholar]
- Ma, H.T., R.L. Patterson, D.B. van Rossum, L. Birnbaumer, K. Mikoshiba, and D.L. Gill. 2000. Requirement of the inositol trisphosphate receptor for activation of store-operated Ca2+ channels. Science. 287:1647–1651. [DOI] [PubMed] [Google Scholar]
- Marty, I., M. Robert, M. Villaz, K. De Jongh, Y. Lai, W.A. Catterall, and M. Ronjat. 1994. Biochemical evidence for a complex involving dihydropyridine receptor and ryanodine receptor in triad junctions of skeletal muscle. Proc. Natl. Acad. Sci. USA. 91:2270–2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakai, J., R.T. Dirksen, H.T. Nguyen, I.N. Pessah, K.G. Beam, and P.D. Allen. 1996. Enhanced dihydropyridine receptor channel activity in the presence of ryanodine receptor. Nature. 380:72–75. [DOI] [PubMed] [Google Scholar]
- Patterson, R.L., D.B. van Rossum, and D.L. Gill. 1999. Store-operated Ca2+ entry: evidence for a secretion-like coupling model. Cell. 98:487–499. [DOI] [PubMed] [Google Scholar]
- Putney, J.W., Jr., L.M. Broad, F.J. Braun, J.P. Lievremont, and G.S. Bird. 2001. Mechanisms of capacitative calcium entry. J. Cell Sci. 114:2223–2229. [DOI] [PubMed] [Google Scholar]
- Sledz, C.A., M. Holko, M.J. de Veer, R.H. Silverman, and B.R.G. Williams. 2003. Activation of the interferon system by short–interfering RNAs. Nat. Cell Biol. 5:834–839. [DOI] [PubMed] [Google Scholar]
- Smith, R.J., L.M. Sam, J.M. Justen, G.L. Bundy, G.A. Bala, and J.E. Bleasdale. 1990. Receptor-coupled signal transduction in human polymorphonuclear neutrophils: effects of a novel inhibitor of phospholipase C-dependent processes on cell responsiveness. J. Pharmacol. Exp. Ther. 253:688–697. [PubMed] [Google Scholar]
- Solovyova, N., P. Feryhough, G. Glazner, and A. Verkhratsky. 2002. Xestospongin C empties the ER calcium store but does not inhibit IP3-induced Ca2+ release in cultured dorsal root ganglia neurons. Cell Calcium. 32:49–52. [DOI] [PubMed] [Google Scholar]
- Sugiyama, T., A. Furuya, T. Monkawa, M. Yamamoto-Hino, S. Satoh, K. Ohmori, A. Miyawaki, N. Hanai, K. Mikoshiba, and M. Hasegawa. 1994. Monoclonal antibodies distinctively recognizing the subtypes of inositol 1,4,5-trisphosphate receptors: application to the studies on inflammatory cells. FEBS Lett. 354:149–154. [DOI] [PubMed] [Google Scholar]
- Tang, J., Y. Lin, Z. Zhang, S. Tikunova, L. Birnbaumer, and M.X. Zhu. 2001. Identification of a common binding site for calmodulin and inositol 1,4,5-trisphosphate receptor on the carboxyl termini of trp channels. J. Biol. Chem. 276:21303–21310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trebak, M., G. St. Bird, R.R. McKay, and J.W. Putney. 2002. Comparison of human TRPC3 channels in receptor-activated and store-operated modes. J. Biol. Chem. 277:21617–21623. [DOI] [PubMed] [Google Scholar]
- Treves, S., G. Feriotto, L. Moccagatta, R. Gambari, and F. Zorzato. 2000. Molecular cloning, expression, functional characterization, chromosomal localization, and gene structure of junctate, a novel integral calcium binding protein of sarco(endo)plasmic reticulum membrane. J. Biol. Chem. 275:39555–39568. [DOI] [PubMed] [Google Scholar]
- Xu, W., F.J. Longo, M.R. Wintermantel, X. Jiang, R.A. Clark, and S. DeLisle. 2000. Calreticulin modulates capacitative Ca2+ influx by controlling the extent of inositol 1,4,5-trisphosphate-induced Ca2+ store depletion. J. Biol. Chem. 275:36676–36682. [DOI] [PubMed] [Google Scholar]
- Zhang, L., J. Kelley, G. Schmeisser, Y.M. Kobayashi, and L.R. Jones. 1997. Complex formation between junctin, triadin, calsequestrin, and the ryanodine receptor. Proteins of the cardiac junctional sarcoplasmic reticulum membrane. J. Biol. Chem. 272:23389–23397. [DOI] [PubMed] [Google Scholar]
- Zhu, X., M. Jiang, M. Peyton, G. Boulay, R. Hurst, E. Stefani, and L. Birnbaumer. 1996. Trp, a novel gene family essential for agonist-activated capacitative calcium entry. Cell. 85:661–671. [DOI] [PubMed] [Google Scholar]
- Zhu, X., M. Jiang, and L. Birnbaumer. 1998. Receptor-activated Ca2+ influx via human Trp3 stably expressed in human embryonic kidney (HEK)293 cells. Evidence for a non-capacitative Ca2+ entry. J. Biol. Chem. 273:133–142. [DOI] [PubMed] [Google Scholar]