

Abstract
A rapid increase in the tyrosine phosphorylation of the non-receptor tyrosine kinase FAK is a prominent early event in fibroblasts stimulated by a variety of signaling molecules. However, a variety of epithelial cells, including intestinal epithelial cells, show a high basal level of tyrosine phosphorylated FAK that is only slightly further increased by addition of G protein-coupled receptors (GPCR) agonists or growth factors. In this study, we determined whether these stimuli could elicit FAK phosphorylation at serine residues, including Ser-910 and Ser-843. Our results show that multiple agonists including angiotensin II (ANGII), lysophosphatidic acid (LPA), phorbol esters and EGF induced a striking stimulation of FAK phosphorylation at Ser-910 in rat intestinal epithelial IEC-18 cells via an ERK-dependent pathway. In striking contrast, none of these stimuli promoted a significant further increase in FAK phosphorylation at Tyr-397 in these cells. These results were extended using cultures of polarized human colonic epithelial T84 cells. We found that either carbachol or EGF promoted a striking ERK-dependent phosphorylation of FAK at Ser-910, but these agonists caused only slight stimulation of FAK at Tyr-397 in T84 cells. In addition, we demonstrated that GPCR agonists also induced a dramatic increase of FAK phosphorylation at Ser-843 in either IEC-18 or T84 cells. Our results indicate that Ser-910 and Ser-843, rather than Tyr-397, are prominent sites differentially phosphorylated in response to neurotransmitters, bioactive lipids, tumor promoters and growth factors in intestinal epithelial cells.
INTRODUCTION
A large body of evidence has demonstrated that a rapid increase in the tyrosine phosphorylation of the non-receptor tyrosine kinase p125 focal adhesion kinase (FAK) is a prominent early event in fibroblasts stimulated by diverse signaling molecules that regulate cell proliferation, migration and survival, including mitogenic agonists that act via G protein-coupled receptors (GPCRs), growth factors, integrin clustering induced by cell adhesion, bacterial toxins, and activated variants of pp60src [1-6]. Autophosphorylation of FAK at Tyr-397 [7, 8], located N-terminal to the catalytic domain, creates a binding site for the tyrosine kinase Src and other downstream signaling effectors, including PI 3-kinase and phospholipase Cγ.[6]. Subsequent Src-mediated phosphorylation of FAK at Tyr-576, Tyr-577, Tyr-861 and Try-925, is important for the maximal activation of FAK and phosphorylation at Tyr-397 [4, 9, 10]. This model of FAK activation and Tyr-397 phosphorylation is thought to be of central importance for FAK-dependent signaling. The biological importance of FAK-mediated signal transduction is underscored by the fact that this tyrosine kinase plays a fundamental role in embryonic development [11, 12] and in the control of cell migration [9, 12-19], cell cycle progression [20] and apoptosis [21-24]. Furthermore, there is increasing evidence linking overexpression of FAK to the invasive properties of cancer cells [25-31].
More recently, it has become clear that FAK is also phosphorylated at multiple serine residues [32-37]. The proximity of these phosphorylated serine residues to sites at which FAK interacts with other proteins suggests a possible function in the regulation of the assembly of FAK signaling complexes [6]. Recent results from our laboratory demonstrated that FAK phosphorylation at Ser-910 is strikingly stimulated by GPCR agonists, tumor promoting phorbol esters and growth factors through an ERK-dependent pathway in Swiss 3T3 fibroblasts [34-36]. In another study, we demonstrated that stimulation of 3T3 fibroblasts with bombesin, bradykinin or vasopressin induced a rapid and transient increase in FAK phosphorylation at Ser-843 through a Ca2+/calmodulin/CaMKII-dependent pathway [37]. These findings support the hypothesis that FAK phosphorylation at both, tyrosine and serine residues is modulated by a variety of stimuli but do not exclude the possibility that FAK phosphorylation at tyrosine and serine residues could be regulated differentially in some cell types.
FAK is expressed in intestinal epithelial cells and re-plating suspended cultures of these cells onto extracellular matrix proteins induces tyrosine phosphorylation of this enzyme and its association with focal adhesions [38, 39]. However, many groups noticed that the basal level of tyrosine phosphorylated FAK is high in epithelial cells attached to a substratum, eg T84 cells [40], IEC-18 cells [41], SW 620 [42] and liver epithelial cells [43]. These results imply that FAK tyrosine phosphorylation is constitutively active in attached epithelial cells and thus, it might not be further enhanced in response to receptor activation by external soluble stimuli. These considerations prompted us to consider that stimulation of epithelial cells with GPCR agonists or ligands of tyrosine kinase receptors could increase FAK phosphorylation differentially at serine rather than at tyrosine residues.
The results presented here show that stimulation of undifferentiated intestinal epithelial IEC-18 cells with multiple agonists, including angiotensin II (ANGII), lysophosphatidic acid (LPA), phorbol-12,13-dibutyrate (PDB) or EGF induced a striking stimulation of FAK phosphorylation at Ser-910 via an ERK-dependent pathway. In contrast, none of these stimuli promoted a significant increase of FAK phosphorylation at Tyr-397 in these cells. These results were extended using polarized human colonic epithelial T84 cells. We found that either carbachol or EGF promoted a striking ERK-dependent phosphorylation of FAK at Ser-910, but only slight stimulation of FAK at Tyr-397 in these cells. In addition, we demonstrated that GPCR agonists also induced a dramatic increase of FAK phosphorylation at Ser-843 in either IEC-18 or T84 cells. Our results indicate that FAK phosphorylation at Ser-910 and Ser-843, rather than at Tyr-397, is strikingly increased in response to neurotransmitters, bioactive lipids, tumor promoters and growth factors in rat and human intestinal epithelial cells.
EXPERIMENTAL PROCEDURES
Cell culture
Both IEC-18 cells and T84 cells were purchased from American Type Culture Collection. Stock cultures of IEC-18 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 5% fetal bovine serum (FBS) in a humidified atmosphere containing 10% CO2 and 90% air at 37°C. For experimental purposes, cells were plated into 35-mm dishes at 1 × 105 cells per dish, in DMEM containing 5% FBS and were allowed to grow to confluency (6-8 days) before use.
The colonic epithelial cell line T84, was cultured in Petri dishes in DMEM/Ham's F12 media (1:1) supplemented with 10% fetal bovine serum. Cells were cultured in an atmosphere of 5% CO2 at 37°C with medium changes every 3-4 days. For experiments involving western blotting, cells were plated on either 35mm Petri dish at approximately 5×105 per dish or BD Falcon cell culture inserts. Cells were seeded onto 23mm BD Falcon cell culture inserts(0.4μm pore) at approximately 5×105 cells per well, or onto 10.5mm BD Falcon inserts at 1×105 cells per well. Transepithelial resistance was routinely monitored using the EVOM apparatus (Millipore) and cells seeded onto filters were cultured for 6-7 days prior to use. Under these conditions, T84 cells develop the polarized phenotype of native intestinal epithelial cells and transepithelial resistance at the time of experimentation was typically around 1000-1200 cm2.
Cell stimulation and cell lysates
Cultures of IEC-18 or T84 cells were washed twice with serum-free DMEM or DMEM/Ham's F12, equilibrated in the same medium at 37°C for 3 h, and then treated with ANG II, LPA, Carbachol, or other factors as described in the individual experiments. Inhibitors were added to the medium at appropriate times before stimulation. The stimulation was terminated by aspirating the medium and solubilaizing the cells in 200μl of 2x SDS-polyacrylamide gel electrophoresis sample buffer (20 mM Tris/HCl, pH 6.8, 6% SDS, 2 mM EDTA, 4% 2-mercaptoethanol, 10% glycerol).
Western Blotting
After SDS-PAGE, proteins were transferred to immobilon membranes. After transfer, membranes were blocked using 5% nonfat dried milk in PBS, pH 7.2 and incubated overnight at 4°C with the anti-FAK-Tyr-397 (0.1 μg/ml), anti-FAK-Ser(P)-843 Ab (0.1 μg/ml), anti-FAK-Ser(P)-910 Ab (0.1 μg/ml), anti-phospho-p44/42 MAP kinase(0.1 μg/ml) or anti-ERK2 (0.1 μg/ml) as indicated. The membranes were washed three times with PBS-0.1% Tween 20 and then incubated with secondary antibodies (horseradish peroxidase-conjugated donkey antibodies to rabbit, NA 934V or Mouse, NA931V) (1:5000) for 1 h at 22°C. After washing three times with PBS-0.1% Tween 20, the immunoreactive bands were visualized using enhanced chemilumiescence (ECL) detection reagents. Autoradiograms were scanned using the GS-710 Calibrated Imaging Densitometer (Bio-Rad), and the labeled bands were quantified using the Quantity One software program (Bio-Rad).
siRNA transfection
The SMART pool siRNA duplexes were purchased from Dharmacon (Lafayette, CO). For siRNA transfection, IEC-18 cells were plated at a density of 2×105 cells/35 mm dish 24 h before transfection. The following day, cells were transfected with siRNA using TransIT-TKO Reagent (Mirus, Madison, WI) as previously described [39]. 48 h after transfection, cells were used for experiments and subsequent Western blot analysis.
Wound healing assay
Forty-eight hours after transfection, cells were washed twice, and changed to serum-free DMEM overnight before wounding. The cell monolayers were wounded by applying a razor blade to the dish and scraping perpendicularly to the plane of the blade (40 mm long × 6-8 mm wide). Cells were then incubated in serum-free DMEM in the presence or absence of agonists as described in the individual experiments. After 16 h, experiments were terminated by washing cells twice in PBS, followed by fixing in 10% buffered formalin phosphate at 25°C for 20 min. To measure cell migration, cells were stained with Wright-Giemsa (Sigma, St. Louis, MO, U.S.A.) and observed under phase contrast with a ×10 lens (Plan-Neo, Carl Zeiss Inc., Jena, Germany) mounted on an upright microscope (Axioskop2, Carl Zeiss Inc., Jena, Germany). Images were collected with a high-resolution digital camera and software (Spot, Diagnostic Instruments) from 5-8 representative wounded areas per condition. Migration was calculated as the number of cells across the cut margin per high-powered field and is presented as means ± SE. Differences between groups were analyzed with the unpaired Student's t-test, with the significance level defined as P < 0.05.
Materials
ANG II, LPA, ionomycin, EGF and GF-109203X were obtained from Sigma (St. Louis, Mo). Carbachol, PD-98059, U-0126, and Ro-31-8220 were purchased from Calbiochem (San Diego, CA). Horseradish peroxidase-conjugated donkey antibodies to rabbit (NA 934V) or mouse (NA931V) and ECL reagents were from Amersham-Pharmacia, (Piscataway, NJ). Anti-ERK2 polyclonal antibody and FAK polyclonal Ab C-20 were from Santa Cruz Biotechnology Inc (Santa Cruz, Ca). The phosphospecific polyclonal Abs to Tyr-397, Ser-843 and Ser-910 of FAK were obtained from BioSource International (Camarillo, CA). Anti-phospho-ERK1/2 MAb was obtained from Cell Signaling Technology (Beverly, MA). All other reagents used were of the purest grade available.
RESULTS
ANG II induces FAK phosphorylation at Ser-910 in IEC-18 cells
ANG II binds to specific endogenous receptors in intestinal epithelial IEC-18 cells [44] and induces multiple signaling pathways and biological responses in these cells, including DNA synthesis and proliferation [45-49]. As shown by the results presented in Fig.1 A (control), stimulation with ANGII also markedly increased the migration of IEC-18 cells into the denuded area of a razor blade wound. In order to determine whether FAK is necessary for the migration of IEC-18 cells in response to ANGII, cultures of these cells were transfected with targeting or non-targeting FAK siRNA and incubated for 48h, using conditions that decrease FAK protein in IEC-18 cells by ∼80% [39]. Then, we measured basal and ANGII-mediated migration in wounded monolayers of these cells. As shown in Fig. 1A, siRNA-mediated knockdown of FAK markedly reduced the migration of ANG II-stimulated IEC-18 cells across the wound margin, as compared with either control or non-targeting siRNA-transfected cells. Having established that FAK is necessary for ANG II-induced IEC-18 cell migration, we next examined the effect of ANG II on the phosphorylation of FAK at serine and tyrosine residues.
Figure 1.
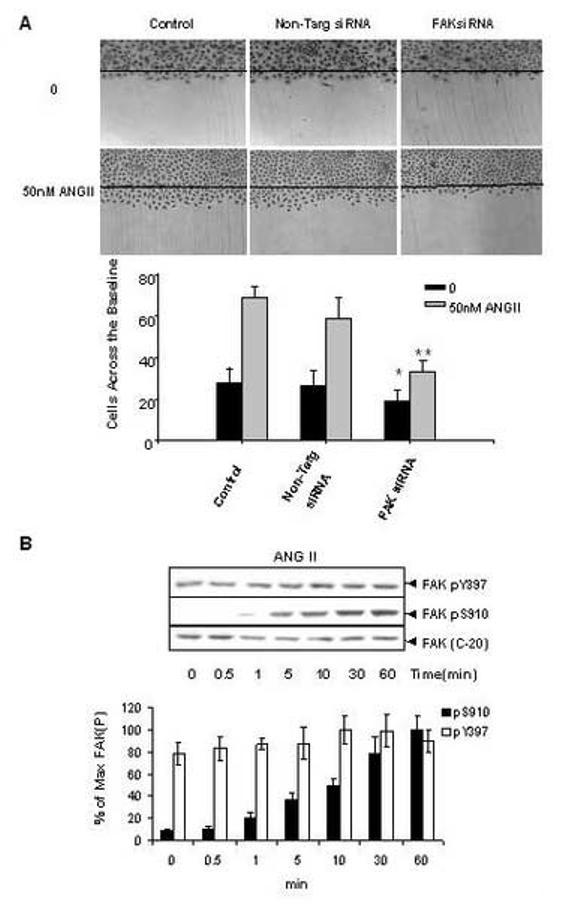
ANGII stimulates FAK phosphorylation at Ser-910 in IEC-18 cells. (A). Knockdown FAK expression by siRNA inhibits ANGII induced-cell migration in IEC-18 cells. IEC-18 cells were transfected with Trans IT-TKO alone, non-targeting negative control or 75nM FAK siRNA. After 48 h cells were wounded as described in Materials and Methods and incubated with or without 50nM ANGII for 16h. Experiments were terminated by washing cells twice in PBS, followed by fixing in 10% buffered formalin phosphate for 20 min. Cells were stained with Wright-Giemsa and observed under phase contrast with a ×10 lens mounted on an upright microscope. Upper Panel: Photographs representative of control, non-targeting siRNA or FAKsiRNA transfectd cells. Results are typical of five independent experiments. Lower panel: Values are means ± SE of at least 8 fields from five independent experiments and are expressed as the number of cells observed across the wound margin at 16 h. * P < 0.01 vs. control, ** P<0.01 vs. control with ANGII treatment. (B). Time-course of ANG II in stimulation of FAK phosphorylation at Ser-910 in IEC-18 cells. Upper Panel: Confluent and quiescent IEC-18 cells were treated at 37°C with 50nM of ANG II for various times as indicated and subsequently lysed in 2×sample buffer. FAK phosphorylation at Ser-910 or Tyr-397 was analyzed by Western blotting with pSer-910 Ab or pTyr-397 Ab. The membranes were further analyzed by Western blotting using anti-FAK Ab. The autoradiograms shown are representative of at least three independent experiments. Lower Panel: Quantification of FAK phosphorylation at Ser-910 and Tyr-397 was performed by scanning densitometry. Values shown as bars are the mean of at least three independent experiments and are expressed as the percentage of the maximal increase of phosphorylation above control (unstimulated) values.
To determine whether the phosphorylation of endogenous FAK at Ser-910 is regulated by GPCR agonists in intestinal epithelial cells, cultures of IEC-18 cells were incubated with 50 nM ANG II for various times and lysates of these agonist-treated cells were analyzed by Western blotting using a site-specific antibody that specifically detects the phosphorylated state of FAK at Ser-910. The Western blot analysis illustrated in Fig. 1B shows that prior to stimulation, pS910 immunoreactivity was virtually absent, indicating that Ser-910 of FAK was not phosphorylated in unstimulated cultures of IEC-18 cells. Upon ANGII stimulation, FAK phosphorylation at Ser-910 increased gradually. A small increase was noticeable after 1 min of exposure to ANG II with maximal stimulation of FAK phosphorylation at Ser-910 evident at 60 min of incubation. The maximal increase of FAK phosphorylation at Ser-910 induced by ANGII was 15.2±1.5 fold, as compared with the unstimulated level.
We also examined whether ANGII regulates FAK phosphorylation at Tyr-397 by Western blotting using a site-specific antibody that detects the phosphorylated state of this residue of FAK. As shown in Fig. 1 B, Tyr-397 of FAK was phosphorylated in unstimulated IEC-18 cells and addition of ANGII did not produce any significant increase in the phosphorylation of this residue of FAK. Thus, ANG II, under conditions that promoted FAK-dependent migration in IEC-18 cells, elicited striking FAK phosphorylation at Ser-910 but did not change the phosphorylation of FAK Tyr-397 in these cells.
ANG II induces FAK phosphorylation at Ser-910 through a PKC/ERK-dependent pathway
ANGII, acting through the Gq-coupled AT1 receptor, stimulates activation of ERK, protein kinase D and Pyk2 through a protein kinase C (PKC)-dependent pathway [46, 48]. Here, we examined whether the increase in FAK Ser-910 phosphorylation in response to ANGII in IEC-18 cells is mediated through a PKC/ERK-dependent pathway.
To determine whether direct activation of PKC increases FAK phosphorylation at Ser-910, quiescent IEC-18 cells were treated with the biologically active tumor promoting agent PDB for various times and lysed. The lysates were analyzed by Western blotting using the site-specific antibody that recognizes the phosphorylated state of FAK at Ser-910. As shown in Fig. 2A, treatment with PDB caused a dramatic, time-dependent, increase in FAK phosphorylation at Ser-910. To further characterize the role of PKC in FAK phosphorylation at Ser-910, cultures of IEC-18 cells were treated with the selective PKC inhibitors GF I (also known as GF 109203X or bisindolylmaleimide I) or Ro 31-8220 prior to ANG II or PDB stimulation. As shown in Fig. 2B, treatment of the cells with GF I or Ro 31-8220 potently blocked FAK Ser-910 phosphorylation induced by subsequent addition of either ANG II or PDB. These results indicate that FAK phosphorylation at Ser-910 in response to ANG II or PDB is mediated through a PKC-dependent pathway.
Figure2.
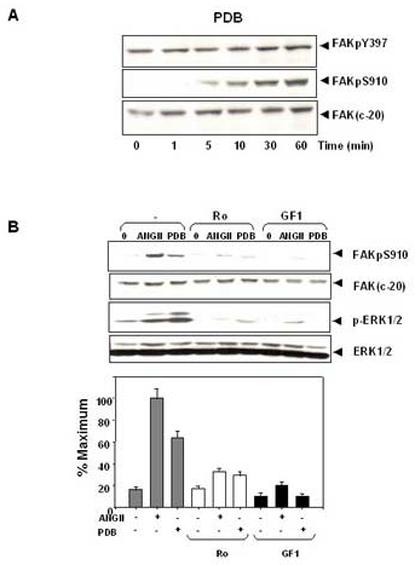
ANG II induces FAK phosphorylation at Ser-910 through a PKC-dependent pathway. (A). Time course of FAK phosphorylation at Ser-910 induced by PDB in IEC-18 cells. Confluent and quiescent cells were treated at 37° C with 100nM PDB for various times as indicated and were subsequently lysed. FAK phosphorylation at Ser-910 or Tyr-397 was analyzed by Western blotting with pSer-910 Ab or pTyr-397 Ab. The membranes were further analyzed by Western blotting using anti-FAK Ab. (B). PKC inhibitors prevent FAK phosphorylation at Ser-910 induced by either ANGII or PDB in IEC-18 cells. Upper Panel: Confluent and quiescent cells were treated for 1 h either in the absence (−) or presence of 2.5 μM Ro-31-8220 or 5μM GF-109203X at 37° C. Cells were then incubated for further 10 min either with 50 nM ANGII or 100 nM PDB. The cells were lysed and the extracts were analyzed by Western blotting with pSer-910 Ab. The membranes were further analyzed by Western blotting using anti-FAK Ab. An aliquot of the lysates was analyzed by Western blotting with phospho-p44/42 MAP Kinase Ab (pERK1/2). These membranes were further analyzed by Western blotting using anti-ERK1/ 2 Ab. Lower Panel: Quantification of the inhibition of Ser-910 phosphorylation by these two PKC inhibitors was performed by scanning densitometry. Values shown as bars are the mean of at least three independent experiments and are expressed as the percentage of the maximal increase of Ser-910 phosphorylation above control (unstimulated) values. In all cases, the autoradiograms shown are representative of at least three independent experiments.
As indicated above, ANG II induces ERK activation through a PKC-dependent pathway in IEC-18 cells [46, 48]. Accordingly, treatment of IEC-18 cells with the selective PKC inhibitors GF I or Ro 31-8220 blocked FAK Ser-910 phosphorylation as well as prevented ERK activation in response to ANGII treatment (Fig 2B). To substantiate that FAK Ser-910 phosphorylation is mediate by the ERKs, cultures of IEC-18 cells were treated for 1 h in the absence or presence of the MEK inhibitor U0126 (5 μM) and subsequently stimulated with either 50nM ANGII or 100nM PDB. As shown in Fig. 3A, exposure to U0126 completely abrogated FAK Ser-910 phosphorylation induced by either ANGII, or PDB in IEC-18 cells. Thus, these results indicate that ANG II induces FAK phosphorylation at Ser-910 via a PKC/ERK pathway in intestinal epithelial IEC-18 cells.
Figure 3.
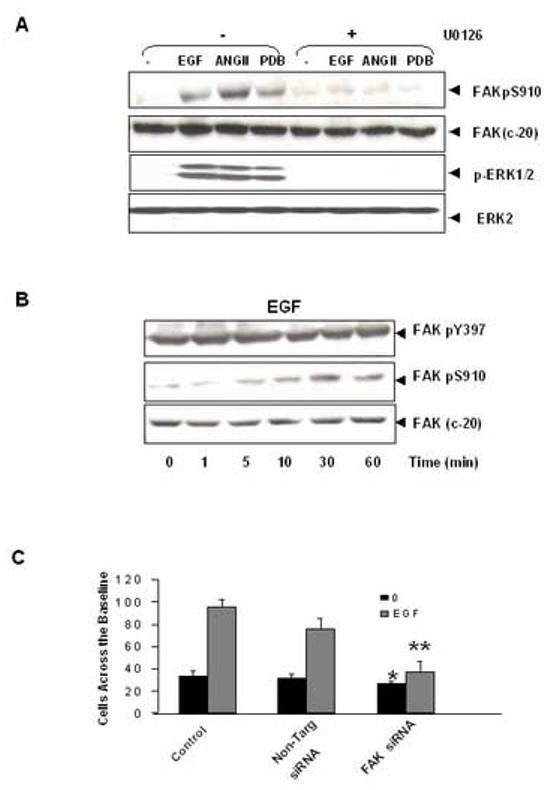
ANGII and EGF induce FAK phosphorylation at Ser-910 through an ERK-dependent pathway. (A). The MEK inhibitor U0126 prevents the phosphorylation of FAK at Ser-910 in IEC-18 cells. Cells were incubated with 10μM U 0126 for 1 h at 37°C and then stimulated with either 5ng/ml EGF or 50 nM ANGII or 100nM PDB for 10 min. The cells were lysed and the extracts were analyzed by Western blotting with pSer-910 Ab. The membranes were further analyzed by Western blotting using anti-FAK Ab. A portion of the lysates was analyzed by Western blotting with pERK1/2 Ab. These membranes were further analyzed by Western blotting using ERK2 Ab. (B). Time-course of EGF in stimulation of FAK phosphorylation at Ser-910 in IEC-18 cells. Confluent and quiescent cells were treated at 37° C with 5 ng/ml EGF for various times as indicated and were subsequently lysed. FAK phosphorylation at Ser-910 or Tyr-397 was analyzed by Western blotting with pSer-910 or pTyr-397. The membranes were further analyzed by Western blotting using anti-FAK Ab. In all cases, the autoradiograms shown are representative of at least three independent experiments. (C). Knockdown FAK expression by siRNA inhibits EGF induced-cell migration in IEC-18 cells. IEC-18 cells were transfected with Trans IT-TKO alone, non-targeting negative control or 75nM FAK siRNA. After 48 h cells were wounded as described in Materials and Methods and incubated with or without 5ng/ml EGF for 16h. Experiments were terminated by washing cells twice in PBS, followed by fixing in 10% buffered formalin phosphate for 20 min. Cells were stained with Wright-Giemsa and observed under phase contrast with a ×10 lens mounted on an upright microscope. Values are means ± SE of at least 8 fields from five independent experiments and are expressed as the number of cells observed across the wound margin at 16 h. * P < 0.01 vs. control, ** P<0.01 vs. control with EGF treatment.
EGF and LPA induce FAK phosphorylation at Ser-910 through ERK-dependent pathway
EGF and LPA stimulate the ERK pathway via Ras-dependent but PKC-independent pathways. In order to generalize the results obtained with ANG II, we examined whether these agonists induce FAK Ser-910 phosphorylation via an ERK-dependent but PKC-independent pathway in IEC-18 cells.
In order to determine whether EGF promotes FAK Ser-910 phosphorylation, cultures of IEC-18 cells were treated with EGF at 5ng/ml for various times (1-60 min) and then FAK phosphorylation at Ser-910 was determined by Western blotting. As shown in Fig. 3B, EGF induced a striking increase in FAK Ser-910 phosphorylation, which was detectable 5 min after the addition of the factor and was nearly maximal after 30 min. Treatment with U0126 prior to EGF completely prevented FAK Ser-910 phosphorylation induced by this growth factor in IEC-18 cells (Fig. 3A). These results indicate that EGF-induced FAK Ser-910 phosphorylation is mediated through an ERK-dependent pathway in IEC-18 cells. In contrast, EGF stimulation had no effect on FAK phosphorylation at Tyr-397 in IEC-18 cells (Fig. 3B). We verified that treatment with FAK siRNA markedly reduced the migration of IEC-18 cells across the wound margin induced by stimulation with EGF (Fig. 3C), substantiating that FAK is required for EGF-induced migration of intestinal epithelial cells.
Subsequently, we examined differential FAK phosphorylation at Ser-910 and Tyr-397 in response to LPA, a major bioactive lipid growth factor present in serum. LPA induced FAK phosphorylation at Ser-910 in IEC-18 cells in a time-dependent manner. The increase of Ser-910 phosphorylation was detected at 0.5 min and reached maximum within 60 min. However, treatment of these cells with LPA had no effect on FAK phosphorylation at Tyr-397 (Fig. 4A). The results presented in Fig.1B and 4A show that stimulation of IEC-18 cells with the GPCR agonists ANG II or LPA induces a striking increase in FAK phosphorylation at Ser-910, but not Tyr-397.
Figure 4.
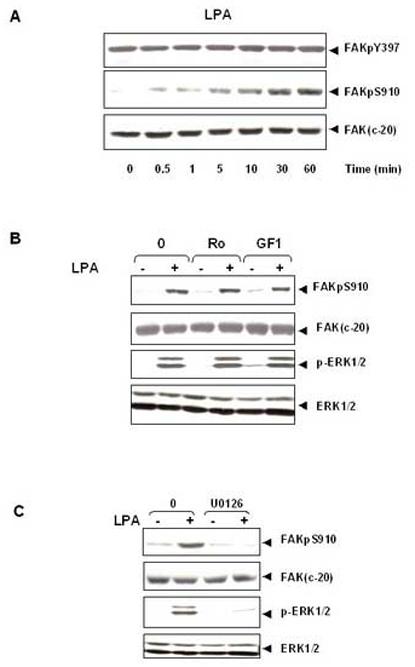
LPA induces FAK phosphorylation at Ser- 910 through PKC-independent, ERK-dependent pathway. (A). Time-course of LPA in stimulation of FAK phosphorylation at Ser-910 in IEC-18 cells. Confluent and quiescent cells were treated at 37° C with 10μM LPA for various times as indicated and were subsequently lysed. FAK phosphorylation at Ser-910 or Tyr-397 was analyzed by Western blotting with pSer-910 Ab or pTyr-397Ab. The membranes were further analyzed by Western blotting using anti-FAK Ab. The autoradiograms shown are representative of at least three independent experiments. (B).LPA stimulates FAK phosphorylation at Ser-910 independent of PKC. Confluent and quiescent cells were treated for 1 h either in the absence (−) or presence of 2.5 μM Ro-31-8220 or 5μM GF-109203X at 37° C. Cells were then incubated for further 10 min with 10μM LPA. The cells were lysed and the extracts were analyzed by Western blotting with pSer-910 Ab. The membranes were further analyzed by Western blotting using anti-FAK Ab. An aliquot of the lysates was analyzed by Western blotting with phospho-p44/42 MAP Kinase Ab (pERK1/2). These membranes were further analyzed by Western blotting using anti-ERK1/ 2 Ab. (C). The MEK inhibitor U0126 prevents LPA induced phosphorylation of FAK at Ser-910 in IEC-18 cells. Cells were incubated with 10μM U 0126 for 1 h at 37°C and then stimulated with 10μM LPA for 10 min. The cells were lysed and the extracts were analyzed by Western blotting with pSer-910 Ab. The membranes were further analyzed by Western blotting using anti-FAK Ab. A portion of the lysates was analyzed by Western blotting with pERK1/2 Ab. These membranes were further analyzed by Western blotting using ERK1/2 Ab. In all cases, the autoradiograms shown are representative of at least three independent experiments.
In contrast to ANGII, LPA induces Ras activation leading to stimulation of the ERKs via a PKC-independent pathway. Accordingly, treatment of the cells with the selective PKC inhibitors GF I or Ro 31-8220 did not have any effect on FAK phosphorylation at Ser-910 induced by LPA (Fig. 4B). Conversely, exposure to U0126 completely prevented both FAK phosphorylation at Ser-910 and ERK activation induced by LPA (Fig. 4C). These results indicate that stimulation with the GPCR agonist LPA strikingly increases the phosphorylation of endogenous FAK at Ser-910 through a PKC-independent but ERK-dependent pathway in IEC-18 cells.
Carbachol and EGF induce FAK phosphorylation at Ser-910 through ERK-dependent pathway in T84 cells
To examine whether Ser-910 phosphorylation of FAK is also regulated in differentiated intestinal epithelial cells, we extended our experiments using cultures of the human colonic epithelial cell line, T84. These cells express the subtype 3 muscarinic acetylcholine receptor (mAChR), a GPCR that induces PLC-mediated hydrolysis of membrane phosphoinositides leading to Ca2+ mobilization and PKC activation. Stimulation of non-polarized T84 cells with the muscarinic agonist carbachol induced FAK phosphorylation at Ser-910 in a time-dependent fashion (Fig. 5A). A small increase was observed after 2.5 min of exposure to carbachol with maximal stimulation of FAK phosphorylation at Ser-910 evident after 30 min of incubation. The maximal increase of FAK phosphorylation at Ser-910 induced by carbachol was 9.1±1.1 fold, as compared with the unstimulated level. In contrast, carbachol caused only a relative small increase in the phosphorylation of Tyr-397 as compared with that of Ser-910. The maximal increase of FAK phosphorylation at Tyr-397 was 45.4±7.9%.
Figure 5.
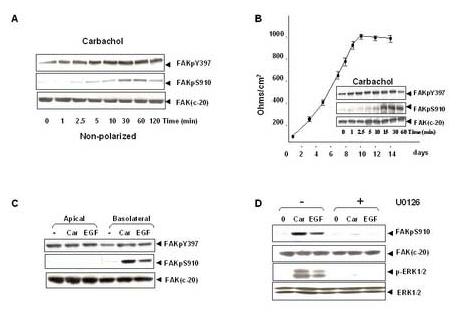
Carbachol and EGF induce FAK phosphorylation at Ser-910 through ERK-dependent pathway in T84 cells. (A). Time-course of carbachol in stimulation of FAK phosphorylation at Ser-910 and Tyr-397 in un-polarized T84 cells. Cultures of T84 (about 2×106 cells in 35mm Petri dish) were incubated with 100μM carbachol for various times and lysates of these agonist-treated cells were extracted and analyzed by Western blotting using FAK pSer-910 Ab or pTyr-397 Ab. The membranes were further analyzed by Western blotting using anti-FAK Ab. (B). Time-course of carbachol in stimulation of FAK phosphorylation at Ser-910 and Tyr-397 in polarized T84 cells. T84 cells (∼5×105) were plated on 23 mm Falcon Transwells and the transepithelial resistance was measured at various times Inserts, Polarized cultures formed 7 days after plating were stimulated basolaterally with 100μM carbachol for various times. The cells were then lysed and the phosphorylation of FAK at Ser-910 and Tyr-397 was detected by Western blot using FAK pS910Ab or pTyr397 Ab. The same membrane was stripped and reprobed with anti-FAK to verify equal loading. (C). EGF stimulates FAK phosphorylation at Ser-910 in T84 cells. Polarized cultures formed 7 days after plating were stimulated apically or basolaterally with either 100μM carbachol or 50ng/ml EGF for 10 min. The cells were then lysed and the phosphorylation of FAK at Ser-910 or Tyr-397 was detected by Western blot using FAK pSer-910 Ab or pTyr-397 Ab. The same membrane was stripped and reprobed with anti-FAK to verify equal loading. (D). The MEK inhibitor U0126 prevents both carbachol and EGF induced phosphorylation of FAK at Ser-910 in T84 cells. Polarized cultures of cells were incubated with 20μM U 0126 for 1 h at 37°C and then stimulated with either 100μM carbachol or 50ng/ml EGF basolaterally for 10 min. The cells were lysed and the extracts were analyzed by Western blotting with pSer-910 Ab. The membranes were further analyzed by Western blotting using anti-FAK Ab. A portion of the lysates was analyzed by Western blotting with pERK1/2 Ab. These membranes were further analyzed by Western blotting using ERK1/2 Ab. In all cases, the autoradiograms shown are representative of at least three independent experiments.
Subsequently, we performed similar experiments with polarized T84 cells. To monitor the development of polarization in T84 cells, we plated 5×105 cells on Falcon Transwells and measured transepithelial resistance at various times (Fig. 5B). Under these conditions, T84 cells develop the polarized phenotype of native intestinal epithelial cells and transepithelial resistance reached ∼ 1,000 Ω/cm2 6-7 days after plating. As shown in Fig 5B, basolateral stimulation of polarized T84 cells with carbachol induced striking increase in FAK phosphorylation at Ser-910, the maximal increase was 14.2±3.8 fold. However, similar to the non-polarized cells, carbachol only slightly increased FAK phosphorylation at Tyr-397, the maximal increase was 65.3±17.2%.
T84 cells express EGF receptors that are thought to play a role in promoting restitution in T84 cells [50]. In order to examine whether EGF promotes FAK Ser-910 phosphorylation, monolayers of T84 cells, grown on permeable supports, were stimulated apically or basolaterally with 50ng/ml EGF for 10 min. Cell lysates were then extracted and immunoblotted with FAK Ser-910 antibody. As shown in Fig. 5C, basolateral stimulation of polarized T84 cells with 50ng/ml EGF dramatically induced FAK phosphorylation at Ser-910 while apical stimulation did not induce any significant effect. In contrast, treatment with EGF in these cells caused small changes in the phosphorylation of Tyr-397 as compared with that of Ser-910. Furthermore, in the same culture condition, apical stimulation of carbachol did not have any effect, while basolateral stimulation caused marked increase in FAK phosphorylation at Ser-910 (Fig. 5C).
To determine whether FAK Ser-910 phosphorylation by carbachol or EGF in T84 cells is mediated through an ERK-dependent pathway, T84 cells grown on permeable supports were treated for 1 h in the absence or presence of U0126 and subsequently stimulated with either 100 μM carbachol or 50ng/ml EGF for another 10 min. As shown in Fig. 5D, treatment with U0126 completely abrogated FAK Ser-910 phosphorylation induced by either carbachol or EGF in T84 cells. The results presented in Fig. 5 indicate that stimulation with either GPCR or growth factor agonists also strikingly increases the phosphorylation of endogenous FAK at Ser-910 through an ERK-dependent pathway in human colonic epithelial T84 cells.
Ionomycin and GPCR agonists induce a rapid and transient increase phosphorylation of FAK at Ser-843 in IEC-18 cells
Recently, we demonstrated FAK phosphorylation at Ser-843 through a Ca2+, calmodulin and CaMKII-dependent pathway in mouse fibroblasts [37]. As a first step to examine whether an increase in [Ca2+]i could trigger FAK phosphorylation at Ser-843 in epithelial cells, cultures of IEC-18 cells were treated with the Ca2+ ionophore ionomycin for various times, at a concentration (500 nM) that rapidly increases [Ca2+]i in these cells (results not shown). As shown in Fig. 6A, FAKpS843 immunoreactivity was virtually absent prior to stimulation, indicating that Ser-843 of FAK was not phosphorylated in unstimulated IEC-18 cells. Addition of ionomycin induced a rapid increase in FAK phosphorylation at Ser-843 which was already striking within 15s, peaked by 30-60s followed by a gradual decline toward baseline levels. These results suggest that an increase in [Ca2+]i is a potential pathway leading to the rapid FAK phosphorylation at Ser-843.
Figure 6.
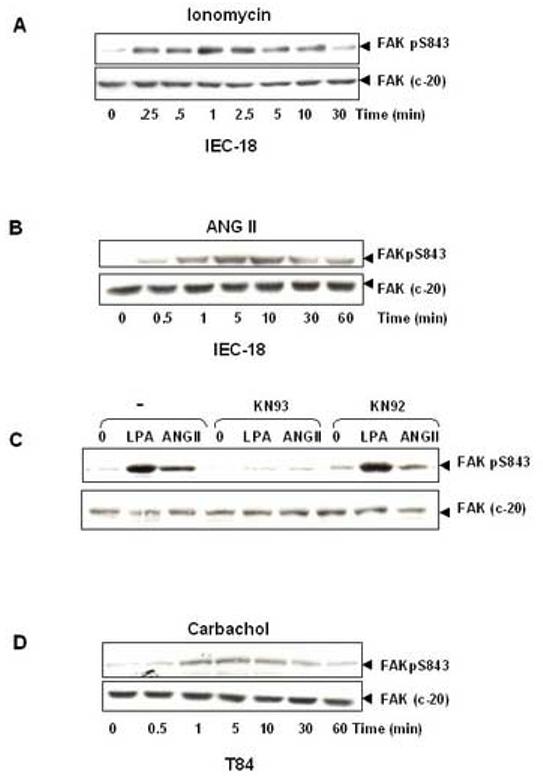
GPCRs induce FAK phosphorylation at Ser-843 in intestinal epithelial cells. (A). The Ca2+ ionophor ionomycin induces FAK phosphorylation at Ser-843 in IEC-18 cells. Confluent and quiescent IEC-18 cells were treated at 37°C with 500 nM ionomycin for the indicated times. Cells were then lysed and FAK phosphorylation at Ser-843 was analyzed by Western blotting with pSer-843 Ab. The membranes were reanalyzed by Western blotting using anti-FAK Ab (C-20), to demonstrate equal loading of total FAK. The autoradiograms shown are representative of at least three independent experiments. (B). ANGII induces FAK phosphorylation at Ser-843 in IEC-18 cells. Confluent and quiescent IEC-18 cells were treated at 37°C with 50 nM ANGII for the indicated times. Cells were then lysed and analyzed by Western blotting with pSer-843 Ab. The membranes were reanalyzed by Western blotting using anti-FAK Ab, to demonstrate equal loading of total FAK. The autoradiograms shown are representative of at least three independent experiments. (C). CaMKII inhibitor (KN 93) prevents bombesin-induced FAK phosphorylation at Ser-843. Confluent and quiescent IEC-18 cells were preincubated with 40μM KN 93 or KN 92 for 30 min and subsequently stimulated with 50 nM ANGII or 10μM LPA for an additional 1 min. Cells were then lysed and analyzed by Western blotting with pSer-843 Ab. The membranes were reanalyzed by Western blotting using anti-FAK Ab, to demonstrate equal loading of total FAK. The autoradiograms shown are representative of at least three independent experiments. (D). Carbachol induces FAK phosphorylation at Ser-843 in T84 cells. Polarized T84 cells were treated at 37°C with 100μM carbachol basolaterally for the indicated times. Cells were then lysed and analyzed by Western blotting with pSer-843 Ab. The membranes were reanalyzed by Western blotting using anti-FAK Ab, to demonstrate equal loading of total FAK. The autoradiograms shown are representative of at least three independent experiments.
To determine whether GPCR activation leads to FAK phosphorylation at Ser-843 in IEC-18 cells, cultures of these cells were stimulated with 50nM ANGII for various times, lysed and extracts of these agonist-treated cells were analyzed by Western blotting using a site-specific antibody that detects the phosphorylated state of FAK at Ser-843. As illustrated in Fig. 6B, Ser-843 of FAK was not phosphorylated in unstimulated IEC-18 cells. Upon ANGII stimulation, FAK pS843 phosphorylation increased dramatically in a sharply time-dependent manner. An increase in FAK phosphorylation at Ser-843, was detected as early as 30 s after the addition of the agonist, reached a maximum within 5 min, and declined rapidly toward baseline. The maximal increase of FAK phosphorylation at Ser-843 induced by ANG II was 14.5±3.8 fold, as compared with the unstimulated level. Furthermore, we also showed that LPA induced striking increase of FAK phosphorylation at Ser-843 in IEC-18 cells (data not shown).
In order to determine whether the increase in FAK Ser-843 phosphorylation in response to ANGII or LPA in IEC-18 cells is mediated by Ca2+ /calmodulin-dependent protein kinases (CaMKs), we examined a role for CaMKs in FAK phosphorylation at Ser-843 using KN-93, a selective cell-permeable inhibitor of CaMK activity, and KN-92, a structurally related but inactive compound [51]. KN 93 selectively binds to the calmodulin binding site of the enzyme and prevents the association of calmodulin. IEC-18 cells were pretreated with either KN 93 or KN 92 followed by stimulation with ANGII or LPA. As shown in Fig. 6C, pretreatment with KN 93 virtually abolished phosphorylation of FAK at Ser-843 induced by ANGII or LPA. In contrast, KN 92, tested at identical concentrations in parallel cultures of IEC-18 cells, had no significant effect on the phosphorylation of FAK at Ser-843 induced by GPCR agonists in IEC-18 cells.
To examine whether FAK can be phosphorylated at Ser-843 by carbachol in T84 cells, monolayers of T84 cells grown on permeable supports were stimulated with 100μM carbachol for various times, lysed and extracts of these agonist-treated cells were analyzed by Western blotting. As illustrated in Fig. 6D, carbachol stimulation induced FAK Ser-843 phosphorylation in a time-dependent manner. An increase in FAK phosphorylation at Ser-843, was detected as early as 0.5 min after the addition of carbachol, reached a maximum within 5 min, and declined rapidly toward baseline within 60 min. Thus, GPCR agonists elicited a rapid and transient increase in FAK phosphorylation at Ser-843 in intestinal epithelial IEC-18 and T 84 cells.
Kinetics of Ser-910, Ser-843 and Tyr-397 in response to GPCR agonists in intestinal epithelial cells
The combination of the results obtained in this study, enabled us to compare the kinetics of FAK phosphorylation at Ser-910, Ser-843 and Tyr-397 in both IEC-18 and T 84 cells under identical experimental conditions.
In IEC-18 cells, ANG II stimulated FAK phosphorylation at Ser-843 within 30 s, reached a maximum within 5-10 min and decreased rapidly (Fig 7A). FAK phosphorylation at Ser-910 in response to ANGII demonstrated a more gradual increase, detectable after 1 min treatment and reaching a maximal stimulation 60 min after the addition of the agonist. In contrast, ANG II did not have any significant effect on FAK phosphorylation at Tyr-397 in IEC-18 cell. Thus, the salient feature of the kinetics shown in Fig 7A in IEC-18 cells, is that ANG II induces a striking increase in FAK phosphorylation at Ser-910 and Ser-843, but not at Tyr-397.
Figure 7.
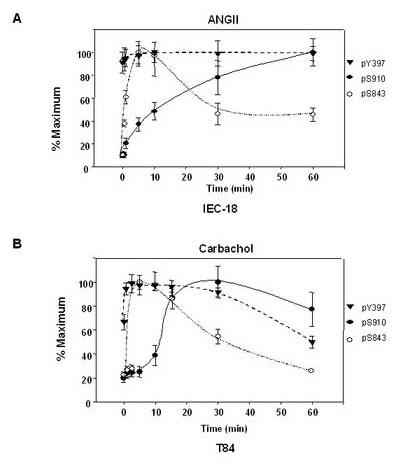
Time-course of GPCRs-induced FAK phosphorylation at Ser-910, Ser-843, and Tyr-397 in intestinal epithelial cells. (A). Time-course of ANGII-induced FAK phosphorylation at Ser-910, Ser-843, and Tyr-397 in IEC-18 cells. Confluent and quiescent IEC-18 cells were treated at 37°C with 50 nM ANGII for various times as indicated in Figs. 1 and 6. Cell lysates were analyzed by Western blotting with pSer-910Ab, pSer-843 Ab and pY-397Ab. Quantification of phosphorylation at Ser-910 (solid line), Tyr-397 (dashed line) and Ser-843 (dotted line) were determined by densitometric scanning of the bands. Values shown are the mean ± S.E. of at least three independent experiments and are expressed as the percentage of the maximal increase in FAK phosphorylation at Ser-910, Tyr-397 or Ser-843 above control (unstimulated) values. (B). Time-course of carbachol-induced FAK phosphorylation at Ser-910, Ser-843, and Tyr-397 in T84 cells. Polarized T84 cells were treated at 37°C with 100μM carbachol basolaterally for various times as indicated in Figs. 5 and 6. Cell lysates were analyzed by Western blotting using pSer-910Ab, pSer-843 Ab and pY-397Ab. Quantification of phosphorylation at Ser-910 (solid line), Tyr-397 (dashed line) and Ser-843 (dotted line) were determined by densitometric scanning of the bands. Values shown are the mean ± S.E. of at least three independent experiments and are expressed as the percentage of the maximal increase in FAK phosphorylation at Ser-910, Tyr-397 or Ser-843 above control (unstimulated) values.
We also compared the kinetics of FAK phosphorylation at Ser-910, Ser-843 and Tyr-397 in T84 cells under identical experimental conditions. As shown in Fig. 7B, FAK phosphorylation at Ser-843, was detected as early as 30s after the addition of carbachol, reached a maximum within 5 min, and declined rapidly toward baseline within 60 min. Carbachol increased FAK phosphorylation at Ser-910 within 5 min after the addition of the agonist, reached a maximum within 30 min and remained relatively constant up to 60 min. However, carbachol only slightly stimulated FAK phosphorylation at Tyr-397. Thus, the kinetics shown in Fig 7B, indicate that carbachol dramatically induces FAK phosphorylation at Ser-910 and Ser-843, but only slightly increases FAK phosphorylation at Try-397 in T84 cells. Collectively, our results show that stimulation with GPCR agonists markedly stimulates FAK phosphorylation at Ser-910 and Ser-843, but not Tyr-397 in both undifferentiated IEC-18 cells and differentiated T84 cells.
DISCUSSION
Numerous studies have demonstrated that a rapid increase in the tyrosine phosphorylation of the non-receptor tyrosine kinase FAK is a prominent early event in cells stimulated by multiple signaling molecules that regulates cell proliferation, migration, and apoptosis [3, 6, 52]. On the contrary, much less is known about the regulation of FAK phosphorylation at serine residues by external stimuli including GPCR agonists and growth factors. Interestingly, both Ser-910 and Ser-843, located in the COOH-terminal region of FAK, have been identified as prominent phosphorylation sites during mitosis [33]. Recent results from our laboratory demonstrated that FAK phosphorylation at Ser-910 was strikingly stimulated by GPCR agonists, tumor promoting phorbol esters and growth factors in Swiss 3T3 fibroblasts [34-36]. Furthermore, phosphorylation of FAK at Ser-843 was induced by GPCR agonists, including bombesin, vasopressin and bradykinin in Swiss 3T3 fibroblasts [37]. These results indicate that phosphorylation of FAK at serine residues can be regulated by external stimuli including GPCRs and growth factors. Interestingly, the basal level of tyrosine phosphorylated FAK is high in many epithelial cells attached to a substratum, including T84 cells [40], IEC-18 cells [41], SW 620 [42] and liver epithelial cells [43]. These considerations prompted us to test the possibility that stimulation of epithelial cells with GPCR agonists or ligands of tyrosine kinase receptors that promote FAK-dependent migration, increases FAK phosphorylation preferentially at serine rather than at tyrosine residues. Very little was known about the regulation of phosphorylation of FAK on serine residues, including Ser-910 and Ser-843, in epithelial cells.
In the present study, we demonstrate that stimulation of either undifferentiated rat IEC-18 or polarized human T84 intestinal epithelial cells with GPCR agonists induces striking increase in FAK phosphorylation at Ser-910 and Ser-843, without producing any major increase in the phosphorylation of FAK at Tyr-397, the major autophosphorylation site. Specifically, stimulation of IEC-18 cells with the GPCR agonist ANGII induced a rapid and dramatic increase in FAK phosphorylation at Ser-910 and Ser-843 but did not elicit any further enhancement of the high basal level of FAK phosphorylation at Tyr-397. Similarly, the mAChR agonist carbachol induced a marked increase in FAK phosphorylation at both Ser-910 and Ser-843, but produced only a slight increase of FAK phosphorylation at Tyr-397 in T84 cells. Collectively, our results show, for the first time, that stimulation of rat or human intestinal epithelial cells with agonists of GPCRs endogenously expressed by these cells induce a striking increase in the phosphorylation of Ser-910 and Ser-843 of endogenous FAK, rather than producing any major change of autophosphorylation at Tyr-397.
Gq-coupled GPCRs activate phospholipase C-mediated production of inositol 1,4,5-trisphosphate, which triggers the release of Ca2+ from internal stores, and diacylglycerol, which directly activates classic and novel PKCs. In fibroblasts, we proposed that FAK serves as a point of integration of signals transmitted via Gαq, leading to PKC/ERK-dependent FAK phosphorylation at Ser-910 [34-36] and to Ca2+/calmodulin/CaMKII-dependent FAK phosphorylation at Ser-843 [37]. Here, we examined whether these pathways also operate in intestinal epithelial cells. We showed that treatment of IEC-18 cells with the potent tumor promoting phorbol ester PDB, a direct activator of PKCs, induced a striking increase in FAK Ser-910 phosphorylation and treatment with selective PKC inhibitors prevented FAK phosphorylation at Ser-910 in response to ANGII or PDB in these cells. Thus, these results indicate that FAK phosphorylation at Ser-910 in response to ANG II or PDB is mediated through a PKC-dependent pathway in IEC-18 cells.
Given that PKCs lead to ERK activation in response to ANGII in IEC-18 cells and that activated ERKs directly phosphorylate FAK at Ser-910 in vitro [34], we determined whether FAK phosphorylation at Ser-910 in GPCR-stimulated intestinal epithelial cells is mediated also through an ERK-dependent pathway. In line with this possibility, treatment of IEC-18 cells with U0126, a potent inhibitor of MEK-mediated ERK activation, prevented FAK phosphorylation at Ser-910 induced by subsequent addition of either ANGII or PDB to these cells. Furthermore, the U0126 also prevented FAK phosphorylation at Ser-910 in response to carbachol, an agonists that elicits ERK activation in polarized T84 cells [40, 53].
In order to substantiate the hypothesis that ERK activation mediates FAK phosphorylation at Ser-910, we examined whether this residue of FAK becomes phosphorylated in response to other stimuli that induce ERK activation through different signal transduction pathways. LPA binds to ubiquitous seven-transmembrane domain receptor(s) of the Edg/LP subfamily of GPCRs [54, 55] and activates multiple heterotrimeric G proteins which are responsible for transducing LPA signals into a broad spectrum of biological responses [56, 57]. Our results show that LPA, which promotes Ras-dependent but PKC-independent stimulation of the Raf/MEK/ERK pathway, induces dramatic FAK phosphorylation at Ser-910 in IEC-18 cells, without producing any change in the phosphorylation of FAK at Tyr-397. Similarly, EGF which induces ERK pathway activation via Ras [58, 59], also stimulates FAK phosphorylation at Ser-910 in both IEC-18 and T84 cells. In addition, the increase in the phosphorylation of FAK at Ser-910 induced by either LPA or EGF was abrogated by treatment with the MEK inhibitor U0126. These results support the notion that FAK phosphorylation at Ser-910 is stimulated by a variety of GPCR agonists, including peptides (ANGII), bioactive lipids (LPA) or neurotransmitters (carbachol) and by the tyrosine kinase receptor agonist EGF through an ERK-dependent pathway in intestinal epithelial cells.
Previous studies from our laboratory demonstrated that ANGII induces a rapid increase in [Ca2+]i in IEC-18 cells [48] and that agonists of Gq-coupled receptors elicit FAK Ser-843 phosphorylation through a Ca2+-dependent pathway in Swiss 3T3 fibroblasts[37]. Here, we demonstrate that treatment of IEC-18 cells with the Ca2+ ionophore ionomycin induced a striking, rapid and transient increase in FAK phosphorylation at Ser-843. Furthermore, we showed similar striking increases in the phosphorylation of this residue in response to ANGII in IEC-18 cells and carbachol in polarized T84 cells. These results imply that an elevation [Ca2+]i leads to FAK Ser-843 phosphorylation in these cells. We conclude that FAK serves as a point of integration of signals transmitted via Gαq in epithelial cells, leading to PKC/ERK-dependent FAK phosphorylation at Ser-910 and to Ca2+-dependent FAK phosphorylation at Ser-843.
A recent study from our laboratory using siRNA-mediated knockdown of FAK demonstrated that this enzyme plays a critical role in GPCR-induced migration, lamellipodia formation and assembly of focal adhesions in intestinal epithelial cells [39]. Here, we extended these findings demonstrating that in addition to LPA, stimulation of IEC-18 cells with ANGII or EGF also elicited cell migration in a FAK-dependent manner. Cell migration is a cyclical process that requires the activation and deactivation of many components in a spatial and temporal manner. Other results suggested that phosphorylation of FAK at Ser-910 regulates the association of FAK with paxillin [34] and that the phosphorylation of FAK at Ser-843 reduces the phosphorylation of Tyr-397 [37]. It is tempting to speculate that the phosphorylation of these serine residues plays a role in modulating the function and/or localization of FAK in epithelial cells. The elucidation of the precise role of FAK phosphorylation at Ser-910 and Ser-843 in epithelial cells challenged with a variety of GPCR agonists, phorbol esters or growth factors warrants further experimental work.
In conclusion, the results presented in this study show that stimulation with GPCR agonists strikingly stimulates FAK phosphorylation at Ser-910 and Ser-843, but do not further enhance the high basal level of Tyr-397 phosphorylation in both undifferentiated rat IEC-18 cells and polarized human T84 cells.
Footnotes
This work was supported by NIH Grant DK 56930, DK 55003, NCI Grant P50 CA90388 and P30 DK41301, Xiaohua Jiang was supported by NCI Grant CA09056.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Zachary I, Rozengurt E. Focal adhesion kinase (p125FAK): a point of convergence in the action of neuropeptides, integrins, and oncogenes. Cell. 1992;71:891–894. doi: 10.1016/0092-8674(92)90385-p. [DOI] [PubMed] [Google Scholar]
- 2.Rozengurt E. Convergent signalling in the action of integrins, neuropeptides, growth factors and oncogenes. Cancer Surv. 1995;24:81–96. [PubMed] [Google Scholar]
- 3.Hanks SK, Polte TR. Signaling through focal adhesion kinase. Bioessays. 1997;19:137–145. doi: 10.1002/bies.950190208. [DOI] [PubMed] [Google Scholar]
- 4.Schlaepfer DD, Hauck CR, Sieg DJ. Signaling through focal adhesion kinase. Prog Biophys Mol Biol. 1999;71:435–478. doi: 10.1016/s0079-6107(98)00052-2. [DOI] [PubMed] [Google Scholar]
- 5.Schaller MD. Biochemical signals and biological responses elicited by the focal adhesion kinase. Biochim. Biophys. Acta. 2001;1540:1–21. doi: 10.1016/s0167-4889(01)00123-9. [DOI] [PubMed] [Google Scholar]
- 6.Parsons JT. Focal adhesion kinase: the first ten years. J. Cell Sci. 2003;116:1409–1416. doi: 10.1242/jcs.00373. [DOI] [PubMed] [Google Scholar]
- 7.Eide BL, Turck CW, Escobedo JA. Identification of Tyr-397 as the primary site of tyrosine phosphorylation and pp60src association in the focal adhesion kinase, pp125FAK. Mol. Cell. Biol. 1995;15:2819–2827. doi: 10.1128/mcb.15.5.2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Calalb MB, Polte TR, Hanks SK. Tyrosine phosphorylation of focal adhesion kinase at sites in the catalytic domain regulates kinase activity: a role for Src family kinases. Mol. Cell. Biol. 1995;15:954–963. doi: 10.1128/mcb.15.2.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Owen JD, Ruest PJ, Fry DW, Hanks SK. Induced focal adhesion kinase (FAK) expression in FAK-null cells enhances cell spreading and migration requiring both auto- and activation loop phosphorylation sites and inhibits adhesion-dependent tyrosine phosphorylation of Pyk2. Mol. Cell. Biol. 1999;19:4806–4818. doi: 10.1128/mcb.19.7.4806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Salazar EP, Rozengurt E. Bombesin and platelet-derived growth factor induce association of endogenous focal adhesion kinase with Src in intact Swiss 3T3 cells. J. Biol. Chem. 2001;276:17788–17795. doi: 10.1074/jbc.274.40.28371. [DOI] [PubMed] [Google Scholar]
- 11.Ilic D, Furuta Y, Kanazawa S, Takeda N, Sobue K, Nakatsuji N, Nomura S, Fujimoto J, Okada M, Yamamoto T. Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature. 1995;377:539–544. doi: 10.1038/377539a0. [DOI] [PubMed] [Google Scholar]
- 12.Ilic D, Damsky CH, Yamamoto T. Focal adhesion kinase: at the crossroads of signal transduction. J. Cell Sci. 1997;110:401–407. doi: 10.1242/jcs.110.4.401. [DOI] [PubMed] [Google Scholar]
- 13.Gilmore AP, Romer LH. Inhibition of focal adhesion kinase (FAK) signaling in focal adhesions decreases cell motility and proliferation. Mol. Cell. Biol. 1996;7:1209–1224. doi: 10.1091/mbc.7.8.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cary LA, Chang JF, Guan JL. Stimulation of cell migration by overexpression of focal adhesion kinase and its association with Src and Fyn. J. Cell Sci. 1996;109:1787–1794. doi: 10.1242/jcs.109.7.1787. [DOI] [PubMed] [Google Scholar]
- 15.Sieg DJ, Iliâc D, Jones KC, Damsky CH, Hunter T, Schlaepfer DD. Pyk2 and Src-family protein-tyrosine kinases compensate for the loss of FAK in fibronectin-stimulated signaling events but Pyk2 does not fully function to enhance FAK- cell migration. EMBO J. 1998;17:5933–5947. doi: 10.1093/emboj/17.20.5933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sieg DJ, Hauck CR, Schlaepfer DD. Required role of focal adhesion kinase (FAK) for integrin-stimulated cell migration. J. Cell Sci. 1999;112:2677–2691. doi: 10.1242/jcs.112.16.2677. [DOI] [PubMed] [Google Scholar]
- 17.Sieg DJ, Hauck CR, Ilic D, Klingbeil CK, Schaefer E, Damsky CH, Schlaepfer DD. FAK integrates growth-factor and integrin signals to promote cell migration. Nat Cell Biol. 2000;2:249–256. doi: 10.1038/35010517. [DOI] [PubMed] [Google Scholar]
- 18.Webb DJ, Donais K, Whitmore LA, Thomas SM, Turner CE, Parsons JT, Horwitz AF. FAK-Src signalling through paxillin, ERK and MLCK regulates adhesion disassembly. Nat. Cell Biol. 2004;6:154–161. doi: 10.1038/ncb1094. [DOI] [PubMed] [Google Scholar]
- 19.Palazzo AF, Eng CH, Schlaepfer DD, Marcantonio EE, Gundersen GG. Localized stabilization of microtubules by integrin- and FAK-facilitated Rho signaling. Science. 2004;303:836–839. doi: 10.1126/science.1091325. [DOI] [PubMed] [Google Scholar]
- 20.Zhao J, Bian ZC, Yee K, Chen BP, Chien S, Guan JL. Identification of transcription factor KLF8 as a downstream target of focal adhesion kinase in its regulation of cyclin D1 and cell cycle progression. Mol Cell. 2003;11:1503–1515. doi: 10.1016/s1097-2765(03)00179-5. [DOI] [PubMed] [Google Scholar]
- 21.Hungerford JE, Compton MT, Matter ML, Hoffstrom BG, Otey CA. Inhibition of pp125FAK in cultured fibroblasts results in apoptosis. J. Cell Biol. 1996;135:1383–1390. doi: 10.1083/jcb.135.5.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu LH, Owens LV, Sturge GC, Yang X, Liu ET, Craven RJ, Cance WG. Attenuation of the expression of the focal adhesion kinase induces apoptosis in tumor cells. Cell Growth Differ. 1996;7:413–418. [PubMed] [Google Scholar]
- 23.Gervais FG, Thornberry NA, Ruffolo SC, Nicholson DW, Roy S. Caspases cleave focal adhesion kinase during apoptosis to generate a FRNK-like polypeptide. J Biol Chem. 1998;273:17102–17108. doi: 10.1074/jbc.273.27.17102. [DOI] [PubMed] [Google Scholar]
- 24.Golubovskaya VM, Gross S, Kaur AS, Wilson RI, Xu LH, Yang XH, Cance WG. Simultaneous inhibition of focal adhesion kinase and SRC enhances detachment and apoptosis in colon cancer cell lines. Mol. Cancer Res. 2003;1:755–764. [PubMed] [Google Scholar]
- 25.Owens LV, Xu L, Craven RJ, Dent GA, Weiner TM, Kornberg L, Liu ET, Cance WG. Overexpression of the focal adhesion kinase (p125FAK) in invasive human tumors. Cancer Res. 1995;55:2752–2755. [PubMed] [Google Scholar]
- 26.Owens LV, Xu L, Dent GA, Yang X, Sturge GC, Craven RJ, Cance WG. Focal adhesion kinase as a marker of invasive potential in differentiated human thyroid cancer. Annals Surgical Oncol. 1996;3:100–105. doi: 10.1007/BF02409059. [DOI] [PubMed] [Google Scholar]
- 27.Mukai M, Togawa A, Imamura F, Iwasaki T, Ayaki M, Mammoto T, Nakamura H, Tatsuta M, Inoue M. Sustained tyrosine-phosphorylation of FAK through Rho-dependent adhesion to fibronectin is essential for cancer cell migration. Anticancer Res. 2002;22:3175–3184. [PubMed] [Google Scholar]
- 28.Gabarra-Niecko V, Schaller MD, Dunty JM. FAK regulates biological processes important for the pathogenesis of cancer. Cancer Metastasis Rev. 2003;22:359–374. doi: 10.1023/a:1023725029589. [DOI] [PubMed] [Google Scholar]
- 29.Hsia DA, Mitra SK, Hauck CR, Streblow DN, Nelson JA, Ilic D, Huang S, Li E, Nemerow GR, Leng J, Spencer KS, Cheresh DA, Schlaepfer DD. Differential regulation of cell motility and invasion by FAK. J Cell Biol. 2003;160:753–767. doi: 10.1083/jcb.200212114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sood AK, Coffin JE, Schneider GB, Fletcher MS, DeYoung BR, Gruman LM, Gershenson DM, Schaller MD, Hendrix MJ. Biological significance of focal adhesion kinase in ovarian cancer: role in migration and invasion. Am J Pathol. 2004;165:1087–1095. doi: 10.1016/S0002-9440(10)63370-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schlaepfer DD, Mitra SK, Ilic D. Control of motile and invasive cell phenotypes by focal adhesion kinase. Biochim. Biophys. Acta. 2004;1692:77–102. doi: 10.1016/j.bbamcr.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 32.Yamakita Y, Totsukawa G, Yamashiro S, Fry D, Zhang X, Hanks SK, Matsumura F. Dissociation of FAK/p130(CAS)/c-Src complex during mitosis: role of mitosis-specific serine phosphorylation of FAK. J Cell Biol. 1999;144:315–324. doi: 10.1083/jcb.144.2.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ma A, Richardson A, Schaefer EM, Parsons JT. Serine phosphorylation of focal adhesion kinase in interphase and mitosis: a possible role in modulating binding to p130(Cas) Mol. Biol. Cell. 2001;12:1–12. doi: 10.1091/mbc.12.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hunger-Glaser I, Salazar EP, Sinnett-Smith J, Rozengurt E. Bombesin, lysophosphatidic acid, and epidermal growth factor rapidly stimulate focal adhesion kinase phosphorylation at Ser-910: requirement for ERK activation. J Biol Chem. 2003;278:22631–22643. doi: 10.1074/jbc.M210876200. [DOI] [PubMed] [Google Scholar]
- 35.Sinnett-Smith J, Zhukova E, Hsieh N, Jiang X, Rozengurt E. Protein kinase D potentiates DNA synthesis induced by Gq-coupled receptors by increasing the duration of ERK signaling in swiss 3T3 cells. J Biol Chem. 2004;279:16883–16893. doi: 10.1074/jbc.M313225200. [DOI] [PubMed] [Google Scholar]
- 36.Hunger-Glaser I, Fan RS, Perez-Salazar E, Rozengurt E. PDGF and FGF induce focal adhesion kinase (FAK) phosphorylation at Ser-910: dissociation from Tyr-397 phosphorylation and requirement for ERK activation. J Cell Physiol. 2004;200:213–222. doi: 10.1002/jcp.20018. [DOI] [PubMed] [Google Scholar]
- 37.Fan RS, Jacamo RO, Jiang X, Sinnett-Smith J, Rozengurt E. G protein-coupled receptor activation rapidly stimulates focal adhesion kinase phosphorylation at Ser-843. Mediation by Ca2+, calmodulin, and Ca2+/calmodulin-dependent kinase II. J Biol Chem. 2005;280:24212–24220. doi: 10.1074/jbc.M500716200. [DOI] [PubMed] [Google Scholar]
- 38.Basson MD, Emenaker NJ, Sanders MA. Alpha integrin subunits regulate human (Caco-2) intestinal epithelial proliferation and phenotype. Cell Physiol Biochem. 2000;10:27–36. doi: 10.1159/000016332. [DOI] [PubMed] [Google Scholar]
- 39.Jiang X, Jacamo R, Zhukova E, Sinnett-Smith J, Rozengurt E. RNA interference reveals a differential role of FAK and Pyk2 in cell migration, leading edge formation and increase in focal adhesions induced by LPA in intestinal epithelial cells. J Cell Physiol. 2006;207:816–828. doi: 10.1002/jcp.20629. [DOI] [PubMed] [Google Scholar]
- 40.Calandrella BK, Keely SJ. Transactivation of the epidermal growth factor receptor mediates muscarinic stimulation of focal adhesion kinase in intestinal epithelial cells. J Cell Physiol. 2005;203:103–110. doi: 10.1002/jcp.20190. SO. [DOI] [PubMed] [Google Scholar]
- 41.Lunn JA, Rozengurt E. Hyperosmotic stress induces rapid focal adhesion kinase phosphorylation at tyrosines 397 and 577. Role of Src family kinases and Rho family GTPases. J Biol Chem. 2004;279:45266–45278. doi: 10.1074/jbc.M314132200. [DOI] [PubMed] [Google Scholar]
- 42.Thamilselvan V, Basson MD. The role of the cytoskeleton in differentially regulating pressure-mediated effects on malignant colonocyte focal adhesion signaling and cell adhesion. Carcinogenesis. 2005;26:1687–1697. doi: 10.1093/carcin/bgi135. [DOI] [PubMed] [Google Scholar]
- 43.Earp HS, Huckle WR, Dawson TL, Li X, Graves LM, Dy R. Angiotensin II activates at least two tyrosine kinases in rat liver epithelial cells. Separation of the major calcium-regulated tyrosine kinase from p125FAK. J Biol Chem. 1995;270:28440–28447. doi: 10.1074/jbc.270.47.28440. [DOI] [PubMed] [Google Scholar]
- 44.Young SH, Rozengurt E. Qdot nanocrystal conjugates conjugated to bombesin or ANG II label the cognate G protein-coupled receptor in living cells. Am J Physiol Cell Physiol. 2006;290:C728–732. doi: 10.1152/ajpcell.00310.2005. [DOI] [PubMed] [Google Scholar]
- 45.Chiu T, Rozengurt E. PKD in intestinal epithelial cells: rapid activation by phorbol esters, LPA, and angiotensin through PKC. Am J Physiol Cell Physiol. 2001;280:C929–942. doi: 10.1152/ajpcell.2001.280.4.C929. [DOI] [PubMed] [Google Scholar]
- 46.Chiu T, Santiskulvong C, Rozengurt E. ANG II stimulates PKC-dependent ERK activation, DNA synthesis, and cell division in intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2003;285:G1–11. doi: 10.1152/ajpgi.00419.2002. [DOI] [PubMed] [Google Scholar]
- 47.Chiu T, Santiskulvong C, Rozengurt E. EGF receptor transactivation mediates ANG II-stimulated mitogenesis in intestinal epithelial cells through the PI3-kinase/Akt/mTOR/p70S6K1 signaling pathway. Am J Physiol Gastrointest Liver Physiol. 2005;288:G182–194. doi: 10.1152/ajpgi.00200.2004. [DOI] [PubMed] [Google Scholar]
- 48.Wu SS, Chiu T, Rozengurt E. ANG II and LPA induce Pyk2 tyrosine phosphorylation in intestinal epithelial cells: role of Ca2+, PKC, and Rho kinase. Am J Physiol Cell Physiol. 2002;282:C1432–1444. doi: 10.1152/ajpcell.00323.2001. [DOI] [PubMed] [Google Scholar]
- 49.Wu SS, Yamauchi K, Rozengurt E. Bombesin and angiotensin II rapidly stimulate Src phosphorylation at Tyr-418 in fibroblasts and intestinal epithelial cells through a PP2-insensitive pathway. Cell Signal. 2005;17:93–102. doi: 10.1016/j.cellsig.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 50.Uribe JM, McCole DF, Barrett KE. Interferon-gamma activates EGF receptor and increases TGF-alpha in T84 cells: implications for chloride secretion. Am J Physiol Gastrointest Liver Physiol. 2002;283:G923–931. doi: 10.1152/ajpgi.00237.2002. [DOI] [PubMed] [Google Scholar]
- 51.Ishida A, Kameshita I, Okuno S, Kitani T, Fujisawa H. A novel highly specific and potent inhibitor of calmodulin-dependent protein kinase II. Biochem Biophys Res Commun. 1995;212:806–812. doi: 10.1006/bbrc.1995.2040. [DOI] [PubMed] [Google Scholar]
- 52.Rozengurt E. Signal transduction pathways in the mitogenic response to G protein-coupled neuropeptide receptor agonists. J Cell Physiol. 1998;177:507–517. doi: 10.1002/(SICI)1097-4652(199812)177:4<507::AID-JCP2>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 53.Keely SJ, Uribe JM, Barrett KE. Carbachol stimulates transactivation of epidermal growth factor receptor and mitogen-activated protein kinase in T84 cells. Implications for carbachol-stimulated chloride secretion. J Biol Chem. 1998;273:27111–27117. doi: 10.1074/jbc.273.42.27111. [DOI] [PubMed] [Google Scholar]
- 54.Contos JJ, Ishii I, Chun J. Lysophosphatidic acid receptors. Mol Pharmacol. 2000;58:1188–1196. doi: 10.1124/mol.58.6.1188. [DOI] [PubMed] [Google Scholar]
- 55.Hauck CR, Sieg DJ, Hsia DA, Loftus JC, Gaarde WA, Monia BP, Schlaepfer DD. Inhibition of focal adhesion kinase expression or activity disrupts epidermal growth factor-stimulated signaling promoting the migration of invasive human carcinoma cells. Cancer Res. 2001;61:7079–7090. [PubMed] [Google Scholar]
- 56.Goetzl EJ, An S. Diversity of cellular receptors and functions for the lysophospholipid growth factors lysophosphatidic acid and sphingosine 1-phosphate. Faseb J. 1998;12:1589–1598. [PubMed] [Google Scholar]
- 57.Moolenaar WH. Lysophosphatidic acid signalling. Curr Opin Cell Biol. 1995;7:203–210. doi: 10.1016/0955-0674(95)80029-8. [DOI] [PubMed] [Google Scholar]
- 58.Marshall CJ. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell. 1995;80:179–185. doi: 10.1016/0092-8674(95)90401-8. [DOI] [PubMed] [Google Scholar]
- 59.Seger R, Krebs EG. The MAPK signaling cascade. Faseb J. 1995;9:726–735. [PubMed] [Google Scholar]