

Abstract
Activated ras genes have been frequently identified in both benign and malignant human tumors, including keratoacanthoma and squamous cell carcinoma. In this study, we developed two lines of transgenic rabbits in which the expression of EJras has been specifically targeted to the rabbit epidermal keratinocytes, using the upstream regulatory region of cottontail rabbit papillomavirus. All of the F1 transgenic progenies developed multiple keratoacanthomas at about 3 days after birth. The rabbits developed an average of 20 tumors, which usually reached the size of approximately 1 cm in diameter and then spontaneously regressed in about 2 months, similar to keratoacanthoma regression in humans. In addition, up to 18% of the rabbits then developed squamous cell carcinoma at about 5 months of age. The expression of EJras was detectable in all of the keratoacanthomas and squamous cell carcinomas. These results strongly support the involvement of the ras oncogene in both the initiation and regression of keratoacanthoma, and in the development of squamous cell carcinomas. These novel transgenic rabbits, with their consistent tumorigenic phenotype at an early age, high similarity to the human lesions, and easy accessibility for examination, manipulation, biopsy, and treatment, should provide a unique model system for studying ras activation-related tumor initiation, regression, and progression, and for evaluating antitumor therapies.
Ras genes encode a family of plasma membrane-bound proteins (p21ras) that function as intermediates in cell signal transduction pathways. These proteins play a pivotal role in regulating cell growth and differentiation in nearly all studied eukaryotic cell types. 1 The activity of ras proteins as regulating switches is strictly controlled by cycling between active GTP-bound and inactive GDP-bound states. 2 The active and inactive cycling of the p21ras proteins may be broken because of a point mutation at position 12, 13, or 61 of the proteins, which has been found in both experimental and spontaneous tumors. This cellular transformation property is considered to be due to the extended life and resultant accumulation of p21ras proteins in the active GTP state, 3 which leads to tumorigenesis due to loss of regulatory control of cell proliferation and differentiation. However, many aspects of ras mutation-related neoplasia still remain to be elucidated, especially the dual roles of activated ras genes in cell growth stimulation or differentiation, and its correlation with tumor initiation, regression, and progression.
It is well known that carcinogenesis is a complex multistep process that usually involves mutation and/or inappropriate expression of certain cellular genes, such as the mutational activation of proto-oncogenes and the inactivation of tumor suppressor genes. Among the oncogenes, activated ras genes have been found in a wide variety of human tumors, and ranges from 10–20% Ha-ras activation in bladder carcinomas to 85–100% Ki-ras in colorectal and pancreatic tumors. 4 Activated Ha-ras is also frequently found in skin tumors. It was reported that 30% of human keratoacanthomas and 13% of squamous cell carcinomas (SCCs) contained specific mutations (A-to-T transversion) in codon 61 of the c-Ha-ras oncogene. 5 Another study reported that 40% of the human skin cancers studied contained mutations in codon 12 of Ha-ras. 6 These results clearly indicate the involvement of activated Ha-ras genes in the development of both benign and malignant skin tumors. The roles of activated Ha-ras in skin tumor development are further supported by animal studies, including carcinogen-treated animals 7-11 and transgenic animals. 12-17 Mouse skin carcinogenesis as a biological model has been used to study Ha-ras activation-related skin tumors for many years. The Sencar mice are the most widely used model for studying so-called two-stage carcinogenesis because of their sensitivity to the carcinogens 7, 12-dimethylbenz(a)anthracene (DMBA) and 12-O-tetradecanoylphorbol-13-acetate (TPA). 8 At the molecular level, mutation of Ha-ras (A-to-T transversion) in codon 61 has been identified as one initiating event in skin tumors by the carcinogens, most notably DMBA. 7,9,11 Similar results have also been obtained from studies of DMBA-treated rabbits. 10 In most of the present transgenic mouse lines, the animals usually develop hyperplasia, hyperkeratosis, and papillomatosis at sites of wounding or abrasion. 18 Only a few ζ-globin–v–Ha-ras mice 13 and the transgenic rabbits that carried both EJras and the cottontail rabbit papillomavirus (CRPV) genome 16 developed carcinomas. Based upon these studies, it has been generally accepted that activation of ras oncogenes plays important roles in neoplastic induction, and that malignant progression requires the participation of additional cellular and genetic factors. For example, mutation of the tumor suppressor gene p53 confers the single greatest known selective advantage favoring cancer formation, and it has been found in many human cancers, 19 including skin cancer. 20 It has been shown that normal p53 protein can suppress the transformation of primary cells by E1a and ras oncogene. 21 However, it is still not well understood how the activated ras genes initiate neoplasia and are also involved in regression in some cases, but progression in other cases of the initiated tumors. A recent study involving the targeted expression of a mutant Ha-ras in epidermal cells of transgenic mice by a truncated keratin 5 promoter suggests that the nature of cells in which tumor initiation occurs is a major determinant of malignant progression. 17 This study showed that the benign tumors derived primarily from cells located within hair follicles have a risk of malignant conversion. This contrasts with the effects seen in previous studies of targeted Ha-ras expression to other epidermal cells in which malignant conversion is rarely observed. 12,14,15
The relationship between keratoacanthoma and squamous cell carcinoma (SCC) has been debated for several decades. 21 Because of its rapid growth, occasional metastasis, and high histological similarity to SCC, keratoacanthoma has been suggested to be a type of SCC. 22 However, it seems to have been generally accepted that keratoacanthoma is a common benign skin tumor in humans and occurs about one-third to 1.8 times as often as SCC. 23 Although keratoacanthoma resembles SCC histologically and can metastasize in some cases, 22 most of the keratoacanthomas regress spontaneously within a few months after an initial period of rapid growth. The causes of keratoacanthoma and SCC are still uncertain. However, it is known that the incidences of both keratoacanthoma and SCC show parallel increases with increased sun exposure. 24 In correlation with this, a relatively high incidence of the mutated Ha-ras oncogene has been found in keratoacanthomas from both human and experimental models, 5, 10 and in human and animal cutaneous SCC. 16, 25-27 These findings further suggest that ras oncogenes play important roles in all steps of tumor initiation, regression, and progression, and that additional cellular factors appear to be involved in the malignant progression. However, the mechanisms of the ras-related regression of keratoacanthoma and progression of SCC are still not well understood.
In this study, the EJras oncogene was targeted to be expressed in the rabbit epidermal keratinocytes, and all of the transgenic rabbits developed keratoacanthomas at an early age. Approximately 18% of them developed SCC several months later. The ras-associated keratoacanthomas and SCCs that developed in the rabbits have tumor morphology and behavior that closely resemble those of the tumors that develop in humans. These transgenic rabbits should provide an excellent model system for studying the roles of ras oncogenes in tumor initiation, regression, and progression. It may also be useful to study the relationship and differential diagnoses between keratoacanthoma and SCC that have challenged pathologists for many years.
Materials and Methods
Preparation of DNA Constructs
This study was designed to target the expression of activated human T24(EJ) c-Ha-ras (EJras) 28 to the rabbit epidermis by the upstream regulatory region (URR) of cottontail rabbit papillomavirus (CRPV). EJras has a point mutation in codon 12 that results in the substitution of valine for glycine at that position in the p21 protein. 28 To prepare the construct URR-EJras (Figure 1A) ▶ for making transgenic rabbits, a plasmid of pGEM3zf(−) URR-EJras was constructed in our laboratory. The CRPV URR fragment was obtained by polymerase chain reaction (PCR), using the wild CRPV genome contained in the plasmid PLAII-CRPV as template. The fragment covering the CRPV sequence from nucleotide (nt) 7241 to nt 128 is known to contain the regulatory elements required for regulating the replication and transcription of the viral coding genes in rabbit epidermal keratinocytes. 29, 30 The two ends of the URR fragment were designed to have the restriction sites SalI (5′) and BamHI (3′) by the PCR primers: the sense primer 5′-CGCGTCGACAGAAAGTTCCTAAATCAAAG-3′ (underlined is SalI site), and the antisense primer 5′-GGATCCGCGGCAGAAATTCCT GGCAATGC-3′ (underlined is BamHI site). The URR fragment was then cloned into the SalI/BamHI site of the polylinker of pGEM3zf(−) (Promega) to form the plasmid pGEM3zf(−)URR. To create the plasmid pGEM3zf(−)URR-EJras, the fragment of EJras coding region with the ends of BamHI (5′) and EcoRI (3′) was cloned into the BamHI/EcoRI site of pGEM3zf(−)URR. The fragment of EJras coding region was also obtained by PCR, using the wild-type EJras genome as the template, and the BamHI (5′) and EcoRI (3′) ends were introduced by the PCR primers: the sense primer 5′-CGCGGATCCCCCTGAGGAGCGATGACGGA-3′ (underlined is BamHI site), and antisense primer 5′-CGGAATTCCGCCGAAAACCAAGATCAAGA-3′ (underlined is the EcoRI site). The EJras coding region contains the sequence from nt 1652 to nt 3758, which contains all four exons, three introns, and the polyadenylation signal of EJras. A 2.9-kb fragment containing the entire URR-EJras sequences was released by SalI and EcoRI and fractionated by gel electrophoresis on a 0.8% agarose gel. The URR-EJras fragment was then purified by a Gene Clean kit (Bio 101, La Jolla, CA) and resuspended in TE buffer (10 mmol/L Tris and 0.25 mmol/L EDTA) for microinjection.
Figure 1.

A: diagram showing the structure of the URR-EJras transgene. The URR-EJras transgene contains a 0.76-kb regulatory fragment (URR) from CRPV and a 2.1-kb coding sequence of EJras. The fragment of CRPV URR (diagonal box) was ligated to the coding sequence of EJras, which contains all of the exons (black box), introns (open box), and a 3′ flanking sequence carrying the polyadenylation signal as indicated. B: Southern blot analysis of EJras in the transgenic founders. Each lane contains 10 μg DNA, which was isolated from the biopsies of the rabbit ear skin and digested by BamHI and EcoRI. DNAs were isolated from transgenic founders TR-ras1 (lane 1) and TR-ras2 (lane 2) and four nontransgenic rabbits (lanes 3 to 6). DNA in lane 7 was the SalI- and EcoRI-released URR-EJras DNA reconstructed with normal rabbit skin DNA as a control. The two fragments produced by BamHI (EcoRI does not cut unintegrated URR-EJras) digestion in lane 7 indicate the appropriate separation of URR (0.76 kb) and the EJras coding sequence (2.1 kb). The fragment patterns shown in lanes 1 and 2 indicate that the TR-ras 1 rabbit may contain a single copy of integrated URR-EJras, and the TR-ras 2 rabbit contains multiple copies of the integrated URR-EJras, with head-to-tail tandem repeats.
Generation of Transgenic Rabbits
The procedure for producing transgenic rabbits was based on the techniques previously described, 31, 32 with some modifications in our laboratory. 16 New Zealand White rabbits were purchased from Covance Research Products (Denver, PA). For inducing superovulation, 120 U of pregnant mare’s serum gonadotropin (PMSG) and 150 U of human chorionic gonadotropin (hCG) were intravenously injected into female donor rabbits on day 1 and day 4, respectively. The rabbits were mated with a fertile male on day 4 immediately before the administration of hCG. Before harvest of fertilized eggs on day 5, the rabbits were euthanized by intravenous injection of an overdose of pentobarbital (100 mg/kg), and oviducts were collected. One-celled embryos were collected by flushing the oviducts with phosphate-buffered saline (PBS) supplemented with 20% fetal calf serum (FCS). Microinjection of the URR-EJras DNA fragment (5 μg/ml) into the male pronucleus of fertilized eggs was conducted immediately after the collection. The injected eggs were incubated about 1 hour after microinjection and then transplanted into the oviduct of pseudopregnant recipient rabbits that had been mated with a sterile (vasectomized) male at the same time that the donors were mated. The surgery for embryo transplantation was by laparotomy while the rabbits were under halothane anesthesia.
Identification of Transgenic Rabbits
DNA samples (10 μg/ml), isolated from ear biopsies, were digested with appropriate restriction enzymes, electrophoresed on a 0.8% agarose gel, and blotted to Zeta-probe nylon membrane (BioRad, Richmond, CA) for Southern blot analysis, using the manufacturer’s protocol. DNA probe for hybridization was prepared by random hexamer [32P]dATP labeling of the URR-EJras fragment (Figure 1A) ▶ , via the Multiprime DNA Labeling System (Amersham, Arlington, IL). Both the hybridization and washes of blots were performed under stringent conditions, as recommended by the manufacturer’s protocol. Briefly, the blotted membranes were hybridized with 1 × 10 6 cpm of probe/ml in the solution containing 1.5× SSPE (20× SSPE equals 3.5 mol/L NaC1, 0.2 mol/L Na2HPO4, and 0.002 mol/L EDTA), 0.1% SDS, and 10% dextran sulfate at 68°C overnight. After completion of hybridization, the membranes were washed at room temperature for 15 minutes in each of the following washing solutions: 2× SSC (0.3 mol/L NaC1 and 0.03 mol/L trisodium citrate)/0.1% SDS, 0.5× SSC/0.1% SDS and 0.1× SSC/0.1% SDS. A last wash was done in 0.1× SSC/0.1% SDS at 65°C for 30 minutes. Signals were visualized by exposing the hybridized blots to Kodak XAR-5 film with DuPont intensifying screens at −70°C.
Northern Blot Analysis
Total cellular RNA was extracted from homogenized tissue samples by the acid guanidinium isothiocyanate-phenol-chloroform technique. 33 The RNA samples (15 μg) were electrophoresed in 1.2% formaldehyde agarose gel and blotted to the Zeta-probe nylon membrane (BioRad) with 10% SSC buffer by capillary action. The procedures of probe preparation, blot hybridization and washes, and signal visualization were the same as described for the Southern blot.
Western Blot Analysis
Protein isolation and Western blot analysis were carried out essentially as previously described. 34 Total proteins in homogenized tumor and normal tissues were extracted in a lysis buffer supplemented with proteinase inhibitor. The protein content was determined by using the Bio-Rad protein assay. The protein samples (50 μg) were fractionated on 12% SDS-PAGE and transferred to nitrocellulose membrane (Schleicher and Schuell, Nashua, NH). The immunoblotting was carried out using pan-ras (Val12) (Oncogene Research Products), a monoclonal antibody raised against a Val 12 peptide. This antibody reacts specifically with Val 12 Ha-ras but not Gly 12 or Asp 12 ras p21 protein. 35 The blot was blocked with 1% Tween 20 and 5% nonfat dry milk, and then visualized using the enhanced chemilumine-scence (ECL) system with horseradish peroxidase-conjugated goat anti-mouse IgG antibody (Amersham).
Necropsy and Histopathology
A complete gross examination was performed, and all organs were carefully dissected and examined. Gross lesions were recorded and photographed. All organs and tissues were collected and fixed in 10% neutral buffered formalin. Bone structures were briefly decalcified in a mild buffered acid solution. Tissues were embedded in paraplast embedding medium, sectioned at 5 μm, stained with hematoxylin and eosin (H&E), coverslipped, and examined histologically. Selected tissues were embedded in Spur’s Resin, sectioned at 2 μm, and stained with toluidine blue. The following tissues were routinely examined histologically: colon, cecum, ileum, jejunum, duodenum, stomach, pancreas, spleen, liver, gall bladder, kidney, adrenal, urinary bladder, testicles or ovaries, uterus, skeletal muscle, salivary glands, lymph nodes, trachea, esophagus, thyroid and parathyroid, heart, lung, thymus, brain, spinal cord, pituitary gland, bone, bone marrow, external ear canal, middle ear, cochlea, eyes, lacrimal glands, nasal passages, skin, and any other tissue that contained a suspected lesion. Some of the animals were anesthetized by halothane inhalation anesthesia, and tissue specimens were collected from tumors that had been surgically biopsied.
Result
Establishment of EJras Transgenic Lines
To create transgenic founders, the DNA construct of URR-EJras was injected into rabbit embryos at the single-cell stage. Two transgenic founders carrying the intact transgene (Figure 1B) ▶ were detected among 18 rabbits derived from the injected embryos. The transgenic founders, named TR-ras1 and TR-ras2, were born with the same appearance as their normal littermates. Although the two founders have different copies of the transgene and different locations of the transgene insertion as shown by Southern blot analysis, they have similar phenotypes. Multiple, small discrete tumors were observed on the rabbit skin at the age of about 20 days. Most tumors reached the size of about 1 cm in diameter and then spontaneously regressed.
Both of the transgenic founders are male, which provides a major advantage in establishing transgenic lines and producing enough F1 progeny for experiments in a short period. To produce F1 progeny, transgenic founders were mated with normal females. Ninety-one of 200 F1 progenies (46%) derived from the TR-ras1 breeding were positive for the transgene, and 14 of 40 progeny (35%) derived from the TR-ras2 breeding were transgenics. These breeding results indicate that the transgene in both TR-ras1 and TR-ras2 can be transmitted to the F1 generation through germ cells. The transmission rate of TR-ras1 (46%) is consistent with Mendel’s law. The transmission rate of TR-ras2 is obviously lower than 50%. There are two possibilities for this low transmission rate: 1) the rabbit is a chimera for the transgene, or 2) the transmission rate may increase with a larger number of F1 progeny produced by breeding the founder.
Tumor Behavior and Gross Morphology
Although the two transgenic founders were found to have tumors at around 20 days of age, all F1 transgenic progeny had small skin tumors at about 3 days after birth. It is possible that we overlooked small skin tumors in the two transgenic founders before 20 days of age. We have performed necropsies or surgical biopsies of tumors from 38 rabbits (28 females and 10 males) ranging from 18 hours to 438 days of age. The initial tumors could be identified at 1 to 3 days as multiple, small discrete nodules that were smooth and raised to about 1 to 3 mm in diameter. Skin tumors continued to form, and the rabbits usually had 5 to 10 nodules within a week, and carried about 20 tumors by 20 days of age. Although more tumors were usually found on ears, head, and dorsum, the sites for tumor development were random. The tumors occurred on almost all parts of the skin, including the pinna and external ear canal, and adjacent to the nose. The tumor development was independent of the wounding stimulus, because the F1 progeny had observable nodules as early as 1 day of age and there was no tumor development from the ear tagged or biopsied sites.
By 15 to 20 days of age, most tumors reached the size of about 1 cm in diameter and were covered by a crusty red or black exudate due to the occurrence of tumor necrosis. The tumor necrosis always started from the central part of the tumors. By this age, tumors could be found on almost all areas of skin, but they began to be covered by fur growth and were more obvious on the head and ears. On section, the tumors all formed a flat border adjacent to the layer of subcutaneous panniculus muscle. A central necrotic crater was obvious and was surrounded by firm, gray homogeneous tissue. Almost all tumors then spontaneously regressed in about 2 months. With the regression of most initial tumors, the rabbits had only a few persistent papillomas and/or hyperplastic skin lesions.
At this time, approximately 200 rabbits have been born from breeding F1 or F2 transgenic rabbits in line TR-ras1 to normal rabbits. Of those, 91 (46%) contained the EJras transgene, and all spontaneously developed multiple keratoacanthomas. Among 65 of the rabbits that survived more than 2 months, 12 (18.5%) have gone on to develop SCC between 60 days and 270 days of age (average = 140 days). SCC developed on the ear (six animals), back (three animals), abdomen (two animals), shoulder (one animal), hip (one animal), and foot (one animal). Two rabbits, at 60 and 225 days of age, developed SCC on both the ear and back, and on the ear and shoulder, respectively. Three of four rabbits that were examined by necropsy had metastatic disease. Among the 12 rabbits that developed SCC, six were males and six were females.
Histopathology of the Tumors
We examined multiple keratoacanthomas from each of 38 rabbits at the following ages: day 1 (five animals), day 3 (nine animals), days 6–14 (seven animals), days 15–23 (12 animals), and days 30–438 (five animals). The earliest histological examination of the tumors was made 18 hours after birth. At that time, the tumors were associated in length and width with hair follicles (Figure 2A) ▶ . At 3 days of age, the tumors had spread significantly in all directions, effacing normal dermal features (Figure 2B) ▶ . There were also small areas of apoptotic necrosis of neoplastic cells, with acantholysis, acantholytic cells, and red blood cells within cystic areas of the tumor (not shown).
Figure 2.
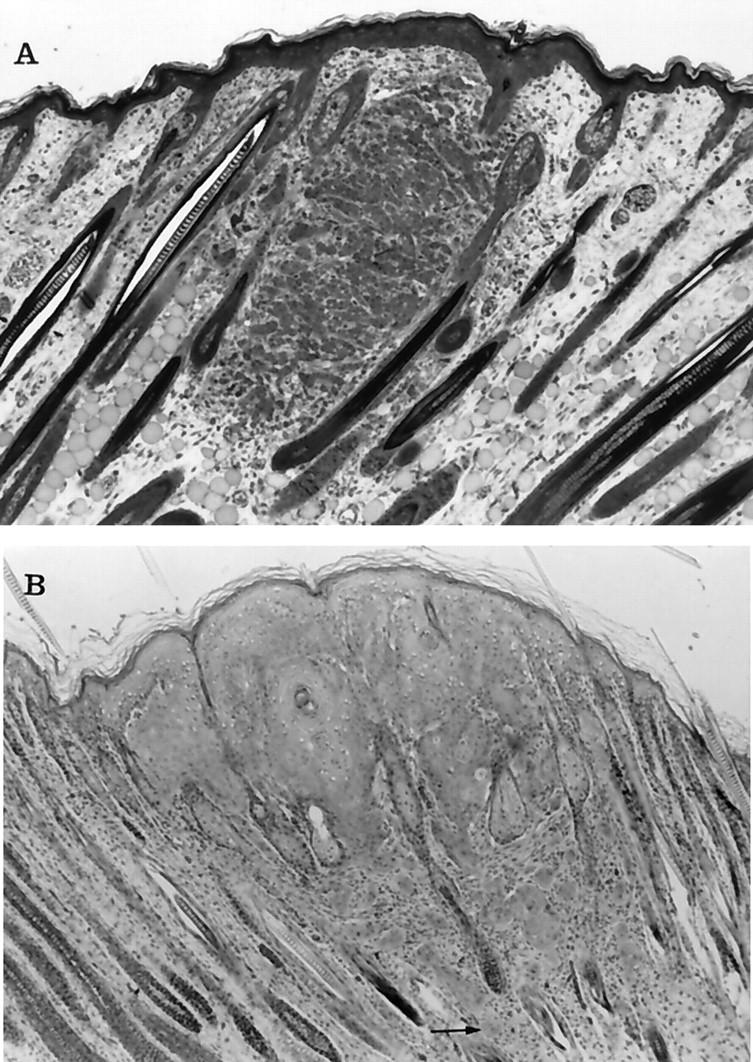
Photomicrographs of keratoacanthomas from transgenic rabbits at 1 and 3 days of age. A: At 18 hours after birth the small mass of epithelial cells appeared to be associated with hair follicles, although the overlying epidermis was also slightly thickened. The cells within the tumor were arranged in small clusters that were separated by blood vessels and connective tissue. Some of the cells contained cytoplasmic vacuoles, suggesting sebaceous differentiation. At higher power (not shown) intercellular bridges were visible between cells. B: At 3 days the tumor cells had spread laterally to replace and compress hair follicles and were contiguous with the overlying epidermis. The neoplastic cells shown squamous differentiation, including clusters of “glassy cells” and keratin horn formation. Notice the clusters of neoplastic cells that have invaded the deeper areas of the dermis separate from the main tumor mass (arrow). Toluidine blue and H&E stains, respectively. Original magnification, ×50.
At 10 days of age the tumors had a well-developed central core of necrosis that was open to the surface. The tumor margins compressed adjacent adnexal structures, forming a characteristic lip at the margins of the tumor. There were numerous heterophils within the areas of necrosis and within the dermis adjacent to neoplastic cells. By 15 to 20 days of age, the tumors were typical of young, rapidly growing keratoacanthomas, having fully developed the characteristic necrotic central core (Figure 3A and B) ▶ . At 60 days of age, the regressing tumors had become flattened and hyperkeratotic and began to resemble cutaneous papilloma (Figure 3C) ▶ . The persistent tumors were usually observed at the areas adjacent to the nose and ear tips, and some resembled pedunculated papillomas. A few of these tumors have persisted up to 6 months. Although some rabbits had both keratoacanthoma as well as SCC at necropsy, we have not yet clearly documented malignant progression directly from the previous tumors.
Figure 3.
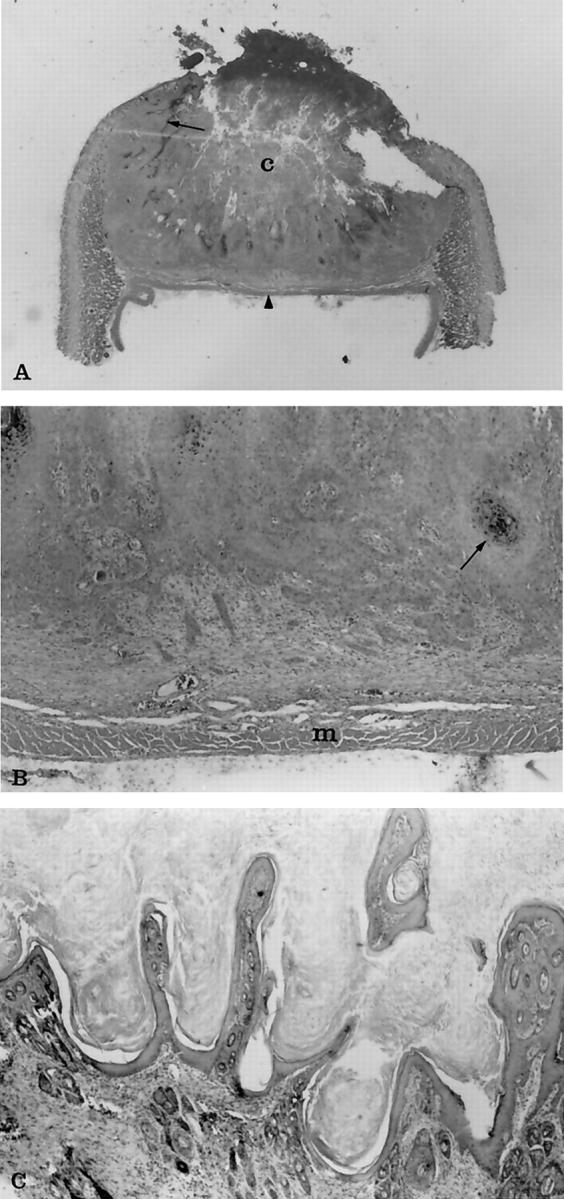
Photomicrographs of histological changes of regressing and almost regressed keratoacanthomas. A: The entire tumor from a 20-day-old transgenic rabbit at ×1.5 original magnification. Notice the characteristic necrotic central core (c) and the wall of epithelial cells that extend laterally under the epidermis (arrow). Typically the tumors terminated at the panniculus muscle (arrowhead), which on section gave the tumors a flattened side that was evident grossly as well as histologically. B: The same tumor at ×30 original magnification. Notice the multiple cords and clusters of neoplastic epithelial cells that have invaded deeper areas of dermis. The neoplastic cells shown squamous differentiation, including keratin production (arrow), dyskeratosis, and intercellular bridges, which are not seen at this magnification. Small clusters of cells at the leading edge of the tumor were associated with a fibrous connective tissue response. The neoplastic cells were not found to invade past the layer of panniculus muscle (m) in any of the specimens examined. C: An almost totally regressed tumor from a 60-day-old transgenic rabbit at ×50 original magnification. Notice the papillary projections of epidermal cells and hyperkeratosis that resembles papilloma. H&E stain.
We examined tumors from 12 rabbits between 60 and 270 days of age (average age = 140 days) that developed SCC confirmed by histopathology. Three of the four rabbits that were examined by necropsy had metastases to lungs (Figure 4A) ▶ , regional lymph nodes (2/3), heart (2/3), urinary bladder (1/3), mediastinum (1/3), spleen (1/3), kidney (1/3), pancreas (1/3), stomach (1/3), small intestine (1/3), large intestine (1/3), abdomen and mesentery (1/3), mammary gland (1/3), salivary gland (1/3), tongue (1/3), and iris and ciliary body within the eye (1/3). Metastases were not found in the two rabbits that each developed two separate SCCs on different areas of the body. The tumors were graded according to a modification of Broders classification 36 and classified into grade 2 (2/12) (Figure 4B) ▶ , grade 3 (9/12) (Figure 4C) ▶ , and grade 4 (2/12) (Figure 4D) ▶ .
Figure 4.
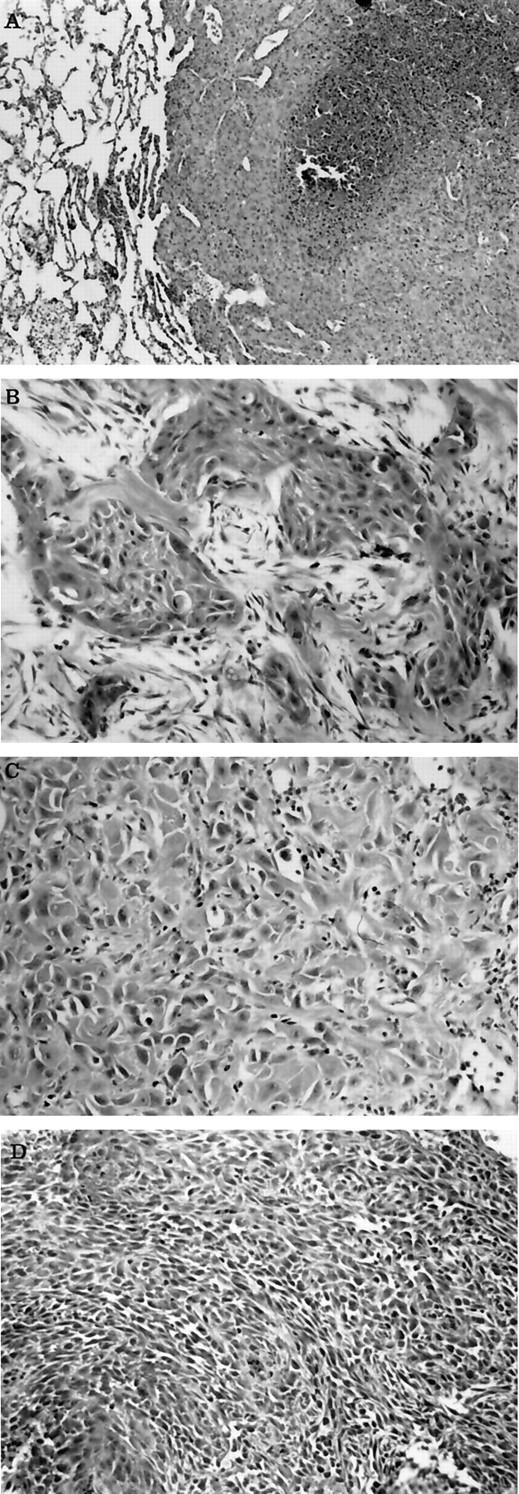
Photomicrographs of metastatic lesion and SCC with different grades. A: Metastatic lesion within lung from a transgenic rabbit that developed SCC. Notice the sheets of poorly differentiated neoplastic epithelial cells that have replaced lung, and the focal tumor necrosis. B: Cutaneous SCC from a transgenic rabbit that resembles grade II squamous cell carcinoma. Notice the clusters and cords of neoplastic epithelial cells with squamous differentiation separated by desmoplastic connective tissue. C: Cutaneous SCC from a transgenic rabbit that resembles grade III squamous cell carcinoma. Notice the sheets of poorly differentiated neoplastic epithelial cells with occasional intracellular keratin production. D: Cutaneous SCC from a transgenic rabbit that resembles grade IV squamous cell carcinoma. Notice the sheets of poorly differentiated neoplastic epithelial cells that show slight whorl patterns. H&E stain. Original magnification, ×50.
Expression of the Transgene
As expected, the URR-EJras transgene induced tumors specifically in the rabbit skin. To confirm the correlation between the expression of EJras and tumor induction, total cellular RNA isolated from keratoacanthomas and other organ tissues was analyzed by Northern blot, using the EJras DNA probe. The transcript of EJras was detected only in the tumors, but not in any other tissues, such as tongue, kidney, spleen, and brain (Figure 5A) ▶ . In addition, the expression of EJras p21 protein in keratoacanthoma and skin was also detected by Western blot, using the monoclonal antibody pan-ras (Val12), which reacts specifically with the Val 12 Ha-ras but not with the normal Gly 12 Ha-ras p21 protein. 35 It has been known that the amino acid sequence of the rabbit Ha-ras p21 protein is almost identical to the sequence of human p21, and both of them contain glycine in codon 12 of the normal sequence. 37 Consistent with the Northern analysis, the expression of EJras protein was also shown in all of the tested keratoacanthomas, but not normal rabbit skin (Figure 5B) ▶ . These results clearly indicate that tumor development in the rabbit skin was induced because of the expression of EJras oncogene under the regulatory control of CRPV URR.
Figure 5.

Detection, of EJras expression in transgenic rabbits. A: EJras mRNA was analyzed by Northern blot hybridization to 32P-labeled EJras DNA probe. Total cellular RNAs (15 μg/lane) were isolated from keratoacanthoma (lane 1), tongue (lane 2), kidney (lane 3), spleen (lane 4), and brain (lane 5 ) of a F1 progeny in the TR-ras transgenic line. Lane 6 is RNA isolated from the URR-EJras-transfected rabbit epidermal keratinocytes. The EJras transcript (1.2 kb) was detected only in keratoacanthoma (lane 1) and positive control (lane 6), but not in any of the normal organ tissues (lanes 2 to 5). The transcription of GAPDH is shown as loading control of RNA in each lane. The GAPDH hybridization was carried out after the previous probe on the same membrane was stripped off. The mRNA size was determined in comparison to the relative mobility of mRNA standards. B: The p21ras (Val12) protein was detected by Western blot with monoclonal antibody pan-ras (Val12). This antibody specifically recognizes the activated Ha-ras p21 with valine at the position of the codon 12, but not the normal Ha-ras with glycine at the same position. Lanes 1 to 4 are proteins isolated from keratoacanthomas of F1 progeny in the TR-ras1 transgenic line, and lane 5 is protein from normal rabbit skin. The Val 12 p21 Ha-ras protein is shown in all of the tested keratoacanthomas, which is consistent with the results shown by the Northern bolt analysis. Prestained molecular mass standards (kd) are shown on the left.
The transcription of EJras was also detectable in the SCC (Figure 6) ▶ , which indicates the involvement of EJras expression in the development of SCC. However, it appears that there was no significant difference in EJras transcription between keratoacanthomas and SCCs, which will be further studied.
Figure 6.

Northern blot analysis of EJras transcription in tumors of TR-ras1 transgenic rabbits. Each lane contains 15 μg total cellular RNA. Lanes 1 to 3 are RNAs isolated from SCCs of 3 transgenic rabbits, and lanes 4 to 7 are RNAs isolated from keratoacan-thomas of four transgenic rabbits, respectively. RNA in lane 8 was isolated from the normal skin of a rabbit, and RNA in lane 9 was isolated from URR-EJras-transfected rabbit epidermal keratinocytes. The probe and hybridization were the same as described in Figure 5 ▶ . The transcription of GAPDH is shown as loading control of RNA in each lane.
Discussion
The tissue specificity of infectious agents is known to be determined by factors intrinsic to the cell phenotype. It has also been shown, but is not as well recognized, that the cell phenotype within skin plays an important role in the response of skin to carcinogens. Cottontail rabbit papillomavirus (CRPV) is a strict species- and tissue-specific virus that infects the skin of cottontail and domestic rabbits and induces papillomas that may progress to carcinomas in the infected skin. 38 Previous studies suggest that this specificity is determined by regulatory elements within the upstream regulatory region (URR) of the virus genome. 29, 30 The tissue specificity of CRPV URR is further confirmed in our novel transgenic rabbits. Furthermore, an interesting finding in these transgenic rabbits is that the tumor development appears to originate from hair follicles, the origin of keratoacanthoma. This observation is consistent with the result of a recent study, which shows that hair follicle stem cells are the primary target cells of CRPV. 39 It appears that CRPV URR targets the expression of EJras in the follicle cells, which initiates the neoplasia in our transgenic rabbits. It has been shown, in mice and rabbits, that the progress of the hair growth cycle over the trunk created linear bilaterally symmetrical bands of active hair growth, whereas the remainder of the skin remained quiescent. 40 Many differences were found between the active and quiescent zones. Hair follicles in the active zones showed significantly more mitoses than did follicles in the quiescent zones. Proliferative lesions could also be induced more readily in areas of active hair growth. Inoculation of Shope papillomavirus into skin showing active hair growth and quiescent skin resulted in the development of larger and more numerous tumors in the active skin than in quiescent skin. 41 A recent study demonstrates that damage to the hair follicle might initiate the expression of Ha-ras oncogene and result in tumor development originating from the hair follicle of Ha-ras transgenic mice. 42 Moreover, evidence from a number of studies suggests that a population of epithelial cells within hair follicles may be the stem cells, which are more sensitive to chemical carcinogenesis. 17, 42, 43 These studies and the phenotypes shown in our transgenic rabbits support the possibility that follicle epidermal stem cells become capable of giving rise to tumors through acquisition of sufficient genetic alteration and may play a central role in skin carcinogenesis.
In the past, many experimental skin carcinogenesis studies have not considered the effect of the hair follicles and their growth patterns on tumor development. 44 Hair follicle growth activity represents a burst of concentrated proliferative activity compared to the stratum corneum of the epidermis and the sebaceous glands, which produce continuously but require only a low maintenance of mitotic activity. The generation of hair requires rates of proliferation and growth of epithelial cells that are far greater than those of the epidermis. Hair follicle epithelium is normally the most active of epidermal tissues to which cutaneous carcinogenic chemicals have access. By using experimental carcinogenesis studies in rabbits, it was shown that the type of tumor produced is dependent upon the state of the hair growth cycle. 45 Tumors may retain not only some of the appearances, but also some of the properties and functions of the tissue from which they originate. The rapid growth and regression exhibited by the deeper part of the hair follicle during the hair follicular cycle parallels the rapid growth, appearance, and regression of keratoacanthoma. It also indicates that keratoacanthoma is still under the control of the normal forces governing the growth of the hair follicle and the factors that set into motion the process of regression. 41 Therefore, keratoacanthoma within the skin of transgenic rabbits may respond to the effects of experimental stimulation and inhibition of hair follicle growth. Because the most important difference between keratoacanthoma and SCC is spontaneous regression of keratoacanthoma, the identification of mechanisms of keratoacanthoma regression may represent a significant step in understanding the development of SCC and a possible avenue for treatment development. When viewed from this perspective, these transgenic rabbits in which SCC follows keratoacanthoma appear somewhat analogous to the relationship of SCC after the CRPV induction of cutaneous papillomas. The carcinomas that develop after CRPV induced papillomas and the carcinomas that originate in the transgenic rabbits after regression of keratoacanthoma may both represent malignant transformation of hair follicle keratinocytes, which by current definition is squamous cell carcinoma. 22 The occurrence of both benign and malignant keratinocyte tumors within the same animal both associated with EJras provides for excellent controls in an animal model.
Wounding is known to be a fairly powerful stimulus to tumor formation in carcinogen-treated skin and is an efficient initiator of hair regrowth. 41 It has been reported that transgenic mice carrying K10 or HK1 gene promoter controlled mutant Ha-ras had tumor growth only at sites of wounding and scratching. 12, 15 However, tumor development in our transgenic rabbits was independent of wounding stimulus because the tumors were present at birth and there was no tumor growth observed at the ear tagged or biopsied sites. Similarly, skin tumors spontaneously developed in transgenic mice expressing a mutant Ha-ras under the control of truncated keratin 5 gene promoter (K5ras). 17 This variation probably resulted from the different regulatory elements used and the different cells that were targeted. Results from our transgenic rabbits and the K5ras mice indicate that the activated Ha-ras gene is enough to initiate the development of benign lesions in some types of cells without the necessity for a wounding stimulus as a second event. This is also supported by the studies of other transgenic mice carrying activated ras gene that produced lesions in Harderian gland, 46 pancreas, 47 and other organs 14 when the ras gene expression was targeted to the cells of these organs by different promoters. Therefore, the tumorigenesis of mutant ras genes may be strongly related to the targeted cell types and cell growth characteristics. 12, 17 However, it remains to be determined why EJras expression and keratoacanthoma induction spontaneously occurred in some sites of the rabbit skin but not in other sites, and why keratoacanthoma development has been seen only during the first cycle of hair growth in rabbits. One possibility is that, as discussed above, only a small population of follicular epidermal stem cells were involved in neoplasia.
Although the EJras oncogene can initiate the development of benign skin tumors during the rapid growth phase of skin and hair in new born rabbits, it does not appear to be able to maintain the lesions and induce malignant progression by itself. Most of the keratoacanthomas are prone to regression after development of the first hair coat in rabbits. This phenomenon is consistent in both our transgenic rabbits and most ras transgenic mice with skin lesions. 12 Tumor regression was also observed in Ha-ras oncogene-related human and rabbit keratoacanthomas. 5, 10 The phenomenon of spontaneous regression is strong evidence for the involvement of both hair follicle influence and ras oncogenes in the early stages of keratinocyte neoplasia. Because the function of CRPV URR and EJras expression may be related to hair follicle activity during hair growth, it can be imagined that the regression may be related to discontinued expression of EJras because of the recession of the hair follicle activity. Therefore, stimulation of hair follicle activity by hair plucking or other methods may be able to reinduce EJras expression and result in tumor growth again. It is known that many carcinogens stimulate mitotic activity, so that any suitable proliferation stimulus may awaken dormant neoplastic cells to tumor formation. 41
Additional possibilities are the influence of tissue barriers or of the blood supply to the tumors. We observed, in our transgenic rabbits, that there are increased numbers of small-caliber vessels associated with the tumors, and tumor regression usually started with necrosis of the tumor center when the tumors reached a size of about 1/2 cm in diameter in about 10 days. It is possible that ras expression-regulated angiogenesis was not enough to support the rapid tumor growth, and this resulted in tumor necrosis and regression, although it has been reported that oncogenic H-ras can stimulate tumor angiogenesis by up-regulation of vascular endothelial growth factor (VEGF). 48, 49 Another possibility is the induction of tumor necrosis factor-α (TNF-α) expression at a later stage that results in apoptotic cell death. 50 Although it has been reported that expression of H-ras oncogene can inhibit the cellular susceptibility to apoptosis, 51, 52 this might be true only at an early stage of tumor development or during the oncogenic cooperation between ras genes and some other tumor genes. It will be interesting to study the correlation between the ras gene expression and TNF-α expression/tumor necrosis or VEGF expression at various stages of tumor development. Further understanding of these reactions may be important for the prevention of malignant progression.
It is well known that activated Ha-ras genes require the cooperation of additional oncogenic factors for malignant transformation of epidermal cells. For example, our previous transgenic rabbits that carried both CRPV genome and EJras developed SCC, and the rabbits that carried CRPV genome alone developed only papillomas. 16 This has been further confirmed by the regression of keratoacanthomas in our current transgenic rabbits carrying only EJras targeted to epidermis by URR of CRPV. However, SCC also occurred in about 18% of our transgenic rabbits and developed in K5ras transgenic mice, 17 although the origin and development of SCC in these animals remain to be elucidated. Further studies are needed to determine whether the SCCs progress directly from keratoacanthomas or if they are independent of each other. Mapping of keratoacanthomas in a larger number of rabbits may determine if SCC appears as progression of the benign tumors. Northern blot analysis indicates that there is no significant difference in EJras transcription in keratoacanthomas and SCCs that developed in the rabbits. This result suggests that some intrinsic factors, or genetic variation among the animals, may play important roles in ras oncogene-related malignant progression. An important candidate among the possible factors is the inactivated p53 tumor suppressor gene due to mutations, the most common genetic alteration in human cancers. 19 The mutations in p53 are the most frequently found gene in cutaneous SCCs 20, 53, 54 but appear not to be common in keratoacanthomas. 55 In addition, several studies have suggested that mutant p53 can act in cooperation with v-Ha-ras to inhibit negative growth regulation by TGF-α. 56, 57 Therefore, interesting results may be obtained by analysis of p53 status in the rabbits with keratoacanthomas and SCCs.
In summary, these results clearly indicate that the EJras oncogene is sufficient to initiate the development of keratoacanthoma in newborn rabbits and that the tumors regress spontaneously after the development of the first coat of hair. EJras is also clearly associated with the development of a lesser number of SCCs within a few months in the transgenic rabbits. Therefore, these rabbits should provide a useful in vivo system for studying the important mechanisms of mutant Ha-ras gene-related tumor initiation, regression, and progression. They may also become useful in the assessment of tumor prevention and antitumor therapies.
Acknowledgments
We thank Dr. Klaus Helm, a dermatopathologist in the Departments of Pathology and Dermatology of the M. S. Hershey Medical Center, for his assistance in histological evaluation and his helpful discussions.
Footnotes
Address reprint requests to Dr. Xuwen Peng, Department of Comparative Medicine, The Milton S. Hershey Medical Center, The Pennsylvania State University, P.O. Box 850, Hershey, PA 17033. E-mail: xxp1@psu.edu.
This work was partially supported by grant IRG-196A from the American Cancer Society and PSGHS Cancer Center Research grant 1998.
References
- 1.Barbacid M: ras genes. Annu Rev Biochem 1987, 56:779-827 [DOI] [PubMed] [Google Scholar]
- 2.Trahey M, McComick F: A cytoplasmic protein stimulates normal N-ras p21 GTPase, but does not affect oncogenic mutants. Science 1987, 238:542-545 [DOI] [PubMed] [Google Scholar]
- 3.Polakis P, McCormick F: Structural requirements for the interaction of p21ras with GAP exchange factors, and its biological effector target. J Biol Chem 1993, 268:9157-9160 [PubMed] [Google Scholar]
- 4.Bos JL: ras oncogenes in human cancer: a review. Cancer Res 1989, 49:4682-4689 [PubMed] [Google Scholar]
- 5.Corominas M, Kamino H, Leon J, Pellicer A: Oncogene activation in human benign tumors of the skin (keratoacanthoma): is Hras involved in differentiation as well as proliferation? Proc Natl Acad Sci USA 1989, 86:6372-6376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Piercell W, Goldburg LH, Tainsky MA, Mukhopadhyay T, Ananthaswamy HN: Ras gene mutation and amplification in human nonmelanoma skin cancers. Mol Carcinog 1991, 4:196-202 [DOI] [PubMed] [Google Scholar]
- 7.Balmain A, Pragnell B: Mouse skin carcinomas induced in vivo by chemical carcinogens have a transforming Harvey-ras oncogene. Nature 1983, 303:72-74 [DOI] [PubMed] [Google Scholar]
- 8.Slaga TJ: Sencar mouse skin tumorigenesis model versus other strains and stocks of mice. Environ Health Perspect 1986, 68:27-32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Quintanilla M, Brown K, Ramsden M, Balmain A: Carcinogen-specific mutation and amplification of Ha-ras during mouse skin carcinogenesis. Nature 1986, 322:78-80 [DOI] [PubMed] [Google Scholar]
- 10.Leon JL, Kamino H, Steinberg JJ, Pellicer A: H-ras activation in benign and self-regressing skin tumors (keratoacanthomas) in both humans and animal model systems. Mol Cell Biol 1988, 8:786-793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brown K, Buchmann A, Balmain A: Carcinogen-induced mutations in the mouse c-Ha-ras gene provide evidence of multiple pathways for tumor progression. Proc Natl Acad Sci USA 1990, 87:538-542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bailleul B, Surani MA, White S, Barton SC, Brown K, Blessing M, Jorcano J, Balmain A: Skin hyperkeratosis and papilloma formation in transgenic mice expressing a ras oncogene from a suprabasal keratin promoter. Cell 1990, 62:697-708 [DOI] [PubMed] [Google Scholar]
- 13.Leder A, Kuo A, Cardiff RD, Sinn E, Leder P: v-Ha-ras transgene abrogates the initiation step in mouse skin tumorigenesis: effects of phorbol esters and retinoic acid. Proc Natl Acad Sci USA 1990, 87:9178-9182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cardiff RD, Leder A, Kuo A, Pattengale PK, Leder P: Multiple tumor types appear in a transgenic mouse with the ras oncogene. Am J Pathol 1993, 142:1199-1206 [PMC free article] [PubMed] [Google Scholar]
- 15.Greenhalgh DA, Rothnagel JA, Quintanilla MI, Orengo CC, Gagne TA, Bundman DS, Longley MA, Roop D: Induction of epidermal hyperplasia, hyperkeratosis, and papillomas in transgenic mice by a targeted v-Ha-ras oncogene. Mol Carcinog 1993, 7:99-110 [DOI] [PubMed] [Google Scholar]
- 16.Peng X, Olson RO, Christian CB, Lang CM, Kreider JW: Papillomas and carcinomas in transgenic rabbits carrying EJras DNA and cottontail rabbit papillomavirus DNA. J Virol 1993, 63:1698-1701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brown K, Strathdee D, Bryson S, Lambie W, Balmain A: The malignant capacity of skin tumors induced by expression of a mutant H-ras transgene depends on the cell type targeted. Curr Biol 1998, 8:516-524 [DOI] [PubMed] [Google Scholar]
- 18.Brown K, Balmain A: Transgenic mice and squamous multistage skin carcinogenesis. Cancer Metastasis 1995, 14:113-124 [DOI] [PubMed] [Google Scholar]
- 19.Hollstein M, Sidransky D, Vogelstein B, Harris CC: p53 mutations in human cancers. Science 1991, 253:49-53 [DOI] [PubMed] [Google Scholar]
- 20.Tornalletti S, Pfeifer GP: Slow repair of pyrimidine dimers at p53 gene mutation hot spots in skin cancer. Science 1994, 263:1436-1438 [DOI] [PubMed] [Google Scholar]
- 21.LeBoit PE: Is keratoacanthoma a variant of squamous cell carcinoma—new insights into an old controversy/t./t./t.soon? Am J Dermatopathol 1995, 17:319-320 [DOI] [PubMed] [Google Scholar]
- 22.Hodak E, Jones RE, Ackerman AB: Solitary keratoacanthoma is a squamous-cell carcinoma: three examples with metastases. Am J Dermatopathol 1993, 15:332-342 [DOI] [PubMed] [Google Scholar]
- 23.Schwartz RA: Keratoacanthoma. J Am Acad Dermatol 1994, 30:1-19 [DOI] [PubMed] [Google Scholar]
- 24.Schartz RA: The keratoacanthoma: a review. J Surg Oncol 1979, 12:305-317 [DOI] [PubMed] [Google Scholar]
- 25.Bizub D, Wood AW, Shalka AM: Mutagenesis of the Ha-ras oncogene in mouse skin tumors induced by polycyclic aromatic hydrocarbons. Proc Natl Acad Sci USA 1983, 86:6048-6052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ananthaswamy HN, Price JE, Goldberg LH, Bales ES: Detective and identification of activated oncogenesis in human skin cancers occurring on sun-exposed body sites. Cancer Res 1988, 48:3341-3346 [PubMed] [Google Scholar]
- 27.Ananthaswamy HN, Price JE, Goldberg LH, Bales ES: Simultaneous transfer of tumorigenic and metastatic phenotypes by transfection with genomic DNA from a human cutaneous squamous cell carcinoma. J Cell Biochem 1988, 36:137-146 [DOI] [PubMed] [Google Scholar]
- 28.Santos E, Tronick SR, Aaronson SA, Pulciani S, Barbacid M: T24 human bladder carcinoma oncogene is an activated form of the normal human homologue of BALB-and Harvey-MSV transforming genes. Nature 1982, 298:343-347 [DOI] [PubMed] [Google Scholar]
- 29.Peng X, Lang CM, Kreider JW: Methylation of cottontail rabbit papillomavirus DNA and tissue-specific expression in transgenic rabbits. Virus Res 1995, 35:101-108 [DOI] [PubMed] [Google Scholar]
- 30.Giri I, Danos O, Yaniv M: Genomic structure of the cottontail rabbit (Shope) papillomavirus. Proc Natl Acad Sci USA 1985, 82:1580-1584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Knight KL, Spieker-Polet H, Kazdin DS, Oi VT: Transgenic rabbits with lymphocytic leukemia induced by the c-myc oncogene fused with the immunoglobulin heavy chain enhancer. Proc Natl Acad Sci USA 1988, 85:3130-3134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Buhler ThA, Bruyere Th, Went DF, Stranzinger G, Burki K: Rabbit Beta-casein promoter directs secretion of human interleukin-2 into the milk of transgenic rabbits. Biotechnology 1990, 8:140-143 [DOI] [PubMed] [Google Scholar]
- 33.Chomczynski P, Sacchi N: Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 1987, 162:156-159 [DOI] [PubMed] [Google Scholar]
- 34.Zhao J, Buick RN: Relationship of level and kinetics of H-ras expression to transformed phenotype and loss of TGF-β1-mediated growth inhibition in intestinal epithelial cells. Exp Cell Res 1993, 204:82-87 [DOI] [PubMed] [Google Scholar]
- 35.Pullano TG, Sinn E, Carney WP: Characterization of monoclonal antibody R256, specific for activated ras p21 with arginine at 12, and analysis of breast carcinoma of v-Harvey-ras transgenic mouse. Oncogene 1989, 4:1003-1008 [PubMed] [Google Scholar]
- 36.Lever WF, Schaumburg-Lever G: Histopathology of the Skin. 1983:pp 499-503 Lippincott, Philadelphia
- 37.Corominas M, Leon J, Kamino H, Cruz-Alvarez M, Novick SC, Pellicer A: Oncogene involvement in tumor regression: H-ras activation in the rabbit keratoacanthoma model. Oncogene 1991, 6:645-651 [PubMed] [Google Scholar]
- 38.Kreider JW, Bartlett GL: The Shope papilloma-carcinoma complex of rabbits: a model system of neoplastic progression and spontaneous regression. Adv Cancer Res 1981, 35:81-110 [DOI] [PubMed] [Google Scholar]
- 39.Schmitt A, Rochat A, Zeltener R, Borenstein L, Barrandon Y, Wettstein FO, Iftner T: The primary target cells of the high-risk cottontail rabbit papillomavirus colocalize with hair follicle stem cells. J Virol 1996, 70:1912-1922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chase H: Growth of the hair. Physiol Rev 1954, 34:113-126 [DOI] [PubMed] [Google Scholar]
- 41.Ghadially FN: The role of the hair follicle in the origin and evolution of some cutaneous neoplasms of man and experimental animals. Cancer 1961, 14:801-816 [DOI] [PubMed] [Google Scholar]
- 42.Hansen LA, Tennant RW: Follicular origin of epidermal papillomas in v-Ha-ras transgenic TG.AC mouse skin. Proc Natl Acad Sci USA 1994, 91:7822-7826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miller SJ, Wei Z, Wilson C, Dzubow L, Sun T, Lavker RM: Mouse skin is particularly susceptible to tumor initiation during early anagen of the hair cycle: possible involvement of hair follicle stem cells. J Invest Dermatol 1993, 101:591-594 [DOI] [PubMed] [Google Scholar]
- 44.Wolbach SB: The hair cycle of the mouse and its importance in the study of sequences of experimental carcinogenesis. Ann NY Acad Sci 1951, 53:517-536 [DOI] [PubMed] [Google Scholar]
- 45.Whitely HJ: Effect of hair-growth cycle on experimental skin carcinogenesis in rabbit. Br J Cancer 1957, 11:196-205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sinn E, Muller W, Pattengale P, Tepler I, Wallace R, Leder P: Coexpression of MMTV/v-Ha-ras and MMTV/c-myc genes in transgenic mice: synergistic action of oncogenes in vivo. Cell 1987, 49:465-475 [DOI] [PubMed] [Google Scholar]
- 47.Quaife CJ, Pinkert CA, Qrnitz DM, Palmiter RD, Brinster RL: Pancreatic neoplasia induced by ras expression in acinar cells of transgenic mice. Cell 1987, 48:1023-1034 [DOI] [PubMed] [Google Scholar]
- 48.Rak J, Mitsuhashi Y, Bayko L, Filmus J, Shrisawa T, Kerbel RS: Mutant ras oncogenes up-regulate VEGF/VPF expression implications for induction and inhibition of tumor angiogenesis. Cancer Res 1995, 55:4575-4580 [PubMed] [Google Scholar]
- 49.Arbiser JL, Moses MA, Fernandez CA, Ghiso N, Cao Y, Klauber N, Frank D, Brownlee M, Flynn E, Parangi S, Byers HR, Folkman J: Oncogenic Ha-ras stimulates tumor oncogenesis by two distinct pathways. Proc Natl Acad Sci USA 1997, 94:861-866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hagari Y, Budgeon LR, Pickel MD, Kreider JW: Association of tumor necrosis factor-α gene expression and apoptotic cell death with regression of Shope papillomas. J Inverst Dermatol 1994, 104:526-529 [DOI] [PubMed] [Google Scholar]
- 51.Lin HJ, Eviner V, Prendergast GC, White E: Activated H-ras rescue E1A-induced apoptosis and cooperates with E1A to overcome p53-dependent growth arrest. Mol Cell Biol 1995, 15:4536-4544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rak J, Mitsuhashi Y, Erdos V, Huang S, Filmus J, Kerbel RS: Massive programmed cell death in intestinal epithelial cells induced by three-dimensional growth conditions: suppression by mutant c-Ha-ras oncogene expression. J Cell Biol 1995, 131:1587-1598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brash DE, Rudolph JA, Simon JA, Lin A, McKenna GJ, Baden HP, Halperin AJ, Ponten J: A role for sunlight in skin cancer: UV-induced p53 mutations in squamous cell carcinoma. Proc Natl Acad Sci USA 1991, 88:10124-10128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kress S, Sutter Ch, Strickland PT, Mukhtar H, Schweizer J, Schwarz M: Carcinogen-specific mutational pattern in the p53 gene in ultraviolet B radiation-induced squamous cell carcinoma of mouse skin. Cancer Res 1992, 52:6400-6403 [PubMed] [Google Scholar]
- 55.Perez MI, Robins P, Biria S, Roco J, Siegel E, Pellicer A: p53 oncoprotein expression and gene mutations in some keratoacanthomas. Arch Dermatol 1997, 133:189-193 [PubMed] [Google Scholar]
- 56.Gerwin BI, Spillare E, Forrester K, Lehman TA, Kispert J, Welsh JA, Pfeifer AMA, Lechner JF, Baker SJ, Vogelstein B, Harris CC: Mutant p53 can induce tumorigenic conversion of human bronchial epithelial cells and reduce their responsiveness to a negative growth factor β1. Proc Natl Acad Sci USA 1992, 89:2759-2763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Reiss M, Sartorelli AC: Regulation of growth and differentiation of human keratinocytes by type β transforming factor and epidermal growth factor. Cancer Res 1987, 47:6705-6709 [PubMed] [Google Scholar]