

Abstract
Although histopathologic criteria for adrenal cortical nodular hyperplasias (ACNHs) and adenomas (ACAs) have been developed, their kinetics and clonality are virtually unknown. We studied 20 ACNHs and 25 ACAs (based on World Health Organization criteria) from 45 females. Representative samples were histologically evaluated, and the methylation pattern of the androgen receptor alleles was analyzed on microdissected samples. Consecutive sections were selected for slide cytometry, flow cytometry, and in situ end labeling (ISEL). Apoptosis was studied by flow cytometry (nuclear area/DNA content plotter analysis) and by ISEL. Appropriate tissue controls were run in every case. Polyclonal gel patterns were revealed in 14/18 informative ACNHs and in 3/22 informative ACAs, whereas monoclonal gel patterns were observed in 4/18 ACNHs and 19/22 ACAs. Overlapping proliferation rates (PRs) were observed in both clonal groups, and apoptosis was detected only in G0/G1 cells, especially in monoclonal ACNHs (3/4; 75%) and in polyclonal ACAs (2/3; 67%). Significantly higher PRs were observed in ACNHs with polyclonal patterns and G0/G1 apoptosis and in ACAs regardless of clonality pattern and presence of G0/G1 apoptosis. All except one ACNH (19/20; 95%) and 15/25 ACAs (60%) showed diploid DNA content, whereas the remaining cases were hyperdiploid. A direct correlation between PR and ISEL was observed in polyclonal lesions (PR = 29.32 ISEL − 1.93), whereas the correlation was inverse for monoclonal lesions (PR = −9.13 ISEL + 21.57). We concluded that only simultaneous down-regulated apoptosis and high proliferation result in selective kinetic advantage, dominant clone expansion, and unbalanced methylation patterns of androgen receptor alleles in ACNHs and ACAs.
Neoplasms result from the progressive and convergent selection of cell populations, but several factors should be considered. On one hand, selection will determine tumor progression and cellular heterogeneity. On the other hand, cellular selection must be related to cell kinetics process. 1,2 All genetic abnormalities seen in tumors should be fixed on the transformed cell before ending in a fully established malignancy. These genetic changes must be cooperative and resistant to the cellular repair systems, and they must not activate the apoptosis pathway. This process determines a complex network in which a potential genetic marker will be useful when associated with kinetic advantages responsible for cellular outgrowths. In general, proliferation markers are very important for the distinction between benign and malignant endocrine neoplasms and for the analysis of tumor progression. 3,4 Actually, the most important parameter for the diagnosis of adrenal cortical neoplasms is the mitotic figure counting. However, any proliferative advantage resulting in dominant growth could be caused by either a high proliferation rate or abnormally low apoptotic indices. The key kinetic factor is the imbalance between cell proliferation and cell loss resulting in tissue overgrowth. 5,6 In this regard, variable results have been reported for adrenal cortical tumors, 7 and no reference is available for nodular hyperplasias.
Clonal origin is still the hallmark of neoplasms and strongly indicates acquired somatic mutations that give survival advantage to a cell population. 8 The acquisition of additional genetic deletions in certain histological areas favors a molecular progression. 2 However, the molecular events in the transformation pathways are not completely understood and, in many instances, remain essentially unknown. Under those circumstances, clonality assays based on the analysis of X-chromosome inactivation in females represent the best molecular option, although this option is restricted to a subset of informative females. These assays are based on the ability to distinguish the paternally inherited X chromosome from the maternally inherited one, and they do not rely on the presence of any tumor-related genetic alteration. 9,10 Monoclonal patterns suggest neoplasia but are not diagnostic of neoplasia. Yet, clonality offers a better understanding of tumors if it is combined with kinetic features (proliferation and apoptosis). No previous study has focused attention on those parameters (clonality and cell kinetics) in benign proliferative lesions of the adrenal cortex, in which the distinction between nodular hyperplasias and adenomas is sometimes controversial. 11
This study addressed the clonal evaluation of adrenal cortical nodular hyperplasias (ACNHs) and adenomas (ACAs), based on an analysis of methylation patterns of androgen receptor alleles, using microdissected tissue samples. The kinetic features of these lesions were also analyzed by means of proliferation and apoptotic markers.
Materials and Methods
Case Selection and Sampling
Consecutive adrenal cortical proliferative lesions (64) were selected and histologically evaluated. 12 Of these lesions, 53 were detected in female patients, including eight malignant tumors. ACNHs (20) and ACAs (25) were studied and classified by World Health Organization criteria, 11 although evidence of metastases was the main criterion for malignancy and case exclusion. The mean follow-up time in this series was 135 months.
All surgical specimens were serially sectioned and embedded for routine histopathologic diagnosis (at least 1 block/cm). The most cellular areas from the biggest nodule in each case of ACNH and from every ACA were screened and selected for further analysis. The same areas were used in each analysis; hematoxylin and eosin (H&E)-stained sections taken before and after the specimen samples were used to check the cellular composition of each sample.
X-Chromosome Inactivation Assay for Clonality Analysis
Two 20-μm unstained paraffin sections were used for microdissection under microscopic control. Adrenal cortical cells and controls (histologically normal adrenal cortex, adrenal medulla, and periadrenal soft tissue from the same slide) underwent DNA extraction. At least two separate areas of 0.25 mm2, containing about 100 target cells each, were harvested from both peripheral and internal areas of the biggest nodule in ACNH or ACA.
The samples were dewaxed with xylene, cleared with absolute ethanol, and digested with proteinase K; DNA was extracted using a modified phenol-chloroform protocol, as previously described. 13 All samples were divided for restriction endonuclease digestion with HhaI (New England Biolabs, Beverly, MA). Half of each sample underwent enzymatic digestion (0.8 U/μl), while the remaining half was kept as undigested control. The undigested samples were processed like the digested ones but excluding HhaI in the reaction mixture. The samples were digested under appropriate buffer conditions (50 mmol/L potassium acetate, 20 mmol/L Tris acetate, 10 mmol/L magnesium acetate, 1 mmol/L dithiothreitol, pH 8.0, 100 μg/ml bovine serum albumin, 100 μg/ml mussel glycogen) at 37°C for 4 to 16 hours. A mimicker (0.3 μg of double-stranded and XhoI-linearized φX174-RII phage; Life Technologies, Inc., Gaithersburg, MD) was included in each reaction mixture. Complete digestion was checked by gel electrophoresis; incompletely digested samples were phenol chloroform-purified and redigested with higher HhaI concentrations.
HhaI was then inactivated by phenol-chloroform extraction as previously described. 13 DNA was precipitated with ice-cold absolute ethanol in the presence of 0.3 mol/L sodium acetate (pH 5.2) and resuspended in 10 μl of polymerase chain reaction (PCR) buffer (10 mmol/L Tris-HCl, pH 8.4, 50 mmol/L KCl, 1.5 mmol/L MgCl2, 100 μg/ml bovine serum albumin). Both HhaI-digested DNA and undigested DNA were then used for PCR amplification of the CAG repeat in the first exon of the human androgen receptor gene (HUMARA). The PCR products also included a DNA sequence recognized by HhaI, which is consistently methylated in the inactive HUMARA allele only. 14-16 Primers and PCR cycling conditions were designed as previously described. 1,15,17 The reactions were run in duplicate and optimized for a 10-μl reaction in a Perkin-Elmer thermal cycler model 480 (Perkin-Elmer, Norwalk, CT).
The whole PCR volume was electrophoresed into 0.75-mm-thick 8% nondenaturing polyacrylamide gel at 5 V/cm until a xylene cyanol band was located within the bottom inch of the gel. After fixation with 7% acetic acid (5 minutes), the gels were dried under vacuum (80°C, 40 minutes) and put inside a developing cassette containing one intensifying screen and preflashed films (Kodak XAR) facing the intensifying screen (16–48 hours, −70°C). The autoradiograms were developed using an automated processor Kodak-X-Omat 100 (Kodak Co., Rochester, NY).
Interpretation and inclusion criteria in each sample were as previously reported. 1 Allelic imbalance was densitometrically evaluated (EC model 910 optical densitometer, EC Apparatus Corp., St. Petersburg, FL), and evidence of monoclonal proliferation was considered to be allele ratios ≥ 4:1 with the normalized HhaI-digested samples. Sample normalization was done in relation to the corresponding undigested sample and tissue controls. Only informative cases (two different alleles in HhaI-undigested and HhaI-digested samples) were included in the final analysis. 8,15,17
Slide Cytometric Analysis of DNA Content
Feulgen-stained sections were used for DNA quantification. 18 The densitometric evaluation was performed with the cell analysis system model 200 and the quantitative DNA analysis package as software (Becton Dickinson). At least 200 nuclei were measured in every case, beginning in the most cellular area, until completion in consecutive microscopic high-power fields. Only complete, nonoverlapping, and focused nuclei were quantified in each field.
External staining calibration was carried out with complete rat hepatocytes (Becton-Dickinson; one slide per staining holder) to normalize the internal controls; the latter included both lymphocytes and adrenal cortical cells from histologically normal areas present in the same tissue section. The internal controls were used for setting the G0/G1 cell limits and calculating the DNA index of each G0/G1 peak (>10% of measured cells with evidence of G2 + M cells). 19
Proliferation rate (PR = S-phase + G2-phase + M-phase fractions) was calculated from the DNA histogram by subtracting the number of cells within G0/G1 limits from the total number of measured cells. The values were compared with the total cell number and expressed as percentages. 19
Nuclear DNA Quantification by Flow Cytometry
Serial 50-μm-thick sections were microdissected, and nuclear preparations were stained with propidium iodine after RNase A digestion to study DNA ploidy (by the technique of Hedley et al). 20 DNA quantification parameters included DNA indices and PRs as previously described. 19 The scatter analysis of nuclear area and DNA content allowed the identification of apoptotic cells in each cell cycle phase (low nuclear area for a given DNA content in each cell cycle phase). 21 Those results were additionally coupled with in situ end labeling (ISEL) to identify apoptotic cells in terms of DNA fragmentation (see below). External diploid controls from paraffin-embedded tissues (lymphocyte from reactive lymph nodes and adrenal cortical cells from histologically normal adrenal glands) were used to determine DNA indices and to standardize the nuclear area/DNA content analysis (considering only adrenal cortical cells for the last purpose). PR was calculated as described for slide cytometry, using the rectangular model for evaluation of the cell cycle histogram. 19
ISEL of Fragmented DNA
Extensive DNA fragmentation associated with apoptosis was detected by ISEL as previously reported. 22,23 After routine dewaxing and hydration, the sections were incubated in 2× standard saline citrate (20 minutes at 80°C) and digested with proteinase K (100 μg/ml in Tris-HCl, pH 7.6, for 30 minutes at room temperature) in a moist chamber.
DNA fragments were labeled on 5′-protuding termini by incubating the sections with the Klenow fragment of Escherichia coli DNA polymerase I (20 U/ml in 50 mmol/L Tris-HCl, pH 7.5, 10 mmol/L MgCl2, 1 mmol/L dithiothreitol, 250 μg/ml bovine serum albumin, 5 μmol/L each of dATP, dCTP, dGTP, as well as 3.25 μmol/L dTTP and 1.75 μmol/L 11-digoxigenin-dUTP at 0.65/0.35), at 37°C in a moist chamber. The incorporated digoxigenin-dUMPs were immunoenzymatically detected by using anti-digoxigenin Fab fragments labeled with alkaline phosphatase (7.5 U/ml, in 100 mmol/L Tris-HCl, pH 7.6, 150 mmol/L NaCl, 1% bovine serum albumin) for 4 hours at room temperature. The reactions were developed with the mixture nitroblue tetrazolium-X phosphate in 100 mmol/L Tris-HCl (pH 9.5), 100 mmol/L NaCl, 50 mmol/L MgCl2 under microscopic control. Appropriate controls were simultaneously run, including positive (reactive lymph node), negative (same conditions omitting DNA polymerase I), and enzymatic (DNase I digestion before the end labeling). The enzymatic controls were used to reliably establish the positivity threshold in each sample.
The ISEL index was expressed as percentages of positive nuclei compared with the total number of corresponding cells present in the same high-power field, using previously reported methods for mitotic figure counting. 24,25 At least 50 consecutive high-power fields were screened, beginning in the most cellular area (from the biggest nodule for ACNH).
Statistical Analysis of Quantitative Variables
The results of quantitative variables were compared by diagnostic groups (ACNH versus ACA), clonality pattern (polyclonal versus monoclonal), and the presence of G0/G1 apoptotic cells in flow cytometry. Variables showing normal distribution were analyzed using a two-tailed Student’s t-test, whereas analyses of variance were used for variables with nonparametric distribution. Normal distribution was previously tested by the Kolmogorov-Smirnoff test. The results were considered statistically significant if P < 0.05. Regression analyses were also performed to test the correlation between proliferation and apoptosis markers in both polyclonal and monoclonal lesions.
Results
The analysis of the methylation pattern of androgen receptor alleles showed polyclonal patterns in 14/18 (78%) informative ACNHs and in 3/22 (14%) informative ACAs, whereas patterns were monoclonal in 4/18 (22%) informative ACNHs and in 19/22 (86%) informative ACAs (Table 1 ▶ , Figure 1 ▶ ). Females showing unbalanced methylation of androgen receptor alleles in histologically normal adrenal cortex were considered noninformative and excluded from further analyses (two ACNHs and three ACAs). Consistent methylation patterns were detected in both peripheral and internal samples from the same cortical nodule or tumor. However, two samples from internal areas of ACAs were contaminated with host stromal cells and showed pseudopolyclonal gel patterns.
Table 1.
Results of Proliferation, Apoptosis, and Clonality in ACNHs and ACAs
| Case | PR by slide cytom. (%) | PR by flow cytom. (%) | DNA ploidy (DNA index) | ISEL (%) | G0/G1 apoptosis | Methylation of AR alleles |
|---|---|---|---|---|---|---|
| ACNH-1 | 31.61 | 32.45 | DIP (1.08)* | 0.9 | + | Balanced |
| ACNH-2 | 7.15 | 8.76 | DIP (1.01) | 0.4 | − | Balanced |
| ACNH-3 | 16.92 | 16.20 | DIP (1.10) | 0.5 | − | Balanced |
| ACNH-4 | 11.22 | 9.07 | DIP (1.09) | 0.4 | − | Balanced |
| ACNH-5 | 32.57 | 14.77 | DIP (1.05) | 0.8 | + | Balanced |
| ACNH-6 | 22.60 | 21.28 | DIP (0.95) | 0.7 | − | Balanced |
| ACNH-7 | 10.44 | 17.03 | DIP (1.05) | 0.4 | − | Balanced |
| ACNH-8 | 14.44 | 10.28 | DIP (0.99) | 0.5 | − | Balanced |
| ACNH-9 | 7.55 | 9.84 | DIP (0.97) | 0.8 | + | Unbalanced |
| ACNH-10 | 13.21 | 10.39 | DIP (1.00) | 0.9 | + | Unbalanced |
| ACNH-11 | 12.56 | 8.50 | DIP (0.99) | 0.5 | − | Balanced |
| ACNH-12 | 8.76 | 7.08 | DIP (1.02) | 0.4 | − | Balanced |
| ACNH-13 | 10.47 | 11.41 | DIP (1.01) | 0.4 | − | Balanced |
| ACNH-14 | 23.81 | 10.68 | DIP (0.97) | 0.6 | − | Balanced |
| ACNH-15 | 19.13 | 12.17 | DIP (0.99) | 0.6 | − | Balanced |
| ACNH-16 | 8.60 | 13.41 | DIP (0.98) | 0.4 | − | Balanced |
| ACNH-17 | 7.90 | 12.12 | DIP (1.03) | 0.4 | − | Noninform. |
| ACNH-18 | 9.35 | 15.07 | DIP (1.01) | 0.5 | − | Noninform. |
| ACNH-19 | 28.19 | 18.47 | DIP (0.99) | 0.6 | − | Unbalanced |
| ACNH-20 | 36.98 | 13.87 | ANEUP (1.54) | 0.9 | + | Unbalanced |
| ACA-1 | 42.65 | 12.78 | DIP (1.02) | 0.8 | − | Unbalanced |
| ACA-2 | 35.78 | 31.80 | ANEUP (1.50) | 0.7 | − | Unbalanced |
| ACA-3 | 17.39 | 8.30 | DIP (1.03) | 0.7 | − | Unbalanced |
| ACA-4 | 34.71 | 13.68 | DIP (1.02) | 0.8 | − | Unbalanced |
| ACA-5 | 20.74 | 18.26 | ANEUP (1.17) | 0.6 | − | Unbalanced |
| ACA-6 | 28.88 | 13.54 | DIP (1.01)† | 0.7 | − | Unbalanced |
| ACA-7 | 9.84 | 13.95 | DIP (0.99) | 0.8 | − | Unbalanced |
| ACA-8 | 15.59 | 15.87 | ANEUP (1.52) | 0.9 | − | Unbalanced |
| ACA-9 | 46.15 | 30.20 | DIP (1.00) | 2.8 | + | Noninform. |
| ACA-10 | 11.73 | 12.52 | DIP (1.02) | 0.6 | − | Balanced |
| ACA-11 | 22.65 | 7.66 | DIP (0.97)† | 1.8 | + | Unbalanced |
| ACA-12 | 34.63 | 13.73 | POLYP (1 and 2) | 1.0 | − | Unbalanced |
| ACA-13 | 11.94 | 9.52 | ANEUP (1.37) | 1.0 | + | Unbalanced |
| ACA-14 | 38.62 | 12.91 | DIP (1.05) | 1.1 | − | Unbalanced |
| ACA-15 | 22.04 | 6.69 | DIP (1.04) | 1.1 | − | Unbalanced |
| ACA-16 | 9.63 | 18.21 | ANEUP (1.33) | 1.7 | + | Balanced |
| ACA-17 | 5.20 | 4.28 | DIP (0.96) | 1.2 | − | Unbalanced |
| ACA-18 | 11.05 | 10.70 | DIP (1.00) | 1.6 | + | Unbalanced |
| ACA-19 | 12.31 | 6.38 | DIP (1.04) | 1.7 | + | Balanced |
| ACA-20 | 21.06 | 11.86 | DIP (0.97) | 0.6 | − | Unbalanced |
| ACA-21 | 23.57 | 15.46 | ANEUP (1.40) | 0.7 | − | Unbalanced |
| ACA-22 | 20.61 | 13.97 | DIP (0.96) | 0.7 | − | Unbalanced |
| ACA-23 | 19.98 | 11.93 | DIP (1.03) | 0.6 | − | Unbalanced |
| ACA-24 | 23.86 | 12.03 | DIP (1.01) | 0.8 | − | Noninform. |
| ACA-25 | 24.09 | 16.07 | ANEUP (1.45) | 0.8 | − | Noninform. |
Abbreviations: ACNH, adrenal cortical nodular hyperplasia; ACA, adrenal cortical adenoma; PR, proliferation rate; ISEL, in situ end labeling; AR, androgen receptor; DIP, diploid; ANEUP, aneuploid; POLYP, Polyploid; cytom., cytometry; Noninform., noninformative.
*Small proportion of hyperdiploid G0/G1 cells (DNA index = 1.48) also identified.
†Small proportion of hyperdiploid G0/G1 cells (DNA index = 1.60) also identified.
Figure 1.

Methylation patterns of androgen receptor alleles in adrenal cortical nodular hyperplasias (a, ACNH-2) and adenomas (b, ACA-7). The digested sample (D) revealed allelic ratios of normalized bands < 4:1 in polyclonal cases (a) and ≥ 4:1 in monoclonal cases (b). Controls shown on the left revealed balanced methylation pattern of androgen receptor alleles.
The combined analysis of nuclear area and DNA content revealed features of apoptosis (reduced nuclear size for a given DNA content) only in G0/G1 cells (Table 2 ▶ , Figure 2 ▶ ). An inverse relationship between the detection of G0/G1 apoptotic cells and the clonal pattern was also revealed: G0/G1 apoptotic cells were detected in 3/4 (75%) of monoclonal ACNHs and 2/3 (67%) of polyclonal ACAs, using flow cytometric criteria.
Table 2.
Comparative Results of Proliferation Indices by Apoptosis and Clonality Patterns
| Clonality pattern | Proliferation rate slide cytometry (Av ± SD) | Proliferation rate flow cytometry (Av ± SD) | ||
|---|---|---|---|---|
| + G0/G1 apoptosis | − G0/G1 apoptosis | + G0/G1 apoptosis | − G0/G1 apoptosis | |
| Polyclonal ACNH | 32.09 ± 0.68 | 13.84 ± 9.68 | 23.61 ± 12.50 | 12.15 ± 5.66 |
| Monoclonal ACNH | 19.25 ± 15.62 | 28.19 | 11.37 ± 2.19 | 18.47 |
| Polyclonal ACA | 10.97 ± 1.90 | 11.73 | 12.30 ± 8.37 | 12.52 |
| Monoclonal ACA | 15.21 ± 6.46 | 24.46 ± 10.54 | 9.29 ± 1.53 | 13.69 ± 5.95 |
ACNH, adrenal cortical nodular hyperplasia; ACA, adrenal cortical adenoma; Av, average; SD, standard deviation.
Figure 2.

a: Flow cytometric analysis of DNA content in adrenal cortical adenoma (ACA-18). Right: the small square (arrow) in the analysis nuclear area (horizontal FL2- RW axis) versus DNA content (vertical FL2-A axis) points to the apoptotic cells. b: ISEL of DNA fragments in adrenal cortical adenoma (ACA-18). The apoptotic cells in a determine the pre-G0/G1 slope (two arrows) in the DNA histogram (a, left side) and were found positive by using ISEL (b, black nuclei).
Variable proliferation rates defined both polyclonal and monoclonal ACNHs. However, a direct and linear correlation was observed between proliferation and apoptosis in polyclonal ACNHs, although an inverse and linear correlation defined monoclonal ACNHs (Tables 1 and 2 ▶ ▶ , Figure 3 ▶ ). Proliferation rates were significantly higher in polyclonal ACNHs with G0/G1 apoptosis than in polyclonal ACNHs with no G0/G1 apoptosis (P = 0.005) and were higher in monoclonal ACNHs with no G0/G1 apoptosis than in monoclonal ACNHs with G0/G1 apoptosis (Table 2 ▶ , Figure 4 ▶ ; P = 0.009). Likewise, significantly higher ISEL indices were observed in monoclonal ACNHs (0.80 ± 0.14) than in polyclonal ACNHs (0.54 ± 0.16; P = 0.03). The flow cytometric detection of G0/G1 apoptotic cells correlated with the in situ detection; higher ISELs were observed in cases with G0/G1 apoptotic cells (ISEL index = 0.86 ± 0.05) than in those without (ISEL index = 0.49 ± 0.24; P = 0.05).
Figure 3.
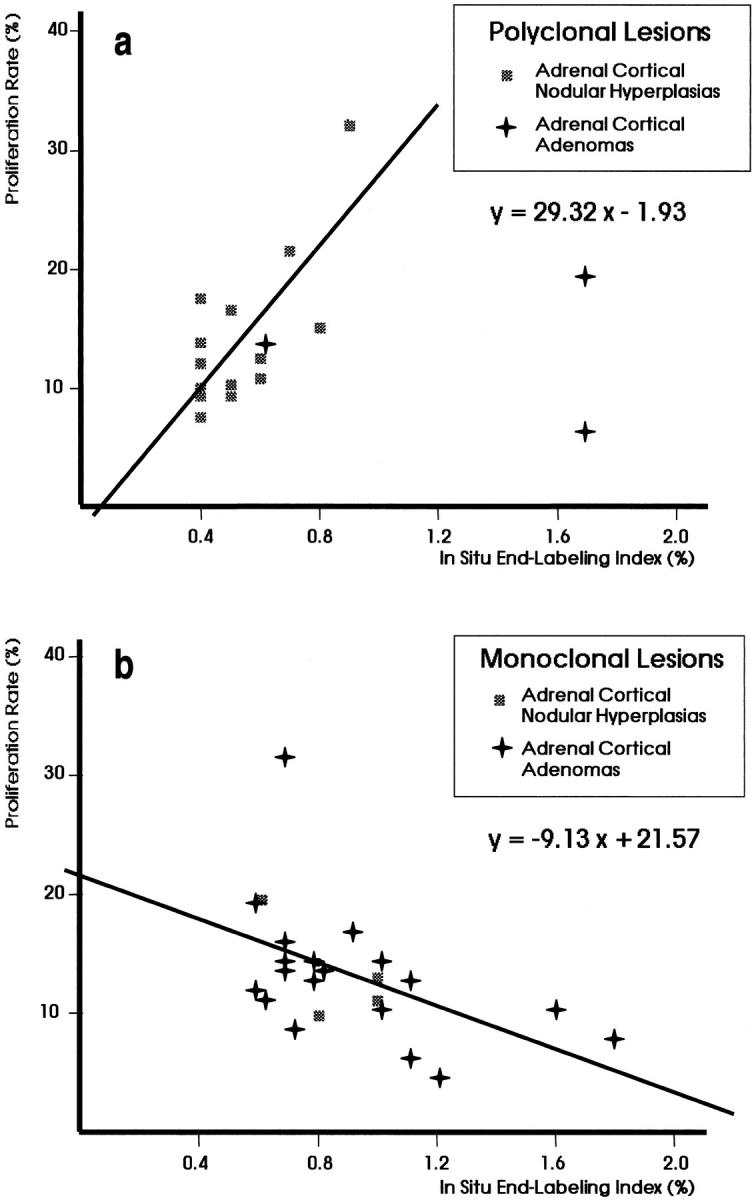
Linear regression analysis of proliferation rate versus in situ end labeling in polyclonal (a) and monoclonal (b) lesions of adrenal cortex. The correlation was direct for polyclonal lesions, but was inverse for monoclonal ones. Only informative cases in clonality analysis were included in this regression analysis.
Figure 4.
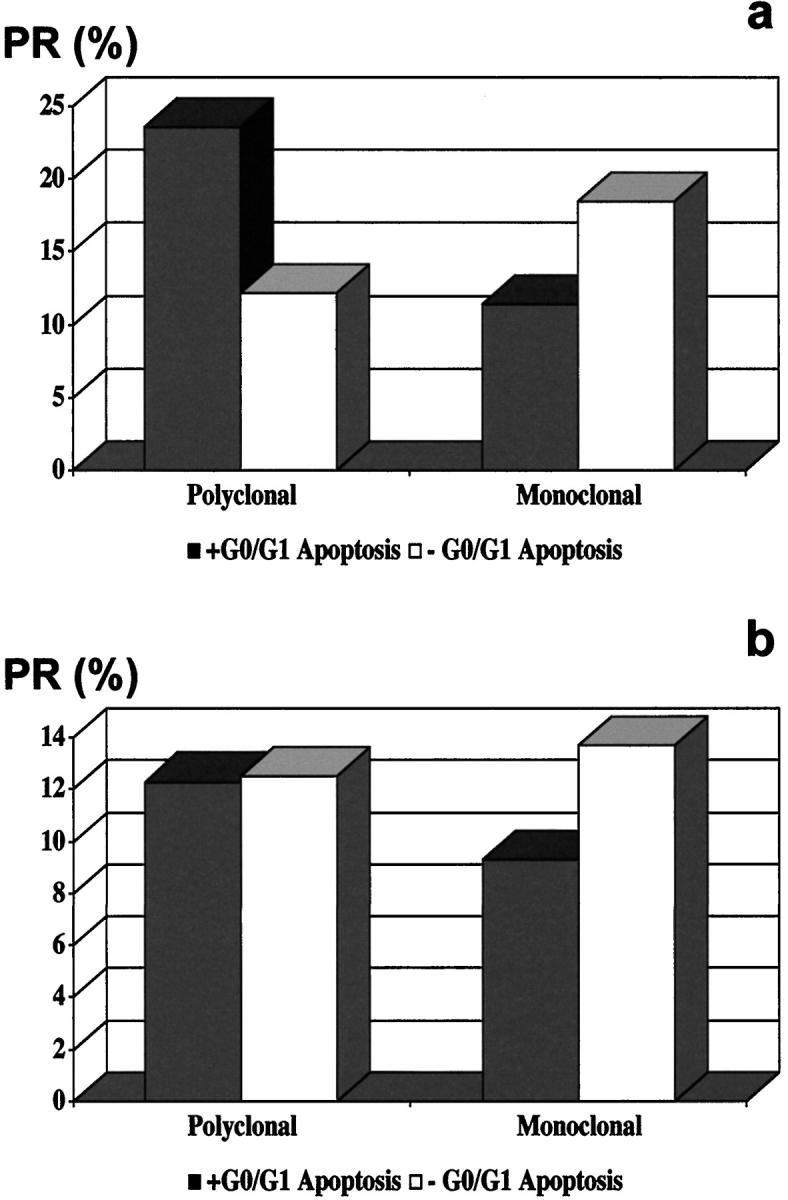
PR in ACNHs (a) and ACAs (b) in the presence (□) or absence (▪) of G0/G1 apoptotic cells. The average PR was significantly higher in polyclonal ACNHs with G0/G1 apoptotic cells, monoclonal ACNHs with no G0/G1 apoptotic cells, and monoclonal ACAs with no G0/G1 apoptotic cells than in their counterparts.
Polyclonal and monoclonal ACAs also showed overlapping and variable proliferation rates, but inversely and linearly correlated with ISEL indices in the monoclonal ACA group (Figure 3) ▶ . Monoclonal ACAs with no G0/G1 apoptotic cells showed significantly higher proliferation rates than monoclonal ACAs with G0/G1 apoptotic cells (P = 0.02; Table 2 ▶ , Figure 4 ▶ ). Again, the ISEL indices directly correlated with the flow cytometric detection of G0/G1 apoptotic cells (Table 1 ▶ , Figure 2 ▶ ); ACAs showing G0/G1 apoptotic cells displayed higher ISEL indices than ACAs without G0/G1 apoptotic cells (1.77 ± 0.58 versus 0.80 ± 0.50; P = 0.03). On the other hand, highly variable ISEL indices were observed in both polyclonal ACAs (1.33 ± 0.64) and monoclonal ACAs (0.92 ± 0.33), with no significant differences (P = 0.14). No significant differences appeared for polyclonal ACAs.
All except one (19/20) ACNHs were diploid, whereas 15/25 (60%) ACAs showed diploid DNA content. One additional ACA was classified as polyploid, and the remaining cases revealed hyperdiploid DNA contents (Table 1) ▶ . No significant differences were found for both proliferation and ISEL indices after DNA-ploidy stratification, although hyperdiploid ACAs tended to show greater scores than diploid ACAs (Table 3) ▶ . Hyperdiploid lesions from informative patients preferentially revealed monoclonal patterns (6/7, 86%, including 5/6 ACAs and 1/1 ACNHs) and G0/G1 apoptotic cells in 3/7 cases (43%, 1 monoclonal ACA, 1 polyclonal ACA, and 1 monoclonal ACNH). Proliferation rates were higher in ACNHs than in ACAs, after stratification by clonality pattern and flow cytometric detection of G0/G1 apoptotic cells (Table 2) ▶ . The homogeneous DNA-ploidy distribution in ACNH precluded additional analyses in this group.
Table 3.
Proliferation and ISEL Indices by DNA Content in ACAs
| ACA DNA content | Proliferation rate flow cytometry (Av ± SD, %) | ISEL (Av ± SD, %) |
|---|---|---|
| Diploid ACA | 12.15 ± 5.84 | 1.06 ± 0.59 |
| Aneuploid ACA | 17.88 ± 6.80 | 1.77 ± 0.58 |
ACA, adrenal cortical adenoma; ISEL, in situ end labeling; Av, average; SD, standard deviation.
Discussion
This study represents the first report on combined features of clonality and cell kinetics in ACNHs and ACAs. The distinctive correlation between apoptosis and proliferation and the heterogeneous clonal patterns revealed by ACNHs and ACAs were the most important findings.
Regarding the correlation between apoptosis and proliferation, polyclonal lesions (14/17 ACNHs, 82%) demonstrated increasing apoptosis as a counterpoise to rising proliferative rates. Monoclonal lesions (19/23 ACAs, 83%), however, had progressively lower apoptotic rates as proliferation increased. That inverted relationship between apoptosis and proliferation in monoclonal adrenal cortical lesions also provides a functional basis for clonal selection and segregates ACNHs from neoplastic ACAs. Cell kinetics represent the basic mechanisms leading to clonal expansions and tumor growths. 2,6 The correlation between proliferation and apoptosis provides rules for cellular selection, ie, clonal expansion or regression. Down-regulated apoptosis (as revealed by ISEL) has been reported in intraepithelial neoplasms of different locations and would allow both survival and replication of genetically damaged cells, giving rise to mutation accumulation in those cells. 26 A maintained cell proliferation would transmit those genetic changes into descendant cells, and a relatively blocked apoptotic process would allow genetically damaged cells to complete the replication cycle, ending in mutation accumulation and tumor promotion. 27 Inversely related proliferation and apoptosis in monoclonal lesions (Figure 3) ▶ would then contribute to clonal progression and would represent additional evidence of the cell cycle dysregulation found in neoplasms. 6,7,28 The opposite scenario would lead to clone regression. A few monoclonal cases showed diploid DNA content with relatively low proliferation and high apoptosis (3/4 monoclonal ACNHs and 4/18 monoclonal ACAs). That index combination provides a negative selection factor that would determine slow tumor growth. Those cases showed more frequently regressive changes and atypical nuclear features (size variation, contour irregularity, and hyperchromatism). Only 2 of 17 polyclonal lesions (one DNA-diploid and one DNA-aneuploid lesion) showed similar kinetic features (Figure 3) ▶ and ACA histopathology. On the other hand, only one monoclonal ACNH displayed kinetic features in the polyclonal domain (Figure 3) ▶ , with higher proliferation than polyclonal ACNH but with a similar ISEL index. Histologically, this monoclonal ACNH showed a coexistent ACA, supporting the close correlation of proliferation, apoptosis, and clonality in these neoplasms.
ACNH and ACA showed a heterogeneous clonal profile as previously reported, 29,30 with polyclonal lesions predominating in ACNHs (78% of informative cases) and monoclonal lesions predominating in ACAs (86% of informative cases). Endocrine hyperplasias have shown polyclonal patterns in hereditary and nonhereditary hyperparathyroidism 30,31 or multinodular goiters. 32,33 Most parathyroid or thyroid adenomas have been found to be monoclonal. 31-33 Those findings support the concept of multistep tumorigenesis and Knudson’s hypothesis. 34 However, monoclonal hyperplasias (parathyroid, in multiple endocrine neoplasia type 1 (MEN-1) and uremic patients, and multinodular goiters) 33,35 and polyclonal adenomas (parathyroid and thyroid) 30 have also been reported. Therefore, it has been concluded that clonality assay itself is of limited utility in differentiating hyperplastic from neoplastic conditions.
Of our ACAs, 14% were polyclonal, a proportion similar to those reported in parathyroid adenomas, thyroid adenomas, and ACAs. 29,30 There are three possible explanations for polyclonal patterns in neoplasms.
1) The selective methylation of an inactive X chromosome is normally demonstrated by using methylation-sensitive restriction endonucleases. Both incomplete endonuclease digestion 2 and aberrant hypermethylation (due to tumor progression or abnormal imprinting) 36,37 would result in false polyclonal patterns in monoclonal tissues. In relation to the enzymatic digestion, our protocol includes a viral DNA in the restriction digestion mixture to verify its completion. Although some DNA denaturation (related to embedding and extraction) should be present, both the long digestion (16 hours) and the activity of HhaI on single-stranded DNA would assure complete digestion. Repeated polyclonal patterns were obtained with unboiled templates. We are currently testing methylation in these tumors.
2) Any significant contamination as a potential cause of pseudopolyclonal patterns 2 could be excluded by both careful microdissections under microscopic control and multiple sampling from a single nodule or tumor. ACAs tend to show myxoid changes in the central areas. Besides its diagnostic utility, that myxoid stromal change can contaminate the samples, giving false polyclonal patterns. It is normally associated with vascular ectasia and hemorrhage, especially in the internal ACA area, and should express the requirements of tumor growth. Preliminary morphometric data (unpublished results) have shown bigger vascular areas of sinusoid-like structures in ACAs than in ACNHs. The dense thin-walled blood vessel network in endocrine organs should certainly contribute to that finding, providing also perivascular stromal cells. Those nonepithelial components have been proposed as a key element of epithelial cell growth either by secretion of stimulatory factors or lack of an inhibitory factor in experimental thyroid nodules. 38
3) Finally, true polyclonal proliferations can explain those results, but additional markers would be required. 2
Similarly, monoclonal patterns can be explained in non-neoplastic conditions. Proliferations of unselected cells from a polymorphic tissue show polyclonal patterns, owing to the mosaic distribution in the random distribution of X-chromosome inactivation. 8,39-41 However, the relative sizes of cell groups sharing the same inactivated X chromosome (patch size concept or contiguous cellular regions of the same lineage) and embryological reasons determine the clonal pattern. Any kinetic advantage in small cell groups sharing the same inactivated X chromosome would result in their preferential growth, thus yielding an overall monoclonal pattern 42 even in early stages. 43,44 These selective growth advantages would also explain monoclonal proliferations in both early neoplasms and precancerous conditions in other locations, such as the female genital tract 17,45-47 or liver, 48 and benign conditions, like epithelial expansion in ovarian endometrial cysts 49 or focal nodular hyperplasias of the liver. 50
DNA-ploidy results also confirmed the differences between ACNHs and ACAs, supporting the neoplastic nature of monoclonal ACAs. Nondiploid DNA contents were found in 40% (10/25) of ACAs and in only 5% (1/20) of ACNHs. Those cases preferentially revealed hyperdiploid G0/G1 cells and monoclonal patterns in 86% of informative cases. DNA content analysis has been reported to be useless as a diagnostic tool to differentiate benign from malignant conditions. 51 Although hyperdiploid DNA content was also found in one ACNH, the close association between DNA aneuploidy and monoclonal proliferation supports a neoplastic nature, especially when they are associated.
In conclusion, both ACNHs and ACAs are kinetically defined by low-apoptotic cell growths. However, a distinctive correlation between proliferation and apoptosis, direct for ACNHs and inverse for ACAs, helps explain clone selection. That inverse correlation of kinetic parameters would provide the best selective mechanism resulting in dominant clone expansion (monoclonal proliferation) in ACAs, whereas direct correlation gives a less selective mechanism, allowing balanced expansion of clones (polyclonal proliferations) in ACNHs.
Footnotes
Address reprint requests to Salvador J. Díaz-Cano, M.D., Ph.D., Dept. of Histopathology & Morbid Anatomy, The Royal London Hospital, Whitechapel, London E1 1BB, United Kingdom. E-mail: s.j.diaz-cano@mds.qmw.ac.uk.
Presented in part as an abstract at the USCAP Meeting, Orlando, FL, March 4, 1997.
References
- 1.Diaz-Cano SJ, Blanes A, Wolfe HJ: PCR-based techniques for clonality analysis of neoplastic progression: bases for its appropriate application in paraffin-embedded tissues. Diagn Mol Pathol (In press)
- 2.Diaz-Cano SJ: Clonality studies in the analysis of adrenal medullary proliferations: application principles and limitations. Endocr Pathol 1998, 9:301-316 [DOI] [PubMed] [Google Scholar]
- 3.DeLellis RA: Proliferation markers in neuroendocrine tumors: useful or useless? A critical reappraisal. Verh Dtsch Ges Pathol 1997, 81:53–61 [PubMed]
- 4.DeLellis RA: Does the evaluation of proliferative activity predict malignancy of prognosis in endocrine tumors? Hum Pathol 1995, 26:131-134 [DOI] [PubMed] [Google Scholar]
- 5.Stewart BW: Mechanisms of apoptosis: integration of genetic, biochemical, and cellular indicators. J Natl Cancer Inst 1994, 86:1286-1296 [DOI] [PubMed] [Google Scholar]
- 6.Salomon RN, Diaz-Cano S: Introduction to apoptosis. Diagn Mol Pathol 1995, 4:235-238 [DOI] [PubMed] [Google Scholar]
- 7.Sasano H, Imatani A, Shizawa S, Suzuki T, Nagura H: Cell proliferation and apoptosis in normal and pathologic human adrenal. Mod Pathol 1995, 8:11-17 [PubMed] [Google Scholar]
- 8.Fialkow PJ: Clonal origin of human tumors. Biochim Biophys Acta 1976, 458:283-321 [DOI] [PubMed] [Google Scholar]
- 9.Fialkow PJ: Primordial cell pool size and lineage relationships of five human cell types. Ann Hum Genet 1973, 37:39-48 [DOI] [PubMed] [Google Scholar]
- 10.Lyon MF: Some milestones in the history of X-chromosome inactivation. Annu Rev Genet 1992, 26:16-28 [DOI] [PubMed] [Google Scholar]
- 11.Williams ED, Siebenmann RE, Sobin LH: Histological Typing of Endocrine Tumours. 1980. World Health Organization, Geneva
- 12.Diaz-Cano SJ, Leon MM, de Miguel M, Galera-Davidson H, Wolfe HJ: Diagnostic and prognostic parameters in adrenal cortical proliferative lesions: a critical review. Lab Invest 1996, 74:47A [Google Scholar]
- 13.Diaz-Cano SJ, Brady SP: DNA extraction from formalin-fixed, paraffin-embedded tissues: protein digestion as a limiting step for retrieval of high-quality DNA. Diagn Mol Pathol 1997, 6:342-346 [DOI] [PubMed] [Google Scholar]
- 14.Allen RC, Zoghbi HY, Moseley AB, Rosenblatt HM, Belmont JW: Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am J Hum Genet 1992, 51:1229-1239 [PMC free article] [PubMed] [Google Scholar]
- 15.Mutter GL, Boynton KA: PCR bias in amplification of androgen receptor alleles, a trinucleotide repeat marker used in clonality studies. Nucleic Acids Res 1995, 23:1411-1418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sleddens HF, Oostra BA, Brinkmann AO, Trapman J: Trinucleotide repeat polymorphism in the androgen receptor gene (AR). Nucleic Acids Res 1992, 20:1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mutter GL, Chaponot ML, Fletcher JA: A polymerase chain reaction assay for non-random X chromosome inactivation identifies monoclonal endometrial cancers and precancers. Am J Pathol 1995, 146:501-508 [PMC free article] [PubMed] [Google Scholar]
- 18.Bibbo M, Bartels PH, Dytch HE, Wied GL: Cell image analysis. Bibbo M eds. Comprehensive Cytopathology. 1991, :pp 965-983 W.B. Saunders, Philadelphia [Google Scholar]
- 19.Dressler LG, Bartow SA: DNA flow cytometry in solid tumors: practical aspects and clinical applications. Semin Diagn Pathol 1989, 6:55-82 [PubMed] [Google Scholar]
- 20.Hedley DW, Friedlander ML, Taylor IW, Rugg CA, Musgrove EA: Method for analysis of cellular DNA content of paraffin-embedded pathological material using flow cytometry. J Histochem Cytochem 1983, 31:1333-1335 [DOI] [PubMed] [Google Scholar]
- 21.Sherwood SW, Schimke RT: Cell cycle analysis of apoptosis using flow cytometry. Methods Cell Biol 1995, 46:77-97 [DOI] [PubMed] [Google Scholar]
- 22.Wijsman JH, Jonker RR, Keijzer R, van de Velde CJ, Cornelisse CJ, van Dierendonck JH: A new method to detect apoptosis in paraffin sections: in situ end-labeling of fragmented DNA. J Histochem Cytochem 1993, 41:7-12 [DOI] [PubMed] [Google Scholar]
- 23.Diaz-Cano SJ, Garcia-Moliner M, Carney W, Wolfe HJ: Bcl-2 expression and DNA fragmentation in breast carcinoma, pathologic and steroid hormone receptors correlates. Diagn Mol Pathol 1997, 6:199-208 [DOI] [PubMed] [Google Scholar]
- 24.Diaz-Cano SJ, Leon MM, de Miguel M, Galera Davidson H, Wolfe HJ: Mitotic index quantification: different approaches and their value in adrenocortical proliferative lesions. Lab Invest 1996, 74:170A
- 25.Simpson JF, Dutt PL, Page DL: Expression of mitoses per thousand cells and cell density in breast carcinomas: a proposal. Hum Pathol 1992, 23:608-611 [DOI] [PubMed] [Google Scholar]
- 26.Kerr JF, Wyllie AH, Currie AR: Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 1972, 26:239-257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Isaacs JT: Role of programmed cell death in carcinogenesis. Environ Health Perspect 1993, 101(suppl. 5):27-34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cordon-Cardo C: Mutations of cell cycle regulators: biological and clinical implications for human neoplasia. Am J Pathol 1995, 147:545-560 [PMC free article] [PubMed] [Google Scholar]
- 29.Beuschlein F, Reincke M, Karl M, Travis WD, Jaursch-Hancke C, Abdelhamid S, Chrousos GP, Allolio B: Clonal composition of human adrenocortical neoplasms. Cancer Res 1994, 54:4927-4932 [PubMed] [Google Scholar]
- 30.Jackson CE, Cerny JC, Block MA, Fialkow PJ: Probable clonal origin of aldosteronomas versus multicellular origin of parathyroid “adenomas.” Surgery 1982, 92:875–879 [PubMed]
- 31.Noguchi S, Motomura K, Inaji H, Imaoka S, Koyama H: Clonal analysis of parathyroid adenomas by means of the polymerase chain reaction. Cancer Lett 1994, 78:93-97 [DOI] [PubMed] [Google Scholar]
- 32.Aeschimann S, Kopp PA, Kimura ET, Zbaeren J, Tobler A, Fey MF, Studer H: Morphological and functional polymorphism within clonal thyroid nodules. J Clin Endocrinol Metab 1993, 77:846-851 [DOI] [PubMed] [Google Scholar]
- 33.Apel RL, Ezzat S, Bapat BV, Pan N, LiVolsi VA, Asa SL: Clonality of thyroid nodules in sporadic goiter. Diagn Mol Pathol 1995, 4:113-121 [DOI] [PubMed] [Google Scholar]
- 34.Knudson AG, Jr: Mutation and cancer: a personal odyssey. Adv Cancer Res 1995, 67:1-23 [DOI] [PubMed] [Google Scholar]
- 35.Friedman E, Sakaguchi K, Bale AE, Falchetti A, Streeten E, Zimering MB, Weinstein LS, McBride WO, Nakamura Y, Brandi ML, Norton JA, Aurbach GD, Spiegel AM, Marx SJ: Clonality of parathyroid tumors in familial multiple endocrine neoplasia type 1. N Engl J Med 1989, 321:213-218 [DOI] [PubMed] [Google Scholar]
- 36.Laird PW, Jaenisch R: DNA methylation and cancer. Hum Mol Genet 1994, 3:1487-1495 [DOI] [PubMed] [Google Scholar]
- 37.Latham KE: X chromosome imprinting and inactivation in the early mammalian embryo. Trends Genet 1996, 12:134-138 [DOI] [PubMed] [Google Scholar]
- 38.Thomas GA, Williams D, Williams ED: The clonal origin of thyroid nodules and adenomas. Am J Pathol 1989, 134:141-147 [PMC free article] [PubMed] [Google Scholar]
- 39.Gale RE, Wheadon H, Linch DC: Assessment of X-chromosome inactivation patterns using the hypervariable probe M27 β in normal hemopoietic cells and acute myeloid leukemic blasts. Leukemia 1992, 6:649-655 [PubMed] [Google Scholar]
- 40.Gale RE, Wainscoat JS: Clonal analysis using X-linked DNA polymorphisms. Br J Haematol 1993, 85:2-8 [DOI] [PubMed] [Google Scholar]
- 41.Gale RE, Linch DC: Interpretation of X-chromosome inactivation patterns. Blood 1994, 84:2376-2378 [PubMed] [Google Scholar]
- 42.Jacoby LB, Hedley-Whyte ET, Pulaski K, Seizinger BR, Martuza RL: Clonal origin of pituitary adenomas. J Neurosurg 1990, 73:731-735 [DOI] [PubMed] [Google Scholar]
- 43.Hicks DG, LiVolsi VA, Neidich JA, Puck JM, Kant JA: Clonal analysis of solitary follicular nodules in the thyroid. Am J Pathol 1990, 137:553-562 [PMC free article] [PubMed] [Google Scholar]
- 44.Nowell PC: The clonal evolution of tumor cell populations. Science 1976, 194:23-28 [DOI] [PubMed] [Google Scholar]
- 45.Mutter GL, Boynton KA: X chromosome inactivation in the normal female genital tract: implications for identification of neoplasia. Cancer Res 1995, 55:5080-5084 [PubMed] [Google Scholar]
- 46.Jovanovic AS, Boynton KA, Mutter GL: Uteri of women with endometrial carcinoma contain a histopathological spectrum of monoclonal putative precancers, some with microsatellite instability. Cancer Res 1996, 56:1917-1921 [PubMed] [Google Scholar]
- 47.Iwabuchi H, Sakamoto M, Sakunaga H, Ma YY, Carcangiu ML, Pinkel D, Yang-Feng TL, Gray JW: Genetic analysis of benign, low-grade, and high-grade ovarian tumors. Cancer Res 1995, 55:6172-6180 [PubMed] [Google Scholar]
- 48.Aihara T, Noguchi S, Sasaki Y, Nakano H, Imaoka S: Clonal analysis of regenerative nodules in hepatitis C virus-induced liver cirrhosis. Gastroenterology 1994, 107:1805-1811 [DOI] [PubMed] [Google Scholar]
- 49.Jimbo H, Hitomi Y, Yoshikawa H, Yano T, Momoeda M, Sakamoto A, Tsutsumi O, Taketani Y, Esumi H: Evidence for monoclonal expansion of epithelial cells in ovarian endometrial cysts. Am J Pathol 1997, 150:1173-1178 [PMC free article] [PubMed] [Google Scholar]
- 50.Gaffey MJ, Iezzoni JC, Weiss LM: Clonal analysis of focal nodular hyperplasia of the liver. Am J Pathol 1996, 148:1089-1096 [PMC free article] [PubMed] [Google Scholar]
- 51.Diaz-Cano S, Gonzalez-Campora R, Rios-Martin JJ, Lerma-Puertas E, Jorda-Heras M, Vazquez-Ramirez F, Bibbo M, Galera-Davidson H: Nuclear DNA patterns in adrenal cortex proliferative lesions: Virchows Arch A Pathol Anat Histopathol 1993, 423:323–328 [DOI] [PubMed]