

Abstract
Cerebrovascular amyloid deposition and microvascular degeneration are frequently associated with Alzheimer’s disease (AD), but the etiology and pathogenetic role of these abnormalities are unknown. Recently, transforming growth factor-β1 (TGF-β1) was implicated in cerebrovascular amyloid formation in transgenic mice with astroglial overproduction of TGF-β1 and in AD. We tested whether TGF-β1 overproduction induces AD-like cerebrovascular degeneration and analyzed how cerebrovascular abnormalities develop over time in TGF-β1-transgenic mice. In cerebral microvessels from 3- to 4-month-old TGF-β1-transgenic mice, which display a prominent perivascular astrocytosis, levels of the basement membrane proteins perlecan and fibronectin were severalfold higher than in vessels from nontransgenic mice. Consistent with this increase, cortical capillary basement membranes of TGF-β1 mice were significantly thickened. These changes preceded amyloid deposition, which began at around 6 months of age. In 9- and 18-month-old TGF-β1 mice, various degenerative changes in microvascular cells of the brain were observed. Endothelial cells were thinner and displayed abnormal, microvilli-like protrusions as well as occasional condensation of chromatin, and pericytes occupied smaller areas in capillary profiles than in nontransgenic controls. Similar cerebrovascular abnormalities have been reported in AD. We conclude that chronic overproduction of TGF-β1 triggers an accumulation of basement membrane proteins and results in AD-like cerebrovascular amyloidosis and microvascular degeneration. Closely related processes may induce cerebrovascular pathology in AD.
The central nervous system (CNS) requires an efficient vasculature to supply energy and oxygen to brain cells. A decrease in cerebrovascular function could change brain perfusion and impair cognitive abilities. In Alzheimer’s disease (AD), progressive dementia is accompanied by neurodegenerative changes and distinct cerebrovascular abnormalities. 1-4 The observations that AD and vascular disease share common risk factors and that stroke may be a risk factor for AD 3,5,6 have sparked new interest in the possible relationships between cerebrovascular pathology, neurodegeneration, and dementia.
Cerebrovascular abnormalities abound in the AD brain, and vascular deformations, and perivascular pseudocalcifications were noted in senile brains several decades ago. 7,8 The most apparent of these abnormalities is cerebral amyloid angiopathy (CAA), the deposition of amyloid in cerebral blood vessel walls. CAA is found in most AD cases and, like other AD-associated lesions (eg, plaques), occurs in the nondemented elderly as well. 9,10 Analysis of cerebrovascular amyloid deposits led to the identification of the amyloid-β (Aβ) peptide, which is generated from the β protein precursor (βPP) by proteolytic cleavage. 11 In its fibrillar form, Aβ is the main component of both vascular and parenchymal amyloid plaques. CAA is a major cause of normotensive intracerebral hemorrhage in the elderly, 9,10,12,13 and may, in some of its inherited forms, cause dementia in the absence of other classic AD lesions. 14,15 AD and CAA are also associated with other changes in the cerebrovasculature, including alterations in smooth muscle cells and pericytes, endothelial cell thinning, and loss of endothelial mitochondria. 1,16,17 Thickening of the vascular basement membrane, probably due to the accumulation of basement membrane proteins, has also been noted in several independent studies, 1,16,18,19 although others did not observe significant differences in AD compared with normal aging. 20 The cause and functional consequences of these microvascular abnormalities are unclear, and the temporal relationships between basement membrane accumulation, amyloid deposition, and other microvascular abnormalities are unknown.
Transforming growth factor-β1 (TGF-β1), a multifunctional cytokine with a central role in tissue injury and repair, 21 has profound effects on vasculogenesis, angiogenesis, and maintenance of vessel wall integrity. 22,23 In the CNS, TGF-β1 organizes injury responses. 24 In AD patients, TGF-β1 levels in plaques, 25,26 cerebrospinal fluid, 27 and serum 28 are higher than in nondemented elderly controls. Furthermore, cortical TGF-β1 messenger RNA (mRNA) levels correlate positively with the degree of cerebrovascular amyloidosis in AD cases, and TGF-β1 immunoreactivity in such cases is elevated along cerebral blood vessels. 29
The expression of a constitutively bioactive form of TGF-β1 in astrocytes of transgenic mice results in perivascular astrocytosis and age-related deposition of amyloid around cerebral blood vessels. 29,30 Overexpression of TGF-β1 in mice expressing human βPP/Aβ in neurons accelerates vascular deposition of human Aβ, suggesting a causal role for TGF-β1 in cerebrovascular amyloidosis. 29 Such a role is supported by other studies in rats, 31 cortical slice cultures, 32 isolated canine cerebral blood vessels, 33 and cultured smooth muscle cells. 34 How TGF-β1 production causes amyloid deposition and whether amyloidosis is associated with other microvascular alterations have not yet been determined. Here, we demonstrate that chronic elevation of TGF-β1 production in the CNS of transgenic mice leads to an accumulation of basement membrane proteins in blood vessels that precedes the formation of cerebrovascular amyloid deposits and is associated with AD-like degenerative changes in cerebrovascular cells.
Materials and Methods
Transgenic Mice and Genotyping
All mice had a BALB/c×SJL F2 genetic background. Nontransgenic breeder mice were obtained from Jackson Laboratories (Bar Harbor, ME). Transgenic mice expressing a constitutively active form of TGF-β1 under control of glial fibrillary acidic protein regulatory sequences have been described previously. 29,30,35 Transgenic mice from lines T64 and T115 express TGF-β1 immunoreactive protein in astrocytes throughout the brain, with increased levels in perivascular locations. In addition, primary astrocytes from TGF-β1 transgenic mice secreted much higher levels of bioactive TGF-β1 than astrocytes from nontransgenic controls. TGF-β1 mice show a prominent perivascular astrocytosis at a young age and increased levels of fibronectin and laminin mRNAs in their brains. 30 High-level overexpression of TGF-β1 results in communicating hydrocephalus; 30 low-level expression, as used in the current study, does not cause this complication. Mouse genotypes were determined by amplification of transgene regulatory sequences from tail DNA by polymerase chain reaction. Nontransgenic littermates of TGF-β1 mice served as controls.
Brain Tissue Preparation
Frontal cortex (midfrontal gyrus) from a patient with AD and severe CAA was obtained from the Alzheimer’s Disease Research Center of the University of California (San Diego, CA). Tissue blocks were fixed in freshly prepared 4% paraformaldehyde in 0.1 mol/L phosphate buffer, pH 7.4, at 4°C for 48 hours. Mice were anesthetized with chloral hydrate and flush-perfused transcardially with 0.9% saline. Brains were removed and divided sagittally. Hemibrains were snap-frozen and stored at −70°C until extraction of RNA or immersion fixation in paraformaldehyde as described above. For electron microscopy (EM), anesthetized mice were perfused with 0.1 mol/L cacodylate buffer, pH 7.4, containing 3% paraformaldehyde, 1% glutaraldehyde, and 3 mmol/L CaCl2 at a rate of 5 ml/min for 3 minutes. This procedure resulted in optimal fixation and tissue preservation. Brains were removed and stored in cacodylate buffer at 4°C until they were embedded in resin (see below).
Histological Analysis
Fixed mouse or human brain specimens were cut with a vibratome into 40-μm free-floating sections and processed essentially as previously described. 29,36 For thioflavin S staining, sections were air-dried overnight on Superfrost slides (Fisher, Pittsburgh, PA), fixed on the slides with 4% formaldehyde in 0.1 mol/L phosphate buffer, and stained with 1% thioflavin S solution for 8 minutes. Sections were rapidly differentiated once in 100% ethanol and twice in 80% ethanol/water, rinsed for 10 minutes with water, and mounted with fluorescence mounting medium (Vectashield, Vector Laboratories, Burlingame, CA). Sections were examined by fluorescence microscopy or by laser scanning confocal microscopy with a Bio-Rad MRC-1024 mounted on a Nikon Optiphot-2 microscope as previously described. 30,36 The following CAA scores (for human brains) and the thioflavin S scores (for murine brains) were determined semiquantitatively by visual inspection of thioflavin S-stained brain sections: 0, no vessels affected; 1, occasional vessels affected; 2–4, multiple vessels affected mildly (Grade 2), moderately (Grade 3), or severely (Grade 4). 37
Preparation of Brain Microvessels
Brain microvessels were isolated from mouse cortex and hippocampus as described by Pardridge et al. 38 Mice were anesthetized, and brains were removed and washed with ice-cold buffer B (103 mmol/L NaCl, 4.7 mmol/L KCl, 2.5 mmol/L CaCl2, 1.2 mmol/L KH2PO4, 1.2 mmol/L MgSO4, 15 mmol/L N-2-hydroxyethylpiperazine-N′-ethane-sulfonic acid (HEPES), pH 7.4). Cortex and hippocampus were dissected and homogenized in freshly prepared buffer A (buffer B plus 25 mmol/L NaHCO3, 10 mmol/L glucose, 1 mmol/L sodium pyruvate, 1 g/100 ml bovine serum albumin) in a Potter-Elvehjem tissue grinder and a rotating (100 rpm) Teflon pestle with a clearance of 0.15–0.23 mm (PCG Scientific, Gaithersburg, MD) using seven up-and-down strokes. The homogenate was transferred to a 30-ml Corex glass tube and mixed with 30% industrial-grade dextran (molecular weight 79,500; Sigma) in buffer B to a final dextran concentration of 17%. After centrifugation for 20 minutes at 2400 × g, the top fatty layer was removed, and the remaining solution was mixed again and recentrifuged as above. The pellet was resuspended in 15 ml of buffer A and passed through a 280-μm metal mesh (Bellco, Vineland, NJ). The filtrate containing microvessels, red blood cells, and nuclei was collected and passed through a 70-μm nylon mesh (Becton Dickinson Labware, Bedford, MA). Microvessels retained on this mesh were washed extensively with buffer A before retrieval from the mesh. The purity of the microvascular preparation was assessed by light microscopic examination of vessels seeded on Superfrost glass slides. Slides were air-dried overnight, and vessels were fixed with 4% formaldehyde in phosphate-buffered saline for staining with thioflavin S and with hematoxylin and eosin (H&E). To assess the purity of brain microvessels, aliquots fixed to glass slides were also stained with a rabbit antibody against glucose transporter 1 (AB1341, 1:1000; Chemicon) as described. 39,40
Processing of EM Samples for Morphometry
Brain tissue, fixed for EM as described above, was processed for thin-section EM as follows. Small blocks (3–5 mm3) were trimmed out of the frontal cortex tissue, washed three times for 15 minutes each in cold 0.1 mol/L sodium cacodylate buffer, pH 7.2 (cacodylate buffer), and postfixed in 1% osmium tetroxide in cacodylate buffer for 45 minutes at room temperature. Excess unreacted osmium tetroxide was removed by washing the blocks three times for 10 minutes each in cold cacodylate buffer. After an initial dehydration in 30% ethanol for 5 minutes, en bloc staining was performed in saturated uranyl acetate in 50% ethanol, diluted with two parts of water, for 1 hour. Tissues were then fully dehydrated in a graded series of ethanol/water solutions, ending in three absolute ethanol steps. Blocks were then transferred with propylene oxide into Polybed 812 epoxy resin (Polysciences, Warrington, PA). After embedding and polymerization at 60°C, large semithin survey sections were cut with glass knives on a Reichert Ultracut E ultramicrotome, stained with heated toluidine blue, and examined by light microscopy to identify cortical regions rich in capillaries. Sections (∼70 nm thick) were cut, stained with saturated, aqueous uranyl acetate for 20 minutes, and 0.4% aqueous lead citrate for 10 minutes, and examined with a JEOL JEM 100CX transmission electron microscope.
Quantitative Morphometry
The capillary profiles were identified as blood vessels 3 to 8 μm in diameter that lacked smooth muscle cells and consisted of a single layer of endothelial cells. In addition, these profiles were chosen not to contain nuclear structures and were centered for electron micrography at a magnification of ×10,000. Sets of 8 to 10 capillary profiles were recorded micrographically for each specimen. Photographic prints were made for each capillary and imported into an image-analysis system (Image1/AT, Universal Imaging Corporation, West Chester, PA) with a black and white video camera. The following morphometric variables were then measured for each capillary profile: capillary profile area, capillary circumference, intimal area, endothelial area (excluding basement membrane area), and pericytic cell area. Results were calculated as follows. Basement membrane area = intimal area − (pericytic area + endothelial area); relative basement membrane thickness = basement membrane area/capillary circumference; capillary diameter = capillary circumference/π; relative endothelial cell area = 100 × endothelial area/capillary profile area; relative pericyte area = 100 × pericyte area/capillary profile area.
RNase Protection Assays
Total RNA from snap-frozen brains was isolated with TRI reagent (Molecular Research Center, Cincinnati, OH) and stored in Formazol buffer (Molecular Research Center) at −20°C. Total RNA was analyzed by solution hybridization RNase protection assay as previously described. 41 Samples were separated on 5% acrylamide, 8 mol/L urea Tris/borate/ethylenediaminetetraacetic acid gels, and dried gels were exposed to X-ray films (Biomax MR, Kodak, Rochester, NY). mRNA levels were quantitated from phosphorimager readings of probe-specific signals corrected for RNA content/loading errors by normalization to the β-actin signals. The following 32P-labeled antisense riboprobes were used to identify specific mRNAs (protected sequences are in brackets): murine β-actin [nucleotides 480–559 of mouse β-actin mRNA (GenBank accession no. M18194)], murine TGF-β1 [nucleotides 500–752 of mouse TGF-β1 mRNA (GenBank accession no. M13177)], and porcine TGF-β1 [nucleotides 999-1412 of porcine TGF-β1 mRNA (M23703)].
Western Blots
Homogenates of isolated cerebral microvessels were prepared with a triple-detergent lysis buffer 42 and protease inhibitors (100 μg/ml phenylmethylsulfonyl fluoride, 1 μg/ml aprotinin, 2× complete inhibitor; Boehringer Mannheim). Insoluble material was removed by centrifugation. The protein concentration in the supernatant was determined with a modified Bradford method (Pierce, Rockford, IL), and sample protein concentrations were equalized with lysis buffer. Sodium dodecyl sulfate loading buffer was added, and the samples were heated to 95°C for 5 minutes. Microvascular preparations and brain homogenates from TGF-β1 mice were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis using 4–15% gradient gels (Biorad, Hercules, CA), electrotransferred at 4°C overnight to polyvinylidene difluroide membranes (Millipore, Bedford, MA), and blocked with Tris-buffered saline containing 5% nonfat dried milk and 0.1% Tween 20. Extracellular matrix proteins secreted by Engelbreth-Swarm-Holm mouse sarcoma cells (Matrigel; Becton Dickinson Labware) were analyzed similarly as a positive control for the presence of basement membrane proteins. The blots were incubated for 5 hours at room temperature in rat anti-heparan sulfate proteoglycan (ie, core protein perlecan) antibodies (clone A7L6, 1:500; Chemicon, Temecula, CA) or mouse anti-fibronectin antibodies (clone TV-1, 1:300; NeoMarkers, Union City, CA). The bound antibodies were detected with horseradish peroxidase-conjugated anti-rat or anti-mouse antibodies (1:5000, Jackson ImmunoResearch, West Grove, PA) and developed with ECL reagents (Amersham, Arlington, IL), and the blots were exposed to X-ray film (Biomax MR; Kodak). For quantitation, exposures of Western blots with densities within the linear range of the film were scanned, and the density of the bands was determined by inflection point analysis with Advanced Quantifier software (BioImage, Ann Arbor, MI).
Statistical Analysis
For all histopathological and morphological analyses and for the RNase protection assay analyses of cortical TGF-β1 mRNA levels, brain tissue samples were coded, and investigators were unaware of the genotype of the mice. Statistical calculations were done with Statview (SAS Institute Inc., Cary, NC).
Results
Accumulation of Amyloid Deposits at the Vascular Basement Membrane
Laser-scanning confocal microscopy of cortical brain sections and isolated cortical microvessels from TGF-β1 brains and nontransgenic controls showed that thioflavin S-positive amyloid deposits were tightly associated with the vascular wall (Figure 1) ▶ . In transverse sections of blood vessels from 12-month-old TGF-β1 mice, discrete deposits were detected on the abluminal side of endothelial cells (Figure 1A) ▶ . Interestingly, cerebrovascular amyloid deposits in AD (Figure 1B) ▶ were detected in a similar location, although deposition was frequently more extensive than in mouse vessels. Thioflavin S-positive deposits were found in 5 to 10% of microvessels extracted from the cortex and hippocampus of 12- to 18-month-old TGF-β1 mice (Figure 1, C–F) ▶ , but were never found in vessels from nontransgenic control mice (data not shown). The degree of amyloid deposition varied widely in different areas of the same vessel (Figure 1E) ▶ and in different vessels from the same brain (compare vessels shown in Figure 1, E and F ▶ ). This observation is in agreement with the distribution of thioflavin S-positive blood vessels in TGF-β1 transgenic brains 29 and in human CAA. 9
Figure 1.
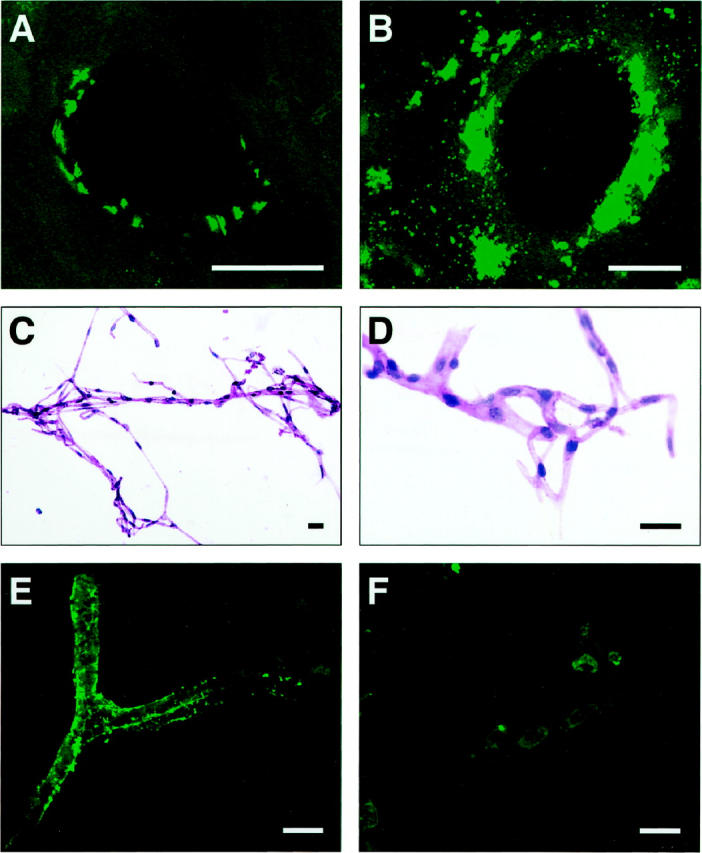
Thioflavin S-positive deposits in cerebral blood vessels of TGF-β1 mice and AD. Cortical brain sections from a 12-month-old TGF-β1 (line T115 heterozygous) mouse (A) and an AD patient with a CAA score of 3 (B) were stained with thioflavin S and analyzed by confocal microscopy. Thioflavin S-positive amyloid deposits are located on the abluminal side of endothelial cells in TGF-β1 mice and the AD case. Isolated cerebral blood vessels stained with H&E from an 18-month-old TGF-β1 mouse were examined at low-power (C) or high-power (D) magnification. No gross structural abnormalities were observed. E and F: Thioflavin S-stained isolated cerebral blood vessels from an 18-month-old TGF-β1 mouse with various degrees of amyloid deposition. Scale bars, 20 μm.
Consistent with the location of thioflavin S-positive amyloid deposits identified by confocal microscopy, electron-dense extracellular deposits were detected within the abluminal basement membrane of TGF-β1 transgenic cortical blood vessels by electron microscopy (Figure 2) ▶ . Such deposits were never observed in nontransgenic control mice (Figure 2) ▶ .
Figure 2.
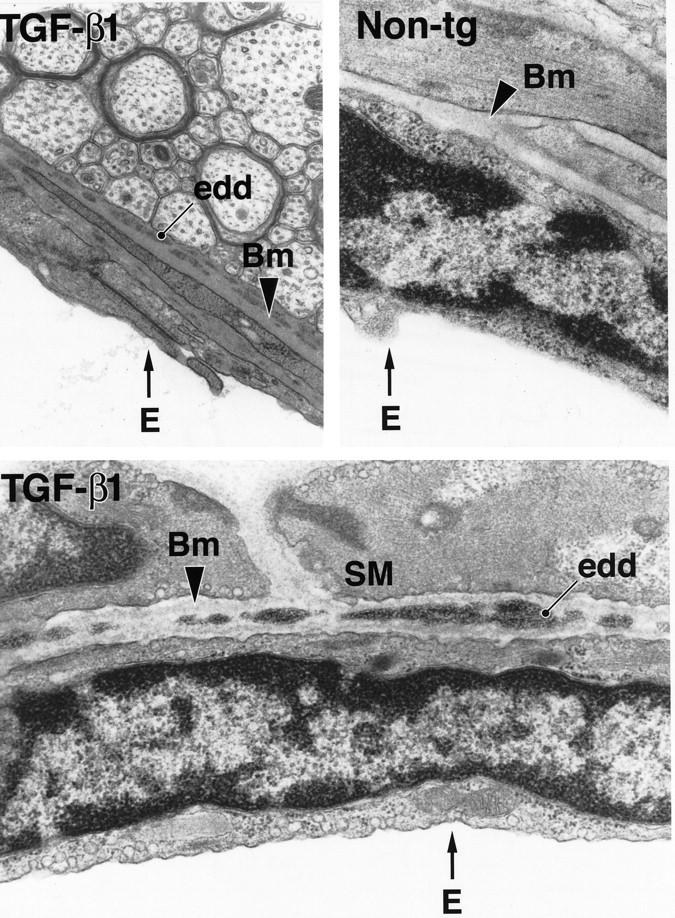
Electron-dense deposits within the basement membrane of cerebral blood vessels in TGF-β1 mice. Ultrathin brain sections from a 9-month-old TGF-β1 mouse (line T64 heterozygous) and a nontransgenic littermate control (Non-tg) were analyzed by electron microscopy. Basement membrane (Bm) of a medium-sized blood vessel from a TGF-β1 mouse shows electron-dense deposits (edd) at medium- and high-power magnification. No such deposits were observed in nontransgenic blood vessels. E, endothelial cell; SM, smooth muscle cell. (Original magnifications: top left, ×31,500; top right and bottom, ×42,500).
Cerebrovascular Amyloid Deposition in TGF-β1 Mice Is Dose- and Age-Dependent
To ascertain whether accumulation of basement membrane proteins precedes amyloid deposition, we first compared the relationship between age, TGF-β1 expression levels, and the development of cerebrovascular amyloid deposits in two independent lines of TGF-β1 transgenic mice (Figure 3A) ▶ . Brain transgene mRNA levels in heterozygous mice from medium-expresser line T115 were approximately twice as high as those in heterozygous mice from the low-expresser line T64. Homozygous line T115 mice showed a further increase in TGF-β1 mRNA levels. In all three groups of mice, transgene (porcine) TGF-β1 levels correlated with the expression of endogenous (murine) TGF-β1 (r = 0.94; P < 0.0001). The amino acid sequences of mature porcine and murine TGF-β1 differ by only a single amino acid, 43 and TGF-β1 can induce its own synthesis in an autocrine fashion. 44 Porcine TGF-β1 mRNA levels were approximately two- and fourfold higher than endogenous TGF-β1 mRNA levels in heterozygous low- and medium-expresser mice, respectively (Figure 3A) ▶ .
Figure 3.
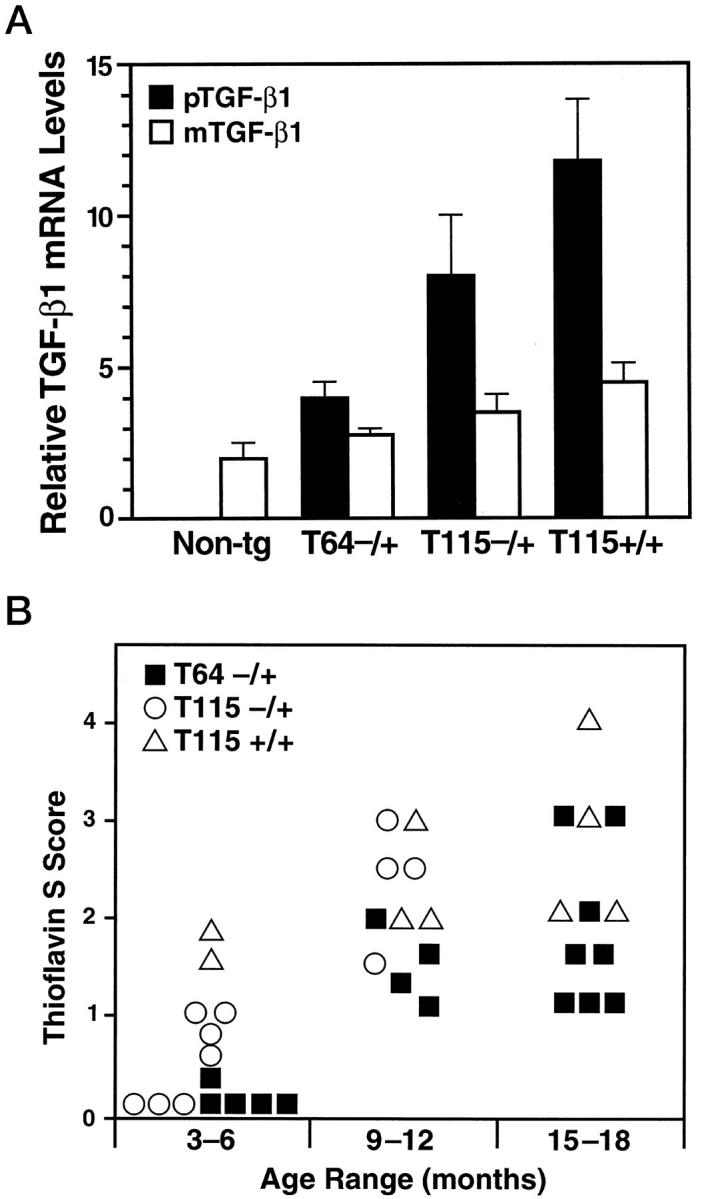
Accumulation of thioflavin S-positive deposits in TGF-β1 mice depends on age and level of TGF-β1 expression. Brains from TGF-β1 line T64 heterozygous mice (T64−/+), line T115 heterozygous mice (T115−/+), and line T115 homozygous mice (T115+/+) and nontransgenic littermate controls (Non-tg) were analyzed for cerebral TGF-β1 mRNA expression levels and for the number of thioflavin S-positive cerebrovascular deposits. A: Total RNA was extracted 3 months postnatally (n = 4 mice per group). The relative levels of porcine (transgene) TGF-β1 (pTGF-β1) and murine (endogenous) TGF-β1 (mTGF-β1) were determined by RNase protection assay. Results are means ± SEMs obtained by phosphorimager analysis and normalization to actin values. B: Sagittal brain sections from the indicated groups of mice (aged 3–6, 9–12, or 15–18 months) were stained with thioflavin S and examined by fluorescence microscopy. Data points represent mean scores from three to six sections per mouse. Older mice had more thioflavin S-positive deposits than younger mice with similar levels of transgene expression, and higher levels of TGF-β1 expression resulted in more amyloid deposits.
The extent of thioflavin S-positive amyloid deposition in cerebral blood vessels was assessed in transgenic mice from lines T64 and T115 at three different age ranges (Figure 3B) ▶ . Thioflavin S-positive deposits were never observed in nontransgenic littermate control mice. In general, older TGF-β1 mice showed more cerebrovascular deposits than younger mice with similar levels of transgene expression. In addition, mice expressing higher levels of TGF-β1 showed more cerebrovascular deposits than age-matched mice expressing lower levels of TGF-β1. Within a given line, transgene TGF-β1 mRNA levels did not significantly increase with age (data not shown). Thus, the formation of thioflavin S-positive deposits in TGF-β1 mice is both dose- and age-dependent.
Accumulation of Basement Membrane Proteins Precedes Amyloid Deposition
Proteins of the vascular basement membrane, including perlecan, laminin, and fibronectin, have been implicated in amyloidosis (reviewed in 45,46 ). Because low-expresser line T64 mice showed no amyloid deposits before 6 months of age, this line was used to study whether TGF-β1 increases basement membrane protein production before amyloid is deposited. Western blot analysis was used to determine semiquantitatively the relative levels of two basement membrane proteins, perlecan and fibronectin, from isolated cortical/hippocampal microvessels of 3- to 4-month-old TGF-β1 mice and nontransgenic controls. The vessel preparations from transgenic brains contained significantly more perlecan and fibronectin than those from controls (Figure 4) ▶ . Light microscopic analysis of H&E-stained microvessel preparations showed very little contaminating tissue debris (Figure 1, C and D) ▶ .
Figure 4.
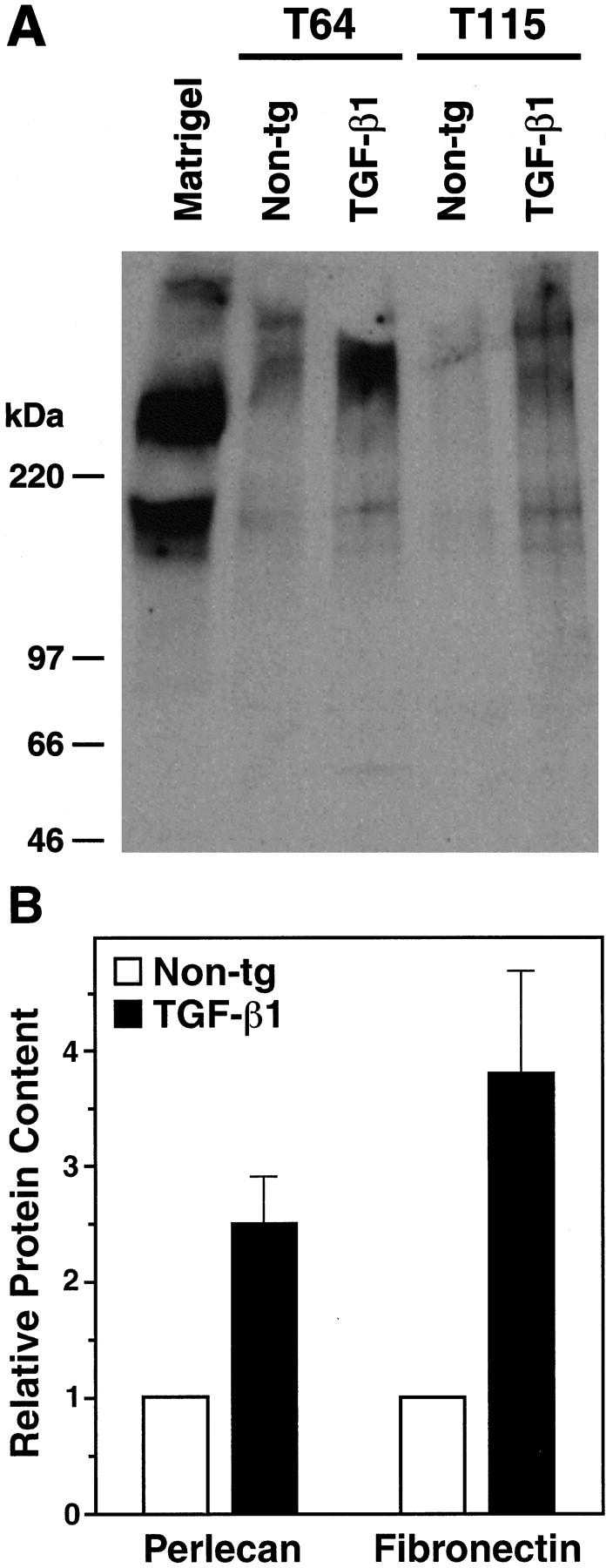
Increased perlecan and fibronectin levels in cerebral microvessels from young TGF-β1 mice. A: Western blot analysis showing perlecan expression in homogenates from isolated cortical and hippocampal microvessels (180 μg per lane) from 4-month-old TGF-β1 mice (line T64 or T115 heterozygous) or nontransgenic littermate controls. A mix of extracellular matrix proteins (Matrigel, 1 μl) served as a positive control. Microvessels prepared from TGF-β1 transgenic or nontransgenic control brains (n = 5 per group) were pooled yielding approximately 300 μg of protein per group. Molecular size standards shown on the left indicate perlecan-immunoreactive products in cerebral microvessels exeeding 400 kd, as well as a smaller-size protein of approximately 200 kd, which is also present in Matrigel. B: Perlecan and fibronectin contents determined semiquantitatively by densitometric analysis in three to four independent experiments were significantly higher in TGF-β1–transgenic (line T64) than in nontransgenic microvessels (*P < 0.05; Mann-Whitney U test). Results are means ± SEMs.
Increased production of basement membrane proteins in the vascular wall can result in a thickening of the basement membrane. 47 Several groups have reported thickening of the basement membrane in AD, 1,16,18,19 although others did not observe significant differences in AD compared with normal aging. 20 Here, we tested whether the increase in basement membrane protein production in TGF-β1–transgenic microvessels would translate into an increased thickness of the capillary basement membrane in TGF-β1 mice. Using electron microscopy and ultrastructural morphometry, we found that cortical capillaries from 4-month-old TGF-β1 mice (line T64) had significantly thicker basement membranes than those from nontransgenic controls (Figures 5 and 6) ▶ ▶ . In 9- to 18- month-old TGF-β1 mice, basement membranes from cortical capillaries were almost twice as thick as those of nontransgenic controls (Figure 6) ▶ . These results suggest that amyloid deposition in TGF-β1 mice was preceded by the accumulation of basement membrane proteins and thickening of the basement membrane.
Figure 5.
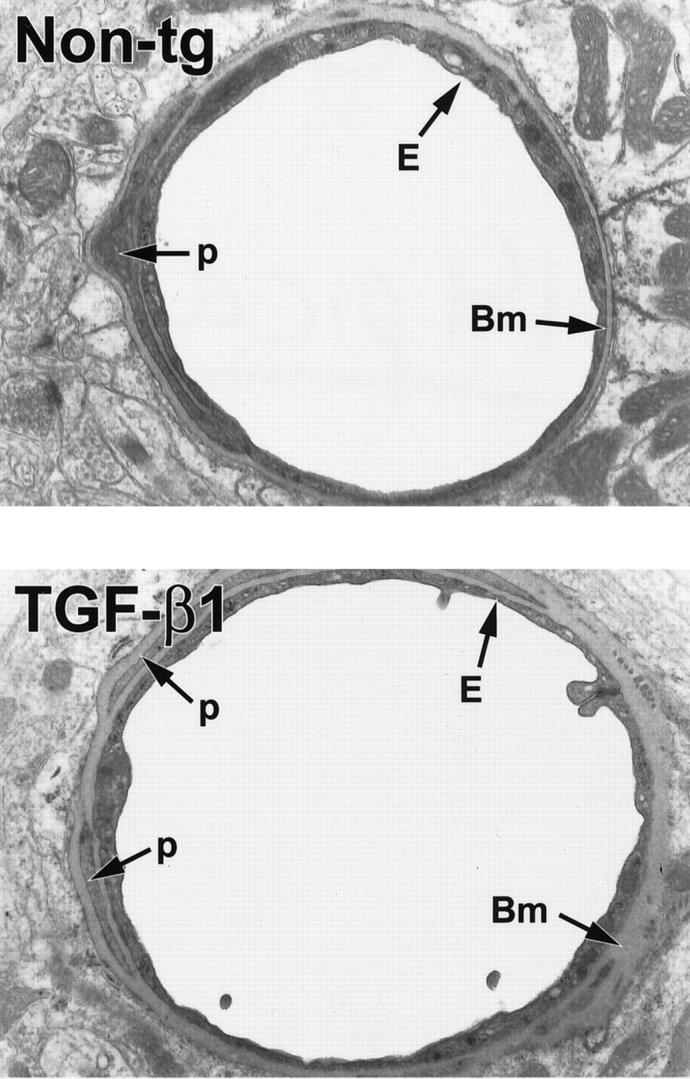
Ultrastructural abnormalities in cerebral blood vessels of TGF-β1 mice. Ultrathin brain sections from 9-month-old TGF-β1 mice (line T64 heterozygous) or nontransgenic littermate controls (Non-tg) were analyzed by electron microscopy. In the TGF-β1 transgenic vessel, the basement membrane (Bm) appears thickened, and the endothelial cell (E) profile is thinner with an irregular luminal surface. The nontransgenic control vessel has a normal basement membrane and a smooth endothelial cell surface. Original magnifications, ×5000. p, pericyte.
Figure 6.

Age-related accumulation of basement membrane proteins in cortical capillaries of TGF-β1 mice. Ultrathin cortical brain sections from TGF-β1 mice (line T64 heterozygous) and nontransgenic littermate controls (Non-tg) at 4, 9, and 18 months of age were analyzed by electron microscopy. The thickness of the basement membrane was measured in seven to nine capillary profiles per brain in a total of three mice per group by computer-assisted morphometry as described in Materials and Methods. Results are means ± SEMs. Basement membranes were significantly thicker in capillaries from TGF-β1 brains than in capillaries from nontransgenic control brains (*P < 0.005; unpaired, two-tailed Student’s t-test).
To test the possibility that the difference in basement membrane thickness simply reflected vascular cell atrophy rather than active thickening, we analyzed the distribution of capillary diameters in TGF-β1–transgenic mice and nontransgenic controls and compared the relative basement membrane thickness with the capillary diameters. Capillary diameters in young TGF-β1 mice were slightly smaller than in littermate nontransgenic controls (14% decrease in diameter), whereas capillary diameters in the older mice were on average 8% greater in TGF-β1 mice then in nontransgenic controls. The relative ratio between basement membrane thickness and diameter was significantly greater in young and old TGF-β1–transgenic than -nontransgenic capillaries (4-month-old: 23.0 ± 2.4 versus 14.2 ± 0.7, P = 0.0012; 18-month-old: 18.8 ± 0.7 versus 14.9 ± 0.5, P < 0.0001, unpaired two-tailed Student’s t-test). These data suggest that, at least in young mice, the increased thickness of the basement membrane is likely due to membrane thickening and not to atrophy of vascular cells. In aged TGF-β1 capillaries, atrophy may contribute to the observed increase in thickness.
Old TGF-β1 Mice Develop Microvascular Injury and Degeneration in Their Brains
To determine whether the accumulation of basement membrane proteins and subsequent amyloid deposition around blood vessels affect vascular integrity, we first examined cerebral microvessels for gross alterations. Isolated microvessels stained with H&E from TGF-β1 and nontransgenic control mice revealed no structural differences (Figure 1, C and D ▶ ; data not shown). To assess whether cerebral overexpression of TGF-β1 might result in vascular cell loss, we counted hematoxylin-stained nuclei in capillaries from cortical microvascular preparations of 9-month-old TGF-β1 mice and nontransgenic controls. No significant loss in vascular cells was observed (data not shown).
Electron microscopic examination of cortical capillaries showed clear morphological alterations in 9-month-old TGF-β1 mice not seen in littermate controls (Figures 5–7) ▶ ▶ ▶ . Frequently, endothelial cells had irregular luminal surfaces with microvilli-like protrusions and bleb-like structures (Figures 5 and 7) ▶ ▶ . Some endothelial cell nuclei from TGF-β1 brains showed evidence of abnormal chromatin condensation not seen in the nuclei from nontransgenic controls. Ultrastructural morphometry demonstrated smaller endothelial cell and pericyte profiles in TGF-β1 brains than in nontransgenic controls (Figure 8) ▶ . In 4-month-old TGF-β1 mice, endothelial cells and pericytes showed no significant changes (data not shown), although capillary basement membranes were already significantly thicker than those in nontransgenic littermate controls (Figure 6) ▶ .
Figure 7.

Microvascular endothelial cell damage in brains of 9-month-old TGF-β1 mice. Ultrathin cortical brain sections from TGF-β1 mice (line T64 heterozygous; A, B, D) and nontransgenic littermate controls (C) were analyzed by electron microscopy. Endothelial cell nuclei (arrows) from TGF-β1 brains showed signs of increased chromatin condensation (A, B), whereas the nucleus from a nontransgenic control appeared normal (C). Frequently, the surface of TGF-β1 transgenic endothelial cells was irregular (A, B, D), with microvilli-like protrusions and bleb-like structures (arrowheads). Original magnifications, ×31,500 (A−C) and ×72,000 (D).
Figure 8.

Abnormalities in brain capillaries from TGF-β1 mice. The relative areas occupied by endothelial cell profiles (left) or pericyte profiles (right) were measured in cross-sections of cortical capillaries from 9-month-old TGF-β1 mice (line T64 heterozygous) and nontransgenic littermate controls. Results are means ± SDs from seven to nine capillaries analyzed in three mice per group. The relative areas of endothelial cell and pericyte profiles in 9-month-old TGF-β1 mice were significantly smaller than those in controls (*P < 0.05; unpaired, two-tailed Student’s t-test).
Discussion
This study demonstrates that chronic elevation of TGF-β1 production in astrocytes of transgenic mice leads to cerebrovascular abnormalities and degeneration. Strong perivascular astrocytosis, as previously reported in TGF-β1 mice, 30 and an accumulation of microvascular basement membrane proteins were followed by amyloid deposition. This accumulation of proteins was associated with degenerative changes in cerebrovascular cells. In many respects, the pathological changes in TGF-β1 mice resembled those found in AD and CAA, including perivascular astrocytosis, basement membrane thickening, amyloid deposition, and degenerative changes in vascular cells (reviewed in 1 ). Therefore, overproduction of TGF-β1 may be an underlying cause of several of these cerebrovascular abnormalities in humans as well.
We proposed recently that TGF-β1 might induce cerebrovascular amyloidosis in AD. 29 The current study further implicates this cytokine as a potential pathogenic factor of other microvascular abnormalities associated with AD and CAA. Interestingly, several reports demonstrated higher TGF-β1 levels in AD brains than in nondemented controls. 25-29 In addition, we recently observed increased levels of the latent form of TGF-β1 in perivascular astrocyte-like cells in AD brains with CAA but not in nondemented controls. 48 Although the reason for this increase is unclear, trauma, ischemia, or age-related oxidative stress and cellular injury may trigger TGF-β1 expression. 24 In chronic conditions, for example in AD, such factors may lead to elevated TGF-β1 levels over long periods.
By studying TGF-β1 mice, we were able to ascertain the earliest pathological cerebrovascular changes preceding amyloid deposition. Unlike other models of cerebral amyloidosis, 49-51 TGF-β1 mice develop amyloid deposits in cerebral blood vessels in the absence of parenchymal amyloid plaques, enabling vascular amyloid deposition to be studied independently of plaque formation. Apart from a previously reported perivascular astrocytosis, 30 one of the earliest changes was the accumulation of basement membrane proteins in the vascular wall (Figure 4) ▶ . Perlecan and fibronectin levels in isolated microvessels were severalfold higher in TGF-β1 mice than in nontransgenic controls, and the basement membrane in TGF-β1 mice was significantly thickened (Figures 5 and 6) ▶ ▶ . Our previous observation that laminin and fibronectin mRNA levels are higher in TGF-β1 brains than in nontransgenic controls 30 suggests that basement membrane protein accumulation is caused by increased gene expression, although there my be decreased basement membrane protein degradation at the same time.
This effect of TGF-β1 on basement membrane protein synthesis and accumulation in TGF-β1 transgenic brains is consistent with its stimulatory effect on extracellular matrix production. 21,52 TGF-β1 induces the transcription of basement membrane proteins, such as perlecan, fibronectin, and collagen in vitro, 53,54 and tissue-specific expression of TGF-β1 in transgenic mice causes fibrosis and excessive production of basement membrane proteins in the skin, 55 pancreas, 56 and kidney. 21 Notably, in the kidney, a similar type of vascular pathology develops regardless of whether TGF-β1 is overproduced within kidney cells or in the serum of transgenic mice (reviewed in 21 ). Although the cerebrovascular phenotype in glial fibrillary acidic protein–TGF-β1 mice results from parenchymal overexpression of TGF-β1, it would be interesting to test whether increased serum TGF-β1 levels produce cerebrovascular abnormalities. Such a finding could have important implications for humans, because AD cases have higher serum TGF-β1 levels than nondemented controls 28 and because serum TGF-β1 levels may be determined genetically. 57
When coinjected with Aβ peptide into rat brains, perlecan precipitates amyloid deposition. 58 However, it has not been shown that accumulation of basement membrane proteins precedes or triggers amyloid deposition in vivo. Our results support the hypothesis that basement membrane proteins initiate cerebrovascular amyloidosis in TGF-β1 mice (Figures 3 and 4) ▶ ▶ . Future studies will need to determine whether basement membrane proteins are necessary for amyloid deposition or fulfill only accessory functions. In addition, it will be important to identify which components of the basement membrane are the main culprits in the amyloidogenic process.
In older TGF-β1 mice, the accumulation of basement membrane proteins and amyloid was associated with degenerative changes in cerebrovascular cells (Figures 5–8) ▶ ▶ ▶ . Although we have not yet identified exactly how the early changes in the vascular wall relate to the degenerative changes in endothelial cells and pericytes, Aβ produced by neurons or vascular cells 59,60 may be trapped by the accumulating basement membrane proteins and injure vascular cells. Aβ causes degeneration of cultured pericytes, 61 endothelial cells, 62,63 and smooth muscle cells. 59 Moreover, human βPP/Aβ-overproducing mice show impaired cerebrovascular functions, although this seems not to be associated with permanent endothelial cell damage. 64 Thioflavin S-positive amyloid deposits in TGF-β1 mice correspond to electron-dense accumulations of Aβ-immunoreactive material located near or within the vascular basement membrane (Figure 2 ▶ and 29 ). Cerebrovascular amyloid deposits in human CAA are also located within the abluminal basement membrane. 65 At least part of the vascular cell degeneration in TGF-β1 mice could be attributed to these deposits. In many instances, however, thickened basement membranes or endothelial cells with morphological abnormalities were not associated with detectable electron-dense basement membrane inclusions in TGF-β1 mice. This suggests that the amyloid in these areas exists in a more soluble form and is not electron dense or that other factors unrelated to amyloid may cause the degenerative changes in these vascular cells. Recent studies support the concept that Aβ may be more toxic in soluble oligomeric form than in polymeric or fibrillar forms. 66
It is intriguing that TGF-β1 overexpression in astrocytes of transgenic mice throughout the brain 30 results in pathological changes that are largely restricted to the cerebrovasculature. One possible explanation is that cells in the cerebrovasculature express a unique TGF-β1 receptor or binding molecule that is not present in other CNS cells and that induces a vascular cell reaction or activation. Among other cells, perivascular astrocytes may become activated or hypertrophied and increase local production of endogenous and transgenic TGF-β1, thus feeding into a vicious cycle. A potential candidate TGF-β1–binding molecule is endoglin, which is expressed mainly in endothelial cells and potentiates TGF-β1 signaling. 67 It is interesting and of possible relevance to the effects of TGF-β1 on the cerebrovasculature that familial mutations in the endoglin gene have been linked to dominant vascular abnormalities in hereditary hemorrhagic telangiectasia type 1, 68 which causes telangiectases and aneurysms. Furthermore, polymorphisms in the endoglin gene have been associated with increased risk for sporadic intracerebral hemorrhage in humans. 69 Abnormal regulation of TGF-β1 signaling in the vasculature may be responsible for the development of hemorrhages in these human cases, and TGF-β1 may contribute similarly to hemorrhages in CAA.
In summary, our analysis of TGF-β1 mice has established a causal link between increased cerebral TGF-β1 production and several AD-like cerebrovascular abnormalities and, together with previous work, 30 has allowed us to establish a temporal relationship among these abnormalities. Chronic overproduction of TGF-β1 in the CNS caused a perivascular astrocytosis, followed by an excessive accumulation of basement membrane proteins and the formation of amyloid deposits. These alterations were associated with the development of degenerative changes in cerebral microvessels. The similarity of the pathological vascular changes in older TGF-β1 mice and in humans with AD and CAA suggests a pathogenic role of TGF-β1 in these human conditions.
Acknowledgments
We thank Eric Huang for helpful comments on the manuscript, Dale Newland for technical assistance with electron microscopy, Stella Sien for quantitative morphometry, Gary Howard and Stephen Ordway for editorial assistance, Maria Martinez for secretarial help, and John C. W. Carroll, Neile Shea, and Stephen Gonzales for graphics and photography.
Footnotes
Address reprint requests to Tony Wyss-Coray, Ph.D., Gladstone Institute of Neurological Disease, P.O. Box 419100, San Francisco, CA 94141-9100. E-mail: twysscoray@gladstone.ucsf.edu.
Supported in part by National Institutes of Health grants AG-15871 (to T. W.-C.), AG-5131 (to E. M.), AG-10689 (to T. W.-C. and E. M.), and AG-11385 (to L. M.) and by the Alzheimer’s Association (to T. W.-C. and E. M.).
References
- 1.Kalaria RN: Cerebral vessels in ageing and Alzheimer’s disease. Pharmacol Ther 1996, 72:193-214 [DOI] [PubMed] [Google Scholar]
- 2.Vinters HV: Cerebral amyloid angiopathy and Alzheimer’s disease: two entities or one? J Neurol Sci 1992, 112:1-3 [DOI] [PubMed] [Google Scholar]
- 3.Plassman BL, Breitner JCS: Recent advances in the genetics of Alzheimer’s disease and vascular dementia with an emphasis on gene-environment interactions. J Am Geriatr Soc 1996, 44:1242-1250 [DOI] [PubMed] [Google Scholar]
- 4.Verbeek MM, Eikelenboom P, De Waal RMW: Differences between the pathogenesis of senile plaques and congophilic angiopathy in Alzheimer disease. J Neuropathol Exp Neurol 1997, 56:751-761 [PubMed] [Google Scholar]
- 5.Slooter AJC, Tang MX, Van Duijn CM, Stern Y, Ott A, Bell K, Breteler MMB, Van Broeckhoven C, Tatemichi TK, Tycko B, Hofman A, Mayeux R: Apolipoprotein E (ε4), and the risk of dementia with stroke: a population-based investigation. J Am Med Assoc 1997, 277:818-821 [DOI] [PubMed] [Google Scholar]
- 6.Snowdon DA, Greiner LH, Mortimer JA, Riley KP, Greiner PA, Markesbery WR: Brain infarction and the clinical expression of Alzheimer disease: the nun study. J Am Med Assoc 1997, 277:813-817 [PubMed] [Google Scholar]
- 7.Hassler O: Vascular changes in senile brains: a micro-angiographic study. Acta Neuropathol 1965, 5:40-53 [DOI] [PubMed] [Google Scholar]
- 8.Vinters HV, Wang ZZ, Secor DL: Brain parenchymal and microvascular amyloid in Alzheimer’s disease. Brain Pathol 1996, 6:179-195 [DOI] [PubMed] [Google Scholar]
- 9.Vinters HV: Cerebral amyloid angiopathy: a critical review. Stroke 1987, 18:311-324 [DOI] [PubMed] [Google Scholar]
- 10.Haan J, Maat-Schieman MLC, Roos RAC: Clinical aspects of cerebral amyloid angiopathy. Dementia 1994, 5:210-213 [DOI] [PubMed] [Google Scholar]
- 11.Checler F: Processing of the β-amyloid precursor protein and its regulation in Alzheimer’s disease. J Neurochem 1995, 65:1431-1444 [DOI] [PubMed] [Google Scholar]
- 12.Itoh Y, Yamada M, Hayakawa M, Otomo E, Miyatake T: Cerebral amyloid angiopathy: a significant cause of cerebellar as well as lobar cerebral hemorrhage in the elderly. J Neurol Sci 1993, 116:135-141 [DOI] [PubMed] [Google Scholar]
- 13.Greenberg SM, Vonsattel JP, Stakes JW, Gruber M, Finklestein SP: The clinical spectrum of cerebral amyloid angiopathy: presentations without lobar hemorrhage. Neurology 1993, 43:2073-2079 [DOI] [PubMed] [Google Scholar]
- 14.Haan J, Lanser JBK, Zijderveld I, van der Does IGF, Roos RAC: Dementia in hereditary cerebral hemorrhage with amyloidosis-Dutch type. Arch Neurol 1990, 47:965-967 [DOI] [PubMed] [Google Scholar]
- 15.Petersen RB, Goren H, Cohen M, Richardson SL, Tresser N, Lynn A, Gali M, Estes M, Gambetti P: Transthyretin amyloidosis: a new mutation associated with dementia. Ann Neurol 1997, 41:307-313 [DOI] [PubMed] [Google Scholar]
- 16.Vinters HV, Secor DL, Read SL, Frazee JG, Tomiyasu U, Stanley TM, Ferreiro JA, Akers M-A: Microvasculature in brain biopsy specimens from patients with Alzheimer’s disease: an immunohistochemical and ultrastructual study. Ultrastruct Pathol 1994, 18:333-348 [DOI] [PubMed] [Google Scholar]
- 17.Wisniewski HM, Wegiel J, Wang KC, Lach B: Ultrastructural studies of the cells forming amyloid in the cortical vessel wall in Alzheimer’s disease. Acta Neuropathol 1992, 84:117-127 [DOI] [PubMed] [Google Scholar]
- 18.Mancardi GL, Perdelli F, Rivano C, Leonardi A, Bugiani O: Thickening of the basement membrane of cortical capillaries in Alzheimer’s disease. Acta Neuropathol 1980, 49:79-83 [DOI] [PubMed] [Google Scholar]
- 19.Perlmutter LS, Chui HC, Saperia D, Athanikar J: Microangiopathy and the colocalization of heparan sulfate proteoglycan with amyloid in senile plaques of Alzheimer’s disease. Brain Res 1990, 508:13-19 [DOI] [PubMed] [Google Scholar]
- 20.Stewart PA, Hayakawa K, Akers MA, Vinters HV: A morphometric study of the blood-brain barrier in Alzheimer’s disease. Lab Invest 1992, 67:734-742 [PubMed] [Google Scholar]
- 21.Border WA, Noble NA: TGF-β in kidney fibrosis: a target for gene therapy. Kidney Int 1997, 51:1388-1396 [DOI] [PubMed] [Google Scholar]
- 22.Pepper MS: Transforming growth factor-β: vasculogenesis, angiogenesis, and vessel wall integrity. Cytokine Growth Factor Rev 1997, 8:21-43 [DOI] [PubMed] [Google Scholar]
- 23.Madri JA, Bell L, Merwin JR: Modulation of vascular cell behavior by transforming growth factors β. Mol Reprod Dev 1992, 32:121-126 [DOI] [PubMed] [Google Scholar]
- 24.Finch CE, Laping NJ, Morgan TE, Nichols NR, Pasinetti GM: TGF-β1 is an organizer of responses to neurodegeneration. J Cell Biochem 1993, 53:314-322 [DOI] [PubMed] [Google Scholar]
- 25.van der Wal EA, Gómez-Pinilla F, Cotman CW: Transforming growth factor-β1 is in plaques in Alzheimer and Down pathologies. Neuroreport 1993, 4:69-72 [DOI] [PubMed] [Google Scholar]
- 26.Flanders KC, Lippa CF, Smith TW, Pollen DA, Sporn MB: Altered expression of transforming growth factor-β in Alzheimer’s disease. Neurology 1995, 45:1561-1569 [DOI] [PubMed] [Google Scholar]
- 27.Chao CC, Hu S, Frey WH, Ala TA, Tourtellotte WW, Peterson PK: Transforming growth factor β in Alzheimer’s disease. Clin Diag Lab Immunol 1994, 1:109-110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chao CC, Ala TA, Hu S, Crossley KB, Sherman RE, Peterson PK, Frey WH, II: Serum cytokine levels in patients with Alzheimer’s disease. Clin Diag Lab Immunol 1994, 1:433-436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wyss-Coray T, Masliah E, Mallory M, McConlogue L, Johnson-Wood K, Lin C, Mucke L: Amyloidogenic role of cytokine TGF-β1 in transgenic mice and Alzheimer’s disease. Nature 1997, 389:603-606 [DOI] [PubMed] [Google Scholar]
- 30.Wyss-Coray T, Feng L, Masliah E, Ruppe MD, Lee HS, Toggas SM, Rockenstein EM, Mucke L: Increased central nervous system production of extracellular matrix components and development of hydrocephalus in transgenic mice overexpressing transforming growth factor-β1. Am J Pathol 1995, 147:53-67 [PMC free article] [PubMed] [Google Scholar]
- 31.Frautschy SA, Yang F, Calderón L, Cole GM: Rodent models of Alzheimer’s disease: rat Aβ infusion approaches to amyloid deposits. Neurobiol Aging 1996, 17:311-321 [DOI] [PubMed] [Google Scholar]
- 32.Harris-White ME, Chu T, Balverde Z, Sigel JJ, Flanders KC, Frautschy SA: Effects of transforming growth factor-β (isoforms 1–3) on amyloid-β deposition, inflammation, and cell targeting in organotypic hippocampal slice cultures. J Neurosci 1998, 18:10366-10374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mazur-Kolecka B, Frackowiak J, Haskea T, Le Vine H, Krzeslowska J, Golabek A, Wisniewski HM: TGFβ potentiates the intracellular aggregation of Aβ in response to apoE: a possible mechanism to form nuclei of Aβ polymerization. Neurobiol. Aging 1998, 19:S130 [Google Scholar]
- 34.Prior R, Urmoneit B, Prikulis I, Whil G, Fischer J, Frank R, D’Urso D: Alzheimer amyloid angiopathy and transforming growth factor β1. Neurobiol. Aging 1998, 19:S113
- 35.Wyss-Coray T, Borrow P, Brooker MJ, Mucke L: Astroglial overproduction of TGF-β1 enhances inflammatory central nervous system disease in transgenic mice. J Neuroimmunol 1997, 77:45-50 [DOI] [PubMed] [Google Scholar]
- 36.Masliah E, Achim CL, Ge N, DeTeresa R, Terry RD, Wiley CA: Spectrum of human immunodeficiency virus-associated neocortical damage. Ann Neurol 1992, 32:321-329 [DOI] [PubMed] [Google Scholar]
- 37.Olichney JM, Hansen LA, Galasko D, Saitoh T, Hofstetter CR, Katzman R, Thal LJ: The apolipoprotein E epsilon 4 allele is associated with increased neuritic plaques and cerebral amyloid angiopathy in Alzheimer’s disease and Lewy body variant. Neurology 1996, 47:190-196 [DOI] [PubMed] [Google Scholar]
- 38.Pardridge WM, Eisenberg J, Yamada T: Rapid sequestration and degradation of somatostatin analogues by isolated brain microvessels. J Neurochem 1985, 44:1178-1184 [DOI] [PubMed] [Google Scholar]
- 39.Vannucci SJ, Clark RR, Koehler-Stec E, Li K, Smith CB, Davies P, Maher F, Simpson IA: Glucose transporter expression in brain: relationship to cerebral glucose utilization. Dev Neurosci 1998, 20:369-379 [DOI] [PubMed] [Google Scholar]
- 40.Kacem K, Lacombe P, Seylaz J, Bonvento G: Structural organization of the perivascular astrocyte endfeet and their relationship with the endothelial glucose transporter: a confocal microscopy study. Glia 1998, 23:1-10 [PubMed] [Google Scholar]
- 41.Bordonaro M, Saccomanno CF, Nordstrom JL: An improved T1/A ribonuclease protection assay. BioTechniques 1994, 16:428-430 [PubMed] [Google Scholar]
- 42.Sambrook J, Fritsch EF, Maniatis T (Eds): Molecular Cloning: A Laboratory Manual, ed 2. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory, 1989
- 43.Kim KJ, Abrams J, Alphonso M, Pearce M, Thorbecke GJ, Palladino MA: Role of endogenously produced interleukin-6 as a second signal in murine thymocyte proliferation induced by multiple cytokines: regulatory effects of transforming growth factor-β. Cell Immunol 1990, 131:261-271 [DOI] [PubMed] [Google Scholar]
- 44.Kim S-J, Angel P, Lafyatis R, Hattori K, Kim KY, Sporn MB, Karin M, Roberts AB: Autoinduction of transforming growth factor β1 is mediated by the AP-1 complex. Mol Cell Biol 1990, 10:1492-1497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kisilevsky R, Fraser PE: Aβ amyloidogenesis: unique, or variation on a systemic theme? Crit Rev Biochem Mol Biol 1997, 32:361-404 [DOI] [PubMed] [Google Scholar]
- 46.Fillit H, Leveugle B: Disorders of the extracellular matrix and the pathogenesis of senile dementia of the Alzheimer’s type. Lab Invest 1995, 72:249-253 [PubMed] [Google Scholar]
- 47.Martinez-Hernandez A, Amenta PS: The basement membrane in pathology. Lab Invest 1983, 48:656-677 [PubMed] [Google Scholar]
- 48.Wyss-Coray T, Lin C, Von Euw D, Masliah E, Mucke L, Lacombe P: Alzheimer’s disease-like cerebrovascular pathology in transforming growth factor β1 transgenic mice and functional metabolic correlates. Ann NY Acad Sci 2000, In press [DOI] [PubMed]
- 49.Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F, Guido T, Hagopian S, Johnson-Wood K, Khan K, Lee M, Leibowitz P, Lieberburg I, Little S, Masliah E, McConlogue L, Montoya-Zavala M, Mucke L, Paganini L, Penniman E, Power M, Schenk D, Seubert P, Snyder B, Soriano F, Tan H, Vitale J, Wadsworth S, Wolozin B, Zhao J: Alzheimer-type neuropathology in transgenic mice overexpressing V717F β-amyloid precursor protein. Nature 1995, 373:523-527 [DOI] [PubMed] [Google Scholar]
- 50.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang FS, Cole G: Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science 1996, 274:99-102 [DOI] [PubMed] [Google Scholar]
- 51.Sturchler-Pierrat C, Abramowski D, Duke M, Wiederhold KH, Mistl C, Rothacher S, Ledermann B, Bürki K, Frey P, Paganetti PA, Waridel C, Calhoun ME, Jucker M, Probst A, Staufenbiel M, Sommer B: Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci USA 1997, 94:13287-13292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Border WA, Ruoslahti E: Transforming growth factor-β in disease: the dark side of tissue repair. J Clin Invest 1992, 90:1-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ignotz RA, Massagué J: Transforming growth factor-β stimulates the expression of fibronectin and collagen and their incorporation into the extracellular matrix. J Biol Chem 1986, 261:4337-4345 [PubMed] [Google Scholar]
- 54.Border WA, Okuda S, Languino LR, Ruoslahti E: Transforming growth factor-β regulates production of proteoglycans by mesangial cells. Kidney Int 1990, 37:689-695 [DOI] [PubMed] [Google Scholar]
- 55.Pierce DF, Jr, Johnson MD, Matsui Y, Robinson SD, Gold LI, Purchio AF, Daniel CW, Hogan BLM, Moses HL: Inhibition of mammary duct development but not alveolar outgrowth during pregnancy in transgenic mice expressing active TGF-β1. Genes Dev 1993, 7:2308-2317 [DOI] [PubMed] [Google Scholar]
- 56.Lee M-S, Gu D, Feng L, Curriden S, Arnush M, Krahl T, Gurushanthaiah D, Wilson C, Loskutoff DL, Fox H, Sarvetnick N: Accumulation of extracellular matrix and developmental dysregulation in the pancreas by transgenic production of transforming growth factor-β1. Am J Pathol 1995, 147:42-52 [PMC free article] [PubMed] [Google Scholar]
- 57.Grainger DJ, Heathcote K, Chiano M, Sneider H, Kemp PR, Metcalfe JC, Carter ND, Spector TD: Genetic control of the circulating concentration of transforming growth factor type β1. Hum Mol Genet 1999, 8:93-97 [DOI] [PubMed] [Google Scholar]
- 58.Snow AD, Sekiguchi R, Nochlin D, Fraser P, Kimata K, Mizutani A, Arai M, Schreier WA, Morgan DG: An important role of heparan sulfate proteoglycan (perlecan) in a model system for the deposition and persistence of fibrillar Aβ-amyloid in rat brain. Neuron 1994, 12:219-234 [DOI] [PubMed] [Google Scholar]
- 59.Davis-Salinas J, Saporito-Irwin SM, Cotman CW, Van Nostrand WE: Amyloid β-protein induces its own production in cultured degenerating cerebrovascular smooth muscle cells. J Neurochem 1995, 65:931-934 [DOI] [PubMed] [Google Scholar]
- 60.Wisniewski HM, Frackowiak J, Mazur-Kolecka B: In vitro production of β-amyloid in smooth muscle cells isolated from amyloid angiopathy-affected vessels: Neurosci Lett 1995, 183:120–123 [DOI] [PubMed]
- 61.Verbeek MM, de Waal RMW, Schipper JJ, Van Nostrand WE: Rapid degeneration of cultured human brain pericytes by amyloid β protein. J Neurochem 1997, 68:1135-1141 [DOI] [PubMed] [Google Scholar]
- 62.Thomas T, Thomas G, McLendon C, Sutton T, Mullan M: Beta-amyloid-mediated vasoactivity and vascular endothelial damage. Nature 1996, 380:168-171 [DOI] [PubMed] [Google Scholar]
- 63.Blanc EM, Toborek M, Mark RJ, Hennig B, Mattson MP: Amyloid β-peptide induces cell monolayer albumin permeability, impairs glucose transport, and induces apoptosis in vascular endothelial cells. J Neurochem 1997, 68:1870-1881 [DOI] [PubMed] [Google Scholar]
- 64.Iadecola C, Zhang F, Niwa K, Eckman C, Turner SK, Fischer E, Younkin S, Borchelt DR, Hsiao KK, Carlson GA: SOD 1 rescues cerebral endothelial dysfunction in mice overexpressing amyloid precursor protein: Nat Neurosci 1999, 2:157–161 [DOI] [PubMed]
- 65.Yamaguchi H, Yamazaki T, Lemere CA, Frosch MP, Selkoe DJ: Beta amyloid is focally deposited within the outer basement membrane in the amyloid angiopathy of Alzheimer’s Disease: an immunoelectron microscopic study. Am J Pathol 1992, 141:249-259 [PMC free article] [PubMed] [Google Scholar]
- 66.Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL: Diffusible, nonfibrillar ligands derived from Aβ1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA 1998, 95:6448-6453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cheifetz S, Bellón T, Calés C, Vera S, Bernabeu C, Massagué J, Letarte M: Endoglin is a component of the transforming growth factor-β receptor system in human endothelial cells. J Biol Chem 1992, 267:19027-19030 [PubMed] [Google Scholar]
- 68.McAllister KA, Grogg KM, Johnson DW, Gallione CJ, Baldwin MA, Jackson CE, Helmbold EA, Markel DS, McKinnon WC, Murrell J, McCormick MK, Pericak-Vance MA, Heutink P, Oostra BA, Haitjema T, Westerman CJJ, Porteous ME, Guttmacher AE, Letarte M, Marchuk DA: Endoglin, a TGF-β binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat Genet 1994, 8:345-351 [DOI] [PubMed] [Google Scholar]
- 69.Alberts MJ, Davis JP, Graffagnino C, McClenny C, Delong D, Granger C, Herbstreith MH, Boteva K, Marchuk DA, Roses AD: Endoglin gene polymorphism as a risk factor for sporadic intracerebral hemorrhage. Ann Neurol 1997, 41:683-686 [DOI] [PubMed] [Google Scholar]