

Summary
To investigate the α-synuclein protein and its role in Parkinson’s disease, we screened a library of random point mutants both in vitro and in yeast to find variants in an unbiased way that could help us understand the sequence-phenotype relationship. We developed a rapid purification method that allowed us to screen 59 synuclein mutants in vitro and discovered two double point mutants that fibrillized slowly relative to wild type, A30P, and A53T α-synucleins. The yeast toxicity of all of these proteins was measured and we found no correlation with fibrillization rate, suggesting that fibrillization is not necessary for synuclein-induced yeast toxicity. We also found that β-synuclein was of intermediate toxicity to yeast and γ-synuclein was non-toxic. Coexpression of Parkinson’s disease related genes DJ-1, parkin, Pink1, UCH-L1, or synphilin, with synuclein, did not affect synuclein toxicity. A second screen, of several thousand library clones in yeast, identified 25 non-toxic α-synuclein sequence variants. Most of these contained a mutation to either proline or glutamic acid that caused a defect in membrane binding. We hypothesize that yeast toxicity is caused by synuclein binding directly to membranes at levels sufficient to non-specifically disrupt homeostasis.
Keywords: synuclein, Parkinson’s disease, fibrillization, membrane binding, yeast toxicity
Introduction
The α-synuclein protein can cause Parkinson’s disease (PD) if it is overexpressed or contains one of the mutations A53T, A30P, or E46K.1 The events leading from α-synuclein expression to the degeneration of dopaminergic neurons, which causes the symptoms of PD, are unknown. In physiological buffer solutions, purified monomeric α-synuclein aggregates into amyloid fibrils.2 The fibrillar form of α-synuclein is the main component of Lewy bodies,3 the characteristic inclusions of PD brain. Monomeric α-synuclein binds negatively charged phospholipid membranes and micelles in vitro, and in doing so takes on significant N-terminal amphipathic α-helical structure.4–6 The A30P mutation inhibits membrane binding by interfering with the α-helix conformation.7 There are two homologs of α-synuclein (β and γ) and both also exhibit α-helical induction upon membrane binding.8 γ-Synuclein fibrillizes more slowly than α, and β-synuclein is much slower to fibrillize than either α or γ.9
A number of model systems in which to study synuclein behavior and possibly the pathogenic events of PD have been described, ranging in complexity from in vitro to yeast, cell culture, worm, fly, mouse, and primate.1,10 In this article we describe screening of an α-synuclein random point mutagenesis library11 in vitro (fibrillization) and in yeast (toxicity), for sequences that behave differently from the wild type protein. The probability of finding an interesting sequence when screening a random mutagenesis library is low, and this makes it impractical to directly screen in most of the more complex and desirable model systems, because the effective throughput is not high enough. One solution to this problem is to “prescreen” in a simple model system such as those described here and then use the selected clones as a very small “library”, for screening in an animal model. The members of such a “library” have a much higher probability of causing an interesting and useful phenotype than blindly chosen library clones.
An alternative to random library screening is to examine a few rationally designed mutants that are expected to yield an interesting phenotype with high probability. This approach is certainly worthwhile12–16 but rational design is restricted and biased in scope by our limited preexisting knowledge17 of α-synuclein structure and biology. Another alternative is to examine natural sequence variants (A53T, A30P, mouse synucleins, etc.), but of course, the pool of natural sequences is limited. Furthermore, variants with extreme properties are unlikely to occur in nature (they would probably be strongly selected against evolutionarily), though at our current state of understanding, extreme variants are more likely to be informative and useful than variants with properties only subtly different from the wt protein.
Our in vitro system consists of purified monomeric α-synuclein in physiological buffer, and fibrillization is measured over time. We are using the yeast Saccharomyces cerevisiae as an in vivo model system, which exhibits toxicity (defined here as slowed growth) and α-synuclein inclusion formation.18,19 Wild type and A53T α-synuclein are both toxic and localize to inclusions and the plasma membrane when expressed from multiple GAL promoters, such as those on 2-micron plasmids (high copy number) or from multiple integrated copies.18,20,21 In contrast, the A30P variant is non-toxic to yeast and is uniformly distributed throughout the cytoplasm.18 All of the synuclein proteins are relatively non-toxic when expressed from a single copy.18 We screened for non-toxic α-synuclein sequences (2-micron plasmid, high copy number expression) and for highly-toxic sequences (centromeric plasmid, low copy number). We were able to screen thousands of sequences in the yeast system, and approximately 60 in the in vitro system. The bottleneck in the former is replica plating, and in the latter, purification and preparation of individual proteins.
The mutations in selected clones cause their altered phenotypes. We used this relationship and our prior knowledge to infer and further test how synuclein functions in these model systems. Examining the selected clones in additional model systems can also yield suggestive results by correlation (or lack thereof). For example, we examined slow fibrillizing mutants from the in vitro screen, in the yeast system, and the results demonstrated that fibrillization is unlikely to be necessary for yeast toxicity. In the future, it will be interesting to examine the sequence variants described here in more complex and realistic PD models.
Results and Discussion
Initial in vitro screen for altered fibrillization rate
Randomly chosen library clones were expressed (131 total), and sufficient protein for screening at at least the 200 μM level was obtained from 59 (52 of these also yielded enough protein for a 400 μM incubation); nonsense mutations, frame-shifts, and improperly ligated inserts account for most of the remainder. Eleven of these sequences turned out to have a wt protein sequence (for reference, the wt α-synuclein protein sequence and its domains are shown at the top of Fig. 4). The in vitro thioflavin T aggregation screen revealed several sequence variants that had increased fibrillization lag times. These candidates were reexamined in secondary trials, finally narrowing the candidates to two double point mutants, both appearing to have a robust increase in fibrillization lag time: V49E/Q79R and A29E/T92S.
Figure 4.
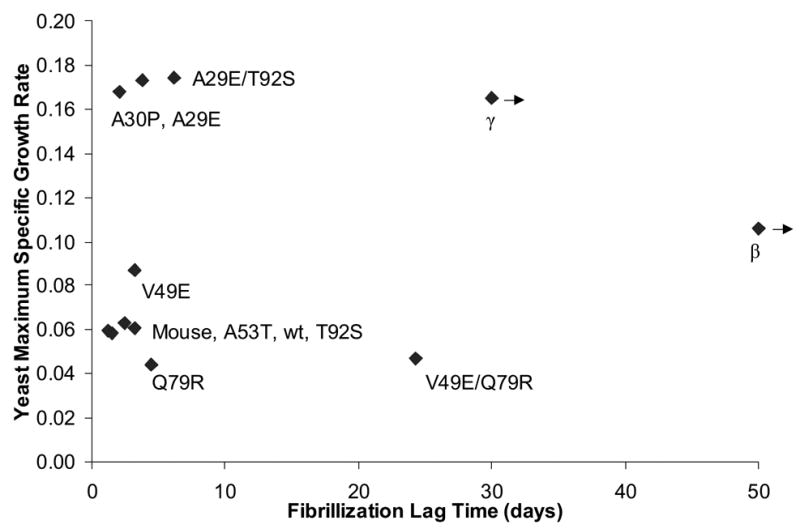
Scatter plot of estimated maximum specific growth rate (data from Fig. 2) versus in vitro synuclein fibrillization lag time (data from Fig. 1), for a number of synuclein variants (labeled). Left-pointing arrows are shown for β and γ-synuclein to indicate that their true (unknown) lag times are actually greater than the value shown; in the case of β-synuclein, the true value is much greater.
No mutants with significantly reduced lag times relative to wt were isolated in this study. Several factors in addition to low-throughput may contribute: fibrillization occurring before or during purification could cause loss of the protein. Our in vitro system itself is less sensitive in detecting fast fibrillizers (control proteins fibrillize within the first few days). Finally, mutations enhancing fibrillization could be less prevalent in a random mutagenesis library than disruptive mutations; there are typically many more ways to disrupt a specific process than to facilitate it.
Detailed kinetic studies of slow fibrillizing sequence variants and their single point mutant derivatives
A series of independent fibrillization time courses were carried out to statistically evaluate the two double point mutants (Fig. 1A). As expected from the initial screening procedures, they were both slow to fibrillize (Fig. 1A) and their soluble concentrations remained elevated (Fig. 1B), compared to the wt protein. β-Synuclein did not fibrillize under these conditions, and only 1 of 4 γ-synuclein incubations fibrillized, within the 30 days of observation (data not shown), in agreement with previous studies.9,22 Six of 8 of the V49E/Q79R samples fibrillized within the 30 days, and every sample of every other protein examined (Fig. 1) fibrillized within 8 days.
Figure 1.
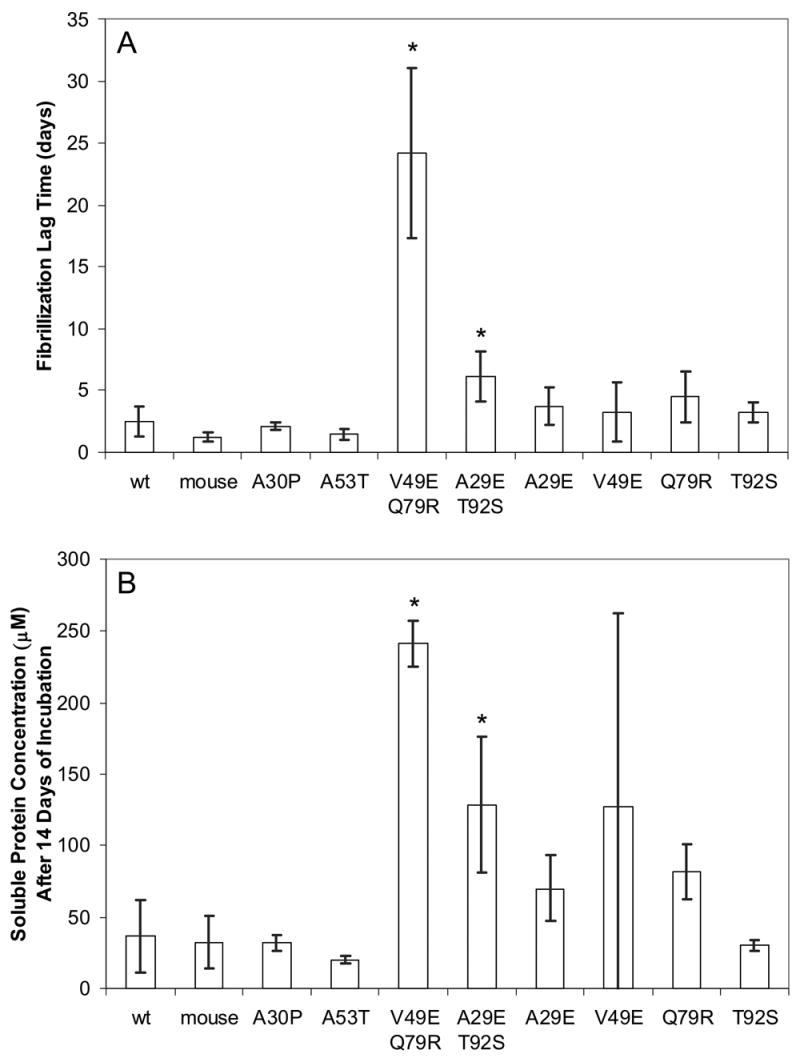
Fibrillization of control α-synuclein sequences, two double point mutant sequence variants, and their single point mutant derivatives. Each bar represents the mean of eight (except single point mutants, where n = 4) trials of independently expressed, purified, and incubated proteins, and the error bars are individual 95% confidence intervals. Single point mutants were created from the double point mutants by subcloning with the aid of enzyme BseRI. (A) Lag time is defined by the crossing of a thioflavin T fluorescence threshold as described in the methods. In 2 of 8 V49E/Q79R incubations, a positive thioflavin T signal had not occurred within the duration of the experiment. In these cases, the day of the final measurement was used as the lag time (32 days). Therefore the value shown for this variant may be somewhat underestimated. (B) Soluble protein concentrations measured after 14 days of incubation. Every non-V49E/Q79R sample had fibrillized within 8 days. Using a pre-planned t-test on the data of (A) or (B), we were able to reject the null hypothesis of no difference in lag time between the wild type protein and both V49E/Q79R and A29E/T92S with a p-value < 0.01 (indicated by an asterisk). A Games-Howell (variance is non-constant; reference51) multiple comparison procedure of (A) did not detect a significant difference between the four derived single point mutants and the wt protein or A29E/T92S. However, comparison of V49E/Q79R with any of the other proteins resulted in rejection of the hypothesis of no difference. Applying the Games-Howell test to the data in (B) gave similar results but was somewhat more liberal in rejecting the null hypothesis.
In addition, the four single point mutants (A29E, V49E, Q79R, T92S) derived from V49E/Q79R and A29E/T92S were studied in order to discover whether a single one of the two point mutations could in either case be responsible for the slow fibrillization (Fig. 1). The delay in fibrillization lag time of double point mutant V49E/Q79R was not due to either one of the mutations acting alone (Fig. 1A). For the mutant A29E/T92S, we can not draw the same conclusion definitively, although the data suggest that this may be the case: both single point mutants showed delayed fibrillization, and neither one was delayed as much as the double point mutant.
Slow fibrillizing variants of α-synuclein have been created before, by rationally introducing hydrophilic point mutations into the center of the hydrophobic NAC region, including mutants A76E and A76R,13 G68R and V74R,12 and V70T/V71T.14 Our Q79R mutation falls within this region and probably acts in the same way. We are unsure how the other three mutations might act, but they do indicate that the hydrophobic core of NAC is not the sole determinant of fibrillization rate. An advantage of random screening is shown here- these three mutations could not have been rationally designed. Similarly, Wurth et al. screened an unbiased library of Aβ peptides, and found mutations slowing aggregation rate that could not have been rationally predicted.17
Synuclein sequences were expressed in a yeast model of Parkinson’s disease
In agreement with previous studies,18,21 we found that wt and A53T but not A30P α-synuclein is toxic to yeast when expressed at high levels (Fig. 2, labels shaded blue). We also found, in agreement with previous work,18,21 that the GFP fusion proteins of wt and A53T form inclusions and localize to the plasma membrane in yeast whereas A30P α-synuclein does not form inclusions, and is predominantly cytoplasmic (Fig. 3 and Supplementary Fig. 1). We extend these results by showing (Figs. 2, 3, and Supplementary Fig. 1) that γ-synuclein, like A30P, was non-toxic and γ-synuclein-GFP mainly localized to the cytoplasm and formed inclusions only rarely. β-Synuclein is estimated to be intermediate in toxicity between wt and A30P, and its GFP fusion protein formed inclusions. Mouse α-synuclein behaved like human wt α-synuclein. The N-terminal 99 amino acids of synuclein were sufficient to produce toxicity at levels comparable to the wt protein (Fig. 2), implying that α-synuclein toxicity is not mediated by the C-terminus. The tendency of wt α, truncated α, A30P, A53T, β, and γ-synucleins to form inclusions in yeast (Fig. 3) is well correlated with their ability to bind lipid droplets in lipid loaded mammalian cells.23
Figure 2.
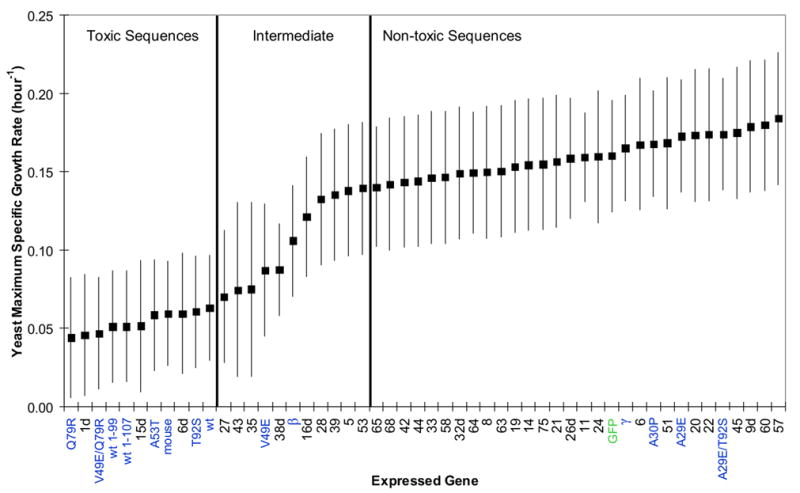
Estimated maximum specific growth rates (proportional to the inverse of doubling time) of the yeast strain W303-1a expressing various synuclein genes or GFP (labeled in green) from 2-micron plasmid p426GAL1. All genes isolated by yeast library screening are labeled numerically; those originally isolated from the yeast screen with co-expressed wt α-synuclein have numbers with the suffix “d”. All other synuclein sequences (those not isolated by yeast screening) have descriptive labels, shaded blue. The synuclein genes were not GFP tagged and all of the synuclein cDNAs were contained in the plasmid within the same non-coding nucleotide sequence context (except wt 1-107, whose C-terminal nucleotides after the stop codon were deleted). The genes are listed on the x-axis in order of decreasing estimated yeast toxicity. Each gene was studied in independent trials (n was typically about 6), independent meaning that a new trial was begun from a different colony picked from the same transformation. An additive 2-way ANOVA model (synuclein genes and blocks) was used for analysis of the maximum specific growth rates obtained by fitting the Gompertz equation. The square points are estimated population marginal means. In 2 of 306 samples sparse data led to a fit near 1.0, which was unreasonably high, and these outliers were set to a more reasonable upper limit of 0.3 to avoid disturbing the statistical analysis. Error bars represent simultaneous Tukey-Kramer comparison intervals: non-overlap between the bars of any two genes indicates that the hypothesis of no difference between the two was rejected at the p<0.05 level. Two vertical bars divide the sequences into three groups: “toxic”, “intermediate”, and “non-toxic”. Every sequence in the non-toxic group is significantly less toxic to yeast than every sequence in the toxic group. Sequences in the intermediate group have comparison intervals that overlap some members of both the toxic and non-toxic sets.
Figure 3.
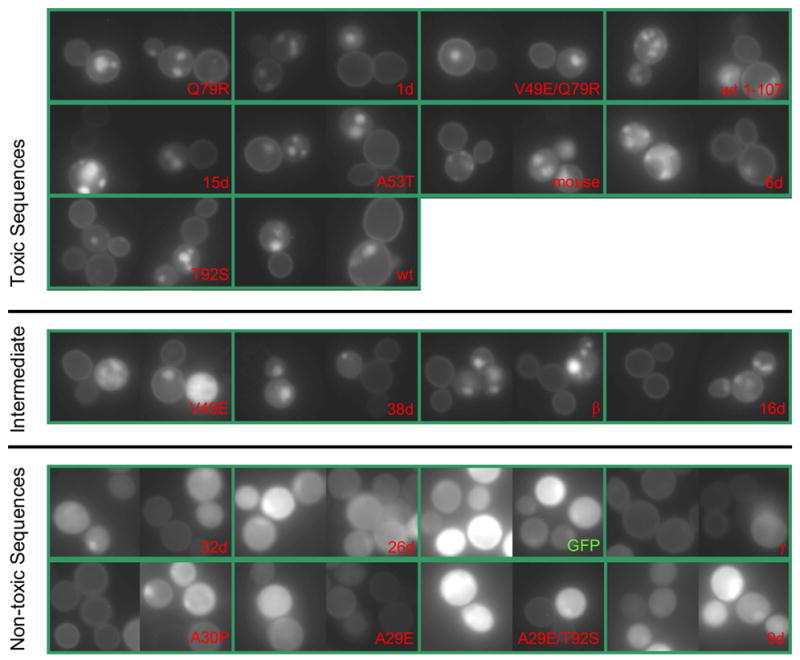
Microscopy of GFP tagged synucleins. Two horizontal bars divide the synuclein variants into three toxicity groups, as defined in Fig. 2. Photographs were taken after 48 hours of growth in synthetic galactose medium lacking uracil, and all were taken with identical excitation intensity and exposure time (100× plan fluor objective; a Gaussian filter was applied to the images; the levels and gamma setting were adjusted identically for all images). Each green box contains two different representative photographic fields and the labels correspond with those used elsewhere in the manuscript. The expression plasmid was p426GAL1 with GFP (the linkage between the last amino acid of synuclein and GFP is described in the methods section, and was identical for α, β, and γ synucleins); synuclein cDNAs were inserted into the plasmid by in vitro ligation.
The broad distribution of 2-micron copy number within a yeast population causes variation in expression level among cells. When a 2-micron plasmid bears a toxic synuclein, cells that happen to have fewer plasmid copies (and therefore express less) selectively propagate. The result is that yeast cultures with toxic synucleins express lower levels of synuclein than those with non-toxic synucleins (Fig. 3, Supplementary Fig. 1, and reference18). This decreases the difference in growth rate between toxic and non-toxic synucleins, compared to what would be observed if a fixed number of copies (≥ 2) were used.18 Thus, our 2-micron assay has less resolution to detect significant differences between toxic and non-toxic species than an assay using multiple integrated copies of synuclein. Despite this limitation, the data collected clearly supported the conclusions below. In compensation for this decreased power, the 2-micron plasmid offers much greater practicality for multicopy expression of a large number of variants.
Interaction of synuclein with PD-related genes
We performed coexpression studies of PD-related genes (DJ-1, parkin, Pink1, UCH-L1, synphilin, or control GFP) and various synuclein sequences to attempt to discover any interactions in the yeast model. For example, in cell culture24 and a rat model of PD,25 parkin overexpression can suppress synuclein toxicity. No obvious and significant interactions either promoting or suppressing synuclein toxicity in yeast were detected in any case (Supplementary Fig. 2), although synphilin itself was somewhat toxic.
Are fibrils necessary for synuclein induced yeast toxicity?
Based on the correlation of Lewy bodies with disease, the aggregation of α-synuclein in vitro, and the observed microscopic inclusions in yeast, one reasonable working hypothesis is that fibrillized synuclein is necessary for yeast toxicity. We measured the yeast toxicity of the slow fibrillizing variants A29E/T92S and V49E/Q79R and their single point mutant derivatives (Fig. 2, labels shaded blue). A scatter plot of fibrillization rate and yeast toxicity for a number of variants is shown in Fig. 4. Whether these data bear on the toxic-fibril hypothesis depends on the relationship between our in vitro fibrillization studies and fibrillization in yeast. To begin with, we must assume a monotonic relationship: slow fibrillizers in vitro are also slow to fibrillize in yeast and those that are faster in vitro are also faster in yeast. Additionally, we have to consider time-scale relevance- is the time scale of fibrillization that we worked with in vitro, relevant to the time scale of fibrillization in yeast? We can roughly divide this into three possible cases and consider the fibril-toxicity hypothesis for each:
Fibrillization is very slow in yeast relative to in vitro. Then, fibrils are not relevant to toxicity; the hypothesis of fibril-induced yeast toxicity is untrue.
Fibrillization in yeast is extremely rapid relative to our in vitro experiments. That is, all of our synuclein proteins actually fibrillize immediately in yeast, compressing the in vitro differences we found into irrelevance. Then, our data do not bear on the hypothesis (in this case differences in toxicity between variants are explained by downstream necessary events affected by the mutations). This seems unlikely: β-synuclein is extremely non-fibrillogenic9 yet exhibited some toxicity to yeast (Fig. 2). It would also imply that the non-toxic protein-GFP fusions such as A30P-GFP are actually fibrillar protein dispersed throughout the cytoplasm. That abundant monomeric synuclein can be purified from Escherichia coli overexpressing the protein also speaks against this.
Fibrillization occurs on a similar time scale as the yeast growth measurements. Then our data (Fig. 4) do not support the toxic-fibril hypothesis because they include V49E/Q79R, which is a toxic, slow fibrillizer. Additionally, β-synuclein is almost non-fibrillogenic yet caused some toxicity to yeast. It would be unlikely that both of these sequences act by a new atypical mechanism (gained by their changes from wt α-synuclein), while the typical form of toxicity is fibril-mediated.
In conclusion, it seems simplest and most consistent with the data to proceed with the hypothesis that non-fibrillar synuclein mediates toxicity. However, if fibrillization is necessary for toxicity then it is not sufficient because proteins with similar fibrillization rates had very different toxicities (Fig. 4). We also conclude that the non-toxic nature of A30P cannot be explained by its slow fibrillization; other sequences (e.g., V49E/Q79R, β-synuclein) that are much slower fibrillizers were more toxic (Fig. 4).
Yeast screening for α-synuclein variants with decreased toxicity
α-Synuclein-GFP fusions were expressed from the 2-micron (high copy number) plasmid p426GAL1. Library transformation yielded 2200 colonies, and we picked 88 that both expressed GFP and appeared to be relatively non-toxic. These had their cDNAs placed into a fresh vector using in vivo homologous recombination.26 A single quantitative growth experiment was carried out, along with fluorescence microscopy of GFP (Supplementary Fig. 1). We carried forward 29 of these that appeared to be the most robustly non-toxic. After ligating the synuclein cDNAs into a fresh vector and removing the GFP tags, we analyzed the candidates and control proteins in a quantitative growth experiment, which allowed us to statistically divide all sequence variants into three classes: toxic, non-toxic, and intermediate (Fig. 2). The protein sequences of these library clones are listed in Fig. 5. A second type of screen (in which a library clone and wt α-synuclein were co-expressed; see methods section) also yielded several non-toxic sequences (these clones are labeled with a numeral and the letter “d”; Figs. 2, 3, and 5).
Figure 5.

α-Synuclein sequences studied by quantitative yeast growth are shown. The division of the α-synuclein sequence into domains is shown at the top. Just below this, the PD-linked mutations are shown. The wild type α-synuclein sequence is the first in the list, and other sequences are listed in order of decreasing estimated yeast toxicity from top to bottom; sequences are divided into three statistical groups as in Figs. 2 and 3. Negatively/positively charged amino-acids are shown in red/blue, and proline is shaded green. As discussed in the methods, only amino acids 8–130 are highly mutatable because of mutation correction at the termini by PCR primers. Stop codons are indicated by an asterisk.
Note the correlation between yeast toxicity, synuclein-containing inclusions, and plasma membrane localization (Fig. 3 and Supplementary Fig. 1; discussed below). Dixon et al.21 showed that mutation of proteins in the yeast secretory pathway (including ER-Golgi transport) can cause α-synuclein to change its localization from plasma membrane to punctate inclusions. It was later shown that α-synuclein itself can disrupt ER-Golgi trafficking.20 Therefore, we suspect that inclusions are downstream, rather than the originator, of yeast toxicity.
Yeast screening for enhanced α-synuclein toxicity
α-Synuclein genes were expressed from the centromeric plasmid p416GAL1. Library transformation yielded 2600 colonies, and 2 of these clones were consistently more toxic to growth on plates. This was confirmed in a quantitative growth experiment (Supplementary Fig. 3). DNA sequencing revealed that both contained 1 bp deletions in nucleotide positions 282 or 323 (numbering from the start codon), resulting in 176 amino-acid proteins, with the final ~80 amino-acids being non-synuclein, and containing three cysteines (Supplementary Fig. 4). We speculated that their enhanced toxicity might be due to loss of the C-terminal portion of α-synuclein, and to test this we examined two truncated α-synuclein sequences (1–99 and 1–107) in our yeast growth experiments. These truncations failed to explain the toxicity of the frame shifted mutants: they were not significantly more toxic than the wt protein (Supplementary Fig. 3). The toxicity of the frame shifted mutants is probably due to some aspect of the severe and irrelevant change they bear and they were not studied further.
There are consensus mutations in the non-toxic α-synuclein sequences
Selected sequences have nucleotide mutations as the cause of their phenotype, and in our case, we can safely assume that the resulting protein mutations are directly responsible for phenotype. We would like to know if some pattern of amino acid mutations can be discerned in the sequencing data. One of the simplest types of pattern is that particular amino acid mutations have been enriched for (or depressed) in the non-toxic sequences. As a measure of this, we use an odds ratio: the number of times a particular type of amino acid mutation (e.g., mutation to E) is observed in our sequences divided by the number of times we would expect to observe it, if there were no selective pressure. For example, an odds ratio of 2 for mutation to E means that E mutations were seen twice as often as would be expected if they were neutral (see methods section for further details).
We detected two types of mutation in the set of 25 non-toxic sequences with estimated odds ratios significantly different from 1: mutation to E (estimated odds ratio = 2.4; p-value = 0.003) and P (estimated odds ratio = 3.0; p = 0.016). Are P and E favored in-of-themselves, or is their abundance simply a result of their being an effective way to remove particular wt amino acids that are essential for toxicity (for example, it could be that removal of lysine and not accumulation of glutamic acid is what is really selected for)? Inspection of Fig. 5 shows that E and P are each favored by mutation from multiple types of wt residue. Additional confirmation of significance and specificity is available for both the P and E odds ratios: single point mutants A30P and A29E are both non-toxic. These results, in conjunction with the mechanistic discussion below, allow us to conclude that E and P are directly favored by selection, on their own merits.
The synuclein sequence variants selected for non-toxicity in yeast have decreased membrane binding ability
The known membrane affinity of wt α-synuclein, its reduction by the A30P mutation,27,28 and the intracellular localization of synuclein-GFP variants (Fig. 3, Supplementary Fig. 1, and reference18), led us to investigate the idea that the non-toxic sequences have decreased affinity for membranes. We determined the ability of 47 synuclein variants to bind negatively charged phospholipid vesicles using a vesicle sedimentation assay (Supplementary Fig. 5). There was a strong correlation between in vitro membrane binding and yeast toxicity (Fig. 6). This result and the additional correlation between toxicity and plasma membrane localization (Fig. 3 and Supplementary Fig. 1) suggests that the plasma membrane localization of synuclein is a result of direct binding to the bilayer.
Figure 6.
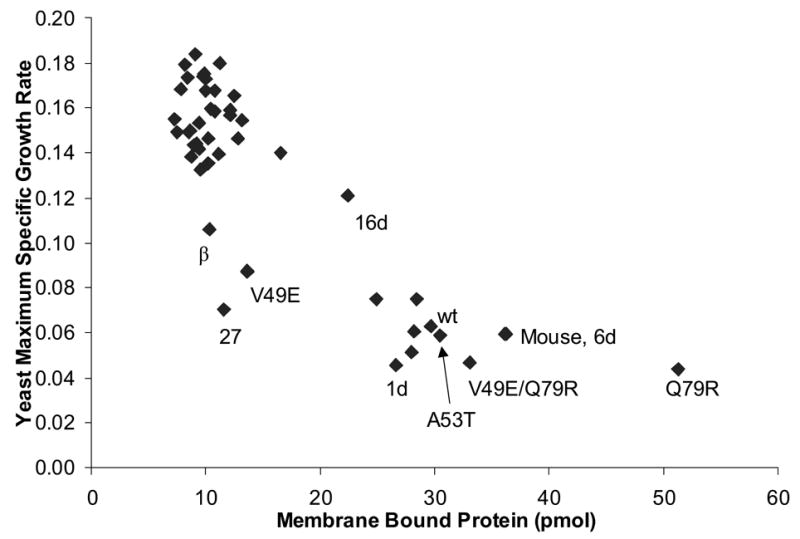
Scatter plot of estimated maximum specific growth rate (data from Fig. 2) versus membrane binding level (data from Supplementary Fig. 5). Membrane binding level is the amount of synuclein that co-sedimented with vesicles during a centrifugation assay (see methods). One point is shown for each of the 47 variants listed in Supplementary Fig. 5 (some are labeled).
The P and E consensus mutations in the N-terminal region of synuclein are consistent with a membrane binding defect and disruption of the amphipathic α-helices in general: proline interferes with α-helix formation7 and based on helical wheel analysis,5,29,30 glutamic acid may interact unfavorably with the negatively charged phospholipid headgroups (especially K→E) or the hydrophobic membrane interior (especially V26E). The P and E paradigm can explain 20 of the 25 non-toxic sequences (Fig. 5). The remaining five (mutants 11, 14, 32d, 45, and 63) also have mutations in the N-terminal region that are consistent with decreased membrane binding: either lysine 12, 23, or 34 (which probably interact with a negatively charged phosphate29) is replaced with an uncharged, polar residue, or leucine 8 (probably located in the membrane interior29) is replaced with a polar or positively charged residue.
Is membrane binding necessary for synuclein induced yeast toxicity?
The nature and distribution of the consensus mutations and the correlation of toxicity with membrane binding ability (Fig. 6) strongly suggests that the causative mechanism of synuclein-induced yeast toxicity requires an amphipathic α-helix. One possibility is that the amphipathic α-helix is used to form a toxic protein-protein interaction, for example, with a yeast protein or with itself. However, the hypothesis most consistent with our previous knowledge of synuclein is that a functional amphipathic helix is necessary because of its role in lipid binding.
Through a mechanism that has no direct analog in (and in fact seems opposite to) the yeast model, it is conceivable that A30P causes PD by interrupting amphipathic α-helix formation. If this is true, then our results indicate that there may exist additional families with autosomal dominant PD that have glutamic acid mutations in the N-terminal half of their α-synuclein genes.
In conclusion, we support a model of yeast toxicity in which overexpressed monomeric synuclein directly coats the plasma and internal membranes via its amphipathic α-helices, disrupting normal membrane processes (including vesicle trafficking19–21), and eventually leading to toxicity. Perhaps the simplest mechanism by which synuclein binding could disrupt membrane processes is a non-specific one. We have previously shown that high concentrations of monomeric synuclein (wt and A53T, but not A30P) can dissolve phospholipid vesicles by a detergent-like mechanism.31 Whether because of such bulk effects or by exclusion of protein or vesicle docking, it would not be surprising if thoroughly coating biomembranes by strongly overexpressing synuclein were toxic, in a broad range of species, including yeast.
Materials and Methods
Random point mutagenesis library
Our error-prone PCR α-synuclein library is contained in the E. coli expression vector pT7-7 and has been described previously.11 The library codes for about 1.3 × 106 unique α-synuclein protein variants with an average of 2 amino acid point mutations per sequence. Amino acids 1–7 are not subject to mutation because this region is complementary to the forward PCR primer.11 The diversity of the library far exceeds the needs of the screens described in this article.
Library transformation of E. coli. Strain BL21-Gold(DE3) (Stratagene) was transformed with DNA, using electroporation in the case of the library,32 and using cells made chemically competent by the Inoue method33 otherwise. All control (non-library) sequences were verified by DNA sequencing and were expressed and purified by the same methods in parallel with the library clones. Our human wt α, β, and γ, and mouse α-synuclein protein sequences match those given by the NCBI reference sequence project34 (GenBank accession nos. NP_000336.1, NP_003076.1, NP_003078.1, and NP_033247.1 respectively). The pT7-7 vector was used to express α-synuclein sequences and pET-30a(+) for β and γ synucleins. We used dilutions of the library DNA stock (~60 μg / mL) of several thousand fold into water for electroporation. We never observed more than one unique plasmid in a transformed colony, by DNA sequencing.
Protein expression
Single colonies of BL21-Gold(DE3) were cut from agar plates using a cork borer, and expelled into 100 mL cultures (LB-ampicillin (150 mg / L) media in 250 mL shake flasks), which were grown at 37 oC with shaking (250 RPM) to an OD600 of approximately 0.7–1.5. For library sequences, glycerol stocks of the cultures were prepared and frozen for future DNA sequence analysis. The cultures were then induced with IPTG (1 mM) and additional ampicillin (100 mg / L) for 3 hours, pelleted, resuspended in 0.75 mL of buffer (50 mM Tris pH 8.0, 10 mM EDTA, 150 mM NaCl), and frozen at −80 oC until purification.
Protein Purification
In order to maximize the throughput of protein purification, a non-chromatographic method was developed. The thermal solubility of synuclein, first pointed out by Jakes et al.,35 is essential to the method. Tubes of the frozen cells were placed directly in a boiling water bath for 7 minutes, and then pelleted at maximum speed in a microfuge for 4 minutes. The supernatant was removed to a fresh tube and streptomycin sulfate (136 μL of a 10% solution / mL supernatant) and acetic acid (glacial, 228 μL / mL supernatant) were added, followed by an additional spin for 2 minutes. The supernatant was again removed and then precipitated with ammonium sulfate (saturated ammonium sulfate at 4oC was used 1:1 vol:vol with supernatant). Precipitated protein was collected by centrifugation (at this stage clones not producing purifiable protein are observed to form no precipitate), and the pellet was washed once with 1 mL ammonium sulfate solution (4 oC; 1:1 vol:vol saturated ammonium sulfate (4 oC) : water). The washed pellet was resuspended in 900 μL 100 mM ammonium acetate (to form a cloudy solution) and precipitated by adding an equal volume of ethanol at room temperature. Ethanol precipitation was repeated twice more, followed by a final resuspension in 100 mM ammonium acetate, freezing in liquid nitrogen, and lyophilization. β-synuclein forms a very large, low density ethanolic pellet which is difficult to recover, and was only subjected to two ethanol precipitations. Lyophilized proteins (100 mL cultures typically yielded 5–10 mg of powder) were stored in darkness, desiccated with P2O5.
Protein resuspension and fibrillization
Typically, 5–6 mg of lyophilized powder was resuspended in 80 μL HBS (10 mM HEPES pH 7.4, 145 mM KCl, 0.04% sodium azide). The necessary amount (several microliters) of 1 M KOH was added in order to titrate the pH to approximately 7.4 (using pH paper), and bring the protein fully into solution. Residual acetate as well as the acidic nature of α-synuclein cause the pH to begin at around 5, where α-synuclein is less soluble. The proteins were dialyzed against 2 L of HBS at 4 oC for 18 h, using pre-rinsed mini-dialysis units (Pierce Slide-A-Lyzer, 10 kDa; typically a 1000 fold excess of solution over sample was used). The proteins were then spun through prerinsed 100 kDa ultrafilters (Microcon) for 7 minutes at RT to sterilize and remove preformed oligo and polymeric species. HBS (30 μL) was added to the filter, followed by another 2 minute spin, and the total filtrate was stored on ice. Gel electrophoresis (SDS-PAGE; colloidal Coomassie stain36) revealed that the protein preparations are essentially homogenous, containing only synuclein. Mass spectral analysis (MALDI-TOF) of wt α-synuclein demonstrated the expected molecular mass of 14.46 kDa. Protein concentrations were determined using the BCA assay (Pierce) and then matched. The BCA assay was standardized against the absorbance of wt α-synuclein using an ε280 of 6500 M−1cm−1 (ε280 was determined by measuring the absorbance of synuclein samples whose absolute concentrations were obtained by amino-acids analysis).
The samples were transferred into either Nunc 384 well plates (used in the initial screen; white polystyrene, optical bottom, non-treated, #242763; 80 μL sample per well at 200 and 400 μM; plates were sterilized with 50% ethanol followed by vacuum; sealed with Corning Costar Thermowell aluminum sealing tape), or sterile 0.5 mL polypropylene tubes (used in secondary, detailed kinetic studies of selected variants, as in Fig. 1; 400 μL at 250 μM). Samples were incubated in darkness at 37 oC with horizontal shaking (3 mm diameter circular orbit, ~550 RPM; Lab-Line Instruments titer plate shaker).
Assays for fibril formation
For the thioflavin T assay, protein samples (2 μL) were added to glycine buffer (100 mM pH 8.5, 40 μL) in a Nunc black polystyrene 384 well plate. To this solution, 50 μL of thioflavin T solution (20 μM thioflavin T in 80 mM glycine pH 8.5) was added. Fluorescence measurements (LJL Analyst; ex: 440 nm bandpass, em: 500 nm bandpass, 475 nm dichroic mirror) were made immediately. Measurements were made daily for incubation days 0 through 8, and then on days 11, 14, 21, and finally at approximately 30 days. Fibrillization lag time was defined as a thioflavin T fluorescence reading (arbitrary units) of greater than 1000. This cutoff was chosen because it is just far enough above the zero plus noise level of the measurement to avoid false positives, while low enough to give sensitive detection of fibrils. Our rationale for using a cutoff value as opposed to fitting the time course is that once fibrils have begun to form, the solution is no longer homogenous and any further ThioT measurements are therefore erratic and of questionable relevance. The onset of fibrillization was often clearly visible by eye as an increase in solution turbidity. As a control, we confirmed that both wt and A53T fibrils contained full length α-synuclein by pelleting and washing fibrils, followed by formic acid solubilization, lyophilization, and SDS-PAGE.
The extent of fibrillization was determined following 14 days of incubation by measuring the amount of soluble protein remaining in solution. An aliquot of the fibrillization reaction (50 μL) was withdrawn from each tube and insoluble material was pelleted at 16000 g. The protein concentration of the supernatant was measured by UV absorbance as described above (except that we used an ε280 of 8125 M−1cm−1 for the mouse protein, as it contains 5 tyrosines versus 4 in the human sequence; neither contain tryptophan or cysteine37).
Library subcloning into yeast vectors
PCR of the library was performed with forward primer 5′gacttctaactagtaaaaaatggatgtattcatgaaaggac and either reverse primer 5′tccggactcgagtcaagaaactgggagcaaagatatttctta for non-fusion proteins or 5′tccggaaagcttggcttcaggttcgtagtcttgatacccttc for preparing α-synuclein-GFP fusions. The forward primer contains a SpeI site and a yeast consensus translation sequence before the start codon.38 The reverse primers contain either XhoI (no fusion) or HindIII (for GFP fusion; no stop codon). The digested, gel-purified PCR products were ligated into the appropriate digested and phosphatased vectors, of which there were three total, one for each screen: For non-GFP-fusions we used p426GAL1 and p416GAL1, for fusion proteins we used p426GAL1 with GFP inserted between the ClaI and XhoI sites. The p426GAL1 vector39 was obtained from ATCC and others were assembled from this and the pRS series vector40 kit from ATCC. The protein sequence at the fusion junction reads synuclein…PEAKLIDSKG…GFP. The last three amino acids of α-synuclein are PEA, the linker is KLID, the N-terminal Met/start of GFP is removed (to help prevent GFP expression if an improper ligation occurs) and SKG are amino acids from GFP (this GFP gene contains the original wild type codons and mutations S65T and V163A). These plasmids were designed to closely emulate those used in Outeiro and Lindquist18 The libraries were transformed into E. coli (chemically competent DH5α) and, following 45 minutes of growth in SOC medium, aliquots were dilution plated to analyze transformation efficiency. The remaining bulk of the transformations were grown in selective media and plasmid prepped for subsequent yeast transformation. About 14,000 independent clones were obtained for each of the three yeast vector libraries, which was sufficient for the throughput of our yeast screening methods.
Yeast library transformation and replica plating
With the intention of finding less-toxic and more-toxic α-synuclein genes, S. cerevisiae haploid strain W303-1a was transformed with the libraries p426GAL1-synuclein-GFP and p416GAL1-synuclein (20 ng each library per 10 mL of 2×YPDA regrown yeast culture) according to reference41 (see also http://www.umanitoba.ca/faculties/medicine/biochem/gietz/method.html). We also attempted to search for clones that were dominantly non-toxic. Although we later found this approach to be inadequate for this purpose, it was successful in discovering non-toxic α-synuclein mutants (see below and results section): W303-1a already containing p424GAL1-(wt α-synuclein) was transformed with the p426GAL1-synuclein library.
Each of these three libraries was plated onto 15 plates of the appropriate synthetic non-inducing (glucose) drop-out media (CSM dropout mixtures were from Qbiogene and contained 20 mg/mL adenine) and grown at 30 oC. After about 30 hours, when small colonies became visible, the plates were replicated onto synthetic inducing (2% galactose) drop-out media using sterile velveteen squares, and all plates were incubated at 30 oC. Directly plating a transformation onto galactose media failed to yield any colonies.
Yeast screening
In each screen, candidate colonies were picked and streaked onto fresh plates to test consistency of phenotype. Candidates were determined by relative colony size and to some extent, color. Non-toxic growth tends to produce reddish colonies (exhaustion of adenine in the media by the growing colony is one explanation; W303-1a is Ade−); toxic synuclein produces smaller, white, colonies. Colonies from the p426GAL1-synuclein-GFP library were also screened to exhibit strong GFP fluorescence in order to eliminate those that were artifactually non-toxic due to lack of expression.
As mentioned above we also employed a second type of screen for dominant non-toxicity. α-Synuclein sequence variants were expressed from the 2-micron plasmid p426GAL1, in a background of W303-1a p424GAL1-wt α-synuclein. That is, all of the clones (9000 transformants were obtained) expressed both wt α-synuclein and one α-synuclein sequence from the library. Our intention was to search for clones that repressed wt α-synuclein toxicity in a dominant fashion. However, the experimental design was flawed: wt α-synuclein toxicity was decreased by the presence of any non-toxic 2-micron plasmid with GAL1 promoter (for this reason screening enriched for non-expressing clones and necessitated that α-synuclein expression be confirmed by immunoblot, as described below). The probable reason is that the total number of 2-micron plasmids (the sum of p424 and p426) is regulated rather than each plasmid individually. This allows the plasmid p424GAL1-wt α-synuclein to exist at a lower copy number (and hence, expression level) than is possible when the plasmid is present by itself. This screen did yield several non-toxic clones, although none were significantly more efficacious than GFP in repressing wt α-synuclein toxicity (data not shown).
It could be that an observed phenotype was due to incidental mutations in the plasmid backbone (for example in the replication origin) and not in α-synuclein itself. Each candidate cDNA was moved into a fresh vector (identical except for a TRP marker instead of URA) by in vivo homologous recombination mediated plasmid construction.26,42 For this procedure, the candidate cDNAs were individually amplified by PCR (from about 100 bp upstream (primer: 5′gcgaagcgatgatttttgatctattaac) and 100 bp downstream (primer: 5′gacctagacttcaggttgtctaactccttcctt non-GFP; 5′ttaagggtaagttttccgtatgttgc for GFP fusion) of the synuclein cDNA), and the products were co-transformed into yeast with the appropriate doubly digested (SpeI and XhoI or ClaI) TRP plasmid (lacking any synuclein cDNA). In vivo double crossover events, one in each of the 100 bp overlap regions, creates a reconstructed TRP version of the candidate, which was selected for on media lacking tryptophan. All of the non-library control plasmids were PCR amplified and prepared in parallel by the same homologous recombination method, to avoid bias. A single quantitative growth trial (see method below) was carried out on the resulting clones, and candidates continuing to display robustness of phenotype were taken forward to the next step. In addition, candidates from the p426GAL1-synuclein library were screened for α-synuclein expression by western blotting following SDS-PAGE of spheroplasts (Sigma Celytic Y Plus).
Finally, we wanted to eliminate all possibility of false positives due to promoter, terminator, or cDNA/plasmid junction mutations (which may have carried through the recombination method above, within the 100 bp of overlap), and GFP tags. Therefore, in the manageable number of remaining candidates, the cDNA inserts in these TRP vectors were PCR amplified and then in vitro ligated into p426GAL1 (5′gacttctaactagtaaaaaatggatgtattcatgaaaggac; 5′tccggactcgagttaggcttcaggttcgtagtcttgatacc; SpeI and XhoI sites directly flank the α-synuclein gene). These vectors were transformed into yeast, and quantitative growth studies for statistical analysis were carried out. At this time, DNA sequencing of the clones was performed. All of the synuclein proteins that we studied that were not selected from the yeast library were also sequence verified. Note that PCR primers will tend to correct any mutations in the regions they anneal to. Therefore, in α-synuclein clones obtained from yeast, only cDNA positions 23–390 (amino acids 8–130) are highly mutatable.
Additional PD genes
We wished to determine whether additional proteins associated with PD might increase or decrease the toxicity of synuclein in yeast. A 6×12 factorial design in randomized blocks43 was carried out to study quantitative growth of every combination of 5 PD-related genes expressed from p426GAL1: DJ-1, synphilin, Pink1, parkin, UCH-L1, and GFP (control) with 5 variants of synuclein: wt, A30P, A53T, β, γ, and GFP. Additionally, the synucleins were all expressed from both a centromeric and a 2-micron plasmid (p414GAL1 or p424GAL1). The inferred protein sequences of DJ-1 and UCH-L1 matched the corresponding NCBI reference sequences (GenBank accession nos. NP_009193.2 and NP_004172.2, respectively). Our parkin sequence contained a P223S mutation relative to GenBank accession no. NP_004553.1 but this change is also found in the human genome (GenBank accession no. NT_007422.13). Pink1 contained mutation P209A relative to the reference sequence (GenBank accession no. NP_115785.1); this mutation is also found in the original mammalian genome collection clone (GenBank accession no. BC028215.1). Synphilin (generously provided by Zhong Pei and Christopher Ross) was identical to the sequence originally given,44 but contains V44A relative to the current NCBI reference sequence (GenBank accession no. NP_005451.2).
Quantitative growth studies
We measured quantitative growth rates in synthetic drop-out galactose media with 100 mg/mL adenine in sterile 12-well polystyrene plates (Falcon 353043). Each well with 1 mL media was inoculated with a small quantity of a yeast colony by picking with a pipet tip. There is no doubt up to several-fold variation in inoculum levels between cultures. However, the parameter we extract from the Gompertz equation (below) is inoculation-level invariant and additionally, the trials were performed in complete randomized blocks, so any inoculation level effects would be unbiased and accounted for statistically.43 The plates were incubated on the plate shaker described above, in darkness at room temperature at an approximate speed of 400 RPM.
Yeast quantity was measured by vertical turbidity through the center of each growth well (no sampling of the culture is required). For this purpose we employed a laser diode from a laser pointer (~650 nm) and a CdS photoresistor. A digital volt meter (Radio Shack 22–812) was used to measure resistance (proportional to 1/intensity) and computer linkage to the meter allowed data recording. By a type of Beer-Lambert law, turbidity is proportional to log(resistance when sample is in the beam / resistance when blank is in the beam), and is roughly proportional to the number of yeast cells. We were able to grow 8 12-well plates per shaker, and the measurement of turbidity in 200 samples can be completed in 30 minutes.
For each sample, the Gompertz growth equation,45,46 τ (t) =α exp(−e−κ (t −γ)) was fitted to the turbidity τ as a function of time t in order to determine the maximum specific growth rate κ. This parameter intrinsically characterizes the growth rate of the yeast/synuclein-variant combination, and was used in the statistical analyses of yeast growth (calculations were performed with Matlab (The MathWorks, Inc.)).
Yeast microscopy
After growth (in liquid galactose media, as described above for quantitative growth studies), a few microliters of yeast in growth media were placed on a slide with hydrophobic adhesion wells (Erie Scientific) and were overlaid with a cover slip. The slides were examined with a Nikon E600 microscope using a 100× plan fluor objective and representative photographs of fluorescence were taken through a FITC-HYQ filter cube with a Diagnostic Instruments RT slider camera in black and white mode.
Membrane binding studies
Yeast clones of interest were subcloned into the pT7-7 E. coli expression vector by PCR (forward primer 5′cgagctctccatatggatgtattcatgaaaggac reverse primer, 5′tccggaaagcttaggcttcaggttcgtagtcttgataccc). All of these pT7-7 clones were verified by sequencing to match their originating yeast clones in protein sequence. Proteins were expressed, purified, resuspended, and concentration matched as described above, except azide was not included in the buffers.
Vesicles were prepared as in reference31 using egg phosphatidyl-glycerol (Avanti Polar Lipids, #841138), with slight changes: After drying, lipid was resuspended in HBS 1 mM EDTA:OptiPrep (OptiPrep is a 60% w/v stock of iodixanol in water; Sigma D-1556) in a 1:2 vol:vol ratio. Membrane extrusion was carried out with 13 passes through a single 200 nm pore-size Whatman Nuclepore membrane. PD-10 chromatography was carried out with HBS 1 mM EDTA as mobile phase. The vesicles were stored in glass at 4 oC, and were used within several days. Optiprep in the vesicles allows them to be pelleted rapidly in a tabletop microcentrifuge (16000 g). This is necessary, considering the large number of samples to be analyzed, and in addition the rapid time scale helps prevent protein-vesicle dissociation during sedimentation. Similar methods have been described previously.47,48
Purified synuclein (25 μL) in HBS 10 mM CaCl2 was mixed with 25 μL of the lipid solution. All reactions were carried out in siliconized 1.7 mL tubes (Research Products International, #145450). After 1 hour of incubation at RT, 10 μL of the solution was removed to a well of a black 96-well plate already containing 15 μL HBS and 50 μL 100 mM borate-triton (pH 9.3; 0.3% Triton X-100). The remaining 40 μL was layered on top of 500 μL of 5% OptiPrep in HBS in a 1.7 mL siliconized tube, which was then centrifuged at 16000 g for 10 minutes at RT. The supernatant was carefully aspirated from the top down, leaving only the final 25 μL of fluid in the tube, and without disturbing the small lipid pellet. The pellet and 25 μL remaining supernatant was dissolved in 50 μL of borate-triton and the entirety was transferred to the 96-well plate. The protein concentrations of samples in the plate were measured using the Invitrogen CBQCA protein quantitation kit. This assay is insensitive to lipids and detergent, and produces a fluorescent signal proportional to the number of primary amines present (lysines and the N-terminus). Fractional adjustments were made in the blank-subtracted measured signals according to the number of primary amines in the protein sequence of each sample, in order to normalize to wt α-synuclein, which has 16 primary amines.
Sequence analysis
We take the 25 non-toxic α-synuclein sequences selected from the library (Figs. 2 and 5) as the set for analysis. We assume position independent nucleotide mutation frequencies in the original, unselected, library; this assumption has empirical justification.11 The null hypothesis is that amino acid mutation Z (Z standing for one of the 20 amino acids, not location specific) is neutral: it has not been enriched for or depressed in our sample. Under this null hypothesis, a nucleotide mutation i→j (e.g., A→T) should be observed at the same frequency at positions where it would and would not cause Z. To examine this, the data (one count for every possible nucleotide mutation at every position of every non-toxic library clone) can be organized into 2×2 contingency tables, with row classification based on whether or not i→j at the particular position under consideration would cause Z, and column classification based on whether or not i→j was actually observed at that position. A separate table is compiled for each of the 12 types of nucleotide mutation, since the different nucleotide mutations occur with different overall frequencies; the Mantel-Haenszel statistical test49,50 is then used to simultaneously analyze these multiple tables. In theory, each library sequence should be separately analyzed into 12 2×2 tables, since different sequences may arise from different generation PCR products and hence have different mutation frequencies. This was not found to alter the conclusions in our case, and the simpler treatment combining all of the sequences was therefore used. In the analysis, we eliminated any count which would produce a premature stop codon, since these were effectively eliminated during the screening procedure. Furthermore, only the mutatable nucleotides were considered (cDNA positions 23–390).
As a test of the procedure, we used a Monte Carlo method to produce 5000 sets of 25 sequences, with overall nucleotide mutation frequencies identical to those found in the set of 25 non-toxic clones (Fig. 5). Those with nonsense mutations were rejected, as they are in our screen. These sets of artificial "selected" sequences exactly follow the null hypothesis, because the 12 nucleotide mutation frequencies used to produce them are completely position independent; there is no selective pressure at all. Using an alpha level of 0.05 for the Mantel-Haenszel statistic, we obtained on average 126 “significant results” (false positives) from the 5000 sets of sequences. The Mantel-Haenszel statistic is referenced to a chi-squared distribution with one degree of freedom, and if that were the correct reference for this problem, we would have obtained ~250 false positives (0.05 × 5000). Because significantly fewer false positives were actually obtained, we conclude that p-values from the Mantel-Haenszel procedure are conservative in this application. On the other hand, the p-values reported for analysis of our experimental data (Fig. 5) also have a liberal contribution: this particular type of analysis was suggested by visual inspection of the data itself and the p-values, which were determined for all 20 mutation types simultaneously, do not include a multiple comparison adjustment.
In this analysis we have tacitly assumed that only amino acid Z, if any, could have been enriched or depressed. If other non-Z amino acids have been selected for or against, this might bias the statistic. None-the-less the test is quite practical. First, we note that selection for or against any particular amino acid will only alter the occurrence of a rather small subset of possible i→j mutations (for example, if methionine mutations via A→T are strongly selected against, there are still numerous other locations where A→T can occur without causing a methionine mutation, so its influence is not excessive). Second, visual inspection of the sequences shows that there is no obvious bias for or against most amino acids.
Supplementary Material
Acknowledgments
This work was supported by a Morris K. Udall Parkinson’s Disease Research Center of Excellence grant (NS038375) and National Institutes of Health grants AG008470 and NS051243. We thank Allegra Petti for assistance with subcloning the single point mutants of Fig. 1 into yeast vectors. We thank Alina Vr bioiu and Katherina Vamvaca for helpful discussion and comments on the manuscript. We thank Sarah Luchansky, Chris Rochet, and Jeff Kessler for many useful discussions as well as advice with molecular biology, and Craig Justman for advice regarding protein fibrillization in plates.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cookson MR. The biochemistry of Parkinson’s disease. Annu Rev Biochem. 2005;74:29–52. doi: 10.1146/annurev.biochem.74.082803.133400. [DOI] [PubMed] [Google Scholar]
- 2.Conway KA, Harper JD, Lansbury PT., Jr Accelerated in vitro fibril formation by a mutant alpha-synuclein linked to early-onset Parkinson disease. Nat Med. 1998;4:1318–20. doi: 10.1038/3311. [DOI] [PubMed] [Google Scholar]
- 3.Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–40. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 4.Bussell R, Jr, Ramlall TF, Eliezer D. Helix periodicity, topology, and dynamics of membrane-associated alpha-synuclein. Protein Sci. 2005;14:862–72. doi: 10.1110/ps.041255905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davidson WS, Jonas A, Clayton DF, George JM. Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J Biol Chem. 1998;273:9443–9. doi: 10.1074/jbc.273.16.9443. [DOI] [PubMed] [Google Scholar]
- 6.Bisaglia M, Tessari I, Pinato L, Bellanda M, Giraudo S, Fasano M, Bergantino E, Bubacco L, Mammi S. A topological model of the interaction between alpha-synuclein and sodium dodecyl sulfate micelles. Biochemistry. 2005;44:329–39. doi: 10.1021/bi048448q. [DOI] [PubMed] [Google Scholar]
- 7.Bussell R, Jr, Eliezer D. Effects of Parkinson’s disease-linked mutations on the structure of lipid-associated alpha-synuclein. Biochemistry. 2004;43:4810–8. doi: 10.1021/bi036135+. [DOI] [PubMed] [Google Scholar]
- 8.Sung YH, Eliezer D. Secondary structure and dynamics of micelle bound beta- and gamma-synuclein. Protein Sci. 2006;15:1162–74. doi: 10.1110/ps.051803606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yamin G, Munishkina LA, Karymov MA, Lyubchenko YL, Uversky VN, Fink AL. Forcing nonamyloidogenic beta-synuclein to fibrillate. Biochemistry. 2005;44:9096–107. doi: 10.1021/bi048778a. [DOI] [PubMed] [Google Scholar]
- 10.Dauer W, Przedborski S. Parkinson’s disease: mechanisms and models. Neuron. 2003;39:889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- 11.Volles MJ, Lansbury PT., Jr A computer program for the estimation of protein and nucleic acid sequence diversity in random point mutagenesis libraries. Nucleic Acids Res. 2005;33:3667–77. doi: 10.1093/nar/gki669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Du HN, Tang L, Luo XY, Li HT, Hu J, Zhou JW, Hu HY. A peptide motif consisting of glycine, alanine, and valine is required for the fibrillization and cytotoxicity of human alpha-synuclein. Biochemistry. 2003;42:8870–8. doi: 10.1021/bi034028+. [DOI] [PubMed] [Google Scholar]
- 13.Giasson BI, Murray IV, Trojanowski JQ, Lee VM. A hydrophobic stretch of 12 amino acid residues in the middle of alpha-synuclein is essential for filament assembly. J Biol Chem. 2001;276:2380–6. doi: 10.1074/jbc.M008919200. [DOI] [PubMed] [Google Scholar]
- 14.Sode K, Usuzaka E, Kobayashi N, Ochiai S. Engineered alpha-synuclein prevents wild type and familial Parkin variant fibril formation. Biochem Biophys Res Commun. 2005;335:432–6. doi: 10.1016/j.bbrc.2005.07.100. [DOI] [PubMed] [Google Scholar]
- 15.Kessler JC, Rochet JC, Lansbury PT., Jr The N-terminal repeat domain of alpha-synuclein inhibits beta-sheet and amyloid fibril formation. Biochemistry. 2003;42:672–8. doi: 10.1021/bi020429y. [DOI] [PubMed] [Google Scholar]
- 16.Griffioen G, Duhamel H, Van Damme N, Pellens K, Zabrocki P, Pannecouque C, van Leuven F, Winderickx J, Wera S. A yeast-based model of alpha-synucleinopathy identifies compounds with therapeutic potential. Biochim Biophys Acta. 2006;1762:312–8. doi: 10.1016/j.bbadis.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 17.Wurth C, Guimard NK, Hecht MH. Mutations that reduce aggregation of the Alzheimer’s Abeta42 peptide: an unbiased search for the sequence determinants of Abeta amyloidogenesis. J Mol Biol. 2002;319:1279–90. doi: 10.1016/S0022-2836(02)00399-6. [DOI] [PubMed] [Google Scholar]
- 18.Outeiro TF, Lindquist S. Yeast cells provide insight into alpha-synuclein biology and pathobiology. Science. 2003;302:1772–5. doi: 10.1126/science.1090439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Willingham S, Outeiro TF, DeVit MJ, Lindquist SL, Muchowski PJ. Yeast genes that enhance the toxicity of a mutant huntingtin fragment or alpha-synuclein. Science. 2003;302:1769–72. doi: 10.1126/science.1090389. [DOI] [PubMed] [Google Scholar]
- 20.Cooper AA, Gitler AD, Cashikar A, Haynes CM, Hill KJ, Bhullar B, Liu K, Xu K, Strathearn KE, Liu F, Cao S, Caldwell KA, Caldwell GA, Marsischky G, Kolodner RD, Labaer J, Rochet JC, Bonini NM, Lindquist S. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science. 2006;313:324–8. doi: 10.1126/science.1129462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dixon C, Mathias N, Zweig RM, Davis DA, Gross DS. Alpha-synuclein targets the plasma membrane via the secretory pathway and induces toxicity in yeast. Genetics. 2005;170:47–59. doi: 10.1534/genetics.104.035493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Biere AL, Wood SJ, Wypych J, Steavenson S, Jiang Y, Anafi D, Jacobsen FW, Jarosinski MA, Wu GM, Louis JC, Martin F, Narhi LO, Citron M. Parkinson’s disease-associated alpha-synuclein is more fibrillogenic than beta- and gamma-synuclein and cannot cross-seed its homologs. J Biol Chem. 2000;275:34574–9. doi: 10.1074/jbc.M005514200. [DOI] [PubMed] [Google Scholar]
- 23.Cole NB, Murphy DD, Grider T, Rueter S, Brasaemle D, Nussbaum RL. Lipid droplet binding and oligomerization properties of the Parkinson’s disease protein alpha-synuclein. J Biol Chem. 2002;277:6344–52. doi: 10.1074/jbc.M108414200. [DOI] [PubMed] [Google Scholar]
- 24.Petrucelli L, O’Farrell C, Lockhart PJ, Baptista M, Kehoe K, Vink L, Choi P, Wolozin B, Farrer M, Hardy J, Cookson MR. Parkin Protects against the Toxicity Associated with Mutant alpha- Synuclein. Proteasome Dysfunction Selectively Affects Catecholaminergic Neurons. Neuron. 2002;36:1007–19. doi: 10.1016/s0896-6273(02)01125-x. [DOI] [PubMed] [Google Scholar]
- 25.Lo Bianco C, Schneider BL, Bauer M, Sajadi A, Brice A, Iwatsubo T, Aebischer P. Lentiviral vector delivery of parkin prevents dopaminergic degeneration in an alpha-synuclein rat model of Parkinson’s disease. Proc Natl Acad Sci U S A. 2004;101:17510–5. doi: 10.1073/pnas.0405313101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ma H, Kunes S, Schatz PJ, Botstein D. Plasmid construction by homologous recombination in yeast. Gene. 1987;58:201–16. doi: 10.1016/0378-1119(87)90376-3. [DOI] [PubMed] [Google Scholar]
- 27.Jensen PH, Nielsen MS, Jakes R, Dotti CG, Goedert M. Binding of alpha-synuclein to brain vesicles is abolished by familial Parkinson’s disease mutation. J Biol Chem. 1998;273:26292–4. doi: 10.1074/jbc.273.41.26292. [DOI] [PubMed] [Google Scholar]
- 28.Jo E, Fuller N, Rand RP, St George-Hyslop P, Fraser PE. Defective membrane interactions of familial Parkinson’s disease mutant A30P alpha-synuclein. J Mol Biol. 2002;315:799–807. doi: 10.1006/jmbi.2001.5269. [DOI] [PubMed] [Google Scholar]
- 29.Bussell R, Jr, Eliezer D. A structural and functional role for 11-mer repeats in alpha-synuclein and other exchangeable lipid binding proteins. J Mol Biol. 2003;329:763–78. doi: 10.1016/s0022-2836(03)00520-5. [DOI] [PubMed] [Google Scholar]
- 30.Segrest JP, Jones MK, De Loof H, Brouillette CG, Venkatachalapathi YV, Anantharamaiah GM. The amphipathic helix in the exchangeable apolipoproteins: a review of secondary structure and function. J Lipid Res. 1992;33:141–66. [PubMed] [Google Scholar]
- 31.Volles MJ, Lansbury PT., Jr Vesicle permeabilization by protofibrillar alpha-synuclein is sensitive to Parkinson’s disease-linked mutations and occurs by a pore-like mechanism. Biochemistry. 2002;41:4595–602. doi: 10.1021/bi0121353. [DOI] [PubMed] [Google Scholar]
- 32.Dower WJ, Miller JF, Ragsdale CW. High efficiency transformation of E. coli by high voltage electroporation. Nucleic Acids Res. 1988;16:6127–45. doi: 10.1093/nar/16.13.6127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sambrook J, Russell DW. Molecular cloning: a laboratory manual. 3. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, N.Y.: 2001. [Google Scholar]
- 34.Pruitt KD, Tatusova T, Maglott DR. NCBI Reference Sequence (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2005;33:D501–4. doi: 10.1093/nar/gki025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jakes R, Spillantini MG, Goedert M. Identification of two distinct synucleins from human brain. FEBS Lett. 1994;345:27–32. doi: 10.1016/0014-5793(94)00395-5. [DOI] [PubMed] [Google Scholar]
- 36.Neuhoff V, Arold N, Taube D, Ehrhardt W. Improved staining of proteins in polyacrylamide gels including isoelectric focusing gels with clear background at nanogram sensitivity using Coomassie Brilliant Blue G-250 and R-250. Electrophoresis. 1988;9:255–62. doi: 10.1002/elps.1150090603. [DOI] [PubMed] [Google Scholar]
- 37.Pace CN, Vajdos F, Fee L, Grimsley G, Gray T. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 1995;4:2411–23. doi: 10.1002/pro.5560041120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Romanos MA, Scorer CA, Clare JJ. Foreign gene expression in yeast: a review. Yeast. 1992;8:423–88. doi: 10.1002/yea.320080602. [DOI] [PubMed] [Google Scholar]
- 39.Funk M, Niedenthal R, Mumberg D, Brinkmann K, Ronicke V, Henkel T. Vector systems for heterologous expression of proteins in Saccharomyces cerevisiae. Methods Enzymol. 2002;350:248–57. doi: 10.1016/s0076-6879(02)50967-8. [DOI] [PubMed] [Google Scholar]
- 40.Christianson TW, Sikorski RS, Dante M, Shero JH, Hieter P. Multifunctional yeast high-copy-number shuttle vectors. Gene. 1992;110:119–22. doi: 10.1016/0378-1119(92)90454-w. [DOI] [PubMed] [Google Scholar]
- 41.Gietz RD, Woods RA. Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol. 2002;350:87–96. doi: 10.1016/s0076-6879(02)50957-5. [DOI] [PubMed] [Google Scholar]
- 42.Oldenburg KR, Vo KT, Michaelis S, Paddon C. Recombination-mediated PCR-directed plasmid construction in vivo in yeast. Nucleic Acids Res. 1997;25:451–2. doi: 10.1093/nar/25.2.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cochran WG, Cox GM. Experimental designs. 2. Wiley classics library; Wiley, New York: 1992. [Google Scholar]
- 44.Engelender S, Kaminsky Z, Guo X, Sharp AH, Amaravi RK, Kleiderlein JJ, Margolis RL, Troncoso JC, Lanahan AA, Worley PF, Dawson VL, Dawson TM, Ross CA. Synphilin-1 associates with alpha-synuclein and promotes the formation of cytosolic inclusions. Nat Genet. 1999;22:110–4. doi: 10.1038/8820. [DOI] [PubMed] [Google Scholar]
- 45.Zwietering MH, Jongenburger I, Rombouts FM, van ’t Riet K. Modeling of the Bacterial Growth Curve. Appl Environ Microbiol. 1990;56:1875–1881. doi: 10.1128/aem.56.6.1875-1881.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Seber GAF, Wild CJ. Nonlinear regression. Wiley series in probability and mathematical statistics; Wiley, New York: 1989. [Google Scholar]
- 47.Cho W, Bittova L, Stahelin RV. Membrane binding assays for peripheral proteins. Anal Biochem. 2001;296:153–61. doi: 10.1006/abio.2001.5225. [DOI] [PubMed] [Google Scholar]
- 48.Diakowski W, Sikorski AF. Interaction of brain spectrin (fodrin) with phospholipids. Biochemistry. 1995;34:13252–8. doi: 10.1021/bi00040a041. [DOI] [PubMed] [Google Scholar]
- 49.Ramsey FL, Schafer DW. The statistical sleuth: a course in methods of data analysis. 2. Duxbury/Thomson Learning; Australia and Pacific Grove, CA: 2002. [Google Scholar]
- 50.Kuritz SJ, Landis JR, Koch GG. A general overview of Mantel-Haenszel methods: applications and recent developments. Annu Rev Public Health. 1988;9:123–60. doi: 10.1146/annurev.pu.09.050188.001011. [DOI] [PubMed] [Google Scholar]
- 51.Hochberg Y, Tamhane AC. Multiple comparison procedures. Wiley series in probability and mathematical statistics; Wiley, New York: 1987. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.