

Abstract
Bacterial L-ASNases (L-asparaginases) catalyse the conversion of L-asparagine into L-aspartate and ammonia, and are widely used for the treatment of ALL (acute lymphoblastic leukaemia). In the present paper, we describe an efficient approach, based on protein chemistry and protein engineering studies, for the construction of trypsin-resistant PEGylated L-ASNase from Erwinia carotovora (EcaL-ASNase). Limited proteolysis of EcaL-ASNase with trypsin was found to be associated with a first cleavage of the peptide bond between Lys53 and Gly54, and then a second cleavage at Arg206-Ser207 of the C-terminal fragment, peptide 54–327, showing that the initial recognition sites for trypsin are Lys53 and Arg206. Site-directed mutagenesis of Arg206 to histidine followed by covalent coupling of mPEG-SNHS [methoxypoly(ethylene glycol) succinate N-hydroxysuccinimide ester] to the mutant enzyme resulted in an improved modified form of EcaL-ASNase that retains 82% of the original catalytic activity, exhibits enhanced resistance to trypsin degradation, and has higher thermal stability compared with the wild-type enzyme.
Keywords: L-asparaginase, enzyme engineering, hydrolase, leukaemia, PEGylation, poly(ethylene glycol) (PEG), proteolytic resistance
Abbreviations: EcaL-ASNase, L-asparaginase from Erwinia carotovora; GDH, glutamate dehydrogenase; IPTG, isopropyl β-D-thiogalactoside; L-ASNase, L-asparaginase; MALDI–TOF, matrix-assisted laser-desorption ionization–time-of-flight; mPEG-SNHS, methoxypoly(ethylene glycol) succinate N-hydroxysuccinimide ester; PEG, poly(ethylene glycol); Tm, temperature at which 50% of the initial enzyme activity is lost after heat treatment
INTRODUCTION
L-Asparaginase (L-ASNase) catalyses the hydrolysis of the non-essential amino acid L-asparagine to L-aspartate and ammonia. L-ASNase is widely used for the treatment of haemopoietic diseases such as ALL (acute lymphoblastic leukaemia). The anti-neoplastic activity results from depletion of the circulating pools of L-asparagine by L-ASNase [1,2]. Unlike normal cells, malignant cells can only synthesize L-asparagine slowly and are dependent on an exogenous supply [1,2]. In contrast, normal cells are protected from asparagine starvation by their ability to produce this amino acid [3]. Thus, unlike conventional cancer therapy, L-ASNase therapy is highly selective.
The main restrictions to the use of L-ASNase as a therapeutic agent include its premature inactivation and rapid clearance, thus necessitating frequent injections to maintain therapeutic levels, and several types of side effects ranging from mild allergies and development of immune responses to anaphylactic shock, which may be life-threatening [4–7]. To date, the most promising approach to extend the half-life and reduce the immunogenicity and antigenicity of therapeutic proteins is to covalently couple them to PEG [poly(ethylene glycol)] [8–10]. In this process (PEGylation), PEG is attached to the therapeutic protein by covalent linkage to residues located at the surface of the native protein [11]. PEGylation of proteins usually results in masking of some surface sites, increasing the molecular size and enhancing steric hindrance. Thus attachment of PEG to proteins reduces their immunogenicity, improves pharmacokinetics [12,13] and plasma half-life, and protects the proteins against proteolytic cleavage [14–17]. The in vivo activity of the modified protein can sometimes be enhanced owing to longer half-life in the circulation after conjugation, even though there may be some reduction in in vitro activity.
PEGylation has been used extensively to modify various proteins such as adenosine deaminase [18], superoxide dismutase [19], asparaginase [20], urokinase [21], hirudin [22], haemoglobin [23] and interferon [24]. In the U.S.A., there are currently six polymer-modified proteins that have already been approved by the FDA (Food and Drug Administration) and about a dozen more in various phases of clinical development [25].
The toxicity of L-ASNase is also partially attributable to the glutaminase activity of these enzymes [26]. Thus L-ASNases with high specificity for L-asparagine and low-to-negligible activity against L-glutamine are reported to be less troublesome during the course of anticancer therapy [27]. The interest in L-ASNase from Erwinia carotovora (EcaL-ASNase) arose from the fact that it shows reduced glutaminase activity, and it is therefore believed to have fewer side effects when used in anticancer therapy [28].
All asparaginases are homotetramers with 222-symmetry and a molecular mass in the range 140–150 kDa, with a highly conserved overall fold [29–31]. They are composed of four identical subunits denoted A, B, C and D [32]. Each monomer consists of about 330 amino acid residues that form 14 β-strands and eight α-helices, which are arranged in two easily identifiable domains: the larger N-terminal domain and the smaller C-terminal domain. These are connected by a linker consisting of ∼20 residues. Each of the four active sites is located between the N- and C-terminal domains of two adjacent monomers. Thus the asparaginase tetramer can be treated as a dimer of dimers, as the active site is derived from subunits A and C or from subunits B and D.
For many pharmaceutical proteins, enzymatic proteolysis makes maintaining appropriate serum levels problematic. The use of L-ASNase in therapy is limited by its premature inactivation. For example, it has been reported that serum trypsin levels increase significantly 10 and 20 days after the start of L-ASNase therapy [33,34]. In the present paper, we report an efficient approach for the construction of water-soluble and trypsin-resistant PEGylated EcaL-ASNase, based on protein chemistry and protein engineering.
MATERIALS AND METHODS
Materials
L-Asparagine and L-glutamine were obtained from Serva. α-Oxoglutaric acid (α-ketoglutaric acid), Sepharose CL6B, trypsin and mPEG-SNHS [methoxypoly(ethylene glycol) succinate N-hydroxysuccinimide ester] (molecular mass ∼5000 Da) were from Sigma. NADH (disodium salt, grade II, approx. 98% pure) and crystalline BSA (fraction V) were purchased from Boehringer Mannheim. Nessler's reagent and GDH (glutamate dehydrogenase) were obtained from Fluka, and Sephadex G-25 was from GE Healthcare. All primers were synthesized and purified by MWG-Biotech AG. TOPO cloning kit and all other molecular biology reagents were from Invitrogen.
Cloning, expression, and purification of EcaL-ASNase
L-ASNase from Erwinia carotovora 1526 (EcaL-ASNase) was cloned into a T7 expression vector (pCR®T7/CT-TOPO®), as described by Kotzia and Labrou [35]. The resulting expression construct pT7ASNase was used to transform competent BL21(DE3) pLysS Escherichia coli cells, in which the synthesis of EcaL-ASNase was induced by 1 mM IPTG (isopropyl β-D-thiogalactoside). The produced enzyme was purified from the other proteins using affinity chromatography on L-asparagine–Sepharose CL6B column, following the method of Kotzia and Labrou [35].
Kinetic analysis of mutants and PEGylated enzymes
Enzyme assays were performed at 37 °C at a Hitachi U-2000 double-beam UV/visible spectrophotometer carrying a thermostatically controlled cell holder (10 mm pathlength). Activities were measured by determining the rate of ammonia formation, by coupling with GDH, using the method of Balcao et al. [36]. The final assay volume of 1 ml contained 71 mM Tris/HCl buffer, pH 8.0, 1 mM asparagine, 0.15 mM α-oxoglutaric acid, 0.15 mM NADH, 4 units of GDH and the sample containing EcaL-ASNase activity. Alternatively, the rate of ammonia formation was measured at 37 °C using Nessler's reagent. One unit of L-ASNase activity is defined as the amount of enzyme that liberates 1 μmol of ammonia from asparagine per min at 37 °C. Protein concentration was determined at 25 °C using the method of Bradford [37] with BSA (fraction V) as a standard.
Steady-state kinetic measurements were performed in 0.1 M Tris/HCl buffer, pH 8.0 (or 8.2 for L-glutamine), by varying the concentration of the substrate (L-asparagine and L-glutamine). The kinetic parameters were calculated by non-linear regression analysis of experimental steady-state data. Kinetic data were analysed using the computer program GraFit, version 3 (Erithacus Software). Kinetic experiments were repeated three times to verify the results.
Limited proteolysis of EcaL-ASNase by trypsin
EcaL-ASNase, in 10 mM Tris/HCl, pH 7.4, was subjected to trypsin digestion. Proteolysis was initiated by adding trypsin (dissolved in 1 mM HCl, containing 1 mM CaCl2) to EcaL-ASNase at a L-ASNase/trypsin ratio of 1:35 (w/w). Samples were withdrawn at selected incubation intervals and trypsinolysis was stopped by freezing the samples with liquid nitrogen. All samples were freeze-dried and then analysed by SDS/PAGE (see below).
Site-directed mutagenesis, expression and purification of mutants
Site-directed mutagenesis of Arg206 was carried out by overlap extension using PCR [39]. The pairs of oligonucleotide primers used in the PCRs for the R206H or R206Q mutations were as follows: the first pair was 5′-ATGTTTAACGCATTATTCGTTGTTGTTTTTG-3′ (P1) and 5′-AAACACGGANTGCGTGGTGTGAAC-3′ (P2), and the second pair was 5′-GTTCACACCACGCANTCCGTGTTT-3′ (P3) and 5′-TTAAGCTTTTAATAAGCGTGGAAGTAATCC-3′ (P4). Sites of mutation are indicated in italics. The expression construct pT7ASNase [35] encoding the wildtype EcaL-ASNase was used as template DNA in all mutagenesis reactions. After completion of the PCRs (using primers P1 and P2, and P3 and P4), the PCR products were digested with DpnI to eliminate parental DNA, and purified by agarose gel electrophoresis. Following gel extraction, the fragments containing either mutation (R206H or R206Q) were used in another PCR as templates using P1 and P4 primers to amplify the entire mutated gene. The latter was TOPO-ligated into a T7 expression vector (pCR®T7/CT-TOPO®), and recombinant plasmids were isolated. Site-directed mutagenesis of Lys30 was carried out as described above. The pairs of oligonucleotide primers used in the PCRs for the K30A mutation were as follows: 5′-ACCACTGGGTATGCAGCGGGTGCGC-3′ and 5′-CGCACCCGCTGCATACCCAGTGGT-3′. All mutations were verified by DNA sequencing.
Mutated expression constructs were used to transform competent BL21(DE3) pLysS E. coli cells. The E. coli cells were grown at 37 °C in 1 litre of LB (Luria–Bertani) medium containing 100 μg/ml ampicillin and 34 μg/ml chloramphenicol. The synthesis of the mutated enzymes was induced by the addition of 1 mM IPTG when the absorbance at 600 nm was 0.6–0.8. At 5 h after induction, cells were harvested by centrifugation at 8000 g and for 20 min at 4 °C, resuspended in 5 mM potassium phosphate buffer, pH 7.5, sonicated and left at 4 °C under agitation. After 1 h, cells were centrifuged at 10000 g for 5 min and the supernatants were collected. Cell-free extracts of E. coli transformants showed high L-ASNase activity. The collected supernatants were applied to a L-asparagine–Sepharose CL6B affinity column (2 ml, 1.5 cm long×1.5 cm internal diameter) previously equilibrated with 5 mM potassium phosphate buffer, pH 7.5 [35]. Non-adsorbed protein was washed off with 20 ml of equilibration buffer. Bound mutant L-ASNase (R206H or R206Q) was eluted with 6 ml of equilibration buffer containing 10 mM L-asparagine. Collected fractions (1 ml) were assayed for L-ASNase activity and protein (A280 or Bradford [37]). The results from a typical purification run showed 75.4-fold purification with a 95.3% yield for R206Q, 119.7-fold purification with a 96.5% yield for the R206H mutant, and 110-fold purification with a 97% yield for K30A.
Modification of the EcaL-ASNase and R206H mutant with mPEG-SNHS and purification of the PEGylated enzymes
Purified L-ASNase (EcaL-ASNase and/or its mutant, R206H) (25 μg), in 50 mM potassium phosphate buffer, pH 8.5, was mixed with different amounts (0.28, 0.7, 1.15, 2.14 and 4.28 mg) of mPEG-SNHS in order to determine the amount necessary for complete modification. Modification reactions were allowed to proceed for 22 h at 4 °C before they were stopped by the addition of 12 mM glycine, pH 8. The effect of PEGylation on enzyme activity was evaluated by taking samples at various times during the modification reaction and measuring the residual activity, using the coupled enzyme assay method described above. The purification of PEG–EcaL-ASNase and/or PEG–R206H was carried out using size-exclusion chromatography on a Sephadex G-25 column (9.1 ml, 5 cm long×1.5 cm internal diameter), equilibrated with 10 mM Tris/HCl buffer, pH 7.4, and eluted with the same buffer at a flow rate of 0.5 ml/min. The samples were analysed by SDS/PAGE and MALDI–TOF (matrix-assisted laser-desorption ionization–time-of-flight) MS as described below. Residues’ accessibility was determined in silico using the WHAT IF software package [40].
Succinylation
EcaL-ASNase was succinylated with succinic anhydride as described in [41] with the following modifications: succinic anhydride crystals were added to a 10 mM final concentration of EcaL-ASNase over a period of 1 h to a solution of 0.2 mg of the enzyme in 0.1 M potassium phosphate buffer, pH 8.5, at 4 °C. The solution was constantly stirred and maintained at pH 8.5 by the addition of 0.01 M NaOH. The modification reaction was allowed to proceed for 6 h at 4 °C, before it was stopped by the addition of 12 mM glycine, pH 9. The extent of reaction with lysine residues was determined from titration of free amino groups with trinitrobenzenesulfonic acid [42].
Limited proteolysis with trypsin of R206H and PEGylated enzymes
R206H, PEG–EcaL-ASNase and PEG–R206H, previously equilibrated in 10 mM Tris/HCl, pH 7.4, were subjected to trypsin digestion. Proteolysis was carried out using a L-ASNase/trypsin ratio of 1:35 (w/w). All samples were freeze-dried and then analysed by SDS/PAGE (see below). Gels were run with a Laemmli buffer system [43]. The intensities of bands were quantified with the 1D Image analysis software version 3.5 (Kodak) analysis software. The intensity change against time was fitted to the first-order rate eqn (1):
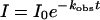 |
(1) |
where I is the intensity at time t, I0 is the intensity at t=0, and kobs is a first-order rate constant.
Thermal stability of EcaL-ASNase, R206H and PEGylated enzymes
Irreversible thermal inactivation of EcaL-ASNase, R206H and PEGylated enzymes was monitored by activity measurements. Samples of the enzymes, in 50 mM potassium phosphate buffer, pH 8.5, were incubated at different temperatures (35–50 °C) for 7.5 min. Subsequently, the samples were assayed for residual activity, using the coupled enzyme assay method described above. The Tm (temperature at which 50% of the initial enzyme activity is lost after heat treatment) values were determined from the plots of relative inactivation (%) against temperature (°C).
The time course of thermal inactivation of EcaL-ASNase, R206H and PEGylated enzymes, in 50 mM potassium phosphate buffer, pH 8.5, was studied at 37 °C. The rate of inactivation was followed by periodically removing samples for assay of enzymatic activity. Observed rates of inactivation (kin) were deduced from plots of log (% of remaining activity) against time (h). Thermal inactivation experiments were repeated three times to verify the results.
MALDI–TOF MS
The samples were desalted with ZipTip μ-C18 (Millipore). A saturated solution of sinapinic acid in water/acetonitrile (1:1, v/v) and 0.1% trifluoroacetic acid was used as the matrix solution. A 1 μl aliquot of the sample and matrix mixture was spotted on to a well of the sample plate and dried on the sample holder. Mass spectra were taken with a Voyager System 6322 (Applied Biosystems). Mass calibration was performed using BSA, apomyoglobin and cytochrome c.
N-terminal amino acid sequence analysis
N-terminal amino acid sequence analysis of the proteolytic products resulting from the trypsin digestion of EcaL-ASNase was carried out on a gas-phase Applied 542 Biosystems Protein sequencer, model 470A, equipped with an online phenylthiohydantoin analyser.
Electrophoresis
SDS/PAGE was performed according to the method of Laemmli [43] on a slab gel containing 12.5% (w/v) polyacrylamide (running gel) and 2.5% (w/v) stacking gel. The protein bands were stained with Coomassie Brilliant Blue R-250 or with silver nitrate.
RESULTS AND DISCUSSION
Limited proteolysis of EcaL-ASNase
To obtain information about the protease-sensitive cleavage sites in EcaL-ASNase, limited proteolysis with trypsin was carried out. To follow the time course of formation and disappearance of proteolytic fragments, samples were withdrawn from the tryptic digestion mixture at intervals and subjected to SDS/PAGE for separation of the proteolytic products (Figure 1A). After 30 min of incubation with trypsin, one main fragment (∼31.5 kDa) was easily identifiable by SDS/PAGE. As the digestion proceeded, the amount of this fragment decreased progressively over time, with concomitant formation of another smaller fragment with a molecular mass corresponding to ∼25 kDa. N-terminal amino acid sequencing analysis of the two fragments gave two sequences, Gly-Glu-Gln-Val-Ala and Ser-Val-Phe-Asp-Val, corresponding to residues 54–58 and 207–211 of EcaL-ASNase for the 31.5 and 25 kDa fragments respectively. Trypsin cleavage resulted in peptides with C-terminal lysine or arginine residues. These results indicate that trypsin initially cleaves EcaL-ASNase at Lys53-Gly54, and then the C-terminal fragment, peptide 54–327, is cleaved again at Arg206-Ser207. The loss of enzyme activity following trypsin treatment is a direct result of this cleavage, since the peptide fragment released contains the main part of the active site of the enzyme [35].
Figure 1. Proteolysis of EcaL-ASNase.

(A) Time course of tryptic digestion of EcaL-ASNase. Analysis was carried out using SDS/PAGE. Proteolysis was initiated by adding trypsin to the enzyme at an L-ASNase/trypsin ratio of 1:35 (w/w). Conditions are shown above lanes. A sample taken immediately after the addition of trypsin to EcaL-ASNase was used as control. The experiment was performed three times to verify the results. Molecular masses are shown in kDa. (B) Structural representations of limited proteolysis sites and critical PEGylation site. Lys30, Lys53 and Arg206 are shown in space-filling mode and are labelled. The Figure was created based on the crystal structure of EcaL-ASNase (PDB code 1ZCF) [29].
The locations of the two cleavage sites in EcaL-ASNase are illustrated in Figure 1(B). Both cleavage sites are positioned at the external surface of EcaL-ASNase. In particular, Lys53 is located at the short β-sheet formed by residues Asn51–Val57, and Arg206 is located at a flexible segment, formed by Asp201–Asp221, which connects the N-terminal and C-terminal domains (Figure 1B).
Our results thus showed that, in order to engineer resistance to trypsin cleavage into EcaL-ASNase, two main amino acid targets should be considered: Lys53 and Arg206. Taking into account that Lys53 is located at a surface accessible to the solvent and that this residue can be efficiently protected through chemical modification with succinimide esters of PEG, Arg206 was selected for site-directed mutagenesis.
Site-directed mutagenesis of EcaL-ASNase
On the basis of limited proteolysis and computational analysis studies, Arg206 was selected for site-directed mutagenesis. This residue was mutated to the conserved residues histidine and glutamine, which are not recognized targets of trypsin. Kinetic analysis of the mutants was carried out to evaluate the effect of each mutation on substrate binding and catalysis, and the results are given in Table 1. R206H showed differences in Km and specific activity values for L-asparagine when compared with the wild-type enzyme. In particular, mutant R206H showed an approx. 6.4-fold increase in Km for L-asparagine and a slight decrease in specific activity (∼9%) compared with the wild-type enzyme. R206Q gave low expression levels and worse kinetic properties compared with R206H (results not shown), thus it was not considered further.
Table 1. Kinetic parameters of PEGylated and non-PEGylated enzymes.
Steady-state kinetic measurements were performed at 37 °C. All initial velocities were determined in triplicate. The kinetic parameter Km was calculated by non-linear regression analysis of experimental steady-state data using the GraFit program. The specific activity was estimated relative to the wild-type EcaL-ASNase [35].
| Substrates | Enzymes | Specific activity (% of EcaL-ASNase) | Km (mM) |
|---|---|---|---|
| L-Asparagine | EcaL-ASNase | 100 | 0.085±0.02* |
| R206H | 90.9±1.9 | 0.55±0.09 | |
| PEG–EcaL-ASNase | 78.0±5.0 | 0.31±0.05 | |
| PEG–R206H | 74.5±2.7 | 0.36±0.07 | |
| L-Glutamine | EcaL-ASNase | 100 | 6.80±1.32* |
| R206H | 137.5±3.5 | 4.94±0.58 | |
| PEG–EcaL-ASNase | 92.0±2.6 | 24.69±5.55 | |
| PEG–R206H | 83.6±1.7 | 24.84±4.90 |
*Data from Kotzia and Labrou [35].
The mutant R206H was subjected to limited proteolysis with trypsin, under the same conditions as those used for the wild-type enzyme, in order to verify the absence of the ∼25 kDa fragment resulting from trypsin digestion. SDS/PAGE analysis of the tryptic digestion mixture showed that, after 90 min of incubation, no band corresponding to ∼25 kDa was formed (Figure 2).
Figure 2. Time course of tryptic digestion of R206H mutant enzyme.

Analysis was carried out using SDS/PAGE. Proteolysis was initiated by adding trypsin to the mutant enzyme at R206H/trypsin ratio of 1:35 (w/w). Conditions are shown above lanes. A sample taken immediately after the addition of trypsin to the R206H mutant enzyme was used as a control. The experiment was performed three times to verify the results. Molecular masses are shown in kDa.
PEGylation of EcaL-ASNase and the R206H mutant enzyme
PEGylation has become one of the most extensively used techniques, and has attracted increasing attention for enhancement of the therapeutic potential of protein-based pharmaceuticals [18–25]. Recent advances in recombinant technology and the development of functional group-selective reactive PEG derivatives have led to the creation of PEGylated proteins or mutants with markedly enhanced in vitro and in vivo characteristics and with minimal loss of bioactivity compared with their unmodified equivalents [13].
Various factors, including inherent nucleophilicity, solvation, protein conformation and also local electronic and pKa effects, determine the actual reactivity of the potential PEGylation sites [44]. The reaction chemistry and the length of PEG chains are the minimum set of variables that can determine the biological activity and stability of the modified protein [7,13]. In the present study, the N-hydroxysuccinamide derivative of PEG (mPEG-SNHS) was employed. mPEG-SNHS allows linkage between PEG and exposed lysine residues and/or the N-terminal amino group in proteins under conditions of mild pH and temperature [45]. This approach is suitable for EcaL-ASNase because the enzyme contains 20 lysine residues in addition to the N-terminal amino acid. None of these lysine residues is located in regions of the protein that are directly involved in substrate binding and catalysis [39].
Purified L-ASNases (wild-type and the R206H mutant) were subjected to PEGylation using different amounts (0.28–4.28 mg) of mPEG-SNHS in order to determine the optimum [enzyme]/[mPEG-SNHS] ratio for complete enzyme modification. The reactions were allowed to proceed for a long period of time (22 h, 4 °C) in order to make sure that complete PEGylation would occur. SDS/PAGE analysis of the products of the modification reactions showed that the amount of mPEG-SNHS necessary for complete modification of the enzymes was 4.28 mg of mPEG-SNHS/25 μg of protein. This ratio promotes the formation of a single modified form of the enzyme with subunit molecular mass of approx. 70.5 kDa, as determined by SDS/PAGE analysis (Figure 3, see ‘Control’ lanes). The other concentrations used gave a mixture of different forms with molecular masses between 36.5 and 70.5 kDa, owing to incomplete modification (results not shown). The lowest amount used, 0.28 mg of mPEG-SNHS, had no significant effect on the enzyme's molecular mass. MALDI–TOF MS was used to determine precisely the number of PEG molecules attached to the wild-type and mutant enzymes. Analysis of the mass spectra gave m/z ratios of 69492.3 and 69472.4 for the modified wild-type and mutant enzymes (subunit molecular mass) respectively. Given the molecular mass of PEG and L-ASNase, the molecular masses of the modified enzymes corresponded to hepta-PEG-modified L-ASNases.
Figure 3. Time course of tryptic digestion of PEGylated enzymes.

Analysis was carried out using SDS/PAGE. Conditions are shown above lanes. Samples taken immediately after the addition of trypsin to each purified PEGylated enzyme were used as controls. The experiment was performed three times to verify the results. Molecular masses are shown in kDa.
The effect of PEGylation (at 4.28 mg of mPEG-SNHS/25 μg of protein) on the enzyme activity of both the wild-type enzyme and the R206H mutant was evaluated by measuring the enzyme activity after the completion of the modification reaction. The PEG–EcaL-ASNase retained 78% of its initial activity and the PEG–R206H retained 82%. The tendency to lose activity can be attributed to the steric hindrance caused by the conjugation of PEG to the free amino groups on the protein surface, which prevents L-asparagine from approaching the active site of the enzyme [46].
The modification of EcaL-ASNase and R206H with mPEG-SNHS led to a 3.6–5-fold increase in Km for L-asparagine and L-glutamine. The increased Km for L-asparagine, although undesirable, is still within the range of acceptability for therapeutic applications [47]. On the other hand, the 5-fold increase in Km (24.8 mM) for L-glutamine is desirable, since the ability of therapeutic preparations of L-ASNases to hydrolyse L-glutamine has been implicated to cause several side effects [27].
To analyse whether the effect of PEGylation on Km (0.09±0.03 mM for L-asparagine) is the result of modification of a specific lysine residue or from the structure/chemistry of PEG, we employed protein chemistry and site-directed mutagenesis studies. Complete chemical modification of EcaL-ASNase by succinic anhydride results in a succinylated enzyme (15 residues succinylated per subunit) which has an unaltered Km, in agreement with the results reported by Shifrin and Grochowski [41]. It is apparent that extensive succinylation does not destroy enzyme function, and therefore this finding indicates that none of the lysine residues is essential for catalytic activity or substrate binding.
In silico analysis of residues' accessibility in EcaL-ASNase showed that the seven, out of 20, lysine residues that exhibit high accessible surface area are: Lys30, Lys45, Lys53, Lys145, Lys216, Lys257 and Lys269. The other lysine residues either are more buried or lie at the subunit interface. These seven residues are probably the targets of chemical modification by mPEG-SNHS. Analysis of the crystal structure of the enzyme showed that none of these lysine residues is located in regions of the protein implicated in substrate binding and catalysis, with the exception of Lys30, which lies at the flexible loop (residues 10–40, Figure 1B) near to the enzyme active site [29,31,35]. Site-directed mutagenesis of Lys30 to alanine gave a mutant enzyme with kinetic parameters very close to the wild-type enzyme (Km=0.075±0.03 mM for L-asparagine). Therefore the results from site-directed mutagenesis and from extensive chemical modification by succinic anhydride point to the conclusion that the influence of PEGylation on Km is an indirect effect and may be described as originating from the overall shielding effect of the PEG chains rather than from the modification of a specific lysine residue. This conclusion is also supported by the observation that R206H mutant enzyme, after PEGylation, exhibits Km values or L-asparagine and L-glutamine comparable with that of the wild-type enzyme.
Trypsin digestion of PEGylated enzymes
To evaluate the effect of modification with PEG on the rate of trypsinolysis of PEG–EcaL-ASNase and the PEG–R206H mutant enzyme, limited proteolysis experiments were carried out under the same conditions as those reported for the unmodified enzymes. SDS/PAGE analysis of the products of the trypsinolysis reaction showed that, after 90 min of reaction, trypsin had completely digested PEG–EcaL-ASNase, whereas PEG–R206H showed considerable resistance to proteolysis (Figure 3). The rate of breakdown of both enzymes was monitored by quantifying the amount of intact enzyme on SDS/PAGE gels as a function of incubation time. The decrease in intensity of the enzyme band was fitted to a first-order rate equation. The observed rates of digestion (kobs) were found to be 1.54±0.18 and 0.1±0.12 for PEG–EcaL-ASNase and PEG–R206H respectively. The protective effect of PEGylation has been described as originating from a shell of PEG chains surrounding the protein, with the shielding effect of the PEG chains contributing to the inaccessibility of the enzyme to trypsin [12,13].
Thermal stability of EcaL-ASNase, R206H and PEGylated enzymes
To determine the structural stability of EcaL-ASNase, R206H and the PEGylated enzymes, heat-inactivation studies were carried out. The enzymes were incubated at different temperatures and subsequently assayed for residual activity (Figure 4). The corresponding Tm values are listed in Table 2.
Figure 4. Thermal-inactivation curves of PEGylated and non-PEGylated enzymes.

The residual activities of EcaL-ASNase (●), R206H mutant enzyme (□), PEG–EcaL-ASNase (■) and PEG–R206H mutant enzyme (○) were measured after heat treatment at various temperatures for 7.5 min. Results are means±S.D. for three independent experiments.
Table 2. Tm (°C) and kin (h−1, at 37 °C) values of PEGylated and non-PEGylated enzymes.
| Enzyme | Tm (°C) | kin (×10−3 h−1) |
|---|---|---|
| EcaL-ASNase | 38.9±0.45 | 347.2±44.6 |
| R206H | 38.8±0.84 | 361.6±45.7 |
| PEG–EcaL-ASNase | 42.9±0.25 | 11.3±0.8 |
| PEG–R206H | 43.1±0.45 | 6.5±0.9 |
The time course of thermal inactivation of EcaL-ASNase, R206H and the PEGylated enzymes was also investigated at the temperature of therapeutic importance, i.e. 37 °C. The results indicated that the PEGylated enzymes are particularly stable at 37 °C, whereas the EcaL-ASNase and R206H mutant enzyme have significantly lower thermostability. Kinetic analysis of the thermal inactivation at 37 °C gave linear plots (Figure 5), from which the first-order inactivation rate constant kin could be calculated according to eqn (2):
 |
(2) |
The results are summarized in Table 2. The PEGylated enzymes showed a dramatic improvement in thermal stability at 37 °C, which is approx. 30.7- and 55.6-fold higher (for PEG–EcaL-ASNase and PEG–R206H respectively) than that for non-PEGylated enzymes. It is believed that PEGylation usually enhances the thermal stability of proteins by sterically blocking degradation pathways induced by hydrophobic interactions [48], and by causing non-specific steric hindrance of the intermolecular interactions that are involved in thermal instabilities [49].
Figure 5. Time course of inactivation of PEGylated and non-PEGylated enzymes at 37 °C.

Time course of inactivation at 37 °C for EcaL-ASNase (□), R206H mutant enzyme (▲), PEG–EcaL-ASNase (■) and PEG–R206H mutant enzyme (●). The rate of inactivation was followed by periodically removing samples for assay of enzymatic activity. Results are means±S.D. for three independent experiments.
Conclusion
A short half-life in serum is a common trait of most proteins being used or developed for therapeutic purposes. Proteolysis is being increasingly acknowledged to play a significant role in the clearance of therapeutic proteins. Thus the design of novel proteins that are resistant to digestion represents an important strategy for the creation of next-generation therapeutic products with improved pharmacological properties. In the present study, the EcaL-ASNase was genetically modified by site-directed mutagenesis and chemically modified with mPEG-SNHS. The modified form of the mutant enzyme (PEG–R206H) was found to have improved proteolytic stability, good catalytic activity for L-asparagine and high structural stability. This enzyme may be tested further on animals and/or humans in order to create a new drug for future therapeutic use.
Acknowledgments
This work was financially supported by the Hellenic General Secretariat for Research and Technology: Operational Program for Competitiveness, Joint Research and Technology Program.
References
- 1.Lee S. M., Wroble M. H., Ross J. T. L-Asparaginase from Erwinia carotovora: an improved recovery and purification process using affinity chromatography. Appl. Biochem. Biotechnol. 1989;22:1–11. doi: 10.1007/BF02922693. [DOI] [PubMed] [Google Scholar]
- 2.Keating M. J., Holmes R., Lerner S., Ho D. H. L-Asparaginase and PEG asparaginase: past, present, and future. Leuk. Lymphoma. 1993;10:153–157. doi: 10.3109/10428199309149129. [DOI] [PubMed] [Google Scholar]
- 3.Duval M., Suciu S., Ferster A., Rialland X., Nelken B., Lutz P., Benoit Y., Robert A., Manel A. M., Vilmer E., et al. Comparison of Escherichia coli-asparaginase with Erwinia-asparaginase in the treatment of childhood lymphoid malignancies: results of a randomized European Organization for Research and Treatment of Cancer-Children's Leukemia Group phase 3 trial. Blood. 2002;99:2734–2739. doi: 10.1182/blood.v99.8.2734. [DOI] [PubMed] [Google Scholar]
- 4.Mashburn L. T., Wriston J. C., Jr Tumor inhibitory effect of L-asparaginase from Escherichia coli. Arch. Biochem. Biophys. 1964;105:450–452. doi: 10.1016/0003-9861(64)90032-3. [DOI] [PubMed] [Google Scholar]
- 5.Marlborough D. I., Miller D. S., Cammack K. A. Comparative study on conformational stability and subunit interactions of two bacterial asparaginases. Biochim. Biophys. Acta. 1975;386:576–589. doi: 10.1016/0005-2795(75)90301-3. [DOI] [PubMed] [Google Scholar]
- 6.Moola Z. B., Scawen M. D., Atkinson T., Nicholls D. J. Erwinia chrysanthemi L-asparaginase: epitope mapping and production of antigenically modified enzymes. Biochem. J. 1994;302:921–927. doi: 10.1042/bj3020921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Soares A. L., Guimaraes G. M., Polakiewicz B., de Moraes Pitombo R. N., Abrahao-Neto J. Effects of polyethylene glycol attachment on physicochemical and biological stability of E. coli L-asparaginase. Int. J. Pharm. 2002;237:163–170. doi: 10.1016/s0378-5173(02)00046-7. [DOI] [PubMed] [Google Scholar]
- 8.Burnham N. L. Polymers for delivering peptides and proteins. Am. J. Hosp. Pharm. 1994;51:210–218. [PubMed] [Google Scholar]
- 9.Veronese F. M. Peptide and protein PEGylation: a review of problems and solutions. Biomaterials. 2001;22:405–417. doi: 10.1016/s0142-9612(00)00193-9. [DOI] [PubMed] [Google Scholar]
- 10.Veronese F. M., Pasut G. PEGylation: successful approach to drug delivery. Drug Discov. Today. 2005;10:1451–1458. doi: 10.1016/S1359-6446(05)03575-0. [DOI] [PubMed] [Google Scholar]
- 11.Matsushima A., Nishimura H., Ashihara Y., Yokota Y., Inada Y. Modification of E. coli asparaginase with 2,4-bis(O-methoxypolyethyleneglycol)-6-chloro-S-triazine (activated PEG2) Chem. Lett. 1980;103:773–776. [Google Scholar]
- 12.Veronese F. M., Morpurgo M. Bioconjugation in pharmaceutical chemistry. Farmaco. 1999;54:497–516. doi: 10.1016/s0014-827x(99)00066-x. [DOI] [PubMed] [Google Scholar]
- 13.Harris J. M., Chess R. B. Effect of PEGylation on pharmaceuticals. Nat. Rev. Drug Discov. 2003;2:214–221. doi: 10.1038/nrd1033. [DOI] [PubMed] [Google Scholar]
- 14.Inada Y., Furukawa M., Sasaki H., Kodera Y., Hiroto M., Nishimura H., Matsushima A. Biomedical and biotechnological applications of PEG- and PM-modified proteins. Trends Biotechnol. 1995;13:86–91. doi: 10.1016/S0167-7799(00)88912-X. [DOI] [PubMed] [Google Scholar]
- 15.Matsushima A., Kodera Y., Hiroto M., Nishimura H., Inada Y. Bioconjugates of proteins and polyethylene glycol: potent tools in biotechnological processes. J. Mol. Catal. B: Enzym. 1996;2:1–17. [Google Scholar]
- 16.Bonora G. M., Ivanova E., Zarytova V., Burcovich B., Veronese F. M. Synthesis and characterization of high-molecular mass polyethylene glycoL-conjugated oligonucleotides. Bioconjug. Chem. 1997;8:793–797. doi: 10.1021/bc970082p. [DOI] [PubMed] [Google Scholar]
- 17.Baran E. T., Ozer N., Hasirci V. Solid-phase enzyme modification via affinity chromatography. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2003;794:311–322. doi: 10.1016/s1570-0232(03)00487-2. [DOI] [PubMed] [Google Scholar]
- 18.Hershfield M. S. PEG-ADA: an alternative to haploidentical bone marrow transplantation and an adjunct to gene therapy for adenosine deaminase deficiency. Hum. Mutat. 1995;5:107–112. doi: 10.1002/humu.1380050202. [DOI] [PubMed] [Google Scholar]
- 19.Beauchamp C. O., Gonias S. L., Menapace D. P., Pizzo S. V. A. A new procedure for the synthesis of polyethylene glycol–protein adducts: effects on function, receptor recognition, and clearance of superoxide dismutase, lactoferrin, and α2-macroglobulin. Anal. Biochem. 1983;131:25–33. doi: 10.1016/0003-2697(83)90131-8. [DOI] [PubMed] [Google Scholar]
- 20.Kodera Y., Sekine T., Yasukohchi T., Kiriu Y., Hiroto M., Matsushima A., Inada Y. Stabilization of L-asparaginase modified with comb-shaped poly(ethylene glycol) derivatives, in vivo and in vitro. Bioconjug. Chem. 1994;5:283–286. doi: 10.1021/bc00028a001. [DOI] [PubMed] [Google Scholar]
- 21.Kajihara J., Shibata K., Nakano Y., Nishimuro S., Kato K. Physicochemical characterization of PEG–PPG conjugated human urokinase. Biochim. Biophys. Acta. 1994;1199:202–208. doi: 10.1016/0304-4165(94)90116-3. [DOI] [PubMed] [Google Scholar]
- 22.Humphries J., Lattimer C., Smith A., McGuinness C. L., Whitton C., Gaffney P. J., Burnand K. G. High and constant plasma levels of tissue plasminogen activator and PEG–hirudin can be achieved by subcutaneous delivery. Thromb. Res. 1997;87:123–129. doi: 10.1016/s0049-3848(97)00111-4. [DOI] [PubMed] [Google Scholar]
- 23.Ajisaka K., Iwashita Y. Modification of human hemoglobin with polyethylene glycol: a new candidate for blood substitute. Biochem. Biophys. Res. Commun. 1980;97:1076–1081. doi: 10.1016/0006-291x(80)91485-0. [DOI] [PubMed] [Google Scholar]
- 24.Wedemeyer H., Wiegand J., Cornberg M., Manns M. P. Polyethylene glycol–interferon: current status in hepatitis C virus therapy. J. Gastroenterol. Hepatol. 2002;17:344–350. doi: 10.1046/j.1440-1746.17.s3.26.x. [DOI] [PubMed] [Google Scholar]
- 25.Pavlou A. K., Reichert J. M. Recombinant protein therapeutics: success rates, market trends and values to 2010. Nat. Biotechnol. 2004;22:1513–1519. doi: 10.1038/nbt1204-1513. [DOI] [PubMed] [Google Scholar]
- 26.Howard J. B., Carpenter F. H. L-Asparaginase from Erwinia carotovora: substrate specificity and enzymatic properties. J. Biol. Chem. 1972;247:1020–1030. [PubMed] [Google Scholar]
- 27.Hawkins D. S., Park J. R., Thomson B. G., Felgenhauer J. L., Holcenberg J. S., Panosyan E. H., Avramis V. I. Asparaginase pharmacokinetics after intensive polyethylene glycol-conjugated L-asparaginase therapy for children with relapsed acute lymphoblastic leukemia. Clin. Cancer Res. 2004;10:5335–5341. doi: 10.1158/1078-0432.CCR-04-0222. [DOI] [PubMed] [Google Scholar]
- 28.Krasotkina J., Borisova A. A., Gervaziev Y. V., Sokolov N. N. One-step purification and kinetic properties of the recombinant L-asparaginase from Erwinia carotovora. Biotechnol. Appl. Biochem. 2004;39:215–221. doi: 10.1042/BA20030138. [DOI] [PubMed] [Google Scholar]
- 29.Wikman L. E., Krasotkina J., Kuchumova A., Sokolov N. N., Papageorgiou A. C. Crystallization and preliminary crystallographic analysis of L-asparaginase from Erwinia carotovora. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2005;61:407–409. doi: 10.1107/S1744309105008249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aghaiypour K., Wlodawer A., Lubkowski J. Do bacterial L-asparaginases utilize a catalytic triad Thr-Tyr-Glu? Biochim. Biophys. Acta. 2001;1550:117–128. doi: 10.1016/s0167-4838(01)00270-9. [DOI] [PubMed] [Google Scholar]
- 31.Aghaiypour K., Wlodawer A., Lubkowski J. Structural basis for the activity and substrate specificity of Erwinia chrysanthemi L-asparaginase. Biochemistry. 2001;40:5655–5664. doi: 10.1021/bi0029595. [DOI] [PubMed] [Google Scholar]
- 32.Kozak M., Borek D., Janowski R., Jaskolski M. Crystallization and preliminary crystallographic studies of five crystal forms of Escherichia coli L-asparaginase II (Asp90Glu mutant) Acta Crystallogr. Sect. D Biol. Crystallogr. 2002;58:130–132. doi: 10.1107/s0907444901016663. [DOI] [PubMed] [Google Scholar]
- 33.Shimizu T., Yamashiro Y., Igarashi J., Fujita H., Ishimoto K. Increased serum trypsin and elastase-1 levels in patients undergoing L-asparaginase therapy. Eur. J. Pediatr. 1998;157:561–563. doi: 10.1007/s004310050878. [DOI] [PubMed] [Google Scholar]
- 34.Guo L., Wang J., Qian S., Yan X., Chen R., Meng G. Construction and structural modeling of a single-chain Fv-asparaginase fusion protein resistant to proteolysis. Biotechnol. Bioeng. 2000;70:456–463. [PubMed] [Google Scholar]
- 35.Kotzia G. A., Labrou N. E. Cloning, expression and characterisation of Erwinia carotovora L-asparaginase. J. Biotechnol. 2005;119:309–323. doi: 10.1016/j.jbiotec.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 36.Balcao V. M., Mateo C., Fernandez-Lafuente R., Malcata F. X., Guisan J. M. Structural and functional stabilization of L-asparaginase via multisubunit immobilization onto highly activated supports. Biotechnol. Prog. 2001;17:537–542. doi: 10.1021/bp000163r. [DOI] [PubMed] [Google Scholar]
- 37.Bradford M. A. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 38. Reference deleted.
- 39.Ho S. N., Hunt H. D., Horton R. M., Pullen J. K., Pease L. R. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 40.Vriend G. WHAT IF: a molecular modeling and drug design program. J. Mol. Graphics. 1990;8:52–56, 29. doi: 10.1016/0263-7855(90)80070-v. [DOI] [PubMed] [Google Scholar]
- 41.Shifrin S., Grochowski B. J. L-Asparaginase from Escherichia coli B: succinylation and subunit interactions. J. Biol. Chem. 1972;247:1048–1054. [PubMed] [Google Scholar]
- 42.Snyder S. L., Sobocinski P. Z. An improved 2,4,6-trinitrobenzenesulfonic acid method for the determination of amines. Anal. Biochem. 1975;64:284–288. doi: 10.1016/0003-2697(75)90431-5. [DOI] [PubMed] [Google Scholar]
- 43.Laemmli U. K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 44.Roberts M. J., Bentley M. D., Harris J. M. Chemistry for peptide and protein PEGylation. Adv. Drug Delivery Rev. 2002;54:459–476. doi: 10.1016/s0169-409x(02)00022-4. [DOI] [PubMed] [Google Scholar]
- 45.Matsuyama H., Taguchi R., Ikezawa H. Phospholipase D modified with a polyethylene glycol derivative. Chem. Pharm. Bull. (Tokyo) 1991;39:743–746. doi: 10.1248/cpb.39.743. [DOI] [PubMed] [Google Scholar]
- 46.Zhang J. F., Shi L. Y., Wei D. Z. Chemical modification of L-asparaginase from Escherichia coli with a modified polyethyleneglycol under substrate protection conditions. Biotechnol. Lett. 2004;26:753–756. doi: 10.1023/b:bile.0000024100.49716.3d. [DOI] [PubMed] [Google Scholar]
- 47.Asselin B. L., Lorenson M. Y., Whitin J. C., Coppola D. J., Kende A. S., Blakley R. L., Cohen H. J. Measurement of serum L-asparagine in the presence of L-asparaginase requires the presence of an L-asparaginase inhibitor. Cancer Res. 1991;51:6568–6573. [PubMed] [Google Scholar]
- 48.Bailon P., Berthold W. Poly(ethylene glycol)-conjugated pharmaceutical proteins. Pharm. Sci. Technol. Today. 1998;1:352–356. [Google Scholar]
- 49.Hinds K. D., Kim S. W. Effects of PEG conjugation on insulin properties. Adv. Drug Deliv. Rev. 2002;54:505–530. doi: 10.1016/s0169-409x(02)00025-x. [DOI] [PubMed] [Google Scholar]