

Abstract
The periplasmic iron-binding protein, FbpA (ferric-ion-binding protein A), performs an essential role in iron acquisition from transferrin in Haemophilus influenzae. A series of site-directed mutants in the metal-binding amino acids of FbpA were prepared to determine their relative contribution to iron binding and transport. Structural studies demonstrated that the mutant proteins crystallized in an open conformation with the iron atom associated with the C-terminal domain. The iron-binding properties of the mutant proteins were assessed by several assays, including a novel competitive iron-binding assay. The relative ability of the proteins to compete for iron was pH dependent, with a rank order at pH 6.5 of wild-type, Q58L, H9Q>H9A, E57A>Y195A, Y196A. The genes encoding the mutant FbpA were introduced into H. influenzae and the resulting strains varied in the level of ferric citrate required to support growth on iron-limited medium, suggesting a rank order for metal-binding affinities under physiological conditions comparable with the competitive binding assay at pH 6.5 (wild-type=Q58L>H9Q>H9A, E57A>Y195A, Y196A). Growth dependence on human transferrin was only obtained with cells expressing wild-type, Q58L or H9Q FbpAs, proteins with stability constants derived from the competition assay >2.0×1018 M−1. These results suggest that a relatively high affinity of iron binding by FbpA is required for removal of iron from transferrin and its transport across the outer membrane.
Keywords: ferric-ion-binding protein A (FbpA), Haemophilus influenzae, iron-binding protein, iron transport, transferrin
Abbreviations: FbpA, ferric-ion-binding protein A; IPTG, isopropyl β-D-thiogalactoside; LA, Luria agar; PEG, poly(ethylene glycol); Tbp, transferrin-binding protein; WT, wild-type
INTRODUCTION
Due to the essential role that iron plays in biological systems, higher organisms have developed elaborate mechanisms for its procurement and internal distribution [1]. Vertebrates utilize the glycoprotein transferrin for distribution of iron throughout the body, and mammals produce a related glycoprotein, lactoferrin, capable of complexing iron on mucosal surfaces and at sites of infection.
Structural analysis of lactoferrin and transferrin revealed that they possess a conserved structure, consisting of two globular lobes, each containing an iron-binding site located in a cleft between two domains that comprise the individual lobes, with ligands on either side of the cleft participating in iron co-ordination [2]. There is a substantial movement of the individual domains relative to one another upon iron release, which has been described as rigid-body ‘jaw-like’ movement between ‘open’ (apo) and ‘closed’ (iron-bound) states [3,4].
The iron-sequestering effects of transferrin and lactoferrin restrict the availability of iron for bacterial pathogens so that they require high-affinity transport mechanisms for growth and survival within the host [5]. Pathogenic bacteria of the Neisseriaceae and Pasteurellaceae have adapted to this environment by utilizing a mechanism of iron acquisition involving direct binding of the host transferrin by a cell-surface receptor that mediates iron removal and transport across the outer membrane [6,7]. The receptor is comprised of two outer-membrane proteins, transferrin-binding proteins A and B (TbpA and TbpB), which exclusively bind to transferrin from the host. Transport of iron across the outer membrane is dependent on energization by an inner membrane complex composed of TonB and the associated proteins ExbB and ExbD [8,9].
There have been extensive studies on the interaction between TonB and TonB-dependent outer-membrane receptors, including recent structural studies with complexes of a TonB subfragment and BtuB and FhuA [10,11]. Nevertheless, it has not been fully resolved whether the mechanism of substrate translocation involves removal of the N-terminal plug domain from the C-terminal barrel of the receptor or whether a channel for transport is provided by conformation changes in the plug domain [12].
Transport of iron from the periplasmic space into the cell interior is mediated by an ABC-type ferric-iron-transporter system, comprising a periplasmic ferric-binding protein and an inner-membrane permease complex, consisting of transmembrane and ATPase components [13]. Mutants lacking the periplasmic iron-binding protein, FbpA (ferric-ion-binding protein A), are deficient in iron acquisition from transferrin, lactoferrin and other ferric iron sources [14,15]. The periplasmic iron-binding protein is approximately the size of a single lobe of transferrin or lactoferrin and has a similar binding affinity for iron [16]. The structure of FbpA from Haemophilus influenzae has been determined and, to a large degree, it resembles a single lobe of transferrin or lactoferrin [17]. In analogy with the transferrins, there are four amino acids (His, Glu, Tyr and Tyr) that act as ligands for complexing the iron atom. The anion completing the octahedral co-ordination complex is a phosphate in place of the carbonate anion present in transferrin. The transition between the iron-loaded and apo forms of the protein involves movement of relatively rigid domains [18] which are reminiscent of the conformational changes observed in transferrins and lactoferrins.
The process of removal of iron from transferrin and its subsequent transport of iron across the outer membrane has not been fully characterized. As with other TonB-dependent uptake systems, it is quite probable that TonB plays a major role in mediating the transport of iron across the outer membrane, but its contribution to the iron removal process is less clear. Although it is evident that FbpA is ultimately required for transport of iron into the cell [14,15], it has not been established whether it plays a role in the transport of iron across the outer membrane. The present study was established to address this question by the preparation and biochemical analyses of a series of site-directed mutants of FbpA from H. influenzae that were tested for their ability to reconstitute the iron uptake pathway.
EXPERIMENTAL
Strains, media and growth assays
Escherichia coli strain BL21(DE3)/pLysS and the pT7-7 vector [19] were used for the expression and production of the WT (wild-type) and mutant FbpAs. H. influenzae strain DL63 (from Professor Eric Hansen, Department of Microbiology, University of Texas Southwestern Medical Center, Dallas, TX, U.S.A.; H36 in our strain collection) was the parent strain for the mutants prepared for the present study. H. influenzae and E. coli strains were both stored in 30% glycerol at −70 °C. Fresh H. influenzae cultures were grown on chocolate agar plates and incubated overnight at 37 °C in a 5% CO2 atmosphere. Fresh E. coli cultures were prepared on LA (Luria agar) plates (Gibco BRL) supplemented with 100 μg/ml ampicillin (Sigma). Liquid cultures of H. influenzae were routinely prepared in BNH broth [BHI broth base (Difco), 3.32 μg/ml NAD+ (Sigma) and 10 μg/ml haemin (Sigma)]. Liquid E. coli cultures were prepared in LA broth (Luria broth base and 100 μg/ml ampicillin). H. influenzae strains were tested for their ability to use holo human transferrin, holo human lactoferrin, haemin and ferric citrate for growth on iron-limited anaerobic agar medium, buffered with 50 mM Hepes (pH 7.4; Sigma), as described previously [14].
Construction of site-directed FbpA mutants
The fbpA gene was amplified by PCR with primers 369 (5′-CAGGCTTAACTGAATAATTTGCAC-3′) and 370 (5′-AACATTTCAGTTCACGCACG-3′) using genomic DNA from H. influenzae strain DL63 (H36) as a template and cloned into the pCR2 vector, and the resulting EcoR1 fragment was cloned into the EcoR1 site of the pT7-7 vector. The resulting expression plasmid contained the native gene with its original ribosome-binding site downstream of the T7 promoter and an artificial promoter generated from the cloning process, and also included a portion of the intergenic region between fbpA and fbpB. The various site-directed mutants of the H. influenzae fbpA gene were prepared either by the Stratagene QuikChange® site-directed mutagenesis kit or by a two-step PCR mutagenesis protocol using this expression plasmid as the starting template. Sequence analysis confirmed that the desired mutations were the only changes in the coding sequence.
A gene replacement vector for H. influenzae was prepared by cloning a 1.26 kb region immediately upstream of the fbpA gene adjacent to a 1.36 kb region containing the fbpAB intergenic region and a portion of the coding sequence for the fbpB gene into the pUC4K vector backbone. Restriction sites added between these two regions were used to insert a chloramphenicol-resistance cassette, which was derived from the pTnMax4 plasmid [20], or the kanamycin-resistance determinant, which was derived from the pUC4K plasmid. The WT or mutant H. influenzae fbpA genes were subcloned from the pT7-7 vector immediately upstream of the kanamycin-resistance cassette in the gene replacement vector. The plasmids were linearized by digestion with Sst1 and used directly to transform H. influenzae using M-IV-competence-inducing medium [21]. The chloramphenicol-resistant strain, H306, was generated by transformation with the plasmid containing the chloramphenicol-resistant determinant and was used as a parent for all subsequent transformations with the fbpA genes.
Expression and purification of FbpA proteins
The FbpA proteins were expressed from a recombinant pT7-7 plasmid after induction with 0.5 mM IPTG (isopropyl β-D-thiogalactoside) in the E. coli BL21(DE3)/pLysS host strain with protein overexpression at 25 °C overnight (see Figure 1). A modified osmotic shock procedure provided relatively pure preparations of protein [22]. For crystallization studies and spectral studies with small molecular chelators, the proteins were further purified by cation-exchange chromatography on a BioCad HPLC system after extenisive dialysis against 10 mM Tris/HCl (pH 8.0) at 4 °C. The column was washed with 10 vol of 20 mM Tris/HCl (pH 7.5) to remove unbound proteins, and the bound FbpAs were eluted with a gradient of 0–1.5 M NaCl. For the competition assays, the FbpAs were purified from the osmotic shock fluid by anion-exchange chromatography with Q-Sepharose (Amersham Biosciences). After concentration, the samples were dialysed extensively against 10 mM Tris/HCl (pH 8.0) at 4 °C. Following dialysis, the samples were then concentrated using Microsep™ 10 K microconcentrators to 10 mg/ml.
Figure 1. Pellets of E. coli strain BL21(DE3) expressing different FbpA proteins.

E. coli BL21(DE3) were freshly transformed with a pT7-7 plasmid carrying WT or mutant H. influenzae fbpA genes for expression of the indicated FbpA protein (WT, Q58L, H9Q, H9A, E57A, Y196A or Y195A). The cultures were corrected to A600=1.0 and the cells from the cultures were collected by centrifugation prior to photographing (A, B, D and E). (A and D) Bacterial pellets from cultures with exogenous iron added for 2 h before centrifugation. (B and E) Bacterial pellets from cultures without exogenous iron added. Periplasmic proteins were isolated from pellets in (B) and (E) by the modified osmotic procedure described in the Experimental section and a 10 μl sample of the periplasmic fraction was subjected to SDS/PAGE and stained with Coomassie Blue. The results are shown in (C) and (F).
Iron removal, addition of phosphate anion and iron-loading were all accomplished by additions to the concentrated sample followed by several rounds of dilution with the standard buffer (10 mM Tris/HCl, pH 8.0) and concentration with the microconcentrator. EDTA was added to give a final concentration of 1 mM and sodium citrate was added at a 4000-fold molar excess in order to generate the apo form of the protein. Sodium phosphate was added to give a concentration of 5 mM phosphate to ensure that there was sufficient anion to occupy the synergistic anion site. After buffer exchange and concentration, samples were retained in the apo form. For iron-loading of the proteins, a 5-fold molar excess of iron was provided by addition of freshly prepared ferric citrate solutions [ferric chloride, FeCl3·6H2O (Amachem), dissolved in 100 mM sodium citrate/100 mM sodium bicarbonate buffer]. After buffer exchange and concentration, samples were retained as the iron-loaded form of the proteins for spectral and crystallization studies.
Crystallization of E57A FbpA
Crystallization was performed as described previously [22], with purified protein preparations concentrated to >30 mg/ml. Crystals of E57A FbpA were grown at 4 °C by the hanging-drop technique from 4 μl drops containing 15 mg/ml protein, 18% PEG [poly(ethylene glycol)] 550 monomethyl ether and 0.05 M Tris/HCl (pH 8.5). The drops were equilibrated against a 1 ml reservoir containing 36% PEG 550 monomethyl ether and 0.1 M Tris/HCl (pH 8.5). Diffraction-quality pink crystals typically grew after 4 days. For cryocrystallography, the crystals were placed in a cryoprotectant solution identical with the reservoir solution with a final concentration of 20% (v/v) ethylene glycol for a short time before being placed in the nitrogen gas stream to be cooled to 100 K.
Data measurement and structure solution
Initial X-ray analysis revealed that the crystals belonged to the orthorhombic space group P21212 with unit cell dimensions of a=106.06 Å, b=75.23 Å, c=33.37 Å, a=b=γ=90o for E57A FbpA. The recording of crystallographic data and the approaches and programs used in determining the high-resolution E57A FbpA structure were identical with those described previously [23]. The refined co-ordinates of H. influenzae E57A FbpA have been deposited in the Protein Data Bank under accession code 2O6A.
Spectral studies
Scans for UV–visible spectra (310–600 nm) were performed on the iron-loaded WT and mutant FbpA proteins using the wavescan software package provided with the Ultrospec 2000 (Pharmacia Biotech). All experiments were performed in 1.5 ml quartz cuvettes, maintained at 20 °C. Calculations were conducted using the visible absorbance maxima for each respective protein.
The rate of iron removal from iron-loaded proteins exposed to a 100-fold molar excess of EDTA was determined as described previously [24]. The time of mixing in the EDTA solution was recorded as time zero. The difference spectra were derived by subtracting a spectrum of the apo form of the protein from the spectrum recorded from the sample cuvette.
A citrate competition assay was used to estimate the iron-binding affinity (logKS1) as described previously [25]. Iron-loaded samples of FbpA at a concentration of 50 μM in 10 mM Tris/HCl (pH 8.0) buffer were exposed to increasing concentrations of citrate (0–384 mM, final concentration) and each absorbance was recorded after incubation for 20 min at room temperature.
Competitive iron-binding assay
Iron-loaded FbpA was coupled to activated Sepharose by standard methods (10 mg of FbpA/ml of resin) and then exposed to a 5:1 molar excess of iron from a freshly prepared solution of FeCl3·6H2O (Amachem) dissolved in 0.1 M sodium citrate/0.1 M sodium bicarbonate (pH 7.0) to ensure iron saturation of the immobilized FbpA. The column preparation was exposed to 80 mg of purified bovine lactoferrin (Agennix Inc., Houston, TX, U.S.A.) dissolved in 50 mM Tris/150 mM NaCl (pH 8.0) buffer to remove the non-FbpA-associated iron and washed with 3 column volumes of buffer alone before and after the lactoferrin treatment. The column preparations were shown to be free of bovine lactoferrin by Western-blot analysis of samples obtained by treating resin with SDS/PAGE sample buffer. A final wash with 50 mM Tris/150 mM NaCl (pH 6.5) was performed for experiments to evaluate exchange at pH 6.5.
The apo forms of the various FbpAs were prepared by adding 0.1 M citrate/0.1 M bicarbonate (pH 7.0) buffer at 2200:1 molar excess (mol of citrate/mol of FbpA), concentrating the protein in an Amicon concentrator (30000 Da molecular-mass cut-off; Millipore) and passing the protein preparation through a desalting column equilibrated in 50 mM Tris/150 mM NaCl (pH 8.0 or pH 6.5). The resulting protein preparations were concentrated and passed through a column buffer equilibrated with 50 mM Tris/150 mM NaCl (pH 8.0 or pH 6.5). The A280 and Aλmax (for λmax see Table 1) of the apo protein preparations were determined.
Table 1. Spectral and iron-binding properties of the WT and mutant FbpAs.
The molar absorption coefficients for the WT and mutant proteins were estimated using the ProtParam bioinformatic tool (http://www.expasy.ch/tools/protparam.html). NA, no available data for this assay.
| Competition | ||||||||
|---|---|---|---|---|---|---|---|---|
| FbpA (logKA)‡ | ||||||||
| Protein | λmax (nm) | λmin (nm) | Amax/Amin | Molar absorption coefficient (M−1·cm−1) | EDTA (pH 7.4) k (min−1) | Citrate (pH 8.0) (logKS1)† | pH 8.0 | pH 6.5 |
| Transferrin | 470 | 360 | NA | 80 000 | NA | 21.3 and 20.7 | 21.0 and 20.3 | |
| FbpA | ||||||||
| WT | 481 | 378 | 1.89±0.03 | 39400 | 0.164 | 20.2 | 20.2 | 18.4 |
| Q58L | 469 | 380 | 2.13±0.06 | 39400 | NA | 19.4 | 19.7±0.8 | 18.3±0.7 |
| H9Q | 481 | 380 | 2.17±0.06 | 39400 | NA | 20.0 | 18.4±0.6 | 18.5±0.4 |
| H9A | 481 | 380 | 1.94±0.04 | 39400 | 0.313 | 19.3 | 17.3±0.3 | 17.2±0.6 |
| E57A | 477 | 385 | 1.72±0.04 | 39400 | 0.101 | 19.9 | 18.3±0.5 | 17.1±0.7 |
| Y196A | 466 | 382 | 1.78±0.04 | 38120 | 0.030 | 20.0 | 17.7±0.8 | 16.6±0.6 |
| Y195A | 467 | 415 | 1.25±0.04 | 38120 | 0.133 | 19.7 | 18.2±1.0 | 16.5±0.8 |
Competition binding experiments were performed by mixing 0.5 ml of resin (approx. 5 mg of immobilized holo-FbpA) with 5 mg of soluble apo-FbpA, adding citrate/bicarbonate to a concentration of 0.1 mM and incubating with gentle agitation at room temperature for 48 h. The citrate was added to facilitate the exchange of iron between the immobilized and soluble forms of FbpA, and was shown to result in negligible removal from the immobilized FbpA (0.5% or less; results not shown) in the absence of added apo-FbpA. The soluble FbpA was separated from the resin by pouring the mixture into a 0.5 cm×5 cm column and washing the column with 3 ml of buffer. The soluble protein preparation was measured to determine the A280 (protein content) and Aλmax. The λmax is a measure of the iron content. An excess of ferric citrate was added to the solution to saturate the iron-binding sites and the sample was passed through a desalting column. The A280 and Aλmax of the resulting iron-saturated protein preparation were measured. The ratio of Aλmax/A280 of the sample was compared with the iron-saturated form in order to determine the percentage saturation of the sample.
Statistical analyses and calculations
One-way ANOVA analyses with post hoc tests were performed with Graphpad Prism version 4 for the experiments at pH 8.0 and pH 6.5. These analyses were performed in order to obtain an indication of whether there were significant differences in percentages of iron binding between the different soluble FbpA preparations (see Figure 4).
Figure 4. Expression of mutant FbpAs.

(A) Osmotic shock fractions isolated from E. coli strain Bl21/pLysS containing recombinant pT7-7 expression plasmid with the WT or indicated mutant fbpA gene from H. influenzae strain H36. The osmotic shock fractions were subjected to SDS/PAGE analysis and the proteins were detected by staining with Coomassie Blue. (B) Immunoblot analysis of whole cells of H. influenzae strains expressing WT or the indicated mutant FbpAs.The blot was developed with polyclonal anti-(H. influenzae FbpA) polyclonal rabbit serum (1:2000) and goat anti-rabbit horseradish-peroxidase conjugate (1:3000). Numbers in parentheses indicate the quantity of protein expressed by each mutant (pg/μ per 109 bacterial cells) by densitometry analysis.
Equilibrium binding constants were calculated for the soluble FbpAs from the measured percentage of iron binding in each experiment using the following equations:
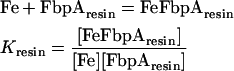 |
(1) |
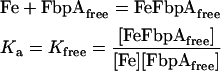 |
(2) |
Combining eqns (1) and (2) with [Fe] being the same in both:
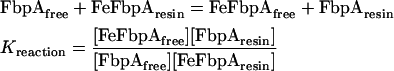 |
(3) |
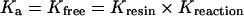 |
(4) |
Since there are experimentally determined equilibrium binding constants available for the WT protein at pH 8.0 (Table 1) and pH 6.5 [26,27], it was possible to derive a value for Kresin from competition experiments performed with WT FbpA using the following equation:
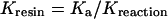 |
(5) |
Thus, for each experiment with the mutant proteins, the measured protein concentration (from A280 using the molar absorption coefficient from Table 1) and the measured absorbance at λmax (and the calculated percentage iron binding), could be used to calculate an equilibrium binding constant. The mean±S.E.M. for three separate experiments is listed in Table 1.
RESULTS
Preparation of site-directed mutants of FbpA
The four amino acids involved in directly co-ordinating the iron atom and one amino acid involved in binding the co-ordinating phosphate anion were targeted for site-directed mutagenesis, with the goal of altering the iron-binding affinity. For simplicity and consistency, we decided to mutate each of the four metal-co-ordinating amino acids (His9, Glu57, Tyr195 and Tyr196) to alanine residues and one of the anion-co-ordinating amino acids (Gln58) to a leucine residue. Mutation of His9 to a glutamine residue was also performed in order to provide a more conservative change of this residue, with the aim that it would result in an intermediate iron-binding affinity.
The recombinant pT7-7 plasmids encoding the WT and mutant proteins were introduced into the E. coli BL21/pLysS host strain, and the production of recombinant protein induced by IPTG was assessed by SDS/PAGE analysis. The production of the WT protein resulted in a red/brown coloration of the cell pellet that was not apparent with production of the site-directed mutant proteins (Figures 1B and 1E), unless additional exogenous iron was added to the medium (Figures 1A and 1D). SDS/PAGE analysis revealed that relatively pure protein preparations of protein were obtained, even after simple isolation of the periplasmic protein fraction by a modified osmotic shock procedure (Figures 1C and 1F). As differences in protein levels could not account for differences in the coloration of the bacterial cell pellets, the results suggesed that the proteins varied in their capacity to acquire iron from the growth medium, reflecting a hierarchy in iron-binding capability under ‘physiological’ conditions (WT>Q58L, H9Q, H9A, E57A>Y196A, Y195A).
Characterization of the mutant proteins
The mutant proteins were purified from the osmotic shock fluid by cation-exchange chromatography, concentrated, converted into the apo form and then iron-loaded prior to performing spectral analysis. Spectral analysis revealed absorbance changes characteristic of members of the transferrin superfamily on addition of iron to the mutant FbpAs. The λmax, λmin and Amax/Amin ratios deduced from scans in the range 310 to 600 nm (Ultrospec 2000, Pharmacia Biotech) are listed in Table 1. The absorption bands for the tyrosine mutants (Y195A and Y196A) and the anion mutant Q58L all display a considerable blue shift, with a λmax value of approx. 467 nm compared with 481 nm for the WT and other mutant proteins. Such a shift indicates an increased Tyr(O)–Fe interaction, with the remaining tyrosine in the case of the tyrosine mutants and an increased interaction with one or both tyrosine residues in the anion mutant.
As we wanted to ensure that any observed changes in the function of the mutant proteins were due solely to changes in iron co-ordination, particularly in the pathway-reconstitution experiments, protein crystallography studies were performed with all the mutant proteins described in the present study. Here, we specifically report on the findings with the E57A protein to illustrate the overall findings. The details for protein production, crystallization and data collection are described in the Experimental section and the crystallographic data are summarized in Table 2.
Table 2. X-ray data collection and refinement statistics for FbpA mutant E57A.
RMSD, root mean square deviation.
| Parameter | Value |
|---|---|
| Data collection | |
| Wavelength (Å) | 1.5418 |
| Resolution (Å) | 100–1.8 |
| Completeness (%)* | 97.7 (81.3) |
| I/σ(I) | 30.2 (3.1) |
| Rsym† | 0.053 (0.237) |
| Redundancy | 6.6 |
| Refinement statistics | |
| Resolution (Å) | 20-1.8 |
| Completeness for range (%) | 97.7 |
| Number of reflections‡ | 23715 |
| Working set | 22451 |
| Free set | 1264 |
| R-factor§, R-free | 0.202, 0.250 |
| No. of protein atoms | 4492 |
| No. of water molecules | 222 |
| Fe atoms | 1 |
| Mean B-factors (Å2) | 20.55 |
| RMSD from ideality | |
| Bond lengths (Å) | 0.008 |
| Bond angles (o) | 1.095 |
* Number in parentheses is the statistic for the highest resolution shell.
† Rsym=Σh(Σj|Ij,h−〈Ih〉|/ΣIj,h), where h=set of Miller indices and j=set of observations of reflection h.
‡ F>2.0σ.
§ R-factor=Σhkl|Fo−Fc|/Σhkl|Fo|.
Crystallization screens were developed around the conditions that were successful for the WT iron-loaded protein [17], and diffraction quality crystals were produced after 4 days of growth. The model for the E57A FbpA protein was derived from the processed data by molecular replacement using the structure of the WT apo protein (Protein Data Bank code 1D9V) as the initial phasing model.
Although the E57A protein crystallized in the iron-loaded state, there was substantial separation between the N-terminal and C-terminal domains (Figure 2). The α-carbon chain for these proteins could easily be superimposed on the apo form of the WT protein, but not the iron-bound form (results not shown). Crystallizing in the ‘open’ conformation is a characteristic that was observed for the iron-loaded forms of all of the site-directed FbpA mutants listed in Table 1.
Figure 2. Structure of E57A mutant FbpA.

Ribbon diagram of the E57A mutant FbpA shows two α/β domains linked by two antiparallel β-strands. α-Helices are displayed in purple and β-strands are shown in cyan. The relative binding modes of the phosphate anion (green and red) and iron atom (orange sphere) are also shown when present.
The implication is that this form of the protein is more prevalent in solution with the mutant proteins than with the WT protein, which suggests that disruption of the normal metal–anion co-ordination complex destabilizes the closed conformation. In addition to the involvement of Tyr195 and Tyr196 in binding iron, a bound phosphate molecule (Figure 2) is found in the same position as in the WT iron-loaded and apoproteins to help co-ordinate the iron atom in the E57A structure. The remaining iron co-ordinating ligands at the present resolution appear to be provided by bound water molecules.
Assessment of iron-binding properties
The initial attempts to evaluate the iron-binding properties of the mutant proteins employed two assays that were used previously to determine the iron-binding properties of transferrins [24] and FbpAs [25].One assay was designed to measure values for the off-rates by incubation in the presence of EDTA [24], and the other assay was designed to provide equilibrium affinity constants by competition with increasing levels of citrate [25]. The results are presented in Table 1. In order to compare the results with those obtained for transferrins and FbpAs reported in the literature, these assays were performed in buffers at pH 8.0 and 7.4. The results with the citrate competition assay yielded a stability constant for WT FbpA that is comparable with that reported for the WT FbpA from Neisseria gonorrhoeae [16,25].
In an attempt to gain a better appreciation of the relative abilities of these proteins to compete for iron, we decided to use direct competition assays. Unfortunately, there were no established assays available and we were unable to develop a format for equilibrium dialysis that was feasible or suitable. Thus we developed a competition assay involving mixtures of soluble and immobilized FbpAs, so that the soluble forms could be readily isolated and analysed for iron content by spectral analysis. In this assay immobilized FbpA was iron-saturated and then mixed with a series of soluble FbpAs in their apo form.
Mixtures of soluble, apo-FbpA and immobilized holo-FbpA were prepared in a buffer containing 0.1 mM sodium citrate and sodium bicarbonate to facilitate the exchange of iron between the soluble and immobilized FbpAs. After incubation for 48 h at room temperature, the soluble FbpAs were separated from the immobilized FbpAs and subjected to spectral analysis. The A280 was measured to determine protein content and Aλmax was determined for each protein (Table 1) to assess iron content. The A280 and Aλmax were also measured for the fully iron-loaded form of each soluble FbpA. The ratio of the Aλmax for a soluble FbpA to the Aλmax for the fully iron-loaded FbpA at equivalent A280 readings is denoted as the percentage iron loading. The iron removal from the immobilized proteins was also evident from a readily detectable bleaching of the resin. The first set of experiments were performed with buffers at pH 8 in order to compare the results with the other assays. A set of experiments was performed with a buffer at pH 6.5 with the aim of reflecting the physiological pH of the periplasm [29].
The experiments were carried out in triplicate and the results were subjected to statistical analysis to provide some indication of whether there were significant differences in the relative binding abilities of the mutant FbpAs. The results from the binding experiments at pH 8.0 and at pH 6.5 are shown in Figure 3. In Figure 3 post hoc tests from one-way ANOVA analyses were used to compare the binding between the WT and mutant FbpAs. At pH 8, all of the mutant proteins, except Q58L, had a lower percentage iron binding than the WT protein. At pH 6.5, only Y196A and Y195A mutants had significant differences in percentage iron binding from WT, but two other proteins, H9A and E57A, had differences nearing significance. The differences in relative binding at the two pHs were largely attributable to two of the FbpA mutants H9Q and H9A that had marked improvement in the percentage iron binding at pH 6.5 (Figure 3).
Figure 3. Competition among WT and mutant FbpAs with immobilized FbpAs at pH 8.0 or pH 6.5.

Percentage iron-binding by soluble WT and mutant FbpAs was determined as described in the Experimental section. Comparisons were made between WT and mutant FbpAs where *P<0.05 is significant.
To gain a better sense of how the results of the new competition binding assay compared with the previous methods, the percentage iron binding values in the experiments were used to calculate the affinity constants for the individual proteins using published affinity constants for WT FbpA at pH 8 [25] and pH 6.5 [26,27] as described in the the Experimental section. The statistical analyses were repeated using these calculated values. This approach was also used to determine the binding constants for human transferrin using results of competition assays performed with human transferrin to evaluate the validity of the approach. The results are presented in Table 1. The values obtained for human transferrin are quite similar to those obtained from citrate competition assays and to values reported in the literature, suggesting that the approach is reasonable. The values for the mutant proteins derived from the direct competition assay suggests a more marked difference in iron-binding abilities between the WT and mutant proteins than were implied from the citrate competition assay.
It is apparent from the results shown in Figure 3 that the relative binding affinities of the mutant proteins differ at pH 8 and pH 6.5. As the results at pH 6.5 are meant to represent the ‘physiological’ conditions in the periplasmic space, it would be expected that the suggested relative ranking of iron-binding capabilities (WT, Q58L, H9Q>H9A, E57A>Y196A, Y195A) would be most relevant for the pathway-reconstitution experiments described below.
Function of the mutant FbpAs in iron acquisition
To evaluate their relative abilities in mediating iron acquisition, the various mutant FbpAs were used to replace the WT protein in H. influenzae and the resulting strains were tested for growth under a variety of different conditions. The WT and mutant genes were introduced into starting strain H. influenzae H306, that contains a chloramphenicol-resistance determinant in place of the fbpA gene, in order to prevent any exchanges between WT and mutant genes. The WT and mutant fbpA genes were cloned into a plasmid that contained the necessary upstream and downstream regions required for gene replacement by homologous recombination. This plasmid also contained a convenient restriction site for introduction of a kanamycin-resistance determinant immediately downstream of the fbpA gene. The resulting plasmids containing the WT or mutant fbpA genes and the selectable antibiotic resistance markers were used to transform the H306 strain, and the selected kanamycin-resistant transformants were tested for the correct genetic organization by a series of colony PCR reactions. A control plasmid without an fbpA gene was also used in the transformation experiments to provide an appropriate fbpA-deficient strain (H374) to compare with the WT FbpA-expressing strain (H317) and mutant FbpA-expressing strains (H361–H364, H368 and H369).
To assess the expression and stability of the mutant FbpAs in H. influenzae, Western-blot analysis was performed with the set of mutant strains grown under iron-restricted conditions. This analysis was performed on the intact cells used for growth experiments and thus truly reflects the levels of FbpA available for supporting growth on exogenous iron sources. A representative blot shown in Figure 4(B) demonstrates that the level of protein in the mutant strains was comparable with that present in the strain containing the WT gene. To quantify the level of protein, the experiment was repeated several times with various concentrations of cells and purified WT protein that was used as a standard. Densitometry was used to quantify each band and the protein concentration was derived from the standard curve. As shown in Figure 4, there was less than a 2.5-fold variation in the level of FbpA protein amongst the various strains.
The set of strains expressing WT and mutant FbpAs were tested for growth on various sources of iron in an anaerobic plate assay, as described previously [14]. In this assay, the growth of an iron-limited culture of cells around discs impregnated with different sources of iron was assessed. The growth of the parent WT strain (H36) and the control WT strain (H317, containing a kanamycin-resistance determinant between the fbpA and fbpBC genes) on different iron sources was virtually identical (results not shown). This confirmed that the presence of the kanamycin-resistance determinant did not perturb the expression of FbpB or FbpC sufficiently to influence growth characteristics in this assay system. Growth of H317 on haem, but not on human lactoferrin (Table 3), confirmed that growth is dependent upon availability of appropriate iron sources, and the growth on transferrin and low levels of ferric citrate confirmed the presence of a functional FbpABC pathway.
Table 3. Growth of WT and FbpA mutant strains using the anaerobic disc-diffusion assay.
Growth of bacteria adjacent to the concentration discs: −, no detectable growth; +, <0.5 mm; ++, 0.5 to 2 mm; +++, >2 mm. Haem, 2.5 nmol of haem; hLf, 2.5 nmol of human lactoferrin; hTf, 2.5 nmol of human transferrin; FC, nmol of ferric citrate.
| Growth on indicated iron source | |||||||
|---|---|---|---|---|---|---|---|
| FC | |||||||
| Strain (FbpA) | Haem | hLf | hTf | 40 | 30 | 20 | 10 |
| H317 (WT) | +++ | − | +++ | +++ | ++ | ++ | + |
| H369 (Q58L) | +++ | − | +++ | +++ | ++ | ++ | + |
| H368 (H9Q) | +++ | − | +++ | +++ | ++ | + | − |
| H362 (H9A) | +++ | − | − | ++ | + | − | − |
| H361 (E57A) | +++ | − | − | ++ | + | − | − |
| H364 (Y195A) | +++ | − | − | + | − | − | − |
| H363 (Y196A) | +++ | − | − | + | − | − | − |
| H374 (none) | +++ | − | − | + | − | − | − |
The most obvious difference between the strains expressing the mutant and WT FbpAs was the ability to grow in the presence of various quantities of added ferric citrate (Table 3) that presumably is a reflection of the relative ability of the FbpAs to acquire iron in vivo. Thus expression of the WT or Q58L proteins permitted growth on 10 nmol of ferric citrate, whereas strains expressing the H9A and E57A proteins required 30 nmol of ferric citrate for growth. The implication that the WT and Q58L proteins were more capable of competing for iron in the growth medium is consistent with the results obtained with the competitive binding assay (Figure 3).The observation that the strain expressing the H9Q protein required 20 nmol of ferric citrate to support growth implies that it has intermediate ability to compete for iron in the growth medium. This protein clustered with the WT and Q58L proteins in the competitive binding assay at pH 6.5 and with the other mutant proteins at pH 8 (Figure 3). This suggests that intermediate pH values may need to be considered for this assay.
The observation that the FbpA-deficient strain required 40 nmol of ferric citrate to support growth implies that a functional FbpABC pathway is required for growth on levels of ferric citrate less than 40 nmol. This infers that the mutant proteins mentioned above are all capable of donating iron to the inner-membrane transport complex comprised of FbpB and FbpC. In contrast, the strains expressing the Y195A and Y196A mutant proteins required 40 nmol of ferric citrate for growth and had the lowest stability constants (Table 1 and Figure 3), suggesting that they either were incapable of acquiring iron from the growth medium or were defective in donating iron to the inner-membrane transport complex. Collectively, these results suggest that the rank order for metal-binding affinities of the FbpA proteins expressed in H. influenzae is WT=Q58L>H9Q>H9A, E57A>Y195A, Y196A.
The mutant strains varied in their ability to grow on human transferrin as a source of iron for growth. Strains expressing the WT FbpA, anion mutant Q58L and the conservative H9Q mutant were capable of utilizing transferrin as an iron source (Table 3). In contrast, strains expressing the non-conservative mutants H9A, E57A, Y195A and Y196A were unable to grow in the presence of transferrin. It is interesting to note that the three proteins that were capable of supporting growth on transferrin (WT, Q58L and H9Q) all had a greater than 75% iron binding in the competition assay at pH 6.5 (Figure 3) and all had calculated affinity constants greater than or equal to 2.0×1018 M−1 (Table 1, logKA>18).
DISCUSSION
Gram-negative pathogenic bacteria in the families Pasteurellaceae and Neisseriaceae have developed an effective means of iron acquisition in the host that relies on direct binding of host transferrin by a receptor complex at the cell surface. The high-affinity binding of transferrin by the bacterial receptor proteins provides an efficient means of capturing transferrin from the external milieu. The subsequent steps that involve iron removal from transferrin and transport of iron across the outer membrane are not well understood, but one or more aspects of this process are dependent upon energy provided through the TonB complex [8,9]. It has been proposed that conformational changes in transferrin upon binding by the receptor proteins facilitates the release of iron by perturbing the tight complex with liganding amino acids [6], but at present there is no direct experimental evidence supporting this model. The subsequent transport of iron across the outer membrane is mediated by the TonB-dependent integral outermembrane protein, TbpA, in a process that likely is similar to other TonB-dependent proteins.
The structures of several TbpA homologues [30–33] have provided insights into the transport process across the outer membrane. The TonB-dependent outer-membrane receptors have a novel structure, consisting of an N-terminal plug region that fills the centre of the C-terminal β-barrel. Interaction with TonB has been proposed to promote formation of a channel for movement of substrate across the outer membrane either by a conformational change of the plug [30] or by removal of the plug [34]. Structural studies have revealed that the normally disordered Ton-box of the outer-membrane receptor adopts a β-sheet conformation in association with the β3 strand of TonB [11], which is consistent with a mechanical pulling model of transport.
Most models for TonB-mediated transport across the outer membrane do not consider the role of the periplasmic binding protein in the process, implying that transport and subsequent capture of ligand by the periplasmic protein are independent processes. In other words, the receptor complex would effectively use energy derived from the TonB complex to ‘pump’ ligand into the periplasmic space where it would subsequently be bound by the cognate binding protein. This would be reasonable for the relatively soluble iron-siderophore complexes or vitamin B12, but the release of a free ferric ion directly into the periplasmic space would potentially present solubility and toxicity problems.
The transport process mediated by the TonB-dependent transferrin and lactoferrin receptors is inherently more complex, since it also involves the removal of ferric ion from the host glycoproteins. This raises the obvious question as to how the TonB interaction could be responsible for the processes of both iron removal and transport. In a previous model we suggested that conformational changes in transferrin mediated by receptor binding would lower its affinity for iron [6] and thus facilitate its removal and transport. In this model the primary role of TonB was considered to be provision of a channel for diffusion of iron across the outer membrane, implying that higher affinity iron binding by FbpA (relative to transferrin in the receptor complex) could drive the transport into the periplasmic space.
The results in the present study suggest that high-affinity binding of iron by FbpA (≥2.0×1018 M−1) is required to drive transport across the outer membrane, which is consistent with our original model for iron removal [6]. In effect, the use of various mutant proteins may be providing an indirect measurement of the affinity of receptor-bound transferrin for iron. Thus transferrin in the receptor complex binds ferric ion more weakly than WT, Q58L and H9Q FbpAs, since strains expressing these proteins are capable of growing on transferrin as the sole iron source (Table 3). In this respect it is interesting to note that published values for the KIII (human transferrin) at pH 6.7 for the N-lobe (1.8×1017 M−1 to 1.4×1018 M−1) [35,36] are near or below the threshold value of 2.0×1018 M−l at pH 6.5 for the WT, Q58L and H9Q FbpAs. Similarly, the published values for human transferrin are greater than the equilibrium affinity constants at pH 6.5 for H9A FbpA (1.6×1017 M−l) and E57A FbpA (1.2×1017 M−l), which is consistent with the inability of these proteins to remove iron from transferrin. Admittedly the actual binding constants may be different in vivo from the values calculated from in vitro experiments, but the relative affinity of receptor-bound transferrin compared with that of the various mutant FbpAs is probably valid.
The inability of strains expressing the H9A and E57A mutants of FbpA to grow on transferrin is proposed to be due to inability to compete with receptor-bound transferrin for iron. The fact that strains expressing these mutant proteins were capable of growing on levels of ferric citrate below that required by the FbpA-negative mutant (Table 3) indicates that there is no defect in the periplasm-to-cytoplasm branch of the pathway. The inability of strains expressing H9A and E57A to support the growth of H. influenzae with transferrin as an exogenous iron source is not primarily due to differences in stability or levels of the protein (Figure 4B), since the levels of these proteins, particularly H9A, were comparable with those in strains proficient in utilization of transferrin iron.
Although spectral studies using competing iron chelators, such as citrate or EDTA, can provide quantitative values to describe the binding properties, they did not seem to provide similar insights to the novel competitive binding assay. The calculated binding constants do not consider interactions between the chelators and FbpA or the formation of higher-order iron complexes, both of which have been demonstrated in structural studies of the mutant proteins [22,23]. In contrast, the competitive binding assay (Figure 3) seemed to be able to distinguish the proteins capable of mediating iron acquisition from transferrin (WT, Q58L and H9Q) from those unable to do so (H9A, E57A, Y196A and Y195A). The reasonable correlation between the results from the competitive binding assay at pH 6.5 (Figure 3) and the growth on exogenous ferric citrate (Table 2) suggest that this assay may be a reasonable assay in vitro for predicting the functional binding properties in vivo.
Transport processes across the outer membrane that require energy derived from the TonB complex are inherently difficult to study, partly due to the limitations in developing systems for reconstituting the transport process. The removal of iron from the host glycoproteins is an added complexity for studying the process mediated by surface transferrin and lactoferrin receptors in Gram-negative bacteria. The retention of host glycoprotein binding activity even by recombinant isolated receptor proteins has aided in characterization of the receptor–ligand interaction [37–41], which would be further enhanced by structural studies with the receptor proteins or preferably with receptor–ligand complexes. These studies could provide insights into potential mechanisms for iron removal and transport, and aid in the design of functional studies directed at determining the mechanisms involved. In the present study we provide evidence suggesting that FbpA also plays a critical role in the process of iron removal and transport across the outer membrane, providing a perspective that the process is to some degree a competition between FbpA and receptor-bound transferrin. The potential to modify the iron-binding properties of transferrin and FbpA, and evaluate their effect on the iron uptake process in vivo and compare it with in vitro competition binding assays, may provide an additional powerful tool for deciphering the iron removal and transport process.
Acknowledgments
This work was supported by grant 49603 from the Canadian Institutes of Health Research. We thank Dr Duncan McRee for assistance in the structural studies, Dennis Pfaff for assiting in generating some of the mutants, and Kent Hecker and Dr Kosta Vasilakos for their insightful discussion about the statistical analysis.
References
- 1.Bergeron R. J. Iron: a controlling nutrient in proliferative processes. Trends Biochem. Sci. 1986;11:133–136. [Google Scholar]
- 2.Baker E. N., Baker H. M., Kidd R. D. Lactoferrin and transferrin: functional variations on a common structural framework. Biochem. Cell Biol. 2002;80:27–34. doi: 10.1139/o01-153. [DOI] [PubMed] [Google Scholar]
- 3.Jeffrey P. D., Bewley M. C., MacGillivray R. T. A., Mason A. B., Woodworth R. C., Baker E. N. Ligand-induced conformational change in transferrins: crystal structure of the open form of the N-terminal half-molecule of human transferrin. Biochemistry. 1998;37:13978–13986. doi: 10.1021/bi9812064. [DOI] [PubMed] [Google Scholar]
- 4.Gerstein M., Anderson B. F., Norris G. E., Baker E. N., Lesk A. M., Chothia C. Domain closure in lactoferrin. Two hinges produce a see-saw motion between alternative close-packed interfaces. J. Mol. Biol. 1993;234:357–372. doi: 10.1006/jmbi.1993.1592. [DOI] [PubMed] [Google Scholar]
- 5.Andrews S. C., Robinson A. K., Rodriguez-Quinones F. Bacterial iron homeostasis. FEMS Microbiol. Rev. 2003;27:215–237. doi: 10.1016/S0168-6445(03)00055-X. [DOI] [PubMed] [Google Scholar]
- 6.Schryvers A. B., Stojiljkovic I. Iron acquisition systems in the pathogenic Neisseria. Mol. Microbiol. 1999;32:1117–1123. doi: 10.1046/j.1365-2958.1999.01411.x. [DOI] [PubMed] [Google Scholar]
- 7.Gray-Owen S. D., Schryvers A. B. Bacterial transferrin and lactoferrin receptors. Trends Microbiol. 1996;4:185–191. doi: 10.1016/0966-842x(96)10025-1. [DOI] [PubMed] [Google Scholar]
- 8.Stojiljkovic I., Srinivasan N. Neisseria meningitidis tonB, exbB, and exbD genes: Ton-dependent utilization of protein-bound iron in Neisseriae. J. Bacteriol. 1997;179:805–812. doi: 10.1128/jb.179.3.805-812.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jarosik G. P., Maciver I., Hansen E. J. Utilization of transferrin-bound iron by Haemophilus influenzae requires an intact tonB gene. Infect. Immun. 1995;63:710–713. doi: 10.1128/iai.63.2.710-713.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pawelek P. D., Croteau N., Ng-Thow-Hing C., Khursigara C. M., Moiseeva N., Allaire M., Coulton J. W. Structure of TonB in complex with FhuA, E. coli outer membrane receptor. Science. 2006;312:1399–1402. doi: 10.1126/science.1128057. [DOI] [PubMed] [Google Scholar]
- 11.Shultis D. D., Purdy M. D., Banchs C. N., Wiener M. C. Outer membrane active transport: structure of the BtuB:TonB complex. Science. 2006;312:1396–1399. doi: 10.1126/science.1127694. [DOI] [PubMed] [Google Scholar]
- 12.Postle K., Kadner R. J. Touch and go: tying TonB to transport. Mol. Microbiol. 2003;49:869–882. doi: 10.1046/j.1365-2958.2003.03629.x. [DOI] [PubMed] [Google Scholar]
- 13.Mietzner T. A., Tencza S. B., Adhikari P., Vaughan K. G., Nowalk A. J. Fe(III) periplasm-to-cytosol transporters of Gram-negative pathogens. Curr. Top. Microbiol. Immunol. 1998;225:113–135. doi: 10.1007/978-3-642-80451-9_7. [DOI] [PubMed] [Google Scholar]
- 14.Kirby S. D., Gray-Owen S. D., Schryvers A. B. Characterization of a ferric binding protein mutant in Haemophilus influenzae. Mol. Microbiol. 1997;25:979–987. doi: 10.1111/j.1365-2958.1997.mmi535.x. [DOI] [PubMed] [Google Scholar]
- 15.Khun H. H., Kirby S. D., Lee B. C. A Neisseria meningitidis fbpABC mutant is incapable of using nonheme iron for growth. Infect. Immun. 1998;66:2330–2336. doi: 10.1128/iai.66.5.2330-2336.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adhikari P., Kirby S. D., Nowalk A. J., Veraldi K. L., Schryvers A. B., Mietzner T. A. Biochemical characterization of a Haemophilus influenzae periplasmic iron transport operon. J. Biol. Chem. 1995;42:25142–25149. doi: 10.1074/jbc.270.42.25142. [DOI] [PubMed] [Google Scholar]
- 17.Bruns C. M., Nowalk A. J., Avrai A. S., McTigue M. A., Vaughan K. A., Mietzner T. A., McRee D. E. Structure of Haemophilus influenzae Fe+3-binding protein reveals convergent evolution within a superfamily. Nat. Struct. Biol. 1997;4:919–924. doi: 10.1038/nsb1197-919. [DOI] [PubMed] [Google Scholar]
- 18.Bruns C. M., Anderson D. S., Vaughan K. G., Williams P. A., Nowalk A. J., McRee D. E., Mietzner T. A. Crystallographic and biochemical analyses of the metal-free Haemophilus influenzae Fe3+-Binding protein. Biochemistry. 2001;40:15631–15637. doi: 10.1021/bi0156759. [DOI] [PubMed] [Google Scholar]
- 19.Studier F. W., Rosenberg A. H., Dunn J. J., Dubendorff J. W. Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol. 1990;185:60–89. doi: 10.1016/0076-6879(90)85008-c. [DOI] [PubMed] [Google Scholar]
- 20.Haas R., Kahrs A. F., Facius D., Allmeier H., Schmitt R., Meyer T. F. TnMax – a versatile mini-transposon for the analysis of cloned genes and shuttle mutagenesis. Gene. 1993;130:23–31. doi: 10.1016/0378-1119(93)90342-z. [DOI] [PubMed] [Google Scholar]
- 21.Barcak G. J., Chandler M. S., Redfield R. J., Tomb J.-F. Genetic systems in Haemophilus influenzae. Methods Enzymol. 1991;204:321–342. doi: 10.1016/0076-6879(91)04016-h. [DOI] [PubMed] [Google Scholar]
- 22.Shouldice S. R., Dougan D. R., Skene R. J., Tari L. W., McRee D. E., Yu R.-H., Schryvers A. B. High resolution structure of an alternate form of the ferric-ion binding protein from Haemophilus influenzae. J. Biol. Chem. 2003;278:11513–11519. doi: 10.1074/jbc.M211780200. [DOI] [PubMed] [Google Scholar]
- 23.Shouldice S. R., Skene R. J., Dougan D. A., McRee D. E., Tari L. W., Schryvers A. B. The Presence of ferric-hydroxide clusters in mutants of the Haemophilus influenzae ferric ion-binding protein A. Biochemistry. 2003;42:11908–11914. doi: 10.1021/bi035389s. [DOI] [PubMed] [Google Scholar]
- 24.He Q. Y., Mason A. B., Woodworth R. C. Iron release from recombinant N-lobe and single point Asp63 mutants of human transferrin by EDTA. Biochem. J. 1997;328:439–445. doi: 10.1042/bj3280439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen C.-Y., Berish S. A., Morse S. A., Mietzner T. A. The ferric iron-binding protein of pathogenic Neisseria spp. functions as a periplasmic transport protein in iron acquisition from human transferrin. Mol. Microbiol. 1993;10:311–318. doi: 10.1111/j.1365-2958.1993.tb01957.x. [DOI] [PubMed] [Google Scholar]
- 26.Taboy C. H., Vaughan K. G., Mietzner T. A., Aisen P., Crumbliss A. L. Fe3+ coordination and redox properties of a bacterial transferrin. J. Biol. Chem. 2001;276:2719–2724. doi: 10.1074/jbc.M004763200. [DOI] [PubMed] [Google Scholar]
- 27.Anderson D. S., Adhikari P., Nowalk A. J., Chen C. Y., Mietzner T. A. The hFbpABC transporter from Haemophilus influenzae functions as a binding-protein-dependent ABC transporter with high specificity and affinity for ferric iron. J. Bacteriol. 2004;186:6220–6229. doi: 10.1128/JB.186.18.6220-6229.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li H., Sadler P. J., Sun H. Unexpectedly strong binding of a large metal ion (Bi3+) to human serum transferrin. J. Biol. Chem. 1996;271:9483–9489. [PubMed] [Google Scholar]
- 29.Bruscella P., Cassagnaud L., Ratouchniak J., Brasseur G., Lojou E., Amils R., Bonnefoy V. The HiPIP from the acidophilic Acidithiobacillus ferrooxidans is correctly processed and translocated in Escherichia coli, in spite of the periplasm pH difference between these two micro-organisms. Microbiology. 2005;151:1421–1431. doi: 10.1099/mic.0.27476-0. [DOI] [PubMed] [Google Scholar]
- 30.Ferguson A. D., Hofmann E., Coulton J. W., Diederichs K., Welte W. Siderophore-mediated iron transport: crystal structure of FhuA with bound lipopolysaccharide. Science. 1998;282:2215–2220. doi: 10.1126/science.282.5397.2215. [DOI] [PubMed] [Google Scholar]
- 31.Ferguson A. D., Chakraborty R., Smith B. S., Esser L., van der Helm D., Deisenhofer J. Structural basis of gating by the outer membrane transporter FecA. Science. 2002;295:1715–1719. doi: 10.1126/science.1067313. [DOI] [PubMed] [Google Scholar]
- 32.Buchanan S. K., Smith B. S., Venkatramani L., Xia D., Esser M., Palnitkar M., Chakraborty R., van der Helm D., Deisenhofer J. Crystal structure of the outer membrane active transporter FepA from Escherichia coli. Nat. Struct. Biol. 1999;6:56–63. doi: 10.1038/4931. [DOI] [PubMed] [Google Scholar]
- 33.Chimento D. P., Mohanty A. K., Kadner R. J., Wiener M. C. Substrate-induced transmembrane signaling in the cobalamin transporter BtuB. Nat. Struct. Biol. 2003;10:394–401. doi: 10.1038/nsb914. [DOI] [PubMed] [Google Scholar]
- 34.Scott D. C., Cao Z., Qi Z., Bauler M., Igo J. D., Newton S. M., Klebba P. E. Exchangeability of N-termini in the ligand-gated porins of Escherichia coli. J. Biol. Chem. 2001;276:13025–13033. doi: 10.1074/jbc.M011282200. [DOI] [PubMed] [Google Scholar]
- 35.Zak O., Leibman A., Aisen P. Metal-Binding Properties of a Single-Sited Transferrin Fragment. Biochim. Biophys. Acta. 1983;742:490–495. doi: 10.1016/0167-4838(83)90266-2. [DOI] [PubMed] [Google Scholar]
- 36.Zak O., Aisen P. Preparation and properties of a single-sited fragment from the C-terminal domain of human transferrin. Biochim. Biophys. Acta. 1985;829:348–353. doi: 10.1016/0167-4838(85)90243-2. [DOI] [PubMed] [Google Scholar]
- 37.Boulton I. C., Yost M. K., Anderson J. E., Cornelissen C. N. Identification of discrete domains within gonococcal transferrin- binding protein A that are necessary for ligand binding and iron uptake functions. Infect. Immun. 2000;68:6988–6996.. doi: 10.1128/iai.68.12.6988-6996.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Masri H. P., Cornelissen C. N. Specific ligand binding attributable to individual epitopes of gonococcal transferrin binding protein a. Infect. Immun. 2002;70:732–740. doi: 10.1128/iai.70.2.732-740.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sims K. L., Schryvers A. B. Peptide-peptide interactions between human transferrin and transferrin binding protein B from Moraxella catarrhalis. J. Bacteriol. 2003;185:2603–2610. doi: 10.1128/JB.185.8.2603-2610.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Retzer M. D., Yu R.-H., Schryvers A. B. Identification of sequences in human transferrin that bind to the bacterial receptor protein, transferrin-binding protein B. Mol. Microbiol. 1999;32:111–121. doi: 10.1046/j.1365-2958.1999.01331.x. [DOI] [PubMed] [Google Scholar]
- 41.Wong H., Schryvers A. B. Bacterial lactoferrin binding protein A binds to both domains of the human lactoferrin C-lobe. Microbiol. 2003;149:1729–1737. doi: 10.1099/mic.0.26281-0. [DOI] [PubMed] [Google Scholar]